

Abstract
Cytotoxic chemotherapy remains to be the first-line therapy for many advanced solid tumors; hence, understanding the underlying mechanisms to overcome chemoresistance remains a top research priority. In the clinic, chemotherapy is administered in multiple cycles that are spaced out to allow the recovery or repopulation of normal tissues and tissue stem cells between treatment cycles. However, residual surviving cancer cells and cancer stem cells can also repopulate tumors during the gap periods between chemotherapy cycles. Tumor repopulation is an understudied phenomenon often overlooked due to current custom experimental study strategies. Recent findings revealed an alarming role for dying cells targeted by chemotherapy in releasing mitogens to stimulate active repopulation of quiescent cancer stem cells. Therefore, new therapeutic options to abrogate tumor repopulation will provide new avenues to improve chemotherapeutic response and clinical outcome.
BACKGROUND
Cytotoxic chemotherapy remains the first-line therapy for many advanced solid cancer types. There are many types of cytotoxic chemotherapy (1). Cell-cycle dependent chemotherapeutic drugs that target dividing cancer cells would concurrently target normal cells that divide rapidly during normal tissue homeostasis. These cause undesirable side effects within normal tissues with a high turnover rate, including myelosuppression (e.g. neutropenia) in bone marrow, mucositis (inflammation) in intestinal tract, and alopecia (or hair loss) in hair follicle. Therefore, to alleviate some of these side effects that can lead to severe complications or potentially life-threatening toxicities, chemotherapeutics are administered in multiple cycles of fractionated doses that are spaced out to allow normal cells and tissue stem cells to recover or repopulate between treatment cycles [reviewed in (2, 3)]. However, residual surviving cancer cells can also repopulate tumors during the gap periods between chemotherapy cycles, which is a major cause of treatment failure that is often overlooked. In the past decade, there are experimental data from preclinical models demonstrating that repopulation of tumor cells occur between and during chemotherapy cycles in many solid tumors [and reviewed in (2, 3)]. The term repopulation is defined as ‘proliferation of surviving tumor cells during or after cytotoxic chemotherapy’. However, most laboratory studies overlook the biologic phenomenon of tumor repopulation, by exposing cancer cells to long-term continuous chemotherapy treatment to select for chemoresistant clones in vitro, followed by high throughput molecular analyses to compare molecular changes that occur between these chemoresistant clones and their parental cells. Similarly, most in vivo studies administer one single dose or continuous treatment of chemotherapy drugs followed by downstream molecular, phenotypic and functional analyses. Such study designs do not take into account the concept of tumor repopulation, the identity of repopulating tumor cells, nor the consequential enrichment of these repopulating clones following multiple chemotherapy treatment cycles (as that administered in the clinic); which would be the main focus of summary and discussions here.
Awakening of dormant or quiescent cancer stem cells to repopulate tumor
An elegant recent study by Dick and colleagues specifically labeled single cancer cells derived from colon cancer patients by lentiviral lineage tracking and examined their repopulation dynamics in response to the chemotherapeutic drug oxaliplatin (4). Under steady state condition without chemotherapy treatment, they observed several different types of clones within these colon patient-derived xenografts: tumor-propagating clone that persisted throughout multiple serial passages, clone that persisted transiently but became undetectable later on, and dormant/quiescent clone that became reactivated to expand following multiple serial transplantation passages (4). Following oxaliplatin chemotherapy treatment, they observed a marked heterogeneity in the response of individual colon cancer clones to chemotherapy. Particularly, there was a significant enrichment of dormant/quiescent clones that became reactivated, verifying a selective response of these dormant/quiescent clones to chemotherapy treatment. On the other hand, while those presumably fast proliferating tumor-propagating clones were sensitive to chemotherapy killing, some tumor-propagating clones did persist through selective pressure of chemotherapy treatment although their growth kinetics became slower (4). Further DNA copy number variation profiling and targeted deep sequencing confirmed that oxaliplatin chemotherapy did not necessarily induce evolvement of new genetically distinct subclones as most studies would presume; in contrary, chemotherapy altered the proportion of pre-existed clonality. Such interesting findings challenged the conventional methodologies to identify “chemoresistance mechanisms or predictive signatures of therapeutic response” by comparing molecular differences at the genomic level, which may not necessarily yield a straightforward answer to understanding chemoresistance. These findings also added another level of intratumoral cellular complexity to understanding chemotherapeutic response, pointing to the existence of dormant or quiescent subpopulations within tumors that could become “awakened” and expanded in response to injury and cell death induced by chemotherapy treatment.
