

Abstract
In this Review, we provide an update on genome-wide association studies (GWAS) in inflammatory bowel disease (IBD). In addition, we summarize progress in defining the functional consequences of associated alleles for coding and non-coding genetic variation. In the small minority of loci where major association signals correspond to non-synonymous variation, we summarize studies defining their functional effects and implications for therapeutic targeting. Importantly, the large majority of GWAS-associated loci involve non-coding variation, many of which modulate levels of gene expression. Recent expression quantitative trait loci (eQTL) studies have established that expression of the large majority of human genes is regulated by non-coding genetic variation. Significant advances in defining the epigenetic landscape have demonstrated that IBD GWAS signals are highly enriched within cell-specific active enhancer marks. Studies in European ancestry populations have dominated the landscape of IBD genetics studies, but increasingly, studies in Asian and African-American populations are being reported. Common variation accounts for only a modest fraction of the predicted heritability and the role of rare genetic variation of higher effects (i.e. odds ratios markedly deviating from one) is increasingly being identified through sequencing efforts. These sequencing studies have been particularly productive in very-early onset, more severe cases. A major challenge in IBD genetics will be harnessing the vast array of genetic discovery for clinical utility, through emerging precision medicine initiatives. We discuss the rapidly evolving area of direct to consumer genetic testing, as well as the current utility of clinical exome sequencing, especially in very early onset, severe IBD cases. We summarize recent progress in the pharmacogenetics of IBD with respect of partitioning patient responses to anti-TNF and thiopurine therapies. Highly collaborative studies across research centers and across subspecialties and disciplines will be required to fully realize the promise of genetic discovery in IBD.
Keywords: Crohn's disease, Ulcerative Colitis, Epigenetics, Autophagy
Genome-wide association studies (gwas) in IBD
The first GWAS using genome-wide SNP chips were published in 20051, 2. The conceptual basis of GWAS is that most complex (i.e., not single-gene Mendelian) genetic disorders are polygenic, being driven by multiple, common genetic polymorphisms. A key methodologic advance enabling GWAS was the development of genome-wide SNP chips that typed a relatively limited number of selected polymorphisms (less than 1 million SNPs for European ancestry individuals) that comprehensively assayed common genetic variation genome-wide. These SNP chips could efficiently test common human genetic variation in a highly accurate and reproducible manner. Furthermore, a variety of analytic approaches were developed (e.g. principal components analysis) which allowed for precise matching of cases and controls; this assured that allele frequency differences would be driven by the disease trait, and not subtle population differences between cases and controls.
The first GWAS in IBD identified many new loci, which were consistently replicated between different studies. Because of the large number of genetic markers and thus statistical tests performed, strict statistical thresholds (p-values less than 5 × 10-8) are required for proof of genome-wide significance. The early GWAS identified the most significant loci; as the sample sizes increased through meta-analyses and inclusion of additional, independent cohorts3, the number of significant loci progressively increased. A major concept recognized early on was the extent to which general genomic regions of association overlapped between distinct chronic immune mediated diseases. This observation formed the basis for the development of the Immunochip4, a cost-effective custom chip with a dense map of markers clustered in 186 regions (i.e., fine-mapping regions) demonstrating genome-wide significant evidence for association in at least one autoimmune or chronic inflammatory disease. In addition to the fine-mapping loci, additional custom content, including genomic regions just missing genome-wide significance by GWAS was added. A meta-analysis combining GWAS with Immunochip data identified 163 loci associated with IBD. Of these loci, 110 confer risk to both IBD subtypes, whereas 30 and 23 loci were unique to Crohn's disease and ulcerative colitis, respectively. These 163 loci explain 13.6% of CD and 7.5% of UC total disease variance, respectively5. More recently a trans-ethnic analysis identified an additional 38 new IBD loci; a striking overlap of directionality of odds ratios is observed between European and Asian ancestry cohorts6. A summary of these loci are provided in Table 1.
Table 1. Summary of significant IBD, CD and UC loci identified by GWAS.
| Chr | Position (Mb) | SNP | Source5, 6 | Type | Candidate Genes |
|---|---|---|---|---|---|
| 1 | 1.24 | rs12103 | Jostins et al., | IBD | TNFRSF18,TNFRSF4,(30) |
| 1 | 2.5 | rs10797432 | Jostins et al., | UC | TNFRSF14,MMEL1,PLCH2,(8) |
| 1 | 8.02 | rs35675666 | Jostins et al., | IBD | TNFRSF9,(6) |
| 1 | 20.15 | rs6426833 | Jostins et al., | UC | (9) |
| 1 | 22.7 | rs12568930 | Jostins et al., | IBD | (3) |
| 1 | 63.05 | rs1748195 | Liu et al., | CD | USP1 |
| 1 | 67.68 | rs11209026 | Jostins et al., | IBD | IL23R,IL12RB2,(4) |
| 1 | 70.99 | rs2651244 | Jostins et al., | IBD | (3) |
| 1 | 78.62 | rs17391694 | Jostins et al., | CD | (5) |
| 1 | 92.55 | rs34856868 | Liu et al., | IBD | BTBD8 |
| 1 | 101.47 | rs11583043 | Liu et al., | UC | SLC30A, EDG1 |
| 1 | 114.3 | rs6679677 | Jostins et al., | CD | PTPN22,DCLRE1B,(7) |
| 1 | 120.45 | rs3897478 | Jostins et al., | CD | ADAM30,(5) |
| 1 | 151.79 | rs4845604 | Jostins et al., | IBD | RORC,(14) |
| 1 | 155.67 | rs670523 | Jostins et al., | IBD | UBQLN4,RIT1,MSTO1,(28) |
| 1 | 160.85 | rs4656958 | Jostins et al., | IBD | CD48,SLAMF1,ITLN1,CD244,F11R,USF1,SLAMF 7,ARHGAP30,(8) |
| 1 | 161.47 | rs1801274 | Jostins et al., | IBD | FCGR2A,FCGR2B,FCGR3A,HSPA6,FCGR3B,FCRL A,(9) |
| 1 | 169.52 | rs6025 | Liu et al., | IBD | SELP, SELE, SELL |
| 1 | 172.85 | rs9286879 | Jostins et al., | CD | FASLG,TNFSF18,(0) |
| 1 | 186.88 | rs10798069 | Liu et al., | CD | PTGS2, PLA2G4A |
| 1 | 197.6 | rs2488389 | Jostins et al., | IBD | C1orf53,(2) |
| 1 | 198.6 | rs7555082 | Liu et al., | CD | PTPRC |
| 1 | 200.09 | rs2816958 | Jostins et al., | UC | (3) |
| 1 | 200.87 | rs7554511 | Jostins et al., | IBD | KIF21B,(6) |
| 1 | 206.93 | rs3024505 | Jostins et al., | IBD | IL10,IL20,IL19,IL24,PIGR,MAPKAPK2,FAIM3,RA SSF5,(3) |
| 2 | 25.12 | rs6545800 | Jostins et al., | IBD | ADCY3,(6) |
| 2 | 27.63 | rs1728918 | Jostins et al., | CD | UCN,(23) |
| 2 | 28.61 | rs925255 | Jostins et al., | IBD | FOSL2,BRE,(1) |
| 2 | 43.81 | rs10495903 | Jostins et al., | IBD | (5) |
| 2 | 61.2 | rs7608910 | Jostins et al., | IBD | REL,C2orf74,KIAA1841,AHSA2,(6) |
| 2 | 62.55 | rs10865331 | Jostins et al., | CD | (3) |
| 2 | 65.67 | rs6740462 | Jostins et al., | IBD | SPRED2,(1) |
| 2 | 102.86 | rs917997 | Jostins et al., | IBD | IL1R2,IL18RAP,IL18R1,IL1R1,IL1RL1,IL1RL2,(3) |
| 2 | 145.49 | rs11681525 | Liu et al., | CD | - |
| 2 | 160.79 | rs4664304 | Liu et al., | IBD | MARCH7, LY75, PLA2R1 |
| 2 | 163.1 | rs2111485 | Jostins et al., | IBD | IFIH1,(5) |
| 2 | 191.92 | rs1517352 | Jostins et al., | IBD | STAT1,STAT4,(2) |
| 2 | 198.65 | rs1016883 | Jostins et al., | UC | RFTN2,PLCL1,(7) |
| 2 | 199.7 | rs17229285 | Jostins et al., | UC | 0 |
| 2 | 204.59 | rs3116494 | Liu et al., | UC | ICOS, CD28, CTLA4 |
| 2 | 219.14 | rs2382817 | Jostins et al., | IBD | SLC11A1,CXCR2,CXCR1,PNKD,ARPC2,TMBIM1, CTDSP1,(8) |
| 2 | 228.66 | rs11178120 3 | Liu et al., | IBD | CCL20 |
