

Abstract
Drosophila melanogaster is an important laboratory model for studies of antiviral immunity in invertebrates, and Drosophila species provide a valuable system to study virus host range and host switching. Here, we use metagenomic RNA sequencing of about 1600 adult flies to discover 25 new RNA viruses associated with six different drosophilid hosts in the wild. We also provide a comprehensive listing of viruses previously reported from the Drosophilidae. The new viruses include Iflaviruses, Rhabdoviruses, Nodaviruses, and Reoviruses, and members of unclassified lineages distantly related to Negeviruses, Sobemoviruses, Poleroviruses, Flaviviridae, and Tombusviridae. Among these are close relatives of Drosophila X virus and Flock House virus, which we find in association with wild Drosophila immigrans. These two viruses are widely used in experimental studies but have not been previously reported to naturally infect Drosophila. Although we detect no new DNA viruses, in D. immigrans and Drosophila obscura, we identify sequences very closely related to Armadillidium vulgare iridescent virus (Invertebrate iridescent virus 31), bringing the total number of DNA viruses found in the Drosophilidae to three.
Keywords: Drosophila, virus, metagenomics, transcriptome, Drosophila X virus, Flock House virus
Introduction
Drosophila melanogaster is an important model system for the study of antiviral immunity in invertebrates1–4 and has been instrumental in defining all of the major insect antiviral immune mechanisms, including the RNAi, IMD, Toll, autophagy, and JAK-STAT pathways, and the antiviral role of Wolbachia.5–10 However, from an evolutionary perspective, the value of D. melanogaster is not only in its experimental tractability but also in its close relationship to many other experimentally tractable species.11 For example, experimental infection studies of more than 50 species of Drosophilidae (representing around 50 million years of evolution) have shown that susceptibility to viral infection has a strong phylogenetic component, such that more closely related host species display more similar viral replication rates and virulence12 and that closer relatives of the virus’ natural host tend to support higher viral replication rates.13 To understand how such phylogenetic patterns relate to host and virus biology in the wild, we need to know the natural host range and frequency of host switching of these viruses. Thus, to capitalize on the value of the Drosophilidae as a model clade, we require a broader perspective on Drosophila viruses than D. melanogaster alone.
Prior to the advent of modern molecular biology, a handful of Drosophila viruses had been described on the basis of traditional virological techniques.14 Starting with the Sigmavirus of D. melanogaster (DMelSV, Rhabdoviridae15), which was initially identified by the failure of infected flies to recover from CO2 anesthesia,16,17 these classical Drosophila viruses also include Drosophila P virus (Picornavirales18), Drosophila C virus (Cripavirus19), Drosophila A virus,20 Drosophila F virus (Reoviridae21), and Drosophila G virus (unclassified21) from adult flies, and Drosophila X virus (DXV, Entomobirnavirus22), Drosophila K virus (Reoviridae23), and unnamed Reoviruses from cell culture.24,25 In broadly the same period, Iota virus (Picornavirales26) was identified from Drosophila immigrans and was shown to be serologically similar to Drosophila P virus, RS virus was identified in Drosophila ananassae and members of the Drosophila montium group21 and shown to be morphologically similar to chronic bee paralysis virus, and Drosophila S virus (Reoviridae27) was identified from Drosophila simulans. Unfortunately, of these classical viruses, only Drosophila A virus, Drosophila C virus, DXV, and DMelSV remained in culture into the era of routine sequencing, and the others have been lost, making their classification tentative and relationships to each other and subsequently discovered viruses uncertain.
