

Abstract
Trelagliptin (SYR-472), a novel dipeptidyl peptidase-4 inhibitor, shows sustained efficacy by once-weekly dosing in type 2 diabetes patients. In this study, we characterized in vitro properties of trelagliptin, which exhibited approximately 4- and 12-fold more potent inhibition against human dipeptidyl peptidase-4 than alogliptin and sitagliptin, respectively, and >10,000-fold selectivity over related proteases including dipeptidyl peptidase-8 and dipeptidyl peptidase-9. Kinetic analysis revealed reversible, competitive and slow-binding inhibition of dipeptidyl peptidase-4 by trelagliptin (t1/2 for dissociation ≈ 30 minutes). X-ray diffraction data indicated a non-covalent interaction between dipeptidyl peptidase and trelagliptin. Taken together, potent dipeptidyl peptidase inhibition may partially contribute to sustained efficacy of trelagliptin.
Introduction
An estimated 347 million people worldwide have diabetes mellitus (diabetes) [1]. Type 2 diabetes (T2DM), a metabolic disease characterized by hyperglycemia caused by impaired insulin secretion (beta cell dysfunction) and insulin resistance of peripheral tissues, represents around 90% of diagnosed diabetes cases. Causal factors include diet, obesity, genetics, and physical inactivity. An estimated 1.5 million deaths were directly caused by diabetes in 2012, and more than 80% of diabetes deaths occur in low- and middle-income countries. Projections for the prevalence of the disease vary and the World Health Organization predicts a 50% increase in cases worldwide in the next decade.
Complications from diabetes include nephropathy, neuropathy, retinopathy, and macrovascular diseases such as stroke or heart disease, and diabetes is a leading cause of blindness, amputation and kidney failure. T2DM is initially managed by life style modifications like increasing exercise and making dietary changes. However, medication may become necessary if these approaches do not lower blood glucose levels sufficiently. As noted elsewhere [2], improving a patient’s adherence to medication during long-term treatment is important in order to maintain favorable glycemic control, which may prevent the onset or lessen the severity of diabetic complications.
Dipeptidyl peptidase-4 (DPP-4), the serine protease responsible for metabolism of the incretin hormones glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide, plays an important role in regulating glucose homeostasis [3]. Thus, DPP-4 is an attractive target for therapeutic intervention, and inhibitors of DPP-4 have been shown to be an effective therapy for treatment of T2DM [4].
Trelagliptin (SYR-472, zafatek) is a novel once-weekly DPP-4 inhibitor approved in Japan. While other current marketed DPP-4 inhibitors are dosed once or more than once per day, trelagliptin showed efficacy as well as a suitable safety profile in T2DM patients by dosing once-weekly in a clinical trial setting [2, 5]. In this article, we characterized the in vitro profile of this unique once-weekly DPP-4 inhibitor, trelagliptin, and examined its contribution to sustained efficacy in the treatment of T2DM.
Materials and Methods
Chemicals
Alogliptin (2-[6-(3(R)-aminopiperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]benzonitrile), trelagliptin (2-[6-(3(R)-aminopiperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluorobenzonitrile), and sitagliptin ((2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine) were synthesized at Takeda Pharmaceutical Company Limited.
Animals
Male Sprague–Dawley rats (7 weeks old, n = 30) and male beagle dogs (2–5 years old, n = 3) were purchased from CLEA Japan (Tokyo, Japan) and Kitayama Labes Co., Ltd. (Nagano, Japan), respectively. All animals were housed in cages in a room with controlled temperature (23°C), humidity (55%) and lighting (lights on from 07:30 am to 07:30 pm) and were maintained on a laboratory chow diet (CE2 [CLEA Japan] for rats and DS-5 [Oriental Yeast Co., Ltd.] for dogs). Rat blood samples were taken from abdominal vein under ether inhalation anesthesia in anesthetized rats before euthanasia by exsanguination. Dog blood samples were collected from anterior limb veins in conscious dogs that were restrained in a retainer. After the blood collection, dogs were returned to their normal housing for other experiments. Plasma samples were prepared and stored at -80°C before use. The care and use of the animals and the experimental protocols used in this research were approved by the Experimental Animal Care and Use Committee of Takeda Pharmaceutical Company Limited.
Human plasma samples
Human plasma was prepared from fresh blood of healthy volunteers who gave written informed consent. The measurement of DPP-4 activity using this human plasma was approved by the research ethics committee in Takeda Pharmaceutical Company Limited.
Enzyme inhibition assays
Human DPP-4 enzyme used in these studies was obtained from several sources. Human DPP-4 partially purified from Caco-2 cells purchased from the ATCC (ATCC No. HTB-37; www.atcc.org), as described previously [6], was used to confirm trelagliptin inhibitor potency. For comparison among the DPP-4 inhibitors, trelagliptin, alogliptin and sitagliptin, commercially available recombinant human DPP-4 (Abnova, Taiwan) was used. For detailed kinetic studies, recombinant human DPP-4 was cloned, expressed and purified as described previously [7]. In addition, inhibition of plasma DPP-4 activity was determined using plasma samples of humans, dogs, and rats. The DPP-4 related proteases, dipeptidyl peptidase-2 (DPP-2) and prolyl endopeptidase (PEP), were prepared from rat kidney and brain, respectively, according to the method previously reported [8, 9]. Human dipeptidyl peptidase-8 (DPP-8), dipeptidyl peptidase-9 (DPP-9), and fibroblast activation protein α (FAPα) were purified by affinity chromatography from 293-F cells expressing each FLAG-tagged protein.