| 2 | 231.09 | rs6716753 | Jostins et al., | CD | SP140,(5) |
| 2 | 234.145 | rs12994997 | Jostins et al., | CD | ATG16L1, INPP5D, (7) |
| 2 | 241.57 | rs3749171 | Jostins et al., | IBD | GPR35,(12) |
| 2 | 242.74 | rs35320439 | Liu et al., | CD | PDCD1, ATG4B |
| 3 | 18.76 | rs4256159 | Jostins et al., | IBD | 0 |
| 3 | 46.46 | rs11301008 1 | Liu et al., | UC | FLJ78302, LTF, CCR1, CCR2, CCR3, CCR5 |
| 3 | 48.96 | rs3197999 | Jostins et al., | IBD | MST1,PFKFB4,MST1R,UCN2,GPX1,IP6K2,BSN,IP 6K1,USP4,(56) |
| 3 | 53.05 | rs9847710 | Jostins et al., | UC | PRKCD,ITIH4,(8) |
| 3 | 101.57 | rs616597 | Liu et al., | UC | NFKBIZ |
| 3 | 141.11 | rs724016 | Liu et al., | CD | - |
| 4 | 3.44 | rs2073505 | Liu et al., | IBD | HGFAC |
| 4 | 26.13 | rs4692386 | Liu et al., | IBD | - |
| 4 | 38.33 | rs6856616 | Liu et al., | IBD | - |
| 4 | 48.36 | rs6837335 | Jostins et al., | CD | TXK,TEC,SLC10A4,(3) |
| 4 | 74.85 | rs2472649 | Jostins et al., | IBD | CXCL5,CXCL1,CXCL3,IL8,CXCL6,PF4,CXCL2,PF4V 1,(3) |
| 4 | 102.86 | rs13126505 | Jostins et al., | CD | (1) |
| 4 | 103.51 | rs3774959 | Jostins et al., | UC | NFKB1,MANBA,(2) |
| 4 | 106.08 | rs2189234 | Liu et al., | UC | - |
| 4 | 123.22 | rs7657746 | Jostins et al., | IBD | IL2,IL21,(2) |
| 5 | 0.59 | rs11739663 | Jostins et al., | UC | SLC9A3,(8) |
| 5 | 10.69 | rs2930047 | Jostins et al., | IBD | DAP,(2) |
| 5 | 38.87 | rs395157 | Liu et al., | IBD | OSMR, FYB, LIFR |
| 5 | 40.38 | rs11742570 | Jostins et al., | IBD | PTGER4,(1) |
| 5 | 55.43 | rs10065637 | Jostins et al., | CD | IL6ST,IL31RA,(1) |
| 5 | 71.69 | rs4703855 | Liu et al., | IBD | - |
| 5 | 72.54 | rs7702331 | Jostins et al., | CD | (4) |
| 5 | 96.24 | rs1363907 | Jostins et al., | IBD | ERAP2,ERAP1,LNPEP,(2) |
| 5 | 130.005 | rs4836519 | Jostins et al., | IBD | (1) |
| 5 | 131.19 | rs2188962 | Jostins et al., | IBD | IRF1,IL13,CSF2,SLC22A4,IL4,IL3,IL5,PDLIM4,SLC 22A5,ACSL6,(8) |
| 5 | 134.44 | rs254560 | Jostins et al., | UC | (6) |
| 5 | 141.51 | rs6863411 | Jostins et al., | IBD | SPRY4,NDFIP1,(5) |
| 5 | 150.27 | rs11741861 | Jostins et al., | IBD | TNIP1,IRGM,ZNF300P1,(8) |
| 5 | 158.8 | rs6871626 | Jostins et al., | IBD | IL12B,(3) |
| 5 | 172.32 | rs564349 | Liu et al., | IBD | C5orf4, DUSP1 |
| 5 | 173.34 | rs17695092 | Jostins et al., | CD | CPEB4,(2) |
| 5 | 176.79 | rs12654812 | Jostins et al., | IBD | DOK3,(17) |
| 6 | 0.38 | rs7773324 | Liu et al., | CD | IRF4, DUSP22 |
| 6 | 3.42 | rs13204048 | Liu et al., | CD | - |
| 6 | 14.71 | rs17119 | Jostins et al., | IBD | 0 |
| 6 | 20.77 | rs9358372 | Jostins et al., | IBD | (2) |
| 6 | 21.42 | rs12663356 | Jostins et al., | CD | (3) |
| 6 | 31.27 | rs9264942 | Jostins et al., | CD | HLA-C,PSORS1C1,NFKBIL1,MICB,(18) |
| 6 | 32.595 | rs6927022 | Jostins et al., | UC | HLA-DQB1,HLA-DRB1,HLA-DQA1,HLA-DRA,(12) |
| 6 | 90.96 | rs1847472 | Jostins et al., | IBD | (1) |
| 6 | 106.43 | rs6568421 | Jostins et al., | IBD | (2) |
| 6 | 111.82 | rs3851228 | Jostins et al., | IBD | TRAF3IP2,FYN,REV3L,(2) |
| 6 | 127.45 | rs9491697 | Jostins et al., | CD | (3) |
| 6 | 128.24 | rs13204742 | Jostins et al., | CD | (2) |
| 6 | 138 | rs6920220 | Jostins et al., | IBD | TNFAIP3,(1) |
| 6 | 143.9 | rs12199775 | Jostins et al., | IBD | PHACTR2,(5) |
| 6 | 149.58 | rs7758080 | Liu et al., | CD | MAP3K7IP2 |
| 6 | 159.49 | rs212388 | Jostins et al., | CD | TAGAP,(5) |
| 6 | 167.37 | rs1819333 | Jostins et al., | IBD | CCR6,RPS6KA2,RNASET2,(3) |
| 7 | 2.78 | rs798502 | Jostins et al., | UC | CARD11,GNA12,TTYH3,(4) |
| 7 | 17.44 | rs1077773 | Liu et al., | UC | AHR |
| 7 | 27.22 | rs4722672 | Jostins et al., | UC | (14) |
| 7 | 28.17 | rs864745 | Jostins et al., | CD | CREB5,JAZF1,(1) |
| 7 | 50.245 | rs1456896 | Jostins et al., | IBD | ZPBP,IKZF1,(4) |
| 7 | 98.75 | rs9297145 | Jostins et al., | IBD | SMURF1,(6) |
| 7 | 100.335 | rs1734907 | Jostins et al., | IBD | EPO,(21) |
| 7 | 107.45 | rs4380874 | Jostins et al., | UC | DLD,(9) |
| 7 | 116.89 | rs38904 | Jostins et al., | IBD | (6) |
| 7 | 128.57 | rs4728142 | Jostins et al., | UC | IRF5,TNPO3,TSPAN33,(11) |
| 7 | 148.22 | rs2538470 | Liu et al., | IBD | CNTNAP2 |
| 7 | 26.88 | rs10486483 | Jostins et al., | CD | (2) |
| 8 | 27.23 | rs17057051 | Liu et al., | IBD | PTK2B, TRIM35, EPHX2 |
| 8 | 49.13 | rs7011507 | Liu et al., | UC | - |
| 8 | 90.87 | rs7015630 | Jostins et al., | CD | RIPK2,(4) |
| 8 | 126.53 | rs921720 | Jostins et al., | IBD | TRIB1,(1) |
| 8 | 129.56 | rs6651252 | Jostins et al., | CD | 0 |
| 8 | 130.62 | rs1991866 | Jostins et al., | IBD | (2) |
| 9 | 4.98 | rs10758669 | Jostins et al., | IBD | JAK2,(4) |
| 9 | 93.92 | rs4743820 | Jostins et al., | IBD | NFIL3,(2) |
| 9 | 117.6 | rs4246905 | Jostins et al., | IBD | TNFSF8,TNFSF15,TNC,(2) |
| 9 | 139.32 | rs10781499 | Jostins et al., | IBD | CARD9,PMPCA,SDCCAG3,INPP5E,(19) |
| 10 | 6.08 | rs12722515 | Jostins et al., | IBD | IL2RA,IL15RA,(6) |
| 10 | 30.72 | rs1042058? | Jostins et al., | IBD | MAP3K8,(3) |
| 10 | 35.295 | rs11010067 | Jostins et al., | IBD | CREM,(3) |
| 10 | 59.99 | rs2790216 | Jostins et al., | IBD | CISD1,IPMK,(2) |
| 10 | 64.51 | rs10761659 | Jostins et al., | IBD | (3) |
| 10 | 75.67 | rs2227564 | Jostins et al., | IBD | (13) |
| 10 | 81.03 | rs1250546 | Jostins et al., | IBD | (5) |
| 10 | 82.25 | rs6586030 | Jostins et al., | IBD | TSPAN14,C10orf58,(4) |
| 10 | 94.43 | rs7911264 | Jostins et al., | IBD | (4) |
| 10 | 101.28 | rs4409764 | Jostins et al., | IBD | NKX2-3,(6) |
| 10 | 104.23 | rs3740415 | Liu et al., | IBD | NFKB2, TRIM8, TMEM180 |
| 11 | 1.87 | rs907611 | Jostins et al., | IBD | TNNI2,LSP1,(17) |
| 11 | 58.33 | rs10896794 | Jostins et al., | IBD | CNTF,LPXN,(8) |
| 11 | 60.77 | rs11230563 | Jostins et al., | IBD | CD6,CD5,PTGDR2,(12) |
| 11 | 61.56 | rs4246215 | Jostins et al., | IBD | C11orf9,FADS1,FADS2,(12) |
| 11 | 64.12 | rs559928 | Jostins et al., | IBD | CCDC88B,RPS6KA4,TRPT1,FLRT1,(20) |
| 11 | 65.65 | rs2231884 | Jostins et al., | IBD | RELA,FOSL1,CTSW,SNX32,(22) |
| 11 | 76.29 | rs2155219 | Jostins et al., | IBD | (5) |
| 11 | 96.02 | rs483905 | Jostins et al., | UC | JRKL,MAML2,(2) |
| 11 | 114.38 | rs561722 | Jostins et al., | UC | FAM55A,FAM55D,(5) |
| 11 | 118.74 | rs630923 | Jostins et al., | IBD | CXCR5,(17) |
| 12 | 6.49 | rs7954567 | Liu et al., | CD | CD27, TNFRSF1A, LTBR |
| 12 | 12.65 | rs11612508 | Jostins et al., | IBD | LOH12CR1,(8) |
| 12 | 40.77 | rs11564258 | Jostins et al., | IBD | LRRK2,MUC19 |
| 12 | 48.2 | rs11168249 | Jostins et al., | IBD | VDR,(8) |
| 12 | 68.49 | rs7134599 | Jostins et al., | IBD | IFNG,IL26,IL22,(1) |
| 12 | 112.01 | rs653178 | Liu et al., | IBD | SH2B3, ALDH2, ATXN2 |
| 12 | 120.15 | rs11064881 | Liu et al., | IBD | PRKAB1 |
| 13 | 27.52 | rs17085007 | Jostins et al., | IBD | (2) |
| 13 | 40.86 | rs941823 | Jostins et al., | IBD | (3) |
| 13 | 43.02 | rs9525625 | Liu et al., | CD | AKAP1, TFSF11 |
| 13 | 44.45 | rs3764147 | Jostins et al., | CD | LACC1,(3) |
| 13 | 99.95 | rs9557195 | Jostins et al., | IBD | GPR183,GPR18,(6) |
| 14 | 69.27 | rs194749 | Jostins et al., | IBD | ZFP36L1,(4) |
| 14 | 75.7 | rs4899554 | Jostins et al., | IBD | FOS,MLH3,(6) |
| 14 | 88.47 | rs8005161 | Jostins et al., | IBD | GPR65,GALC,(1) |
| 15 | 38.89 | rs16967103 | Jostins et al., | CD | RASGRP1,SPRED1,(2) |
| 15 | 41.55 | rs28374715 | Jostins et al., | UC | ITPKA,NDUFAF1,NUSAP1,(8) |
| 15 | 67.43 | rs17293632 | Jostins et al., | IBD | SMAD3,(2) |
| 15 | 91.17 | rs7495132 | Jostins et al., | IBD | CRTC3,(3) |
| 16 | 11.54 | rs529866 | Jostins et al., | IBD | SOCS1,LITAF,RMI2,(10) |
| 16 | 23.86 | rs7404095 | Jostins et al., | IBD | PRKCB,(5) |
| 16 | 28.595 | rs26528 | Jostins et al., | IBD | RABEP2,IL27,EIF3C,SULT1A1,SULT1A2,NUPR1,(9) |
| 16 | 30.47 | rs11150589 | Jostins et al., | UC | ITGAL,(20) |
| 16 | 50.66 | rs2066847 | Jostins et al., | CD | NOD2,ADCY7,(5) |
| 16 | 68.58 | rs1728785 | Jostins et al., | UC | ZFP90,(6) |