Indeed, other studies in animal models of glioblastoma (5) and medullobastoma (6), and patient-derived xenografts from bladder urothelial carcinomas (7) supported the existence of a quiescent tumor subpopulation or cancer stem cells. Parada and colleagues employed in vivo lineage tracing to identify a quiescent tumor subpopulation that coexpressed Sox2 within a mouse model of glioblastoma (hGFAP-Cre; Nf1fl/+; P53fl/fl; Ptenfl/+ mice) (5). The DNA alkylating chemotherapeutic drug temozolomide (TMZ) was able to target and diminish the proportion of proliferating glioblastoma progenitor cells, while repopulation of tumor was driven by residual quiescent cancer stem cells that were recruited to divide after temozolomide treatment was discontinued. Similarly, Dirks and colleagues demonstrated a rare and quiescent Sox2+ tumor subpopulation within a mouse model of medulloblastoma (PTCH1+/− mouse model of SHH subtype) (6). These Sox2+ cells contain cancer stem cell properties with both self-renewal and differentiation potential in vivo. The S phase-specific chemotherapeutic drug cytarabine was used and shown to effectively target proliferating differentiated cells, while Sox2+ cancer stem cells did not respond to drug treatment and instead their frequency was significantly increased following drug treatment (6). Independently, our group employed immortalized cancer cell-derived and patient-derived xenografts from bladder urothelial carcinomas to demonstrate the existence of a quiescent tumor subpopulation that could be enriched by cytokeratin (CK) 14 expression (7). These CK14+ urothelial carcinoma cells exhibited functional cancer stem cell properties as sphere-forming stem cells in vitro and as tumorigenic cells in vivo. Combination chemotherapeutic treatment of the cytidine nucleoside analog gemcitabine and the DNA alkylating agent cisplatin was effective in diminishing proliferating urothelial carcinomas cells, while quiescent CK14+ cancer stem cells were spared and recruited into cell division by chemotherapy treatment, leading to an increased frequency post chemotherapy treatment (7). In additional to the previously proposed intrinsic resistance of cancer stem cells to chemotherapeutic drugs (8–14) [reviewed in (15, 16)], the above intriguing observations collectively confirmed an active response and “reawakening” of quiescent cancer stem cells into cell division, contributing to repopulation of residual tumors following cytotoxic chemotherapy (Fig. 1A).
Figure 1. Schematic diagram illustrating a “wound response” like tumor repopulation driven by cancer stem cells (7).