DPP-4 activity from Caco-2 cells or plasma was assayed using the chromophoric substrate Gly-Pro-p-nitroaniline (GP-pNA) (0.5 mmol/L final concentration) and carried out in pH 7.5 buffer containing 100 mmol/L Tris-HCl, 1 mg/mL bovine serum albumin, and 0.5 mg/mL CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid) for 60 min at 37°C (DPP-4 fraction from Caco-2 cells) or 30°C (plasma). Change in absorbance at 405 nm was measured to determine the reaction rate. Recombinant human DPP-4 (Abnova, Taiwan) activity was assayed using the fluorescent substrate Gly-Pro-7-amido-4-methyl-coumarin (GP-AMC) (90 μmol/L final concentration) and carried out in pH 7.8 buffer containing 25 mmol/L HEPES, 140 mmol/L NaCl, 1 mg/mL bovine serum albumin for 15 min at 37°C. The reaction was stopped by the addition of 100 μL of 25% (v/v) acetic acid, and fluorescence was measured (380 nm excitation/460 nm emission) using Envision 2103 Multilabel Reader (Perkin Elmer Japan, Japan). Reaction conditions for measurement of DPP-2, DPP-8, DPP-9, PEP, and FAPα activities are described in Table 1. Change in absorbance at 405 nm was measured to determine the reaction rate.
Table 1. Reaction conditions for measurement of DPP-2, DPP-8, DPP-9, PEP, and FAPα activities.
| Enzymes | DPP-2 | DPP-8 | DPP-9 | PEP | FAPα |
|---|---|---|---|---|---|
| Assay buffer* | 100 mmol/L DMGA buffer(pH 5.5) | 100 mmol/L Tris-HCl buffer(pH 7.5) | 100 mmol/L Tris-HCl buffer(pH 7.5) | 25 mmol/L glycine,25 mmol/L acetic acid, 23 mmol/LMES,75 mmol/L Tris, and100 mmol/L NaCl(pH 8.0) | 88 mmol/L Na-K phosphate buffer(pH 7.0) |
| Substrates* | 0.5 mmol/L Lys-Ala-pNA | 1 mmol/L Gly-Pro-pNA | 2 mmol/L Gly-Pro-pNA | 2 mmol/L Ala-Pro-pNA | 0.5 mmol/L Suc-Ala-Pro-pNA |
| Trelagliptin * | 0.03, 0.1, 0.3, 1, 3, 10, 30 and 100 μmol/L | ||||
| Temperature and duration | 37°C, 60 min | 37°C, 90 min | 37°C, 90 min | 37°C, 60 min | 37°C, 60 min |
*Final concentrations are listed.
For detailed kinetic studies, GP-pNA was used as substrate and assays carried out in pH 7.4 buffer containing 20 mmol/L HEPES, 20 mmol/L MgCl2, 0.1 mg/ml bovine serum albumin, and 1% (v/v) DMSO at room temperature. In most cases, DPP-4 enzyme (1 nmol/L final concentration) was added last to initiate the enzymatic reaction, except when measuring the recovery of DPP-4 enzyme activity from a preformed DPP-4-inhibitor complex, in which case enzyme was first pre-incubated with trelagliptin for 70 min before initiating the reaction by dilution 50-fold into a reaction buffer containing a large excess (2 mmol/L, ca. 17x Km) of GP-pNA substrate. All assays were conducted as duplicates in 96-well format with total assay volume of 200 uL and absorbance at 405 nm was measured every 10 seconds to determine the reaction time-course. In most cases, the entire reaction progress curve was analyzed as described below. However, for initial rate studies to establish GP-pNA substrate-competitive inhibition by trelagliptin, only absorbance measurements from the first 40 seconds were used.
Slow Binding Inhibition Model
Fig 1 illustrates a simple two-step inhibition model in which the inhibitor I binds to enzyme E in a rapid first step to form a weak EI complex with dissociation constant Ki (Eq 1), followed by a slower second step to yield an EI* complex, where inhibitor is bound more tightly, the overall dissociation is now given by Ki* (Eq 2), and Kisom (Eq 3) is the equilibrium constant for conversion between weak EI and tight EI* complexes, respectively. The observed rate constants for association (kon) and dissociation (koff) of inhibitor from the tight EI* complex in Fig 1 are then given by Eqs 4 and 5, respectively.
Fig 1. Two-step inhibition model.