| 16 | 86 | rs10521318 | Jostins et al., | IBD | IRF8,(4) |
| 17 | 25.84 | rs2945412 | Jostins et al., | CD | LGALS9,NOS2,(3) |
| 17 | 32.59 | rs3091316 | Jostins et al., | IBD | CCL13,CCL2,CCL11,(4) |
| 17 | 37.91 | rs12946510 | Jostins et al., | IBD | IKZF3,ZPBP2,GSDMB,ORMDL3,GSDMA,(12) |
| 17 | 40.53 | rs12942547 | Jostins et al., | IBD | STAT3,STAT5B,STAT5A,(13) |
| 17 | 54.88 | rs3853824 | Liu et al., | CD | - |
| 17 | 57.96 | rs1292053 | Jostins et al., | IBD | TUBD1,RPS6KB1,(9) |
| 17 | 70.64 | rs7210086 | Jostins et al., | UC | (3) |
| 17 | 76.74 | rs17736589 | Liu et al., | UC | - |
| 18 | 12.8 | rs1893217 | Jostins et al., | IBD | (6) |
| 18 | 46.39 | rs7240004 | Jostins et al., | IBD | SMAD7,(2) |
| 18 | 56.88 | rs9319943 | Liu et al., | CD | - |
| 18 | 67.53 | rs727088 | Jostins et al., | IBD | CD226,(2) |
| 18 | 77.22 | rs7236492 | Liu et al., | CD | NFATC1, TST |
| 19 | 1.12 | rs2024092 | Jostins et al., | CD | GPX4,HMHA1,(20) |
| 19 | 10.49 | rs11879191 | Jostins et al., | IBD | TYK2,PPAN-P2RY11,ICAM1,(25) |
| 19 | 33.73 | rs17694108 | Jostins et al., | IBD | CEBPG,(8) |
| 19 | 46.85 | rs4802307 | Jostins et al., | CD | (9) |
| 19 | 47.12 | rs1126510 | Jostins et al., | UC | CALM3,(14) |
| 19 | 49.2 | rs516246 | Jostins et al., | CD | DBP,SPHK2,IZUMO1,FUT2,(22) |
| 19 | 55.38 | rs11672983 | Jostins et al., | IBD | NLRP7,NLRP2,KIR2DL1,LILRB4,(15) |
| 20 | 30.75 | rs6142618 | Jostins et al., | IBD | HCK,(10) |
| 20 | 31.37 | rs4911259 | Jostins et al., | IBD | DNMT3B,(8) |
| 20 | 33.8 | rs6088765 | Jostins et al., | UC | PROCR,UQCC,CEP250,(8) |
| 20 | 43.06 | rs6017342 | Jostins et al., | UC | ADA,HNF4A,(9) |
| 20 | 44.74 | rs1569723 | Jostins et al., | IBD | CD40,MMP9,PLTP,(11) |
| 20 | 48.95 | rs913678 | Jostins et al., | IBD | CEBPB,(5) |
| 20 | 57.82 | rs259964 | Jostins et al., | IBD | ZNF831,CTSZ,(5) |
| 20 | 62.34 | rs6062504 | Jostins et al., | IBD | TNFRSF6B,LIME1,SLC2A4RG,ZGPAT,(23) |
| 21 | 16.81 | rs2823286 | Jostins et al., | IBD | 0 |
| 21 | 34.77 | rs2284553 | Jostins et al., | CD | IFNGR2,IFNAR1,IFNAR2,IL10RB,GART,TMEM50 B,(6) |
| 21 | 40.46 | rs2836878 | Jostins et al., | IBD | (3) |
| 21 | 45.62 | rs7282490 | Jostins et al., | IBD | ICOSLG,(9) |
| 22 | 21.92 | rs2266959 | Jostins et al., | IBD | MAPK1,YDJC,UBE2L3,RIMBP3,CCDC116,(8) |
| 22 | 30.425 | rs2412970 | Jostins et al., | IBD | LIF,OSM,MTMR3,(8) |
| 22 | 39.69 | rs2413583 | Jostins et al., | IBD | ATF4,TAB1,APOBEC3G,(16) |
| 22 | 41.87 | rs727563 | Liu et al., | CD | TEF, NHP2L1, PMM1, L3MBTL2, CHADL |
Parentheses refer to the number of additional genes in the locus.
Functional consequences of missense alleles associated to IBD
Because of the highly correlated nature of human genetic polymorphisms, association signals often span broad genomic regions including multiple genes and transcribed sequences. Thus far, it is often been difficult to assign the likely “causal gene” driving the association signal. The presence of maximal association signals corresponding to missense alleles within a gene in the region, especially when linked to functional studies demonstrating altered gene function, provides strong support for that gene in contributing to disease pathogenesis. Importantly, these studies provide critical, often unexpected insight into disease mechanisms. For example, the finding that Crohn's disease associated missense alleles in the bacterial peptidoglycan sensing leucine rich repeat domain of NOD27, 8 result in impaired activation of NF-κB8, 9 supported the general concept that deficiencies of innate immune cell function represent a central factor in Crohn's disease, distinguishing it from ulcerative colitis.
ATG16L1 and sequence annotation
The association of the Thr300Ala polymorphism within ATG16L1 to Crohn's disease in 2007 represents one of the key advances of the GWAS era10, 11, propelling an intense interest in the role of autophagy in Crohn's disease. However, the functional impact of the alanine allele associated to Crohn's disease defied elaboration for many years following the initial genetic association. Recently, it was discovered that the alanine polymorphism introduces a consensus caspase 3 or 7 cleavage sequence, resulting in more efficient degradation of the Crohn's disease associated ATG16L1 allele. These results clearly establish that the CD risk allele is correlated with impaired autophagy. Therefore, active efforts to induce autophagy to treat a variety of human disease, including Crohn's disease, are ongoing12. These studies also highlight the necessity of more broadly considering the impact of genetic variation on covalent modifications (e.g., gain or loss of glycosylation13, phosphorylation, sumolyation, as well as alterations which alter proteolytic susceptibility such as with ATG16L1) in defining the functional impact of nonsynonymous genetic variation14, 15.
Uncommon protective, loss-of-function allele in IL23R
While there are multiple, independently acting risk alleles in the IL23R gene region, the most significant association is at Arg381Gln, where the minor glutamine allele (observed in approximately one out of seven European ancestry individuals) confers a two to three-fold protection against developing IBD16. This protective effect is greater in Crohn's disease than in ulcerative colitis, similar to many IBD-general loci5. Importantly, multiple groups have established that the protective glutamine allele is a loss-of-function allele17-19, resulting in multiple functional alterations including decreased numbers of IL-23 dependent CD4+ Th17 and CD8+ Tc17 cells19. Thus far, no data that carriage of the protective glutamine allele incurs an increased risk of complications (e.g. infectious diseases) have thus far been reported. Taken together, this would indicate that decreasing IL-23 signaling, such as through monoclonal antibody blockade of anti-p4020 or anti-p1921 (the two subunits of the IL-23 cytokine) may be beneficial in the treatment of IBD. Because IL23R is specific to IL-23 signaling, and not shared with the related IL-12 pathway, it is theoretically possible that selected targeting of the IL-23 pathway through blocking p19 (IL-23 specific) may either be more effective or be associated with fewer side effects than anti-p40 blockade, which blocks both IL-12 and IL-23 signaling.
Crohn's disease associated FUT2 null alleles associated with non-secretor status
Approximately 20% of European ancestry cohorts are homozygous carriers for W134X in FUT2, and this allele represents a Crohn's disease-specific association22. FUT2 encodes for a fucosyltransferase 2 enzyme which creates a soluble precursor for the ABO blood group antigens23. Homozygous carriers of the FUT2 null allele do not express ABO antigens in saliva. Taken together, these studies highlight the potential importance of the mucus layer in Crohn's disease.
Rare missense alleles in PRDM1 and CARD9
Targeted sequencing within GWAS signals to identify rare missense alleles has proved quite productive in identifying likely causal alleles with functional effects. Rare mutations in PRDM1 (PR domain containing 1, with ZNF domain) have been identified, which increase T cell proliferation and cytokine secretion with activation24. In a separate study, resequencing of CARD9 identified a rare splice variant which is highly protective (odds ratio of 0.3) and results in skipping of exon 11 and premature truncation close to the final splice junction with exon 12. If this rare protective splice variant results in a loss-of-function, this would be consistent with the finding that the common IBD risk allele at CARD9 is associated with higher gene expression25. Taken together, this would indicate that, analogous to IL23R, CARD9 loss-of-function alleles are protective against IBD, and so therefore approaches to block CARD9 to treat IBD may be effective.