As large-scale sequencing became routine, it led to the serendipitous discovery of Drosophila viruses in host RNA sequenced for other purposes. Starting with the discovery of Nora virus (unclassified Picornavirales) in a D. melanogaster cDNA library,28 such discoveries have included six viruses from small RNAs of D. melanogaster cell culture and D. melanogaster laboratory stocks (American Nodavirus, D. melanogaster totivirus, D. melanogaster Birnavirus, and Drosophila tetravirus29; Drosophila uncharacterized virus and Drosophila reovirus30), a new Cripavirus in Drosophila kikkawai,31 and a new Sigmavirus in Drosophila montana.32 At the same time, polymerase chain reaction (PCR) surveys of other Drosophila species using primers designed to D. melanogaster viruses were used to detect new Nora viruses in D. immigrans and Drosophila subob-scura,33 and new Sigmaviruses in CO2-sensitive individuals of Drosophila affinis and Drosophila obscura34 and subsequently in D. immigrans, Drosophila tristis, and D. ananassae.35
With the widespread adoption of high-throughput sequencing technologies, the metagenomic (transcriptomic) sequencing of wild-collected flies is now starting to revolutionize our understanding of the drosophilid virome. The first explicitly metagenomic virus study in Drosophila discovered the first DNA virus of a drosophilid, Drosophila innubila Nudivirus.36 Subsequently, RNA and small RNA sequencing of around 3000 D. melanogaster from the United Kingdom and 2000 individuals of several species from Kenya and the USA (primarily D. melanogaster, D. ananassae, Drosophila malerkotliana, and Scaptodrosophila latifasciaeformis) were used to identify more than 20 new RNA virus genomes and genome fragments, and a single near-complete DNA virus (Kallithea virus, an Nudivirus).31 Metagenomic sequencing targeted to CO2-sensitive individuals has also been recently used to identify new Sigmaviruses and other Rhabdoviruses in Drosophila algonquin, Drosophila sturtevanti, Drosophila busckii, D. subobscura, Droso-phila unispina, and Scaptodrosophila deflexa.32
In total, studies using classical virology, serendipitous transcriptomic discovery, and metagenomic sequencing have reported more than 60 viruses associated with the Drosophilidae and Drosophila cell culture (for a comprehensive list, see Supplementary File 1). And, while the lost classical viruses and incomplete metagenomic genomes make the exact number of distinct viruses uncertain, around 50 are currently represented by sequence data in public databases. From these, it is possible to draw some general observations about the virus community of the Drosophilidae. For example, it is clear that RNA viruses substantially outnumber DNA viruses: of approximately 50 viruses with published sequence, only two are DNA viruses (the Nudiviruses of D. innubila36 and D. melanogaster31). However, the extreme sampling bias introduced by targeted virus discovery, such as CO2-sensitivity analysis for Sigmaviruses (Rhabdoviridae32), makes it difficult to draw robust conclusions about the taxonomic composition of the Drosophila viruses. For example, among RNA viruses, positive-sense single-stranded (+ssRNA) viruses are generally more common than other groups, but negative-sense viruses (−ssRNA) constitute around 30% of classifiable Drosophila RNA viruses, and double-stranded (dsRNA) viruses nearly as high a proportion (Supplementary File 1). To generalize such patterns and to gain broader insight into the host range of Drosophila viruses and their relationship to the viruses of other organisms will require further unbiased metagenomic sequencing.
Here, we report the viruses we have discovered through metagenomic sequencing of RNA from around 1600 wild-collected flies of the species D. immigrans, D. obscura, D. subobscura, Drosophila subsilvestris, D. tristis, and S. deflexa. We also report the reanalysis of two putatively virus-like sequences previously identified in a large pool of mixed Drosophila.31 In total, we describe 25 new viruses and place these within the phylogenetic diversity of known viruses and undescribed virus-like sequences from public transcriptomic datasets. Remarkably, in wild D. immigrans, we identify new viruses that are extremely closely related to the laboratory models DXV (previously known only from D. melanogaster cell culture) and Flock House virus (originally isolated from beetles), and we detect the presence of Armadillidium vulgare iridescent virus37 in D. immigrans and D. obscura – only the third DNA virus to be reported in a drosophilid. We find that a few viruses, such as La Jolla virus,31 appear to be generalists, and that many viruses are shared between the closely related members of the D. obscura group, but that viruses are more rarely shared between more distantly related species. We discuss our findings in the context of the Drosophilidae as a model clade for studying host–virus coevolution, and the diversity and host range of invertebrate viruses more generally.
Methods
Sample collections and sequencing
We collected around 1400 adult flies representing five species in the United Kingdom in summer 2011 (D. immigrans, D. obscura, D. subobscura, D. subsilvestris, and D. tristis) and 200 S. deflexa in France in summer 2012. Flies were netted or aspirated from banana/yeast bait in wooded and rural areas at intervals of 24 hours for up to a week at each location. They were sorted morphologically by species, and RNA was extracted using TRI-zol (Ambion) according to the manufacturer’s instructions. Females of the obscura group (including D. obscura, D. subobscura, D. subsilvestris, and D. tristis) are hard to identify morphologically, and for these species, only males were used for RNA extraction and sequencing.
In total, 498 D. immigrans were collected in three groups (63 flies in July 2011 in Edinburgh 55.928N, 3.170W; 285 flies in July 2011 in Edinburgh 55.921N, 3.193W; and 150 flies in July 2011 in Sussex 51.100N, 0.164E). The 502 D. obscura males were collected in four groups (280 flies collected in July 2011 in Edinburgh 55.928N, 3.170W; 52 flies in October 2011 in Edinburgh 55.928N, 3.170W; 115 flies in July 2011 in Sussex 51.100N, 0.164E; and 55 flies in August 2011 in Perthshire 56.316N, 3.790W). The 338 D. subobscura males were collected in four groups (60 flies collected in July 2011 in Edinburgh 55.928N, 3.170W; 60 flies in October 2011 in Edinburgh 55.928N, 3.170W; 38 flies in July 2011 in Sussex 51.100N, 0.164E; and 180 flies in August 2011 in Perthshire 56.316N, 3.790W). The 64 D. subsilvestris were collected in three groups (44 flies collected in July 2011 in Edinburgh 55.928N, 3.19W; 15 flies in October 2011 in Edinburgh 55.928N, 3.19W; and 5 flies in August 2011 in Perthshire 56.316N, 3.790W). The 29 D. tristis were collected in two groups from a single location (21 flies collected in July 2011 in Edinburgh 55.928N, 3.190W; 8 flies in October 2011 in Edinburgh 55.928N, 3.190W), and approximately 200 S. deflexa were collected in a single collection (August 2012 in Les Gorges du Chambon, France 45.66N, 0.556E). Pooled Cytochrome Oxidase I (COI) sequence data subsequently showed that some of these collections may be contaminated with other species. Specifically, around 2% of reads in the D. subobscura sample appear to derive from D. tristis, and around 5% of reads in the D. subsilvestris sample may derive from D. bifasciata.