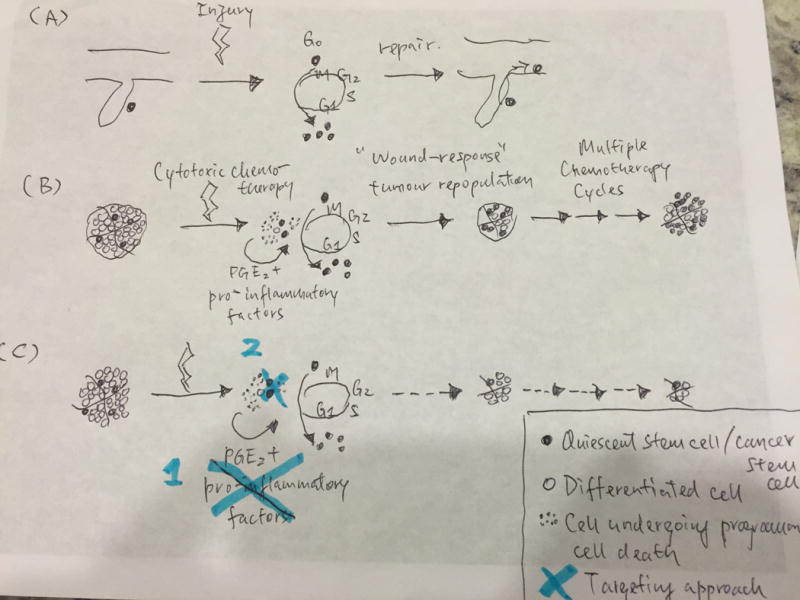
(A) Tissue injuries induce reactivation of quiescent stem cells to repopulate and repair wound sites. (B) Cytotoxic chemotherapy effectively induces cell death in differentiated cancer cells, while dying cells release pro-inflammatory factors (including PGE2) that stimulate reawakening of quiescent cancer stem cells into cell divisions and tumor repopulation. Recurrent wound response-like tumor repopulation following multiple chemotherapy cycles ultimately leads to chemoresistance. (C) Two targeting approaches proposed to abrogate tumor repopulation and chemoresistance: (1) Drugs that effectively block the reawakening of quiescent cancer stem cells, (2) Drugs that directly target and deplete cancer stem cells.
Mitogens released by dying cancer cells “reawaken” quiescent cancer stem cells for tumor repopulation
The next important question to be addressed would be: what are the underlying mechanisms contributing to the “reawakening” of quiescent cancer stem cells into active cell division and tumor repopulation?
During normal tissue homeostasis, quiescent tissue stem cells generally serve as a reserve until being challenged by emergency circumstances such as tissue injury or stress. For instance, quiescent stem cells within the hair follicle bulge region of skin epidermis do not contribute to daily epidermal homeostasis, until being challenged by a physical full thickness wound or incision wound (17, 18). Similarly, quiescent stem cells (lineage-negative eptheilial stem/progenitor cells) within normal distal lung were recently demonstrated to respond by cellular proliferation to repair injury induced by influenza or bleomycin (19). A dormant or quiescent neural stem cell within the subventricular zone of the brain was also demonstrated to be primed and could be recruited in response to brain ischemia (20, 21). Further, two functional distinct phases of quiescent satellite muscle stem cells and long-term hematopoietic stem cells were reported, namely G0 and GAlert phases, that could reversibly transit between these two phases in response to injury-induced signals (22, 23). Genes involved in mitochrondrial metabolism seemed to correlate with the ability of stem cells in G0 state to transit into the GAlert state, priming these GAlert quiescent stem cells into accelerated entry to cell cycle for sustaining proliferation and differentiation necessary to repair tissue injury (23). Since the cancer types discussed above contain functionally distinct quiescent cancer stem cells, they likely retain safeguard mechanisms similar to that as in tissues stem cells for responding to wounding or tissue injuries (24, 25). Therefore, it seems rational for us in drawing a parallel perspective to hypothesize that the molecular mechanisms involved during the process of wound response within an organ may be shared during chemotherapy-induced damages within tumors.
During tissue wound repair, Li and colleagues proposed a role for cells undergoing programmed cell death in releasing mitogens and promoting healing of excision wounds in skin and other organs (26). While the release of cytokines and factors from dying cells were previously thought to elicit an immune response for cell clearance (27), these authors demonstrated that the release of prostaglandin E2 (PGE2) acted downstream of the executor caspases 3 and 7, which acted as a mitogen to promote wound healing by stimulating proliferation in a paracrine manner (26). Particularly, caspase 3 or 7 cleavages led to enhanced activity of calcium-independent phospholipase A2 (iPLA2), which in turn increased synthesis and release of arachidonic acid, which is the upstream phospholipid to be metabolized into PGH2 and finally PGE2 (26). Independently, caspase 3 activated iPLA2 had been implicated to mediate chemotactic activity by recruiting monocytes, likely contributing to recruitment of immune cells to wound sites for clearance of dead cells by phagocytic engulfment (28). Unfortunately, cancer cells undesirably adapted such wound response mechanism following radiation (29) and chemotherapy-induced cell death (7). Results from our group demonstrated that many pro-inflammatory factors including PGE2 were released from urothelial carcinoma cells during chemotherapy-induced cell death (Fig. 1B), which drove these chemotherapy treated tumors into a “wound response” state as demonstrated by global RNA-sequencing analyses following by gene-set enrichment analyses confirming an enrichment of a “wound response” gene-signature, when compared with vehicle treated xenograft tumors (7). The pro-inflammatory phospholipid PGE2 contributed to enhanced ability to generate sphere-forming stem cells in vitro and induced recruitment of quiescent cancer stem cells into cell division for tumor repopulation (7) (Fig. 1B). Since chemotherapy was administered in multiple cycles as clinical regimen, successive rounds of “wound induced” tumor repopulation driven by PGE2 and other pro-inflammatory factors ultimately led to marked expansion of cancer stem cells and treatment failure following multiple chemotherapy cycles (7) (Fig. 1B).