Fine-Mapping Non-Coding Genetic Variation: Integration With Expression and Epigenetic Data Implicates Cell Subtypes In Disease Pathogenesis
It has long been understood that disease-causing alleles are highly enriched for nonsynonymous, coding region variation that modulates protein structure and/or function26. However, for 80-90% of GWAS-identified loci, the maximal association signals do not include a nonsynonymous variant, instead being confined to non-coding variation. Presumably, much of this non-coding variation exerts its pathogenic effects through modulation of gene expression. Expression quantitative trait loci (eQTL) involve DNA polymorphisms which are correlated with inter-individual variation in gene expression (typically mRNA) and early studies reported a marked enrichment of eQTLs overlapping GWAS loci. A particular value to such eQTL-GWAS mappings is that directionality of effect (disease-associated risk allele associated with increased or decreased expression) may be inferred. With the reporting of many more eQTL studies, including more well-powered and highly context specific studies (e.g. time-course expression analysis with cellular stimulation) it is now understood that the vast majority of genes, demonstrate eQTLs under certain conditions27. This underscores the ubiquity of eQTLs, their highly context-specific nature and the potential value of studies focused on maximally relevant contexts, such as directly examining relevant disease tissues, as opposed to a reliance on in vitro systems. In this regard, transcriptome analyses of small mRNA amounts isolated from select immune cell subsets from primary intestinal tissues provides insight into differential gene expression for associated loci28.
A major recent advance has been the demonstration of significant enrichment of GWAS signals with various epigenetic marks, including cell specific active enhancer (e.g. H3K27Ac) or promoter (e.g. H3K4me1) marks as defined by ChIP-Seq (chromatin immunoprecipitation sequencing). Importantly, recent studies have demonstrated enrichment of cell-specific H3K27Ac marks in patterns consistent with present understanding of disease mechanisms. For example, enrichment of precisely-mapped GWAS signals in cardiac disease, autoimmune and metabolic diseases has been observed within heart tissue, immune and liver cell enhancer marks, respectively29. Within IBD, the greatest enrichment for cell specific enhancers has been observed within CD4+ T cell subsets, with lesser enrichment observed for CD8+, B cells and monocytes30. Integrating epigenetic, intestinal expression and eQTL data provides improved capacity to predict likely IBD genes compare to gene association results alone31. Of note, while Crohn's disease demonstrated the greatest enrichment for immune cell epigenetic marks, ulcerative colitis GWAS signals were enriched within both immune cell and colonic mucosal marks30. While ChIP-seq studies of colonic mucosa would involve a highly heterogeneous mixture of cells30, a separate study demonstrated enrichment of ulcerative colitis GWAS signals within H3K27Ac marks in intestinal enteroids32, thereby implicating a primary pathogenic role for altered epithelial function in IBD.
Functional analyses of GWAS signals: variation in pattern recognition receptor signaling
The NOD2 associations to Crohn's disease highlight the importance of host genetics and inter-individual variability in responses to innate microbial stimulation, or pattern recognition receptor (PRR) signaling. Beyond the NOD2 associations which are associated with decreased cytokine secretion with PRR-stimulation9, 33, inter-individual variability in host cytokine secretion from PRR-stimulated macrophages has been reported to a variety of IBD-associated alleles. PRR-initiated cytokine secretion was diminished in ICOSLG (ICOS ligand) risk allele carriers, similar to NOD2, and identified a novel role for ICOSLG signaling in innate immune cells34. In contrast, IBD-associated polymorphisms in IRF5 (interferon regulatory factor 5) are a major determinant of inter-individual variability in PRR-stimulated cytokine secretion, with IRF5 risk alleles associated with increased cytokine secretion35.
Other pathogenic mechanisms of non-coding variation
A functional methylome map of ulcerative colitis has been reported whereby intestinal biopsies from 10 monozygotic twin pairs (20 individuals) were analyzed for variable DNA methylation patterns. Altogether, 61 genomic regions with differential methylation patterns were identified that were also associated with nearby differentially expressed transcripts36. These regions were not identified by GWAS and therefore implicate development or environmental factors regulating gene expression in contributing to UC pathogenesis. Finally, miRNAs are small, non-coding RNAs, 18-23 nucleotides in length which can broadly regulate gene expression37. A number of studies have implicated specific miRNAs in regulating a broad array of IBD-associated genes, including NOD2, ATG16L138 and IL-2339.
Population Differences in IBD
Initially thought to be a disease that affects primarily populations of European ancestry, it is increasingly evident that IBD involves almost all racial and ethnic groups40. Our current understanding of IBD genetics is based on studies done primarily in populations of European ancestry, but increasingly, studies in non-European populations are being reported. Some studies have shown that disease markers identified in European ancestry populations may also be observed in admixed populations. For example, the frameshift NOD2 risk variant found in European populations are also associated with CD in African Americans, albeit at much lower allele frequencies, consistent with the recent European admixture present in African Americans41, 42
IBD genetic studies in Asian populations
The first GWAS published in IBD was in a Japanese population, with an index cohort of only 94 cases, identifying TNFSF15 as a susceptibility gene for IBD2. The ‘dominant’ CD TNFSF15 association in East Asians has been confirmed43, 44, and associations, although more modest, confirmed in European ancestry populations suggesting that the genetic architecture of IBD would be similar in Asians and Europeans45, 46. However, a number of studies have confirmed no role for NOD2 or ATG16L1 variants in East Asian CD suggesting that some susceptibility genes are shared across populations while others appear to have an effect in one population only47-49. Subsequent studies have confirmed this phenomenon with a Japanese GWAS identifying 4 novel non- HLA associations with UC including variation at the FCGR2A50, a finding subsequently confirmed in a European ancestry UC meta-analysis51. More recent Korean and Japanese CD GWAS identified a number of susceptibility regions not implicated in European populations including a CD association with ATG16L2 implicating a role for autophagy in CD in this population despite the lack of association from either NOD2 or ATG16L152, 53. Utilizing the Immunochip a study from Korea recently identified 6 additional loci CD loci shared with European ancestry individuals including NkX2-3, PTPN2, and ZNF365 bringing the total number of CD loci identified in Koreans to 1554.
IBD genetic studies in Ashkenazi Jewish populations
A major goal of genetic studies in IBD is to account for the significantly higher disease prevalence observed in Ashkenazi Jewish compared to non-Jewish European ancestry cohorts. For common variants, the directionality of effects is almost exclusively in the same direction between these two cohorts, indicating a largely similar genetic architecture55. Where Jewish populations may provide particularly important pathogenic insight may be in the interrogation of rare risk alleles that may be relatively population-specific.
Common and Rare IBD Genetic Variation: Sequencing Studies in IBD
Variants that affect protein function are more likely to have greater penetrance and may be more amenable to functional studies. Because natural selection acts against variants, such as Mendelian, or monogenic mutations, that confer large effects on disease susceptibility, such mutations are usually of low to rare frequency (Figure 1). However, rare mutations of higher effects (i.e., high odds ratios) are not only predicted to outnumber common variants in the human genome56, but could also explain a substantial fraction of common complex diseases57. Sequencing is the method of choice to comprehensively ascertain low frequency to rare variants that have effect sizes higher than those detected by GWAS.
Figure 1. Common and rare genetic variation.
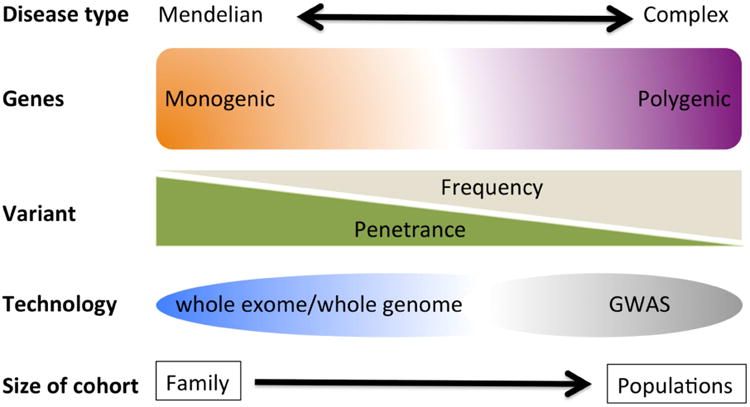
It is estimated that about 85% of mutations with large effect size reside in the 1 to 2% portion of the human that represents the entire protein coding sequence known as the exome58. Currently, sequencing of the whole exome has not only become a practical method but also a cost effective option to identify functionally relevant variants in the protein coding regions of the genome. Whole exome sequencing (WES) has emerged as a hypothesis free and unbiased way of surveying the entire genome for variants (known or novel) that may be at lower frequency and not well covered for detection by GWAS in IBD case-control studies. The first application of WES in a very early onset IBD case revealed a rare mutation affecting the regulatory function of the XIAP (X-linked inhibitor of apoptosis) gene in a boy who presented at 15 months with intractable IBD59. Importantly, XIAP is a positive regulator of NOD2 function60, and so therefore, loss-of-function mutations in XIAP would result in similar functional effects as those observed with NOD2 mutations. Subsequent successful application of WES has identified more rare functional variants in novel genes implicated in the pathogenesis of VEO (FOXP361, IL10RB62;63 and adult onset (GSDMB64 and NDP52 genes65) IBD.
Genetics of Early onset Cases of IBD
While childhood onset IBD represents only 10-25% of all IBD cases, genetic research of pediatric IBD has contributed new knowledge and revealed unsuspected pathways. A substantial proportion of patients with monogenic diseases present with very early onset intestinal inflammation (at less than 10 years of age), that is reminiscent of very early onset IBD. There is also considerable overlap with primary immunodeficiencies and very early onset IBD (Table 2), a topic which has been reviewed recently66.