RNA was treated with DNAse (TURBO DNA-free; Ambion) to reduce DNA contamination and precipitated in RNAstable (Biomatrica) for shipping. All library preparation and sequencing were performed by the Beijing Genomics Institute (BGI Tech Solutions) using the Illumina platform and either 91 or 101 nt paired-end reads. Raw data are available from the sequencing read archive under project accession SRP070549. Initially, two separate sequencing libraries were prepared for D. immigrans, the first used Ribo-Zero (Illumina) depletion of rRNA to increase the representation of viruses and host mRNAs (SRR3178477), and the second used duplex-specific nuclease normalization (DSN) to increase the representation of rare transcripts (SRR3178468). Subsequently, for each of the other species, a single library was prepared, again using DSN normalization (D. obscura SRR3178507, D. subobscura SRR3180643, D. subsilvestris SRR3180644, D. tristis SRR3180646, and S. deflexa SRR3180647). Unfortunately, due to a miscommunication with the sequencing provider, these six libraries were subject to polyA selection prior to normalization. This process substantially increases the amount of virus sequence available for assembly and identification (by excluding rRNA) but will bias viral discovery toward virus genomes and subgenomic products that are polyadenylated (eg, Picornavirales). Sequencing resulted in an average of 48 million read pairs per library, ranging from 47.3 M read pairs for D. subobscura to 52.7 M read pairs for the D. immigrans DSN library.
Virus genome assembly and identification
Raw reads were quality trimmed using sickle (version 1.238) only retaining reads longer than 40 nt, and adapter sequences were removed using cutadapt (version 1.8.139). Paired-end sequences were then de novo assembled using Trinity (version 2.0.640) with default parameters, and the resulting raw unannotated assemblies are provided in Supplementary File 2. In the absence of confirmation (eg, by PCR), such assemblies necessarily remain tentative and may represent chimeras of related sequences or contain substantial assembly errors.
We took two approaches to identify candidate virus-like contigs for further analysis. First, for each nominal gene assembled by Trinity, we identified and translated the longest open reading frame and used these translations to query virus sequences present in the GenBank nonredundant protein database (nr)41 using blastp (blast version 2.2.28+)42 with default parameters and an e-value threshold of 10−5, and retaining the single best hit. Second, for each nominal gene, we used the transcript with the longest open reading frame to query virus sequences in nr using blastx with default parameters, but again using an e-value threshold of 10−5 and retaining the single best hit. These two candidate lists, comprising all the sequences for which the top hit was a virus, were then combined and used to query the whole of nr using blastp, using an e-value threshold of 10−5 and retaining the top 20 hits. Sequences for which the top hit was still a virus, and sequences with a blastx hit to viruses but no other blastp hits in nr, were then treated as putatively viral in origin and subject to further analysis. In parallel with these analyses, raw data that were previously reported from D. melanogaster31 were reassembled and reanalyzed in the same way.
For each putative virus fragment, we selected other virus-like fragments in the same host that showed sequence similarity to the same virus taxonomic group, eg, combining all Negevirus-like sequences in D. immigrans and combining all Rhabdovirus-like sequences in D. obscura. We then manually ordered and orientated these fragments by reference to the closest relatives in GenBank to identify longer contigs that had not been assembled by Trinity. In some cases, we were able to identify very long contigs (ie, near-complete viral genomes) in the GenBank Transcriptome Shotgun Assembly database (“tsa_ nt”) and use these to order, orientate, and join overlapping virus fragments that had remained unjoined in the Trinity assembly. In cases of ambiguity, for example, where fragments failed to overlap and related viruses were present in the same pool, we did not manually join contigs. Where helpful, we used the longer TSA sequences to query our Drosophila metagenomic data using tblastx, thereby identifying further fragments to complete viral genomes. Near-complete genome sequences from Nora viruses of D. immigrans and D. subobscura and Sigmaviruses of D. tristis and S. deflexa were reported previously and are not further analyzed here.32,33 The remaining novel virus contigs are reported here and have been submitted to GenBank under accession numbers KU754504–KU754539.