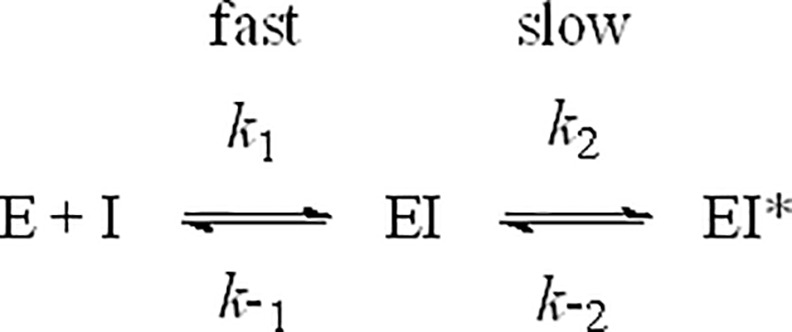
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
Kinetic analysis
To obtain estimates for the association (kon) and dissociation (koff) rate constants for trelagliptin inhibition of DPP-4, reaction progress curve data monitoring the onset of inhibition (DPP-4 enzyme added last to initiate reaction) were analyzed using the procedures outlined by Morrison [10]. Briefly, each progress curve was fitted to Eq 6:
| (6) |
where t is time, A is absorbance, and A0 is absorbance at time t = 0, to determine k', the apparent rate constant for decay from the faster initial rate (vo) to the slower inhibited steady-state rate (vs). An estimate for kon’, the apparent on-rate for trelagliptin binding, was obtained as the slope of the plot of k’ versus trelagliptin concentration according to Eq 7:
| (7) |
The y-intercept of this plot yielded an estimate for koff, and thus an estimate of t1/2 (= (ln2/koff)) for dissociation of trelagliptin. To obtain an estimate for kon, Eq 8 was used to correct for the GP-pNA substrate concentration because trelagliptin is a substrate-competitive DPP-4 inhibitor:
| (8) |
The ratio (koff/kon) yielded a value for Ki*, the steady-state inhibition constant for the tight EI* complex. Ki* was also estimated by plotting 1/vs, where vs is the steady-state rate at long reaction times, versus inhibitor concentration in a standard Dixon plot [11].
To obtain a more accurate estimate for koff, reaction progress curve data monitoring the recovery of DPP-4 enzyme activity following dilution of a preformed DPP-4- inhibitor complex into a reaction mixture containing a large excess of GP-pNA substrate were again fitted to Eq 6 to determine k’, the apparent rate constant for recovery from the slower inhibited initial rate (vo) to the faster steady-state rate (vs). As above, the y-intercept of the plot of k’ versus trelagliptin concentration according to Eq 7, yielded an estimate for koff.
X-ray diffraction data
Wild-type human DPP-4 was purified and crystallized as previously reported [7, 12]. All protein-inhibitor complexes were obtained by soaking preformed DPP-4 crystals in a solution containing the compound of interest. Crystals were then cryo-protected with ethylene glycol and flash frozen in liquid nitrogen. X-ray diffraction data were collected at the Advanced Light Source (ALS) beam line 5.0.3, and processed using the program HKL2000 [13]. The structures of DPP-4 inhibitor complexes were determined by molecular replacement using MOLREP, utilizing the previously determined coordinates of DPP-4 with accession code 1R9M [7, 14]. Subsequent structure refinement and model building were performed utilizing REFMAC and XtalView [14, 15]. Bound inhibitors were clearly visible in the electron density maps.
Results
Potency of DPP-4 inhibition and specificity towards DPP-4-related proteases
Trelagliptin exhibited potent inhibitory activity toward DPP-4 prepared from Caco-2 cells with an IC50 value of 5.4 nmol/L [95% confidence interval (CI) = 5.2–5.7]. Trelagliptin also inhibited human, dog, and rat plasma DPP-4 activity with IC50 values of 4.2 (CI = 4.1–4.3), 6.2 (CI = 6.0 –6.4), and 9.7 (CI = 8.0–11.8) nmol/L respectively (Table 2). When tested under identical experimental conditions using recombinant human DPP-4, trelagliptin exhibited potent inhibition with IC50 values for trelagliptin, alogliptin, and sitagliptin of 1.3 (CI = 1.1–1.5), 5.3 (CI = 5.0 –5.7), and 16.0 (CI = 15.1–16.9) nmol/L, respectively (Fig 2). When tested against DPP-4-related proteases, including DPP-2, DPP-8, DPP-9, PEP, and FAPα (Table 3), trelagliptin displayed IC50 values >100,000 nmol/L corresponding to >10,000-fold selectivity (Table 3). These data indicate that trelagliptin is a highly selective and potent DPP-4 inhibitor, about 4- and 12-fold more potent than alogliptin and sitagliptin, respectively.
Table 2. Inhibitory activity of trelagliptin in human, dog, and rat plasma.
| Species of plasma | human | dog | rat |
|---|---|---|---|
| IC50 (nmol/L) (95% CI) | 4.2 (4.1–4.3) | 6.2 (6.0–6.4) | 9.7 (8.0–11.8) |
Fig 2. Concentration response curves of DPP-4 inhibitory activities by trelagliptin, alogliptin and sitagliptin.

Concentration response curves of DPP-4 inhibitory activities by trelagliptin, alogliptin and sitagliptin. Activity was measured as described under Materials and Methods.