Table 2. Genes associated with disorders where early onset IBD is well recognized. Broader function of the genes / pathways are indicated in the first column.
| Function | Gene | Associated conditions | |
|---|---|---|---|
| Epthelial barrierand epithelial response defects | COL7A1 | Dystrophic epidermolysis bullosa | |
| FERMT1 | Kindler syndrome | ||
| IKBKG | X linked ectodermal dysplasia and immunodeficiency | ||
| ADAM17 | ADAM-17 deficiency | ||
| GUCY2C | Familial diarrhoea | ||
| Neutropenia and defects in phagocyte bacterial killing | CYBB, CYBA, NCF1, NCF2, NCF4 | Chronic granulomatous disease | |
| SLC37A4 | Glycogen storage disease type 1b | ||
| G6PC3 | Congenital neutropenia | ||
| ITGB2 | Leucocyte adhesion deficiency 1 | ||
| Hyper – and autoinflammation | MVK | Mevalonate kinase deficiency | |
| PLCG2 | Phospholipase Cγ2 defects | ||
| MEFV | Familial Mediterranean fever | ||
| STXBP2 | Familial haemophagocytic lymphohistiocytosis type 5 | ||
| XIAP | X linked lymphoproliferative syndrome 2 | ||
| SH2D1A | X linked lymphoproliferative syndrome 1 | ||
| HPS1, HPS4, HPS 6 | Hermansky–Pudlak syndrome | ||
| ICOS | Common variable immunodeficiency type 1 | ||
| LRBA | Common variable immunodeficiency type 8 | ||
| BTK, PIK3R1 | Agammaglobulinaemia | ||
| CD40LG, AICDA | Hyper-IgM syndrome | ||
| WAS | Wiskott–Aldrich syndrome | ||
| DCLRE 1C | Omenn syndrome | ||
| DOCK8 | Hyper IgE syndrome | ||
| SKIV2L, TTC37 | Trichohepatoenteric syndrome | ||
| PTEN | PTEN hamartoma tumour syndrome | ||
| Regulatory T cells and immune regulation | FOXP3, IL2RA | X linked immune dysregulation, polyendocrinopathy, enteropathy | |
| IL10RA, IL10RB, IL10 | IL-10 signalling defects | ||
| Defects in intestinal innervation | RET | Hirschsprung's disease |
Modified from Uhlig66.
The definition of childhood or early onset IBD can be arbitrary because the age at which childhood ends and adulthood begins represents a continuum. The exact determinants of the age of onset and disease course remain largely unexplained. Except for NOD2, common alleles of large effect size have not been reported for IBD, and additional larger GWAS are highly unlikely to reveal more common variants with larger effect. Interestingly, increasing genetic burden was associated with earlier age of onset for CD, but not for UC67. Importantly, most GWAS studies involved mainly adult or adolescent onset IBD cases, and very early onset (VEO) cases were relatively uncommon. The VEO group experiences a more severe disease course and more frequently shows a positive family history for IBD, in support of higher genetic load 68;69;70. This also suggests that VEO cases are likely to display Mendelian-like forms of IBD characterized by highly penetrant variants of higher effect size as shown by mutations identified in the IL10R gene62. It is necessary to study VEO populations to ascertain low frequency variations (major allele frequency of 0.5 to 1%) to expand our understanding of the contribution of genetics to IBD. Children constitute a population with distinct physiology and disease risks, such that studying VEO-IBD offer the opportunity to dissect the initial immune response and the early gut microbiome as well as changes over the time, the natural history of the disease, and the impact of early environmental modifiers. In short, the pediatric population represents a relatively unexplored and fertile source of novel and helpful information that promise to elucidate the triggers and pathogenic mechanisms of IBD.
Association between Monogenetic Disorders and Very Early Onset IBD
Inflammatory bowel disease-undetermined (IBD-U) is a class of IBD seen at higher rate in young children (34% in children under 2 years and 21% in children under 7 years) as opposed to adults (6%)71. The distinct and more severe disease phenotypes seen in VEO cases and often classified as IBD-U71 include manifestations of known monogenic diseases. Recently, a Mendelian form of VEO IBD was confirmed through a mutation in the IL10R gene62 that underlined an association of IBD with primary immunodeficiency. Many monogenic disease genes have been shown to confer overlapping pathology with IBD (known as IBD-like pathogenesis) and are seen more frequently in VEO cases 66. These diseases represent potential targets for identifying additional VEO heritability using exome sequencing. A list of genes underlying known monogenic conditions associated with IBD is detailed in Table 1. This confirms previous reports that single gene disorders also predispose to complex disorders72;73 and suggest that many Mendelian and complex disorders could share a similar genetic architecture, where Mendelian loci may contain common variants with low effect size (detectable by GWAS) characterized by incomplete penetrance. Such common variants are capable of modifier functions and likely contribute to complex diseases alongside Mendelian, high effect size variants, detectable mainly by sequencing. Importantly, the utility of targeted gene panel sequencing in therapy refractory VEOIBD in influencing clinical decision-making has been recently advocated74.
Clinical Applications and Genetic Testing
Of particular relevance to IBD genetics has been the launching of major initiatives in precision medicine involving prevention and treatment strategies that take individual variability into account 75. Inflammatory bowel diseases, because of their heterogeneity with respect to natural history and response to therapy, are, arguably, the prototype immune-mediated disease for incorporating genetic parameters into everyday clinical practice. This, coupled with, the very significant advances in understanding the genetic architecture of IBD, present wonderful opportunities for improved care. However, significant challenges still remain. The history of discovery in genetics has taught researchers the value of a collegial approach to research allowing the generation of large cohorts. These lessons should be heeded as we look to additional discovery with respect to genetic influence on clinical phenotypes and pharmacogenetics.
Genetic testing for diagnosis in common forms of IBD
In retrospect, a naïve, hope for advances in IBD genetics was the potential role of variants to contribute to diagnosing IBD. Early studies suggested however that, as a consequence of the low pre-test probability (i.e. background prevalence of IBD) together with moderate genotype-relative risks of IBD-associated loci, the utility of genetic testing for a diagnosis of IBD is poor 76. This remained the case for a composite genetic and smoking status score 76. Another study suggested that a combination of genetic, serological and inflammatory markers was superior to a serological panel alone for discriminating non-IBD (a combination of healthy, IBS, and inflammatory ‘controls’) from IBD 77. The AUC for IBD increased to 0.87 from 0.80 for the combination score versus the serological only panel although its unclear how much the addition of genetic data from 4 IBD-associated loci (ATG16L1, NKX2-3, ECM1, STAT3) contributed (ECM1 and STAT3 where not independently associated with IBD in this cohort). The study omitted any mention of study cohort ethnicity and any correction for ethnicity or population structure in the analyses. Despite the limitations suggested by such studies, consumers now have the ability, through direct to consumer (DTC) genetic services, to obtain their own data from genetic markers associated with more than ‘254 specific diseases and conditions’ 78. The difficulty of using individual SNP data to ‘predict’ risk of disease was emphasized by one study investigating disease risk assessment by two different DTC genetic testing products 79 in which the direction of risk for Crohn's disease was discordant for 3 of 5 individuals. In fact, for seven of the diseases tested, ‘50% or less of the predictions agreed across the 5 individuals.’ The authors of the study recommended communicating better about risks distributed with the results, observed that markers should be assessed in all ethnicities, and noted that formal studies should be performed to see if receiving genetic information alters behavior. In fact, in a well-chronicled dispute, the Food and Drug Administration (FDA), suggesting that 23andMe™ had failed to show that it had ‘analytically or clinically validated their “Personal Genome Services” (PGS) for its intended uses,’ ordered the company to cease providing information on its disease and condition-associated markers to new consumers. This is likely the first skirmish in an ongoing debate about consent, access, education and other issues related to DTC genetic tests. Nevertheless, the consensus is that consumers will have increased access to their genotype/sequence data and that healthcare providers will need to develop skills and strategies for explaining the limitations and possible benefits of these data when confronted in the clinic.
Genetic testing in very early onset IBD
The genetic testing of common variants as a screening or diagnostic too has little clinical value in the general population. For example, common variants in NOD2 have the highest effect size in IBD, but are found in 10-20% of the general population and in less than 5% of positive cases who have or will develop CD. On the other hand, with monogenetic disorders, the finding a gene variants is almost always associated with a disease. To date, as many as 50 or more single genes causing IBD (mono-genic forms of IBD) are implicated in very early onset IBD, and number of genes is expected to increase. Traditional and individual single gene testing can be cumbersome and expensive for clinical use. Clinicians are often confused about what gene to test first, when they encounter a clinical problem such as very early onset IBD that is polygenic. Clinicians have been waiting for a reliable, fast, accurate and affordable gene testing solution for their young patients with very early onset IBD or IBD-like intestinal inflammation. Although gene panel testing is widely available for many diseases/disorders, commercially available IBD gene panel is offered by few organizations. Any commercial testing that is covered by insurance for patients living in the USA and certify by CLIA (Clinical Laboratory Improvement Amendments) and/or CAP (College of American Pathologists) is necessary. Such testing provides targeted full gene sequencing for the 50+ genes implicated with early onset IBD and provide comprehensive interpretation of the results. Some laboratories offer testing that is scalable to full exome genome sequencing in patients with initial initial results, however, full exome sequencing needs to be requested separately after the first round of results. The genetic testing for IBD not only establishes the molecular diagnosis with the basis of pathogenesis, but it also allows rationale for patient-specific early intervention with emerging or experimental therapeutics and cell based approaches and the opportunity to screen family members for carrier detection and genetic counseling. For a substantial proportion of IBD or IBD-like diseases, these monogenic disorders also overlap with immunodeficiency affecting granulocyte and phagocyte activity, hyper- and autoinflammatory disorders, defects with disturbed T and B lymphocyte selection and activation, and defects in immune regulation affecting regulatory T cell activity and interleukin (IL)-10 signaling. An area of IBD genetic research with significant progress prompting possible clinical use has been the genetics of very early onset IBD (VEOIBD). In a seminal study researchers from Europe and North America identified a homozygous interleukin 10 receptor (IL10R) variant encoding a premature stop codon in a young child with severe IBD resistant to conventional therapy 62. Functional studies suggested a hematopoietic source for the functional defect and a bone marrow transplant resolved the IBD. Further studies have confirmed the role of IL10R variants in VEOIBD, and such VEOIBD findings have prompted the use of whole exome sequencing as a clinical standard in VEOIBD 62, 63, 80-82.
Pharmacogenetics
Predicting response to adverse events from IBD therapies has been a longtime goal of research efforts. The ability to identify individuals likely to respond to, or have adverse effects from a therapy may become increasingly important as the number of IBD treatment agents expand. The limited success of studying pharmacogenetics of anti-TNF therapies has been partially due to significant inter-individual variation in the pharmacokinetics of these agents suggesting that many ‘non-responders’ were likely under-dosed, confounding previous studies. Nevertheless, a number of studies have been reported, the largest suggesting that variants in apoptotic genes can be combined to form an ‘apoptotic pharmacogenetic index.’ The index, combined with clinical factors, helped construct an algorithm for predicting response to infliximab in CD83. Other studies have utilized mucosal gene expression for predicting response to anti-TNF; one study used a combination of gene expression profiles from 5 innate immunity genes to generate a ‘score’ with 95% sensitivity and 85% specificity for response to infliximab in UC84. A second study identified that a renal tissue gene signature associated with graft rejection after renal transplant was similar to that seen in the mucosa of UC patients unresponsive to anti-TNF therapy85. These findings clearly require replication, but ready access to disease tissue may be a unique and not fully explored opportunity for IBD.