Reanalysis of RNA data from D. melanogaster
Blast analysis suggests that two of the putative viral genomes identified during the course of this study (Hermitage virus of D. immigrans and Buckhurst virus of D. obscura; see “Results” section) are close relatives of short virus-like contigs that had previously been identified in D. melanogaster (previous contigs available from Ref. 31). Therefore, we used the new longer contigs from D. immigrans and D. obscura to guide the assembly of (partial) genomes for the D. melogaster viruses. As small RNA data were available for the published D. melanogaster samples (data available under the sequencing read archive accession SRP05612031), we additionally mapped small RNAs to these viral genomes using Bowtie243 to examine their properties.
Phylogenetic analysis
We inferred the phylogenetic placement of each virus using a conserved region of coding sequence. Where possible, this was the RNA polymerase, as these tend to be highly conserved in RNA viruses. We used blastp to query the GenBank nonredundant protein database (nr) and tblastn to query the GenBank Transcriptome Shotgun Assembly database (tsa_nt) to identify potential relatives for inclusion in the phylogenetic analysis. For viruses that could be tentatively assigned by blast to a well-studied group (eg, Iflaviruses and Nodaviruses), we additionally selected key representative members of the clade from the NCBI Viral Genomes Resource database.44 We aligned protein sequences using M-Coffee from the T-Coffee package,45 combining a consensus of alignments from ClustalW,46 T-Coffee,45 POA,47 Muscle,48 MAFFT,49 DIALIGN,50 PCMA,51 and Prob-Cons.52 Consensus alignments were examined by eye, and the most ambiguous regions of alignment at either end removed. Nevertheless, as expected for an analysis of distantly related and rapidly evolving RNA viruses, these alignments retain substantial ambiguity, and more distant relationships within the resulting phylogenetic trees should be treated with caution. Alignments are provided in Supplementary File 3.
Alignments were used to infer maximum-likelihood trees using PhyML (version 20120412)53 with the LG substitution model,54 empirical amino acid frequencies, and a four-category gamma distribution of rates with an inferred shape parameter. Maximum parsimony trees were used to provide the starting tree for the topology search, and the preferred tree was the one with the highest likelihood identified after both nearest-neighbor interchange and subtree prune-and-regraft searches. Support was assessed in two ways: first, using the Shimodaira–Hasegawa-like nonparametric version of an approximate likelihood ratio test,55 as implemented in PhyML, and second, by examining 100 bootstrap replicates.
Origin of RNA sequence reads
To infer the proportion of reads mapping to each virus and to detect potential cross-species contamination in the fly collections, quality-trimmed reads were mapped to all the new and previously published drosophilid virus genomes, and to a 343 nt region of cytochrome oxidase 1 that provides a high level of discrimination between drosophilid species. Mapping was performed using Bowtie 2 (version 2.2.5)43 with default parameters and global mapping, and only the forward read in each read pair was mapped. To reduce the potential for cross-mapping between closely related sequences, we excluded all trimmed reads with fewer than 80 contiguous non-N characters.
Results
In total, we identified 25 new RNA viruses through metagenomic sequencing of wild-caught Drosophilidae (Table 1). Among those viruses that could easily be classified were four members of the Picornavirales, three Rhabdoviruses, two Nodaviruses, two Reoviruses, and an Entomobirnavirus (Fig. 1). Among those lacking a current classification were five viruses distantly related to Negeviruses, four viruses distantly related to Sobemoviruses and Polerovirus, two distantly related to Flaviviruses, and two distantly related to Tombusviruses (Fig. 2). It is striking that among this latter group, there are many viruses that are closely related to unrecognized virus-like sequences in transcriptomic data. Indeed, of the 355 sequences included in our phylogenetic analyses, nearly one-third (29%) was derived from transcriptome data rather than from published viruses, illustrating the undersampling of RNA viruses generally. All phylogenetic trees, including node-support values and GenBank accession numbers, are provided in Supplementary File 4.
Table 1.
New viruses reported here.