Table 3. Inhibitory activity of trelagliptin against DPP-4-related proteases.
| Proteases | DPP-2 | DPP-8 | DPP-9 | PEP | FAPα |
|---|---|---|---|---|---|
| IC50 (nmol/L) | >100,000 | >100,000 | >100,000 | >100,000 | >100,000 |
Demonstration of competitive inhibition
Initial rate (first 40 seconds only) measured across a range of GP-pNA substrate concentrations bracketing the apparent 120 μmol/L Km value at several fixed trelagliptin concentrations and plotted in Lineweaver-Burk format (1/rate versus 1/[GP-pNA]) as shown in Fig 3, demonstrate that trelagliptin competes directly with substrate for binding at the DPP-4 active site, with an apparent Ki value of 61 nmol/L for the initial weak EI complex (see Fig 1).
Fig 3. Double-reciprocal plot showing competitive inhibition of DPP-4 by trelagliptin.

Initial rate (first 40 seconds only) was measured as described under Materials and Methods using a range of GP-pNA substrate concentrations bracketing the apparent Km value at several fixed trelagliptin concentrations.
Slow-binding inhibition
Deviation from linearity observed for reaction progress curves as the trelagliptin concentration was increased when either DPP-4 enzyme (onset of inhibition, Fig 4) or GP-pNA substrate (recovery of activity from preformed DPP-4-inhibitor complex, Fig 5) was added last to initiate the reaction, is consistent with slow-binding inhibition [10]. Kinetic analysis of both sets of progress curves provided estimates for kon (Table 4) and koff (Tables 4 and 5) that are internally consistent, and that correspond to a t1/2 ≈ 30 min for dissociation of trelagliptin from DPP-4. The 1.5 nmol/L value for Ki* estimated as the koff/kon ratio determined from the detailed kinetic analysis was furthermore consistent with the 1.0 nmol/L Ki* value estimated from a Dixon plot of 1/vs versus trelagliptin concentration at long reaction times (replot not shown), where the tight EI* complex makes the major contribution to the inhibition. The roughly 40-fold difference between Ki and Ki* determined implies a Kisom (= k2/k-2) value of approxiately 40, reflecting a 40-fold tighter binding of trelagliptin in the tight EI* complex as opposed to the weak initial EI complex.
Fig 4. Time course of the reaction of DPP-4 in the absence or presence of different concentrations of trelagliptin.

Progress curves at 405 nm for pNA generation were recorded over 2000 sec using a 10 sec interval. Reaction was initiated with 1 nmol/L DPP-4 in the presence of 400 μmol/L GP-pNA substrate (approximately 4x Km) and varying concentrations of trelagliptin. Inset: Replot of apparent association rate constant, k’, against trelagliptin concentration used to estimate kon’ from the slope according to Eq 7.
Fig 5. Time course of the recovery of DPP-4 activity following dissociation of trelagliptin from the preformed DPP-4-inhibitor complex.

A preformed enzyme-inhibitor complex (where [DPP-4] = 50 nmol/L and trelagliptin concentration is as shown in the plot) was diluted 50-fold into a solution containing 2 mmol/L GP-pNA substrate (approximately 17x Km). Absorbance readings were taken every 10 seconds. Inset: Replot of the apparent dissociation rate constant, k’, against trelagliptin concentration used to estimate koff from the Y-intercept according to Eq 7.
Table 4. Kinetic constants for trelagliptin determined from association progress curves.
| parameter | Ki*(nmol/L) | kon (M-1 sec-1) | koff (sec-1) | EI* t1/2 (min) |
|---|---|---|---|---|
| trelagliptin | 1.5 ± 0.1 | (2.6 ± 0.1) x 105 | (4.0 ± 0.5) x 10−4 | 29 |
Table 5. Kinetic constants for trelagliptin determined from dissociation progress curves.
| parameter | koff (sec-1) | EI* t1/2 (min) |
|---|---|---|
| trelagliptin | (3.7 ± 0.5) x 10−4 | 31 |
Similar kinetic studies conducted using sitagliptin did not reveal any evidence for slow-binding inhibition (data not shown), indicating an upper limit for t1/2 < 2 min for dissociation of sitagliptin from DPP-4, which is consistent with the t1/2 ≈ 0.2 min value reported from surface plasmon resonance studies [16].