The ultimate pharmacogenetic approach may be the genetic identification of pathways and processes involved in disease pathogenesis and therefore pathways for therapeutic intervention either through novel development or for re-purposing of existing drugs. Determining whether this approach will be effective in IBD is premature, but a large study in rheumatoid arthritis (RA) suggested, as a proof of principle, that RA genetic associations tagged pathways targeted by existing RA drugs86. Furthermore, this study identified novel pathways for potential therapeutic intervention and drugs, already in use in other areas, that may be beneficial if ‘re-purposed’ for RA. Astra-Zeneca published a retrospective review of their small-molecule drug projects from 2005 to 2010 and identified that 73% of phase II projects with ‘human genetic linkage of the target to the disease indication’ were successful compared to only 43% with no genetic ‘linkage.’ These results suggest that an understanding of the genetic architecture of a trait may help streamline the drug discovery process87.
In addition, testing for thiopurine methyltransferase (TPMT) variation, prior to thiopurine initiation in order to reduce the risk of bone marrow toxicity, has influenced clinical practice and therapeutic choices in IBD. Two recent studies have significantly increased our understanding of genetic factors associated with thiopurine-related adverse events. Recognizing a higher prevalence of thiopurine induced leucopenia in Koreans despite lower TPMT variant frequency, Yang et al. performed an unbiased genome-wide study identifying a non-synonymous (basic arginine to polar cysteine at codon 139) NUDT15 polymorphism that is notably associated with both early and late leucopenia after thiopurine therapy. The effect size of this variant for leucopenia was greater than that for TPMT variants in Koreans and while this variant was rare in caucasians (< 1%), it was still associated with leucopenia in this population88. In addition, a similarly designed study from Heap et al. identified HLA-DQA1-HLA-DRB1 variants associated with developing pancreatitis after thiopurine exposure in Europeans89. These variants being added to TPMT status as constituents of a ‘thiopurine panel,’ enabling clinicians to reduce the risk of serious adverse events related to these useful compounds, is highly likely in the near future.
Prognostics
The heterogeneity seen in the natural history of the IBDs has prompted a number of studies investigating genetic associations with disease severity. These studies have largely been retrospective, underpowered, and lacking replication. Nevertheless, the studies have presented some interesting findings and a methodology that may influence future studies. An unbiased genome-wide approach identified a composite score of 46 SNPs that identified subjects with medically-refractory ulcerative colitis (MRUC)90. These findings clearly require replication but show the potential utility of including a number of SNPs, each of moderate effect size, to develop a composite risk score. Similarly, a study looking at associations with first surgery in Crohn's disease combined genetic variants, serological markers, and clinical characteristics in a multi-modal scoring system91. A variant that remained significant in all models tagged IL12B, a locus, although at a different SNP, that was previously identified with disease severity in CD92. An intriguing recent study suggested that a noncoding FOXO3A SNP was associated with a milder clinical natural history in both Crohn's disease and rheumatoid arthritis but also an increased risk of severe malaria 93. However, this finding was not replicated in a subsequent, independent study94. Functional studies demonstrated that altered inflammatory responses in monocytes with decreased pro-inflammatory and increased anti-inflammatory cytokine production are associated with this variant. The same group also confirmed that a gene expression signature in CD8 positive T cells, previously associated with more severe disease in lupus, identified both UC and CD subjects with more severe disease 95. These studies, also in need of replication, extend the pleiotropic observations of the effect of genetic variation from disease susceptibility to association with disease behavior. Prospective inception cohorts currently being followed96 will be invaluable in confirming these and other findings.
Extra-intestinal manifestations
The study of genetic associations with extra-intestinal manifestations (EIMs) has, on the whole, been hampered by underpowered studies, with the possible exception of the study of primary sclerosing cholangitis (PSC). A number of PSC GWAS and large-scale genotyping studies have been performed with the latest immunochip-based study of nearly 30,000 subjects extending the number of susceptibility loci to 1697-99. A further analysis of these data incorporating associations from immune-mediated diseases associated with PSC suggested additional loci totaling 30 non-HLA associations. Approximately half of these PSC loci are known IBD ‘genes’ and PSC ‘genetically’ is more similar to UC than CD with an AUC of 0.624 and 0.559 for PSC from UC and CD risk alleles respectively97. A functional network analysis of where PSC genes fall in an IBD network disappointingly revealed that the PSC genes were scattered around the network and not localized to one part, which suggests that PSC is not associated with a molecular subset of IBD 97. This may simply reflect limitations of our understanding of the full ‘molecular’ PSC story or limitations in current analytic approaches. Similar to IBD, only a minority of variance is explained by these genetic variants, prompting investigators to examine the role of the environment, including the microbiome in PSC pathogenesis100. An intriguing study identified an association between fucosyltransferase 2 (FUT2) genotype and episodes of cholangitis, fungobilia (biliary Candida infections), and dominant stenosis, suggesting that FUT2 may be a marker of ‘host microbial diversity’ and ‘disease progression’ in PSC. These results support other studies suggesting that FUT2 variants ‘garden’ the microbiome in both healthy and IBD populations101-103.
There has been considerable interest in genetic pleiotropy among the immune-mediated diseases, and IBD ‘shares’ more susceptibility loci with spondyloarthropathy (SpA) than any other trait104. Whether SpA is an EIM of IBD or IBD is an extra-articular manifestation of SpA, or whether they are in fact conditions with similar molecular architecture with different end-organ manifestations remains to be seen105, 106. However, the vast majority of shared susceptibility loci are concordant between IBD and SpA, which is in contrast to other traits, including psoriasis in which approximately one quarter of shared loci show discordance. The relationship between psoriasis and IBD is complex. Recent observations that anti-TNF agents can cause psoriasiform lesions in IBD patients, that this phenomenon may be associated with IL23R variants and respond to anti-IL12/23 antibody therapy107, 108 and that anti-IL17a therapy, so successful in psoriasis, appears to worsen Crohn's disease, only adds to the complexity109. The finding that a subset of CD patients tagged by a TNFSF15 variant improved with anti-IL17a therapy (secukinumab) adds to the complexity and surely makes the case for inclusion of genotyping in clinical trials of novel therapeutic agents109. These observations require further study as they surely reveal insights to underlying mechanisms leading to both IBD and psoriasis.
There have been few ‘within-IBD’ studies searching for genetic variants associated with EIMs, and these studies have largely been underpowered and lacked the depth of genotyping that is now standard in genetic studies. Historical studies identified association with HLA variants and the development of eye and skin manifestations as well as both peripheral and axial arthropathy in IBD110, 111. More recently, an unbiased approach identified interesting genetic associations with pyoderma gangrenosum (including IL8RA, TIMP3). However, no associations achieved genome-wide significance, unsurprising given the small sample size112. This study, in keeping with previous observations, confirmed the co-segregation of the EIMs in IBD, suggesting shared genetic mechanisms. Studies delineating the molecular causes of the EIMs will clearly require collaboration across multiple-sites in order to generate appropriately sized study cohorts. Studying these under-researched traits is a challenge that the international community should embrace, as identification of novel areas for therapeutic targeting for EIMs, which can often be as disabling as the underlying IBD, would potentially have utility beyond IBD.
Future Directions
The advances in our understanding of the genetic variation underlying IBD, particularly in European ancestry individuals, has been spectacular and has provided an outstanding framework for basic and translational research. These advances have also generated significant challenges and posed many additional questions. From an ethical and scientific perspective it is imperative that the resources applied to generating these datasets be applied to non-European ancestry populations. Large-scale genotyping studies are designed to identify loci and particular those tagged by common variants. A natural extension of these studies is large-scale fine-mapping approaches to ‘narrow’ associated regions and to identify ‘causative’ variants thereby greatly facilitating functional studies. Many of these variants are likely to be in non-coding regions and understanding how these alter gene expression is an important challenge the research community will need to address. There have been some interesting clues from recent studies but it is increasingly apparent that these eQTL studies will also need to be performed in single-cell compartments relevant to disease pathogenesis. While this will help overcome some of the heterogeneity of cell types in standard tissue sampling there is an evolving story of heterogeneity even within cell types suggesting that this approach may need to be extended to the single-cell level. Ultimately, the purpose of this research is to improve the lives of people with IBD and therefore increasing migration of these advances to the clinic is imperative. This will likely come, over the next few years, in the arenas of biomarker and drug development for the wider population but more immediately for ‘diagnostics’ in the rare but very serious VEOIBD subjects where these data have the ability to transform clinical approaches and outcomes. There will likely be significant ‘push-back’ to the current attitudes to sharing genetic data with research subjects and also evolution of the regulation of the direct to consumer companies and it is likely that practicing clinicians will be faced with patients armed with their genotype data in the near future.