CLINICAL-TRANSLATIONAL ADVANCES
Current adjuvant therapies developed to enhance chemotherapeutic response focus on targeting the molecular basis adapted by cancer cells to evade drug resistance. These include, but are not limited to, reduced drug uptake by altering expression of nucleoside transporters and copper transporters, enhanced drug export by increased expression of ATP binding cassette protein transporters, enhanced drug metabolism via cytochrome P450 superfamily enzymes, enhanced survival by promoting pro-survival pathways or overexpression of anti-apoptotic proteins, and alterations in the components of DNA repair pathways.
However, with mounting evidences demonstrating the existence of quiescent cancer stem cells in multiple tumor types (presented earlier), that they can either evade cell-cycle dependent chemotherapy by hibernating in their dormant state, or they can be “reawakened” by chemotherapy-induced damages to actively participate in a wound repair type of tumor repopulation; this adds another level of complexity to the existing multifactorial problem of chemoresistance. Many recent clinical trials were initiated to investigate whether modifying the schedule of chemotherapy to reduce the gap periods between treatment cycles may alter therapeutic response and clinical outcome. These does-dense chemotherapy trials were designed to achieve maximum tumor killing by shortening the intervals between chemotherapy treatments without altering the dosage. Such condensed treatment schedule became feasible due to the co-administration of colony-stimulating factors e.g. GM-CSF/G-CSF/CSFs to induce mobilization and differentiation of hematopoietic stem and progenitor cells for enhancing leukocyte recovery (30, 31). While in theory the hypothesis for dose dense chemotherapy was appealing, clinical trials in early breast cancers demonstrated some impact on disease-free survival with no significant benefit on overall survival (32–34). Therefore, new approaches to directly target tumor repopulation may be more efficacious. Here, we propose at least two approaches to target tumor repopulation fueled by quiescent cancer stem cells as adjuvant therapies to enhance chemotherapeutic response:
Drugs that effectively block the reawakening of quiescent cancer stem cells (Fig. 1C);
Drugs that directly target and deplete cancer stem cells (Fig. 1C).
In fact, preclinical studies convincingly demonstrated efficacious results for these approaches.
There are examples in preclinical models for testing drugs that block the reawakening of quiescent cancer stem cells for tumor repopulation. In xenografts from urothelial carcinomas, blocking PGE2 release by a neutralizing antibody or using the FDA approved drug celecoxib was effective in diminishing paracrine effects of neighboring dying cells to promote expansion of cancer stem cells induced by chemotherapy treatment (7). More importantly, celecoxib drug treatment was sufficient to block chemotherapy-treated xenografts from entering into a “wound response” state, thereby preventing subsequent wound-induced tumor repopulation and eventual chemoresistance (Fig. 1C), as measurable by significant reduced volume of xenograft tumors and significant reduction in the frequency and size of distal metastatic foci in lung (7). The fact that celecoxib is efficacious in improving chemotherapeutic response in a primary xenograft model derived from an original patient who failed chemotherapy, this provided convincing proof-of-concept evidence for testing such adjuvant therapeutic approaches with comparable concepts in other preclinical models and subsequent human clinical trials. Indeed, other approaches that block tumor repopulation (although not specifically targeting cancer stem cells) has been proven effective to enhance chemotherapeutic response in xenograft models of ovarian (35–37), glioma (38), breast (39, 40), prostate cancer (40) and mesothelioma (41).