DPP-4 crystal structure with trelagliptin
Both trelagliptin and alogliptin were discovered with the aid of structure based drug design, and we were able to obtain co-complex X-ray crystal structures of both compounds bound to DPP-4. The co-complex structures of alogliptin and trelagliptin in the active site of DPP-4 are shown in Fig 6. Both structures show that the aminopiperidine forms a salt bridge to Glu205/Glu206, while the cyanobenzyl group effectively fills the S1 pocket (formed by Val656, Tyr631, Tyr662, Trp659, Tyr666, and Val711) and interacts with Arg125. The 2-position carbonyl participates in an important hydrogen bond with the backbone NH of Tyr631, and the uracil ring π-stacks with Tyr547. The single chemical structural difference between the two compounds is the presence of a fluorine atom at the 5-position of the cyanobenzyl group. Examination of the environment around the fluorine atom in the DPP-4 active site reveals the proximity of residues Trp659, Tyr631, and Val656 (Fig 7). Of these residues, only hydrogen atoms are within Van der Waals distance of the fluorine. These hydrogen atoms carry a permanent partial positive charge due to the position on the aromatic rings of Trp659 and Tyr631, and an inducible positive charge on the side chain of Val656. Fluorine carries a permanent partial negative change and so these atoms would be expected to produce an additional attractive force between ligand and protein, compared to a hydrogen atom at the 5-position. The magnitude of this attraction would be expected to be small but may be responsible for the 4-fold potency increase observed for trelagliptin versus alogliptin (Fig 2). The structures show that both compounds bind non-covalently to DPP-4.
Fig 6. Comparison of x-ray crystal structures of inhibitors bound to DPP-4.

Comparison of x-ray crystal structure of inhibitors bound to DPP-4 for alogliptin (left panel) and trelagliptin (right panel).
Fig 7. Potential fluorine atom interactions in trelagliptin x-ray crystal structure.

Close-up showing potential differential interaction of F-atom of trelagliptin (right panel) with Trp659 residue in DPP-4 crystal structure as compared to H-atom of alogliptin (left panel).
Discussion
Trelagliptin (SYR-472, zafatek) is a novel once-weekly DPP4 inhibitor that shows sustained efficacy by once-weekly dosing regimen in patients with type 2 diabetes. However, the underlying mechanism for this sustained efficacy remains to be clarified. In this study, we have characterized the in vitro properties for trelagliptin and compared those with alogliptin and sitagliptin.
Trelagliptin exhibits potent inhibition of human DPP-4 with IC50 values in the single digit nanomolar range, using either recombinant enzyme or a partially purified Caco-2 cell extract, that is approximately 4- to 12-fold more potent than either alogliptin or sitagliptin, respectively (Fig 2). The IC50 values for alogliptin and sitagliptin were consistent with those previously reported [17–20]. This enhanced potency carries over to human plasma when one compares the 4.2 nmol/L IC50 value for trelagliptin (Table 2) with the 10 nmol/L IC50 determined previously for alogliptin under similar experimental conditions (unpublished results). Thus, trelagliptin could exhibit a similar DPP-4 inhibition at lower plasma concentration compared with alogliptin.
X-ray diffraction data confirms that trelagliptin binds to DPP-4 in a pose (Fig 6) that is virtually identical to that seen for alogliptin, with the exception of the single fluorine atom at the 5-position of the cyanobenzyl group which may account for the slight potency advantage for trelagliptin. Unlike the covalent DPP-4 inhibitors, saxagliptin [21, 22] and vildagliptin [23, 24], where X-ray crystal structures show formation of a covalent imidate adduct between Ser630 and the cyano moiety of their cyano-pyrrolidine headgroup, X-ray results for both alogliptin and trelagliptin show no evidence for covalent complex formation. Initial velocity kinetic studies also confirm that DPP-4 inhibition by trelagliptin is reversible and substrate-competitive (Fig 3). In addition to enhancing binding affinity, the single fluorine substitution in trelagliptin also has the effect of slowing the dissociation rate for release of inhibitor from the enzyme by approximately 8-fold, if one compares the t1/2 ≈ 30 min for trelagliptin dissociation determined in this study (Tables 4 and 5) with the t1/2 ≈ 3.7 min value reported for alogliptin dissociation as determined by surface plasmon resonance [16]. Although the slower off-rate for trelagliptin compared to alogliptin contributes to more potent DPP-4 inhibitory activity, t1/2 for dissociation of trelagliptin from DPP-4 (≈ 30 min) is too short to entirely account for sustained efficacy of trelagliptin by once-weekly dosing.
What about other possible factors? No meaningful difference in plasma protein binding was observed between trelagliptin and alogliptin to explain differences in in vitro potency and in vivo efficacy (the binding of trelagliptin and alogliptin over the range of 0.1 to 10 μg/mL to human plasma protein were 22.1–27.6% and 28.2–32.3%, respectively (unpublished results)). In terms of microsomal stability using either rat or human microsomal preparations [18], trelagliptin (Compound 27j) showed slightly better stability than did alogliptin (Compound 27b), but again not sufficient to account for the much greater sustained efficacy. Likewise, comparing the pharmacokinetic and pharmacodynamic results obtained when trelagliptin was tested in dogs or cynomolgus monkeys [18] with those obtained using alogliptin [17], other than a slight potency advantage for trelagliptin, there was no indication from these preclinical studies that this compound would be amenable to a once-weekly as opposed to a once-daily dosing regimen. That conclusion was only revealed through human clinical trials [2, 5].