Footnotes
No conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamazaki K, McGovern D, Ragoussis J, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn's disease. Hum Mol Genet. 2005;14:3499–506. doi: 10.1093/hmg/ddi379. [DOI] [PubMed] [Google Scholar]
- 3.Barrett JC, Clayton DG, Concannon P, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009 doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cortes A, Brown MA. Promise and pitfalls of the Immunochip. Arthritis Res Ther. 2011;13:101. doi: 10.1186/ar3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu JZ, van Sommeren S, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015 doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 8.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Moran T, Swanson E, et al. Regulation of IL-8 and IL-1beta expression in Crohn's disease associated NOD2/CARD15 mutations. Hum Mol Genet. 2004;13:1715–25. doi: 10.1093/hmg/ddh182. [DOI] [PubMed] [Google Scholar]
- 10.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 11.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levine B, Packer M, Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125:14–24. doi: 10.1172/JCI73938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vogt G, Chapgier A, Yang K, et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat Genet. 2005;37:692–700. doi: 10.1038/ng1581. [DOI] [PubMed] [Google Scholar]
- 14.Lassen KG, Kuballa P, Conway KL, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A. 2014;111:7741–6. doi: 10.1073/pnas.1407001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murthy A, Li Y, Peng I, et al. A Crohn's disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014;506:456–62. doi: 10.1038/nature13044. [DOI] [PubMed] [Google Scholar]
- 16.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Meglio P, Di Cesare A, Laggner U, et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS One. 2011;6:e17160. doi: 10.1371/journal.pone.0017160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pidasheva S, Trifari S, Phillips A, et al. Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PLoS One. 2011;6:e25038. doi: 10.1371/journal.pone.0025038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarin R, Wu X, Abraham C. Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc Natl Acad Sci U S A. 2011;108:9560–5. doi: 10.1073/pnas.1017854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandborn WJ, Feagan BG, Fedorak RN, et al. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn's disease. Gastroenterology. 2008;135:1130–41. doi: 10.1053/j.gastro.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 21.OP025. A randomized, double-blind placebo-controlled phase 2a induction study of MEDI2070 (anti-p19 antibody) in patients with active Crohn's disease who have failed anti-TNF antibody therapy. J Crohns Colitis. 2015;9(Suppl 1):S15–6. [Google Scholar]
- 22.McGovern DP, Jones MR, Taylor KD, et al. Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn's disease. Hum Mol Genet. 19:3468–76. doi: 10.1093/hmg/ddq248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelly RJ, Rouquier S, Giorgi D, et al. Sequence and expression of a candidate for the human Secretor blood group alpha(1,2)fucosyltransferase gene (FUT2). Homozygosity for an enzyme-inactivating nonsense mutation commonly correlates with the non-secretor phenotype. J Biol Chem. 1995;270:4640–9. doi: 10.1074/jbc.270.9.4640. [DOI] [PubMed] [Google Scholar]
- 24.Ellinghaus D, Zhang H, Zeissig S, et al. Association between variants of PRDM1 and NDP52 and Crohn's disease, based on exome sequencing and functional studies. Gastroenterology. 2013;145:339–47. doi: 10.1053/j.gastro.2013.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rivas MA, Beaudoin M, Gardet A, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–73. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet. 2003;(33 Suppl):228–37. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- 27.Fairfax BP, Humburg P, Makino S, et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science. 2014;343:1246949. doi: 10.1126/science.1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raine T, Liu JZ, Anderson CA, et al. Generation of primary human intestinal T cell transcriptomes reveals differential expression at genetic risk loci for immune-mediated disease. Gut. 2015;64:250–9. doi: 10.1136/gutjnl-2013-306657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roadmap Epigenomics C, Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farh KK, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2014 doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ning K, Gettler K, Zhang W, et al. Improved integrative framework combining association data with gene expression features to prioritize Crohn's disease genes. Hum Mol Genet. 2015 doi: 10.1093/hmg/ddv142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mokry M, Middendorp S, Wiegerinck CL, et al. Many inflammatory bowel disease risk loci include regions that regulate gene expression in immune cells and the intestinal epithelium. Gastroenterology. 2014;146:1040–7. doi: 10.1053/j.gastro.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 33.van Heel DA, Ghosh S, Butler M, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn's disease. Lancet. 2005;365:1794–6. doi: 10.1016/S0140-6736(05)66582-8. [DOI] [PubMed] [Google Scholar]
- 34.Hedl M, Lahiri A, Ning K, et al. Pattern recognition receptor signaling in human dendritic cells is enhanced by ICOS ligand and modulated by the Crohn's disease ICOSLG risk allele. Immunity. 2014;40:734–46. doi: 10.1016/j.immuni.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hedl M, Abraham C. IRF5 risk polymorphisms contribute to interindividual variance in pattern recognition receptor-mediated cytokine secretion in human monocyte-derived cells. J Immunol. 2012;188:5348–56. doi: 10.4049/jimmunol.1103319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasler R, Feng Z, Backdahl L, et al. A functional methylome map of ulcerative colitis. Genome Res. 2012;22:2130–7. doi: 10.1101/gr.138347.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalla R, Ventham NT, Kennedy NA, et al. MicroRNAs: new players in IBD. Gut. 2015;64:504–17. doi: 10.1136/gutjnl-2014-307891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu C, Chen J, Xu HG, et al. MIR106B and MIR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology. 2014;146:188–99. doi: 10.1053/j.gastro.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brain O, Owens BM, Pichulik T, et al. The intracellular sensor NOD2 induces microRNA-29 expression in human dendritic cells to limit IL-23 release. Immunity. 2013;39:521–36. doi: 10.1016/j.immuni.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 40.Jackson JF, 3rd, Dhere T, Repaka A, et al. Crohn's disease in an African-American population. Am J Med Sci. 2008;336:389–92. doi: 10.1097/MAJ.0b013e31816a5c06. [DOI] [PubMed] [Google Scholar]
- 41.Adeyanju O, Okou DT, Huang C, et al. Common NOD2 risk variants in African Americans with Crohn's disease are due exclusively to recent Caucasian admixture. Inflamm Bowel Dis. 2012;18:2357–9. doi: 10.1002/ibd.22944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang MH, Okazaki T, Kugathasan S, et al. Contribution of higher risk genes and European admixture to Crohn's disease in African Americans. Inflamm Bowel Dis. 2012;18:2277–87. doi: 10.1002/ibd.22931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kakuta Y, Kinouchi Y, Negoro K, et al. Association study of TNFSF15 polymorphisms in Japanese patients with inflammatory bowel disease. Gut. 2006;55:1527–8. doi: 10.1136/gut.2006.100297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang SK, Lim J, Chang HS, et al. Association of TNFSF15 with Crohn's disease in Koreans. Am J Gastroenterol. 2008;103:1437–42. doi: 10.1111/j.1572-0241.2007.01752.x. [DOI] [PubMed] [Google Scholar]
- 45.Picornell Y, Mei L, Taylor K, et al. TNFSF15 is an ethnic-specific IBD gene. Inflamm Bowel Dis. 2007;13:1333–8. doi: 10.1002/ibd.20223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tremelling M, Berzuini C, Massey D, et al. Contribution of TNFSF15 gene variants to Crohn's disease susceptibility confirmed in UK population. Inflamm Bowel Dis. 2008 doi: 10.1002/ibd.20399. [DOI] [PubMed] [Google Scholar]
- 47.Guo QS, Xia B, Jiang Y, et al. NOD2 3020insC frameshift mutation is not associated with inflammatory bowel disease in Chinese patients of Han nationality. World J Gastroenterol. 2004;10:1069–71. doi: 10.3748/wjg.v10.i7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inoue N, Tamura K, Kinouchi Y, et al. Lack of common NOD2 variants in Japanese patients with Crohn's disease. Gastroenterology. 2002;123:86–91. doi: 10.1053/gast.2002.34155. [DOI] [PubMed] [Google Scholar]
- 49.Mahurkar S, Banerjee R, Rani VS, et al. Common variants in NOD2 and IL23R are not associated with inflammatory bowel disease in Indians. J Gastroenterol Hepatol. 2011;26:694–9. doi: 10.1111/j.1440-1746.2010.06533.x. [DOI] [PubMed] [Google Scholar]
- 50.Asano K, Matsushita T, Umeno J, et al. A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet. 2009;41:1325–9. doi: 10.1038/ng.482. [DOI] [PubMed] [Google Scholar]
- 51.McGovern DP, Gardet A, Torkvist L, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 2010;42:332–7. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamazaki K, Umeno J, Takahashi A, et al. A genome-wide association study identifies 2 susceptibility Loci for Crohn's disease in a Japanese population. Gastroenterology. 2013;144:781–8. doi: 10.1053/j.gastro.2012.12.021. [DOI] [PubMed] [Google Scholar]
- 53.Yang SK, Hong M, Zhao W, et al. Genome-wide association study of Crohn's disease in Koreans revealed three new susceptibility loci and common attributes of genetic susceptibility across ethnic populations. Gut. 2014;63:80–7. doi: 10.1136/gutjnl-2013-305193. [DOI] [PubMed] [Google Scholar]
- 54.Yang SK, Hong M, Choi H, et al. Immunochip analysis identification of 6 additional susceptibility loci for Crohn's disease in Koreans. Inflamm Bowel Dis. 2015;21:1–7. doi: 10.1097/MIB.0000000000000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kenny EE, Pe'er I, Karban A, et al. A genome-wide scan of Ashkenazi Jewish Crohn's disease suggests novel susceptibility loci. PLoS Genet. 2012;8:e1002559. doi: 10.1371/journal.pgen.1002559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marth GT, Yu F, Indap AR, et al. The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tennessen JA, Bigham AW, O'Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–9. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106:19096–101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 13:255–62. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 60.Krieg A, Correa RG, Garrison JB, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci U S A. 2009;106:14524–9. doi: 10.1073/pnas.0907131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okou DT, Mondal K, Faubion WA, et al. Exome sequencing identifies a novel FOXP3 mutation in a 2-generation family with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2014;58:561–8. doi: 10.1097/MPG.0000000000000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–45. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moran CJ, Walters TD, Guo CH, et al. IL-10R polymorphisms are associated with very-early-onset ulcerative colitis. Inflamm Bowel Dis. 2013;19:115–23. doi: 10.1002/ibd.22974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Melissas J, Sandler AD, Ehsman SR. Spontaneous arteriovenous fistulas of common iliac vessels. A case report. S Afr J Surg. 1987;25:99–102. [PubMed] [Google Scholar]