On the other side of this equation, there have been proposals based on the conceptual assumption that driving dormant stem cells into cell cycle could sensitize them to cell cycle dependent chemotherapy. In fact, experimental evidences based on this concept existed primarily in the context of leukemic stem cells. In a patient-derived xenograft model of human acute myeloid leukemia, dormant cancer stem cells residing in the endosteal bone marrow niche were induced into entering cell cycle by G-CSF and were demonstrated to become sensitized to the chemotherapeutic drug cytarabine in vivo (42). While this strategy was effective in diminishing the frequency of leukemic stem cells, residual stem cells clearly persisted (presumably dormant ones) and were able to re-initiate leukemia upon re-transplantation. Such approach should be highly cautioned, since the molecular mechanisms regulating the balance between driving stem cell exhaustion and uncontrolled expansion were not understood currently.
In certain clinical settings, blocking tumor repopulation alone may be sufficient to improve clinical outcome since the bulk tumor would be surgically removed following neoadjuvant chemotherapy. However, in certain cancer types such as medulloblastomas and gliomas that are highly infiltrating to the normal brain regions, the surgical margins are difficult to be determined and sometimes it is presumably not feasible remove all bulk tumor cells by surgery. Therapeutics that enables direct targeting and effective killing of cancer stem cells may be more desirable in such scenarios. Preclinical studies using the HSV-thymidine kinase suicidal gene approach to ablate glioma cancer stem cells plus temozolomide chemotherapy demonstrated prolonged survival when compared with treatment group receiving temozolomide alone (5). Another preclinical study identified two aureolic acids, dactinomycin and mithramycin, in targeting Sox2+ medulloblastoma quiescent stem cells. These cancer stem cell targeting drugs were effective in killing neural sphere forming cells in vitro and prevented xenograft growth following engraftment of Sox2+ cancer stem cells in vivo (6). It would be predicted that combination of such cancer stem cells targeted approach together with tumor debulking drugs would yield optimal efficacies. In fact, these targeted approaches in combination with tumor debulking chemotherapy also demonstrated success in preclinical models of breast cancer (43–45), and head and neck cancer (46).
While most of these concepts that block quiescent cancer stem cells from tumor repopulation or direct their specific targeting are still in the preclinical phase, they are yielding promising efficacies. The next era of human clinical trials will hopefully unravel their efficacies in modulating therapeutic response and patient survival. Nonetheless, accumulating basic knowledge in understanding the regulation of stem cell quiescence versus emergency-induced proliferation will undoubtedly provide an important foundation for designing more specific anti-cancer strategies to target this “good/evil” switch.
Acknowledgments
Grant Support
K.S. Chan was supported by the NCI (R00CA129640 and R01CA175397), Cancer Prevention and Research Institute of Texas (CPRIT RP140252), and the V Foundation for Cancer Research (V Scholar Award).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflict of interests were disclosed.