In the clinical trial setting, trelagliptin showed sustained inhibition of DPP-4 activity until 7 days after dosing in the phase 2 dose-ranging study in patients with type 2 diabetes mellitus (T2DM) [2]. Further analysis of the results of this phase 2 dose-ranging study using a sigmoid Emax model indicated that the plasma trelagliptin concentration estimated to yield 50% and 70% inhibition of plasma DPP-4 activity was 1.43 ng/mL and 2.31 ng/mL, respectively (Fig 8). 1.43 ng/mL corresponds to about 4.02 nmol/L and is consistent with the IC50 value for DPP-4 inhibition in human plasma (Table 2). Moreover, in the previous phase 1 study [25], where single dose administration of trelagliptin (3.125–800 mg) was carried out in Japanese healthy volunteers, the plasma trelagliptin concentration [mean (SD)] at the 100 mg dose 168 hours after administration was 2.12 (0.679) ng/mL, and this value was almost the same as the plasma trelagliptin concentration estimated to yield 70% inhibition of plasma DPP-4 activity in phase 2 dose-ranging study as explained above (i.e., 2.31 ng/mL). These results indicate that the plasma concentration of trelagliptin even at 168 hours after 100 mg dosing is enough to sustain its pharmacodynamic effect, i.e., 70% inhibition of plasma DPP-4 activity, by weekly dosing of trelagliptin in T2DM patients. For futher insight into the high potency of trelagliptin, we refer to the similar pharmacokinetic/pharmacodynamic modeling performed for alogliptin [26], which is one of the once-daily DPP-4 inhibitors. Based on a simple Emax modeling, the estimated IC50 value of alogliptin was 6.6 ng/mL. The difference in estimated IC50 values (1.43 ng/mL [4.02 nmol/L] vs. 6.6 ng/mL [19.4 nmol/L]) between trelagliptin and alogliptin is consistent with the difference in IC50 values for human plasma DPP-4 inhibition in vitro (4.2 and 10 nmol/L for trelagliptin and alogliptin, respectively). These results suggest that the potent in vitro DPP-4 inhibitory activity of trelagliptin at least partially contributes to the in vivo efficacy of trelagliptin at lower plasma concentration at 7 days after administration. Of note, it has been shown that the efficacy of trelagliptin 100 mg weekly dosing is non-inferior to alogliptin 25 mg daily dosing in terms of reduction of HbA1c in T2DM patients [5].
Fig 8. Relationship between trelagliptin pharmacokinetics and pharmacodynamics in T2DM patients in phase 2 dose-ranging study.

Observed value plotted in “X” and predicted relationship between pharmacokinetics and pharmacodynamics by sigmoid Emax model is indicated by solid line. The plasma trelagliptin concentration estimated to yield 70% and 80% inhibition of human plasma DPP-4 activity was 2.31 ng/mL and 3.13 ng/mL, respectively.
In addition, no major safety concerns for trelagliptin have been confirmed and the overall safety profile of trelagliptin was similar to that of alogliptin in clinical studies [2, 5]. Selectivity for DPP-4 over other related serine proteases including DPP-8 and DPP-9 could be important to avoid potential adverse events. DPP-8 and DPP-9 inhibition was reported to be associated with multiorgan toxicities and immunotoxicity in rats and dogs and attenuation of human T-cell activation in vitro in some [27], but not other [28, 29] studies. In the present study, we demonstrated that trelagliptin shows > 10,000-fold selectivity for DPP-4 over other related serine proteases including DPP-8 and DPP-9. Moreover, no significant response (≥ 50% inhibition or stimulation) was observed at 10 μmol/L in an expansive selectivity counterscreen (47 enzymatic assays and 79 radioligand binding assays) carried out at MDS Pharma (data not shown). Thus, trelagliptin has a favorable target selectivity profile as a selective DPP-4 inhibitor.
Conclusion
Trelagliptin is a novel once-weekly DPP-4 inhibitor that shows sustained efficacy by once-weekly treatment in T2DM patients. The in vitro data described in this paper demonstrated that trelagliptin is a potent and selective DPP-4 inhibitor. Kinetic analysis revealed that trelagliptin is a reversible, substrate-competitive and slow-binding DPP-4 inhibitor (t1/2 for dissociation ≈ 30 minutes). Furthermore, X-ray diffraction data indicated a non-covalent interaction between DPP-4 and trelagliptin. Potent DPP-4 inhibitory activity of trelagliptin may at least partially contribute to the sustained efficacy of trelagliptin.
Acknowledgments
The authors wish to thank Dr. Yusuke Moritoh for preparing human, dog and rat plasma. We wish to thank Dr. Kathleen Aertgeerts and Dr. Jason Yano for their contributions to the DPP-4 crystal structures. The X-ray crystallography data reported here are based on research conducted at the Advanced Light Source (ALS). ALS is supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences Division, of the U.S. Department of Energy under Contract No. DE-AC03-76SF00098 at the Lawrence Berkeley National Laboratory. We thank the staff at ALS for their excellent support in the use of the synchrotron beamlines.
Data Availability
The crystal structure information for trelagliptin bound to human DPP-4 is deposited to the PDB databank. The PDB code for the deposited DPP-4:SYR-472 (aka trelagliptin) co-complex is 5KBY. All relevant information is contained within the paper.