- 65.Robertson CS, Womack C, Morris DL. Does secondary amyloidosis occur with human hydatid disease? Ann Trop Med Parasitol. 1986;80:265. doi: 10.1080/00034983.1986.11812013. [DOI] [PubMed] [Google Scholar]
- 66.Uhlig HH. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut. 2013;62:1795–805. doi: 10.1136/gutjnl-2012-303956. [DOI] [PubMed] [Google Scholar]
- 67.Ananthakrishnan AN, Huang H, Nguyen DD, et al. Differential effect of genetic burden on disease phenotypes in Crohn's disease and ulcerative colitis: analysis of a North American cohort. Am J Gastroenterol. 2014;109:395–400. doi: 10.1038/ajg.2013.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cannioto Z, Berti I, Martelossi S, et al. IBD and IBD mimicking enterocolitis in children younger than 2 years of age. Eur J Pediatr. 2009;168:149–55. doi: 10.1007/s00431-008-0721-2. [DOI] [PubMed] [Google Scholar]
- 69.Ruemmele FM, El Khoury MG, Talbotec C, et al. Characteristics of inflammatory bowel disease with onset during the first year of life. J Pediatr Gastroenterol Nutr. 2006;43:603–9. doi: 10.1097/01.mpg.0000237938.12674.e3. [DOI] [PubMed] [Google Scholar]
- 70.Bianco AM, Zanin V, Girardelli M, et al. A common genetic background could explain early-onset Crohn's disease. Med Hypotheses. 2012;78:520–2. doi: 10.1016/j.mehy.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 71.Prenzel F, Uhlig HH. Frequency of indeterminate colitis in children and adults with IBD -a metaanalysis. J Crohns Colitis. 2009;3:277–81. doi: 10.1016/j.crohns.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 72.Ristow M. Neurodegenerative disorders associated with diabetes mellitus. J Mol Med (Berl) 2004;82:510–29. doi: 10.1007/s00109-004-0552-1. [DOI] [PubMed] [Google Scholar]
- 73.Sinibaldi L, De Luca A, Bellacchio E, et al. Mutations of the Nogo-66 receptor (RTN4R) gene in schizophrenia. Hum Mutat. 2004;24:534–5. doi: 10.1002/humu.9292. [DOI] [PubMed] [Google Scholar]
- 74.Kammermeier J, Drury S, James CT, et al. Targeted gene panel sequencing in children with very early onset inflammatory bowel disease--evaluation and prospective analysis. J Med Genet. 2014;51:748–55. doi: 10.1136/jmedgenet-2014-102624. [DOI] [PubMed] [Google Scholar]
- 75.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–5. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McGovern DP, Butler H, Ahmad T, et al. TUCAN (CARD8) genetic variants and inflammatory bowel disease. Gastroenterology. 2006;131:1190–6. doi: 10.1053/j.gastro.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 77.Plevy S, Silverberg MS, Lockton S, et al. Combined serological, genetic, and inflammatory markers differentiate non-IBD, Crohn's disease, and ulcerative colitis patients. Inflamm Bowel Dis. 2013;19:1139–48. doi: 10.1097/MIB.0b013e318280b19e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Annas GJ, Elias S. 23andMe and the FDA. N Engl J Med. 2014;370:985–8. doi: 10.1056/NEJMp1316367. [DOI] [PubMed] [Google Scholar]
- 79.Ng PC, Murray SS, Levy S, et al. An agenda for personalized medicine. Nature. 2009;461:724–6. doi: 10.1038/461724a. [DOI] [PubMed] [Google Scholar]
- 80.Glocker EO, Frede N, Perro M, et al. Infant colitis—it's in the genes. Lancet. 2010;376:1272. doi: 10.1016/S0140-6736(10)61008-2. [DOI] [PubMed] [Google Scholar]
- 81.Begue B, Verdier J, Rieux-Laucat F, et al. Defective IL10 signaling defining a subgroup of patients with inflammatory bowel disease. Am J Gastroenterol. 2011;106:1544–55. doi: 10.1038/ajg.2011.112. [DOI] [PubMed] [Google Scholar]
- 82.Engelhardt KR, Shah N, Faizura-Yeop I, et al. Clinical outcome in IL-10- and IL-10 receptor-deficient patients with or without hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2013;131:825–30. doi: 10.1016/j.jaci.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 83.Hlavaty T, Ferrante M, Henckaerts L, et al. Predictive model for the outcome of infliximab therapy in Crohn's disease based on apoptotic pharmacogenetic index and clinical predictors. Inflamm Bowel Dis. 2007;13:372–9. doi: 10.1002/ibd.20024. [DOI] [PubMed] [Google Scholar]
- 84.Arijs I, Li K, Toedter G, et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut. 2009;58:1612–9. doi: 10.1136/gut.2009.178665. [DOI] [PubMed] [Google Scholar]
- 85.Halloran B, Chang J, Shih DQ, et al. Molecular patterns in human ulcerative colitis and correlation with response to infliximab. Inflamm Bowel Dis. 2014;20:2353–63. doi: 10.1097/MIB.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–81. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cook D, Brown D, Alexander R, et al. Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13:419–31. doi: 10.1038/nrd4309. [DOI] [PubMed] [Google Scholar]
- 88.Yang SK, Hong M, Baek J, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat Genet. 2014;46:1017–20. doi: 10.1038/ng.3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heap GA, Weedon MN, Bewshea CM, et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat Genet. 2014;46:1131–4. doi: 10.1038/ng.3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haritunians T, Taylor KD, Targan SR, et al. Genetic predictors of medically refractory ulcerative colitis. Inflamm Bowel Dis. 2010;16:1830–40. doi: 10.1002/ibd.21293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dubinsky MC, Kugathasan S, Kwon S, et al. Multidimensional prognostic risk assessment identifies association between IL12B variation and surgery in Crohn's disease. Inflamm Bowel Dis. 2013;19:1662–70. doi: 10.1097/MIB.0b013e318281f275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Henckaerts L, Van Steen K, Verstreken I, et al. Genetic risk profiling and prediction of disease course in Crohn's disease patients. Clin Gastroenterol Hepatol. 2009;7:972–980e2. doi: 10.1016/j.cgh.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 93.Lee JC, Espeli M, Anderson CA, et al. Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway. Cell. 2013;155:57–69. doi: 10.1016/j.cell.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schnitzler F, Friedrich M, Wolf C, et al. The NOD2 p.Leu1007fsX1008 mutation (rs2066847) is a stronger predictor of the clinical course of Crohn's disease than the FOXO3A intron variant rs12212067. PLoS One. 2014;9:e108503. doi: 10.1371/journal.pone.0108503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee JC, Lyons PA, McKinney EF, et al. Gene expression profiling of CD8+ T cells predicts prognosis in patients with Crohn disease and ulcerative colitis. J Clin Invest. 2011;121:4170–9. doi: 10.1172/JCI59255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Haberman Y, Tickle TL, Dexheimer PJ, et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest. 2014;124:3617–33. doi: 10.1172/JCI75436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu JZ, Hov JR, Folseraas T, et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet. 2013;45:670–5. doi: 10.1038/ng.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Melum E, Franke A, Schramm C, et al. Genome-wide association analysis in primary sclerosing cholangitis identifies two non-HLA susceptibility loci. Nat Genet. 43:17–9. doi: 10.1038/ng.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Folseraas T, Melum E, Rausch P, et al. Extended analysis of a genome-wide association study in primary sclerosing cholangitis detects multiple novel risk loci. J Hepatol. 2012;57:366–75. doi: 10.1016/j.jhep.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tabibian JH, O'Hara SP, Lindor KD. Primary sclerosing cholangitis and the microbiota: current knowledge and perspectives on etiopathogenesis and emerging therapies. Scand J Gastroenterol. 2014;49:901–8. doi: 10.3109/00365521.2014.913189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rupp C, Friedrich K, Folseraas T, et al. Fut2 genotype is a risk factor for dominant stenosis and biliary candida infections in primary sclerosing cholangitis. Aliment Pharmacol Ther. 2014;39:873–82. doi: 10.1111/apt.12663. [DOI] [PubMed] [Google Scholar]
- 102.Tong M, McHardy I, Ruegger P, et al. Reprograming of gut microbiome energy metabolism by the FUT2 Crohn's disease risk polymorphism. ISME J. 2014;8:2193–206. doi: 10.1038/ismej.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rausch P, Rehman A, Kunzel S, et al. Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (Secretor) genotype. Proc Natl Acad Sci U S A. 2011;108:19030–5. doi: 10.1073/pnas.1106408108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Parkes M, Cortes A, van Heel DA, et al. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet. 2013;14:661–73. doi: 10.1038/nrg3502. [DOI] [PubMed] [Google Scholar]
- 105.Vander Cruyssen B, Ribbens C, Boonen A, et al. The epidemiology of ankylosing spondylitis and the commencement of anti-TNF therapy in daily rheumatology practice. Ann Rheum Dis. 2007;66:1072–7. doi: 10.1136/ard.2006.064543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Matzkies FG, Targan SR, Berel D, et al. Markers of intestinal inflammation in patients with ankylosing spondylitis: a pilot study. Arthritis Res Ther. 2012;14:R261. doi: 10.1186/ar4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tillack C, Ehmann LM, Friedrich M, et al. Anti-TNF antibody-induced psoriasiform skin lesions in patients with inflammatory bowel disease are characterised by interferon-gamma-expressing Th1 cells and IL-17A/IL-22-expressing Th17 cells and respond to anti-IL-12/IL-23 antibody treatment. Gut. 2014;63:567–77. doi: 10.1136/gutjnl-2012-302853. [DOI] [PubMed] [Google Scholar]
- 108.Sherlock ME, Walters T, Tabbers MM, et al. Infliximab-induced psoriasis and psoriasiform skin lesions in pediatric Crohn disease and a potential association with IL-23 receptor polymorphisms. J Pediatr Gastroenterol Nutr. 2013;56:512–8. doi: 10.1097/MPG.0b013e31828390ba. [DOI] [PubMed] [Google Scholar]
- 109.Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–700. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Orchard TR, Holt H, Bradbury L, et al. The prevalence, clinical features and association of HLA-B27 in sacroiliitis associated with established Crohn's disease. Aliment Pharmacol Ther. 2009;29:193–7. doi: 10.1111/j.1365-2036.2008.03868.x. [DOI] [PubMed] [Google Scholar]
- 111.Orchard TR, Chua CN, Ahmad T, et al. Uveitis and erythema nodosum in inflammatory bowel disease: clinical features and the role of HLA genes. Gastroenterology. 2002;123:714–8. doi: 10.1053/gast.2002.35396. [DOI] [PubMed] [Google Scholar]
- 112.Weizman A, Huang B, Berel D, et al. Clinical, serologic, and genetic factors associated with pyoderma gangrenosum and erythema nodosum in inflammatory bowel disease patients. Inflamm Bowel Dis. 2014;20:525–33. doi: 10.1097/01.MIB.0000442011.60285.68. [DOI] [PMC free article] [PubMed] [Google Scholar]