| PROVISIONAL NAME | HOST | CLASSIFICATION | ACCESSION | DESCRIPTION |
|---|---|---|---|---|
| Blackford virus | Dtri | cf. Negevirus | KU754514 | +ssRNA. Distantly related to Brandeis virus (detected in RNA-seq data from D. melanogaster), to virus-like transcripts from a range of invertebrates, and to Negeviruses.76 [4.5 kbp fragment encoding a single ORF] |
| Bofa virus | (Pool) | cf. Negevirus | KU754515 | +ssRNA. Distantly related to Brandeis virus (detected in RNA-seq data from D. melanogaster31), to virus-like transcripts from a range of invertebrates, and to Negeviruses. Derived from pools E and K of Webster et al.31 and replaces two Negevirus-like sequences (KP757936 KP757935) and a small-RNA rich sequence (KP757975) previously reported there. [10.7 kbp near-complete genome encoding a two ORFs] |
| Braid Burn virus | Dsus | cf. Polerovirus Sobemovirus | KU754508 | +ssRNA. Related to Motts Mill virus of D. melanogaster, to Ixodes scapularis associated viruses 1 and 2 (ref. 79), to Humaita-Tubiacanga virus30 and to virus-like transcripts from a range of invertebrates. Distantly related to plant Poleroviruses and Sobemoviruses. [2.5 kbp fragment encoding two ORFs] |
| Buckhurst virus | Dobs | cf. Negevirus | KU754516 | +ssRNA. Distantly related to Brandeis virus (detected in RNA-seq data from D. melanogaster, see supporting information in ref. 31), to virus-like transcripts from a range of invertebrates, and to Negeviruses. [11.1 kbp near-complete genome encoding a two ORFs] |
| Cherry Gardens virus | Dsub | Rhabdoviridae | KU754524 | −ssRNA. Related to Soybean cyst nematode associated northern cereal mosaic virus.77 [5.7 kbp fragment encoding a partial polymerase] |
| Corseley virus | Dsub | Unclassified | KU754520 | +ssRNA. Very closely related to virus-like transcripts from a range of invertebrates, including near-identical virus-like transcripts from D. pseudoananassae. Distantly related to the Tombusviridae and Diaphorina citri associated C virus.73 [4 kbp fragment encoding three ORFs] |
| Craigmillar Park virus | Dsus | Alphanodavirus | KU754525 KU754526 | +ssRNA Segmented. Closely related to Craigie’s Hill virus of D. melanogaster, and related to Bat guano-associated Nodavirus.87 [Near-complete genome of two segments: RNA1 is 2.8 kbp encoding a polymerase, RNA2 is 1.8 kbp encoding a putative coat-protein precursor] |
| Empeyrat virus | Sdef | Cripavirus | KU754505 | +ssRNA. Very closely related (90% AA identity) to ‘Goose Dicistrovirus’ from goose faeces,64 to a virus-like transcript from Teleopsis dalmanni, and to a Cripavirus present in raw RNAseq data from D. kikkawai supporting material of ref. 31 [9.2 kbp near-complete genome encoding two open reading frames] |
| Eridge virus | Dimm | Entomobirnavirus | KU754527 KU754528 | dsRNA Segmented. Closely related to Drosophila X virus (and similarly present in some D. melanogaster cell cultures. [Near-complete genome of two segments: Segment A is 3.4 kbp, Segment B is 3.2 kbp and encodes a putative polymerase] |
| Grange virus | Dsub | Reoviridae | KU754536–KU754538 | dsRNA Segmented. Related to Bloomfield virus of D. melanogaster (ref., 31 see also refs. 23,25,30), to virus-like transcripts from a range of invertebrates, and to Fijiviruses. By similarity to Bloomfield virus, fragments of segments 1, 2, 6, and 7 are identifiable. [Segment 1 is a 1.7 kbp fragment encoding a partial polymerase, Segment 2 is a 1.9 kbp fragment encoding the partial major core protein, Segment 6 is a 1.1 kbp, fragment Segment 7 is a 1.3 kbp fragment] |
| Grom virus | Dobs | cf. Polerovirus Sobemovirus | KU754506 | +ssRNA. Related to Motts Mill Virus of D. melanogaster, to Ixodes scapularis associated viruses 1 and 2 (ref. 79), to Humaita-Tubiacanga virus30 and to virus-like transcripts from a range of invertebrates. Distantly related to plant Poleroviruses and Sobemoviruses. [3 kbp fragment encoding an ORF] |
| Hermitage virus | Dimm | Unclassified | KU754511 KU754512 | RNA. Related to Gentian Kobu-sho-associated virus (reported to be dsRNA74) and a virus-like transcript from Conwentzia psociformis. Distantly related to Soybean cyst nematode virus 5 and the Flavivirus-like Xinzhou spider virus 2. [Two un-joined contigs of 3.2 kbp and 3.5 kbp encoding a putative polyprotein] |
| Kinkell virus | Dsus | Iflavirus | KU754510 | +ssRNA. Closely related to virus-like transcripts from Ceratitis and Bactrocera, equally distantly related to Deformed Wing virus and Sacbrood Virus. [6.7 kbp fragment encoding a putative incomplete polyprotein] |