Funding Statement
During the time this work was conducted, each of the co-authors was an employee of the global Takeda Pharmaceutical Company Limited or one of its wholly owned affiliates, which provided support in the form of salaries for authors [CEG, AJ, RK, HU, NN, TK, AT, HS, YK, EK, LS, and KT], but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the ‘author contributions’ section.
References
- 1.Organization WH. Diabetes Fact Sheet No 312: World Health Organization; 2015. Available from: http://www.who.int/mediacentre/factsheets/fs312/en/.
- 2.Inagaki N, Onouchi H, Sano H, Funao N, Kuroda S, Kaku K. SYR-472, a novel once-weekly dipeptidyl peptidase-4 (DPP-4) inhibitor, in type 2 diabetes mellitus: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2(2):125–32. Epub 2014/03/14. 10.1016/S2213-8587(13)70149-9 S2213-8587(13)70149-9 [pii]. . [DOI] [PubMed] [Google Scholar]
- 3.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–705. Epub 2006/11/14. 10.1016/S0140-6736(06)69705-5 . [DOI] [PubMed] [Google Scholar]
- 4.Ceriello A, Sportiello L, Rafaniello C, Rossi F. DPP-4 inhibitors: pharmacological differences and their clinical implications. Expert Opin Drug Saf. 2014;13(S1):57–68. Epub 2014/08/30. 10.1517/14740338.2014.944862 . [DOI] [PubMed] [Google Scholar]
- 5.Inagaki N, Onouchi H, Maezawa H, Kuroda S, Kaku K. Once-weekly trelagliptin versus daily alogliptin in Japanese patients with type 2 diabetes: a randomised, double-blind, phase 3, non-inferiority study. The lancet Diabetes & endocrinology. 2015;3(3):191–7. Epub 2015/01/23. 10.1016/S2213-8587(14)70251-7 . [DOI] [PubMed] [Google Scholar]
- 6.Nonaka N, Asai Y, Nishio M, Takahashi K, Okuda T, Tanaka S, et al. TMC-2A, -2B and -2C, novel dipeptidyl peptidase IV inhibitors produced by Aspergillus oryzae A374. I. Taxonomy of producing strain, fermentation, and biochemical properties. The Journal of antibiotics. 1997;50(8):646–52. . [DOI] [PubMed] [Google Scholar]
- 7.Aertgeerts K, Ye S, Tennant MG, Kraus ML, Rogers J, Sang BC, et al. Crystal structure of human dipeptidyl peptidase IV in complex with a decapeptide reveals details on substrate specificity and tetrahedral intermediate formation. Protein science: a publication of the Protein Society. 2004;13(2):412–21. Epub 2004/01/14. 10.1110/ps.03460604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mantle D. Characterization of dipeptidyl and tripeptidyl aminopeptidases in human kidney soluble fraction. Clinica Chimica Acta. 1991;196(2–3):135–42. [DOI] [PubMed] [Google Scholar]
- 9.Toide K, Okamiya K, Iwamoto Y, Kato T. Effect of a novel prolyl endopeptidase inhibitor, JTP-4819, on prolyl endopeptidase activity and substance P- and arginine-vasopressin-like immunoreactivity in the brains of aged rats. J Neurochem. 1995;65(1):234–40. . [DOI] [PubMed] [Google Scholar]
- 10.Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Advances in enzymology and related areas of molecular biology. 1988;61:201–301. Epub 1988/01/01. . [DOI] [PubMed] [Google Scholar]
- 11.Dixon M. The determination of enzyme inhibitor constants. Biochem J. 1953;55(1):170–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hosfield D, Palan J, Hilgers M, Scheibe D, McRee DE, Stevens RC. A fully integrated protein crystallization platform for small-molecule drug discovery. Journal of structural biology. 2003;142(1):207–17. Epub 2003/04/30. . [DOI] [PubMed] [Google Scholar]
- 13.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode In: Carter CW Jr., Sweet RM, editors. Methods in Enzymology. 276: Academic Press; (New York: ); 1997. p. 307–26. [DOI] [PubMed] [Google Scholar]
- 14.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr A. 1994;D50:760–3. [DOI] [PubMed]
- 15.McRee DE. XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density. Journal of structural biology. 1999;125(2–3):156–65. Epub 1999/05/01. 10.1006/jsbi.1999.4094 . [DOI] [PubMed] [Google Scholar]
- 16.Schnapp G, Klein T., Mark M., Fuchs H., Bakker R.A., Nar H. Compartive enzyme kinetic analysis of the launched DPP-4 inhibitors. Diabetes. 2014;63 (Supplement 1)(June 2014):A271. [Google Scholar]