References
- 1.Chabner BA, Roberts TG., Jr Timeline: Chemotherapy and the war on cancer. Nature reviews Cancer. 2005;5:65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 2.Davis AJ, Tannock JF. Repopulation of tumour cells between cycles of chemotherapy: a neglected factor. The Lancet Oncology. 2000;1:86–93. doi: 10.1016/s1470-2045(00)00019-x. [DOI] [PubMed] [Google Scholar]
- 3.Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nature reviews Cancer. 2005;5:516–25. doi: 10.1038/nrc1650. [DOI] [PubMed] [Google Scholar]
- 4.Kreso A, O’Brien CA, van Galen P, Gan OI, Notta F, Brown AM, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339:543–8. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–6. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanner RJ, Remke M, Gallo M, Selvadurai HJ, Coutinho F, Lee L, et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer cell. 2014;26:33–47. doi: 10.1016/j.ccr.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015;517:209–13. doi: 10.1038/nature14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, et al. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer research. 2005;65:4320–33. doi: 10.1158/0008-5472.CAN-04-3327. [DOI] [PubMed] [Google Scholar]
- 9.Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell stem cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. Journal of the National Cancer Institute. 2008;100:672–9. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 11.Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell stem cell. 2009;4:226–35. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pang R, Law WL, Chu AC, Poon JT, Lam CS, Chow AK, et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell stem cell. 2010;6:603–15. doi: 10.1016/j.stem.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell stem cell. 2011;9:50–63. doi: 10.1016/j.stem.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell stem cell. 2009;5:168–77. doi: 10.1016/j.stem.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 15.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 16.Clevers H. The cancer stem cell: premises, promises and challenges. Nature medicine. 2011;17:313–9. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 17.Ito M, Liu Y, Yang Z, Nguyen J, Liang F, Morris RJ, et al. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nature medicine. 2005;11:1351–4. doi: 10.1038/nm1328. [DOI] [PubMed] [Google Scholar]
- 18.Mascre G, Dekoninck S, Drogat B, Youssef KK, Brohee S, Sotiropoulou PA, et al. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature. 2012;489:257–62. doi: 10.1038/nature11393. [DOI] [PubMed] [Google Scholar]
- 19.Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015;517:621–5. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo Y, Coskun V, Liang A, Yu J, Cheng L, Ge W, et al. Single-cell transcriptome analyses reveal signals to activate dormant neural stem cells. Cell. 2015;161:1175–86. doi: 10.1016/j.cell.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Llorens-Bobadilla E, Zhao S, Baser A, Saiz-Castro G, Zwadlo K, Martin-Villalba A. Single-cell transcriptomics reveals a population of dormant neural stem cells that become activated upon brain injury. Cell stem cell. 2015;17:329–40. doi: 10.1016/j.stem.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–29. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 23.Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature. 2014;510:393–6. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324–31. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- 25.Arwert EN, Hoste E, Watt FM. Epithelial stem cells, wound healing and cancer. Nature reviews Cancer. 2012;12:170–80. doi: 10.1038/nrc3217. [DOI] [PubMed] [Google Scholar]
- 26.Li F, Huang Q, Chen J, Peng Y, Roop DR, Bedford JS, et al. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Science signaling. 2010;3:ra13. doi: 10.1126/scisignal.2000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green DR. The end and after: how dying cells impact the living organism. Immunity. 2011;35:441–4. doi: 10.1016/j.immuni.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–30. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- 29.Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nature medicine. 2011;17:860–6. doi: 10.1038/nm.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozer H, Armitage JO, Bennett CL, Crawford J, Demetri GD, Pizzo PA, et al. 2000 update of recommendations for the use of hematopoietic colony-stimulating factors: evidence-based, clinical practice guidelines. American Society of Clinical Oncology Growth Factors Expert Panel. Journal of clinical oncology. 2000;18:3558–85. doi: 10.1200/JCO.2000.18.20.3558. [DOI] [PubMed] [Google Scholar]