| La Tardoire virus | Sdef | cf. Polerovirus Sobemovirus | KU754509 | +ssRNA. Related to Motts Mill Virus of D. melanogaster, to Ixodes scapularis associated viruses 1 and 2 (ref. 79), to Humaita-Tubiacanga virus30 and to virus-like transcripts from a range of invertebrates. Distantly related to plant Poleroviruses and Sobemoviruses. [2.3 kbp fragment encoding two ORFs] |
| Lye Green virus | Dobs | Rhabdoviridae | KU754522 | −ssRNA. Related to Drosophila busckii Rhabdovirus.32 [14.5 kbp near-complete genome encoding five ORFs] |
| Machany virus | Dobs | Picornavirales | KU754504 | +ssRNA. Related to Kilifi virus and Thika virus of D. melanogaster, and to Rosy Apple Aphid virus and Acyrthosiphon pisum virus. [4.9 kbp fragment encoding a putative polyprotein] |
| Marsac virus | Sdef | cf. Negevirus | KU754518 | +ssRNA. Related to Brandeis virus (detected in RNA-seq data from D. melanogaster31), to a virus-like transcript from Ceratitis capitata, and to Negeviruses. [11.1 kbp near-complete genome encoding a two ORFs] |
| Muthill virus | Dimm | cf. Negevirus | KU754517 | +ssRNA. Closely related to Brandeis virus (detected in RNA-seq data from D. melanogaster31), to a virus-like transcript from Ceratitis capitata, and to Negeviruses. [10.6 kbp near-complete genome encoding a two ORFs] |
| Newington virus | Dimm | Alphanodavirus | KU754529 KU754530 | +ssRNA Segmented. Very closely related to Boolarra virus and Bat nodavirus. [Near-complete genome of two segments: RNA1 is 3 kbp encoding a polymerase, RNA2 is 1.2 kbp encoding a putative coat-protein precursor] |
| Pow Burn virus | Dsub | Picornavirales | KU754519 | +ssRNA. Related to Fisavirus 1 and to a virus-like transcript from Anopheles sinensis. [9.3 kbp near-complete genome encoding a single polyprotein] |
| Prestney Burn virus | Dsub | cf. Polerovirus Sobemovirus | KU754507 | +ssRNA. Related to Motts Mill Virus of D. melanogaster, to Ixodes scapularis associated viruses 1 and 2 (ref. 79), to Humaita-Tubiacanga virus30 and to virus-like transcripts from a range of invertebrates. Distantly related to plant Poleroviruses and Sobemoviruses. [3 kbp fragment encoding two ORFs] |
| Soudat virus | Sdef | Cypovirus | KU754531–KU754534 | dsRNA Segmented. Related to Torrey Pines virus of D. melanogaster and to Bombyx mori Cypovirus 1 and Lutzomyia reovirus 2 (ref. 30). By similarity to Torrey Pines virus, fragments of segments 1, 2, 3, and 5 are identifiable. [Segment 1 is near-complete 3.7 kbp encoding a polymerase, Segment 2 is a 0.6 kbp fragment, Segment 3 is near-complete 3.9 kbp encoding the major core protein, Segment 5 is a 1.3 kbp fragment] |
| Takaungu virus | (Pool) | Unclassified | KU754513 KP757925 | RNA. Related to Gentian Kobu-sho-associated virus (reported to be dsRNA74) and a virus-like transcript from Conwentzia psociformis. Distantly related to Soybean cyst nematode virus 5 and the Flavivirus-like Xinzhou spider virus 2 (ref. 75). Derived from pools E and K of Webster et al.31, this virus incorporates Flavivirus-like sequence KP757925 that was previously reported there. [Two un-joined contigs of 2.3 kbp and 3.9 kbp encoding a putative polyprotein] |
| Tartou virus | Sdef | Unclassified | KU754521 | ++ssRNA. Related to Diaphorina citri associated C virus73 and virus-like transcripts from a range of invertebrates. Distantly related to the Tombusviridae. [1.8 kbp fragment encoding a single ORF] |
| Withyham virus | Dobs | Rhabdoviridae | KU754523 | −ssRNA. Very closely related to Drosophila subobscura Rhabdovirus.32 [6.9 kbp fragment encoding the polymerase] |
Figure 1.

Viruses related to well-studied clades. Mid-point rooted maximum-likelihood phylogenetic trees for the viruses reported here, inferred using polymerase protein sequences.
Notes: The gray scale bars represent 0.5 amino acid substitutions per site. In each tree, viruses reported from Drosophilidae are labeled in red, viruses from other taxa are labeled in black, and unannotated virus-like sequences from publicly available transcriptome datasets are labeled in blue. Viruses newly reported here are underlined, and Drosophila species abbreviations are given for the reference sequence (Dimm, D. immigrans; Dobs, D. obscura; Dsub, D. subobscura; Dsus, D. subsilvestris; Dtri, D. tristis; Sdef, S. deflexa). Tree A: viruses near to the Dicistroviridae (Picornavirales); B: putative Cripaviruses (Dicistroviridae, Picornavirales – the corresponding tree in Supplementary File 4 additionally includes Aparaviruses); C: Nodaviruses; D: Birnaviruses; E: unclassified members of the Rhabdoviridae that form the sister clade to the Cytorhabdoviruses and the Nucleorhabdoviruses32; and F: Reoviridae. Alignments are provided in Supplementary File 3, and clade support values and sequence accession identifiers are provided in Supplementary File 4.