- 17.Lee B, Shi L, Kassel DB, Asakawa T, Takeuchi K, Christopher RJ. Pharmacokinetic, pharmacodynamic, and efficacy profiles of alogliptin, a novel inhibitor of dipeptidyl peptidase-4, in rats, dogs, and monkeys. Eur J Pharmacol. 2008;589(1–3):306–14. Epub 2008/06/10. 10.1016/j.ejphar.2008.04.047 S0014-2999(08)00472-X [pii]. . [DOI] [PubMed] [Google Scholar]
- 18.Zhang Z, Wallace MB, Feng J, Stafford JA, Skene RJ, Shi L, et al. Design and synthesis of pyrimidinone and pyrimidinedione inhibitors of dipeptidyl peptidase IV. J Med Chem. 2011;54(2):510–24. Epub 2010/12/29. 10.1021/jm101016w . [DOI] [PubMed] [Google Scholar]
- 19.Kim D, Wang L, Beconi M, Eiermann GJ, Fisher MH, He H, et al. (2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)- yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem. 2005;48(1):141–51. Epub 2005/01/07. 10.1021/jm0493156 . [DOI] [PubMed] [Google Scholar]
- 20.Feng J, Zhang Z, Wallace MB, Stafford JA, Kaldor SW, Kassel DB, et al. Discovery of alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV. J Med Chem. 2007;50(10):2297–300. Epub 2007/04/20. 10.1021/jm070104l . [DOI] [PubMed] [Google Scholar]
- 21.Kim YB, Kopcho LM, Kirby MS, Hamann LG, Weigelt CA, Metzler WJ, et al. Mechanism of Gly-Pro-pNA cleavage catalyzed by dipeptidyl peptidase-IV and its inhibition by saxagliptin (BMS-477118). Arch Biochem Biophys. 2006;445(1):9–18. Epub 2005/12/21. S0003-9861(05)00481-9 [pii] 10.1016/j.abb.2005.11.010 . [DOI] [PubMed] [Google Scholar]
- 22.Metzler WJ, Yanchunas J, Weigelt C, Kish K, Klei HE, Xie D, et al. Involvement of DPP-IV catalytic residues in enzyme-saxagliptin complex formation. Protein Sci. 2008;17(2):240–50. Epub 2008/01/30. 10.1110/ps.073253208 17/2/240 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oefner C, D'Arcy A, Mac Sweeney A, Pierau S, Gardiner R, Dale GE. High-resolution structure of human apo dipeptidyl peptidase IV/CD26 and its complex with 1-[([2-[(5-iodopyridin-2-yl)amino]-ethyl]amino)-acetyl]-2-cyano-(S)-pyrrolidine. Acta Crystallogr D Biol Crystallogr. 2003;59(Pt 7):1206–12. Epub 2003/07/02. S0907444903010059 [pii]. . [DOI] [PubMed] [Google Scholar]
- 24.Brandt I, Joossens J, Chen X, Maes MB, Scharpe S, De Meester I, et al. Inhibition of dipeptidyl-peptidase IV catalyzed peptide truncation by Vildagliptin ((2S)-{[(3-hydroxyadamantan-1-yl)amino]acetyl}-pyrrolidine-2-carbonitrile). Biochem Pharmacol. 2005;70(1):134–43. Epub 2005/05/24. S0006-2952(05)00237-6 [pii] 10.1016/j.bcp.2005.04.009 . [DOI] [PubMed] [Google Scholar]
- 25.Matsuno K, Horayama M, Araki T, et al. Evaluation for sustained DPP-4 inhibitory effect of SYR-472, a novel DPP-4 inhibitor, in Japanese healthy subjects [in Japanese]. J Japan Diabetes Soc. 2012;55(suppl 1):S–117 (abstr I-P-38). [Google Scholar]
- 26.Covington P, Christopher R, Davenport M, Fleck P, Mekki QA, Wann ER, et al. Pharmacokinetic, pharmacodynamic, and tolerability profiles of the dipeptidyl peptidase-4 inhibitor alogliptin: a randomized, double-blind, placebo-controlled, multiple-dose study in adult patients with type 2 diabetes. Clin Ther. 2008. March;30(3):499–512. 10.1016/j.clinthera.2008.03.004 . [DOI] [PubMed] [Google Scholar]
- 27.Lankas GR, Leiting B, Roy RS, Eiermann GJ, Beconi MG, Biftu T, et al. Dipeptidyl peptidase IV inhibition for the treatment of type 2 diabetes: potential importance of selectivity over dipeptidyl peptidases 8 and 9. Diabetes. 2005;54(10):2988–94. . [DOI] [PubMed] [Google Scholar]
- 28.Burkey BF, Hoffmann PK, Hassiepen U, Trappe J, Juedes M, Foley JE. Adverse effects of dipeptidyl peptidases 8 and 9 inhibition in rodents revisited. Diabetes Obes Metab. 2008;10(11):1057–61. 10.1111/j.1463-1326.2008.00860.x . [DOI] [PubMed] [Google Scholar]
- 29.Wu JJ, Tang HK, Yeh TK, Chen CM, Shy HS, Chu YR, et al. Biochemistry, pharmacokinetics, and toxicology of a potent and selective DPP8/9 inhibitor. Biochem Pharmacol. 2009;78(2):203–10. 10.1016/j.bcp.2009.03.032 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The crystal structure information for trelagliptin bound to human DPP-4 is deposited to the PDB databank. The PDB code for the deposited DPP-4:SYR-472 (aka trelagliptin) co-complex is 5KBY. All relevant information is contained within the paper.