- 31.Heuser M, Ganser A, Bokemeyer C, American Society of Clinical O, National Comprehensive Cancer N, European Organization for R et al. Use of colony-stimulating factors for chemotherapy-associated neutropenia: review of current guidelines. Seminars in hematology. 2007;44:148–56. doi: 10.1053/j.seminhematol.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 32.Citron ML, Berry DA, Cirrincione C, Hudis C, Winer EP, Gradishar WJ, et al. Randomized trial of dose-dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. Journal of clinical oncology. 2003;21:1431–9. doi: 10.1200/JCO.2003.09.081. [DOI] [PubMed] [Google Scholar]
- 33.Venturini M, Del Mastro L, Aitini E, Baldini E, Caroti C, Contu A, et al. Dose-dense adjuvant chemotherapy in early breast cancer patients: results from a randomized trial. Journal of the National Cancer Institute. 2005;97:1724–33. doi: 10.1093/jnci/dji398. [DOI] [PubMed] [Google Scholar]
- 34.Jones RL, Walsh G, Ashley S, Chua S, Agarwal R, O’Brien M, et al. A randomised pilot Phase II study of doxorubicin and cyclophosphamide (AC) or epirubicin and cyclophosphamide (EC) given 2 weekly with pegfilgrastim (accelerated) vs 3 weekly (standard) for women with early breast cancer. British journal of cancer. 2009;100:305–10. doi: 10.1038/sj.bjc.6604862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis AJ, Chapman W, Hedley DW, Oza AM, Tannock IF. Assessment of tumor cell repopulation after chemotherapy for advanced ovarian cancer: pilot study. Cytometry A. 2003;51:1–6. doi: 10.1002/cyto.a.10001. [DOI] [PubMed] [Google Scholar]
- 36.Freeburg EM, Goyeneche AA, Telleria CM. Mifepristone abrogates repopulation of ovarian cancer cells in between courses of cisplatin treatment. International journal of oncology. 2009;34:743–55. doi: 10.3892/ijo_00000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gamarra-Luques CD, Goyeneche AA, Hapon MB, Telleria CM. Mifepristone prevents repopulation of ovarian cancer cells escaping cisplatin-paclitaxel therapy. BMC cancer. 2012;12:200. doi: 10.1186/1471-2407-12-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gilbert CA, Daou MC, Moser RP, Ross AH. Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer research. 2010;70:6870–9. doi: 10.1158/0008-5472.CAN-10-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu L, Tannock IF. Effect of the selective estrogen receptor modulator arzoxifene on repopulation of hormone-responsive breast cancer xenografts between courses of chemotherapy. Clinical cancer research. 2005;11:8195–200. doi: 10.1158/1078-0432.CCR-05-1258. [DOI] [PubMed] [Google Scholar]
- 40.Saggar JK, Tannock IF. Chemotherapy rescues hypoxic tumor cells and induces their reoxygenation and repopulation-an effect that is inhibited by the hypoxia-activated prodrug TH-302. Clinical cancer research. 2015;21:2107–14. doi: 10.1158/1078-0432.CCR-14-2298. [DOI] [PubMed] [Google Scholar]
- 41.Wu L, Yun Z, Tagawa T, Rey-McIntyre K, de Perrot M. CTLA-4 blockade expands infiltrating T cells and inhibits cancer cell repopulation during the intervals of chemotherapy in murine mesothelioma. Molecular cancer therapeutics. 2012;11:1809–19. doi: 10.1158/1535-7163.MCT-11-1014. [DOI] [PubMed] [Google Scholar]
- 42.Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nature biotechnology. 2010;28:275–80. doi: 10.1038/nbt.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu D, Chen S, Tan X, Li N, Liu C, Li Z, et al. Fra-1 promotes breast cancer chemosensitivity by driving cancer stem cells from dormancy. Cancer research. 2012;72:3451–6. doi: 10.1158/0008-5472.CAN-11-2536. [DOI] [PubMed] [Google Scholar]
- 44.Wang XK, He JH, Xu JH, Ye S, Wang F, Zhang H, et al. Afatinib enhances the efficacy of conventional chemotherapeutic agents by eradicating cancer stem-like cells. Cancer research. 2014;74:4431–45. doi: 10.1158/0008-5472.CAN-13-3553. [DOI] [PubMed] [Google Scholar]
- 45.Park EY, Chang E, Lee EJ, Lee HW, Kang HG, Chun KH, et al. Targeting of miR34a-NOTCH1 axis reduced breast cancer stemness and chemoresistance. Cancer research. 2014;74:7573–82. doi: 10.1158/0008-5472.CAN-14-1140. [DOI] [PubMed] [Google Scholar]
- 46.Chang CW, Chen YS, Chou SH, Han CL, Chen YJ, Yang CC, et al. Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer research. 2014;74:6291–305. doi: 10.1158/0008-5472.CAN-14-0626. [DOI] [PubMed] [Google Scholar]