Figure 2.

Viruses not closely related to well-studied clades.
Notes: See Figure 1 for a key to the colors and abbreviations. Tree A: unclassified Picornavirales; B: unclassified clade of basally branching Flavi-like viruses75; C: an unclassified clade that branches basally to Poleroviruses and Sobemoviruses79; C: Nodaviruses; D: Iflaviruses, including a new clade that falls within (or close to) the Iflaviruses; E: two unclassified clades related to the Tombusviridae73; and F: two unclassified clades related to the Negeviruses and the Virgaviridae. Alignments are provided in Supplementary File 3, and clade support values and sequence accession identifiers are provided in Supplementary File 4.
Following common practice, we have provisionally named the new Drosophila viruses after localities near to our collection sites. We have chosen this approach as it avoids associating the sequence with higher levels of either the host or virus taxonomy, when both may be uncertain or unstable. The new Drosophila viruses are each represented between 1.8 and 13.7 kbp of sequence (Tartou virus of S. deflexa and Lye Green virus of D. obscura, respectively), and six are likely to be near-complete genomes with more than 9 kbp of sequence each. We have not named, and do not report, virus sequences that were near identical to previously published viruses (ie, KS, < 0.3, or falling within the published diversity of other viruses). See Figure 3 for read numbers of previously published viruses.
Figure 3.
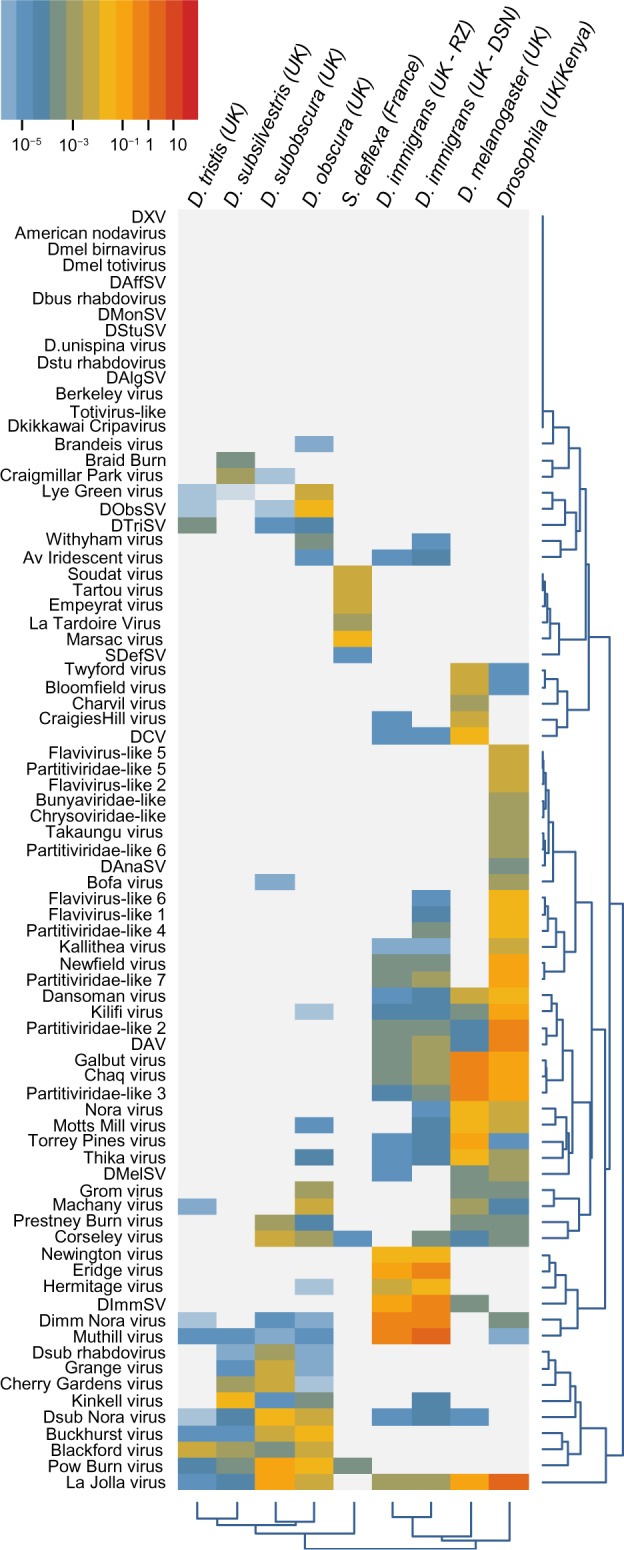
Virus read numbers (relative to host COI, normalized for length).
Notes: A heat map showing the relative number of high-quality (80 nt) forward reads from each library that map to each of the Drosophila viruses. Read numbers are normalized by target sequence length and by the number of reads mapping to a fragment of the host COI gene (so that a value of 1 implies equal read numbers per unit length of the virus and the host cytochrome oxidase 1). Rows and columns are clustered by the similarity in read frequency on a log scale. Note that some viruses may be sufficiently similar for a small proportion of reads to cross-map and that a small level of cross-contamination between fly species means that the data presented here cannot be used to confidently infer host specificity.
New viruses closely related to viruses of D. melanogaster
For around half of the newly discovered viruses (11 of 25), the closest previously reported relative was associated with D. melanogaster. Most striking of these is Eridge virus, a segmented dsRNA Entomobirnavirus closely related to the D. melanogaster laboratory model, Drosophila X virus (Fig. 1D; 78% sequence identity and 83% amino acid identity in Segment A).56 DXV has not been previously observed in wild flies but has been reported from flies injected with fetal bovine serum and has therefore been considered a cell culture contaminant.57 In addition to DXV, we detected sequences that were >98% identical to Eridge virus in some Drosophila cell cultures (eg, ModEn-code dataset SRR1197282 from S2-DRSC cells58), showing that fly cell cultures can harbor both viruses.
Other viruses that are also closely related to a published Drosophila virus include Machany virus of D. obscura (unclassified Picornavirales, close to Kilifi virus and Thika virus of D. melanogaster; Fig. 1A), Grange virus of D. subobscura (a Reovirus close to Bloomfield virus of D. melanogaster; Fig. 1F), Craigmillar Park virus of D. subsilvestris (an Alphanodavirus close to Craigie’s Hill virus of D. melanogaster; Fig. 1C), Grom virus and Prestney Burn virus (of D. obscura and D. subobscura, respectively, both close to Motts Mill virus of D. melanogaster; Fig. 2C), and Muthill virus and Marsac virus (of D. immigrans and S. deflexa, respectively, Fig. 2F; both close to Brandeis virus identified in publicly available D. melanogaster sequence data from laboratory stocks31,59).
New Drosophila viruses closely related to viruses of other species
We identified two new viruses that are extremely closely related to viruses reported from other taxa. Newington virus of D. immigrans is an Alphanodavirus extremely similar to Boolarra virus60 (isolated from the lepidopteran Oncopera intricoides; 84% nucleic acid identity and 89% amino acid identity in the polymerase), the widely used laboratory model Flock House virus60 (from the coleopteran Costelytra zealandica; 79% nucleic acid and 87% amino acid identity) and American Noda virus (ANV, identified from small RNAs of D. melanogaster cell culture29). This clade of closely related nodaviruses also includes Bat Nodavirus (detected in the brain tissue of the insectivorous bat Eptesicus serotinus61) and transcriptome sequences from the flies Bactrocera cucurbitae62 and Ceratitis capitate.63
We further identified a novel Cripavirus in S. deflexa that is very closely related to Goose Dicistrovirus (90% sequence identity and 92% amino acid identity), recently identified from a fecal sample from geese.64 However, given that the next closest relatives to this sequence are a transcriptome sequence from the stalk-eyed fly Teleopsis dalmanni,65 and a Cripavirus present in publicly available transcriptome data from D. kikkawai (supplementary information in Ref. 31), we think it likely that these represent invertebrate viruses. To reflect this, and given the divergence between them, we have decided to consider the S. deflexa-associated sequence as a new virus and have provisionally named it as Empeyrat virus.
New viruses without close relatives
The remaining other new putative viruses (13 of 25) do not have published close relatives, although many are related to unreported viruses present in host transcriptome datasets. Most notable among these are Kinkell virus of D. subsilvestris and Corseley virus of D. subobscura. Kinkell virus, along with transcriptome sequences from the fly genera Bactrocera62,66 and Ceratitis,63 the beetle Colaphellus,67 the thrip Frankliniella,68 and the spider Latrodectus,69 appears to define a major new clade that falls within or close to the Iflaviruses (Fig. 2D). Similarly, Corseley virus, which is almost identical to transcriptome sequences from Drosophila pseudoananassae70 and is related to transcriptome sequences from the bug genus Lygus71 and the beetle genus Anoplophora,72 appears to define an entirely new group of viruses distantly related to Tombusviridae and the recently described Diaphorina citri associated C virus73 (which is itself closely related to the newly identified Tartou virus of S. deflexa; Fig. 2E).
Two other groups are also noteworthy. First, the clade that includes Takaungu virus, which we have identified through reanalyses of mixed drosophilid sequences from Kenya,31 and Hermitage virus of D. immigrans. These viruses are most closely related to a transcriptome sequence from the neuropteran Conwentzia psociformis, and the enigmatic Gentian Kobu-sho-associated virus, which is reported to be an extremely large dsRNA relative of the Flaviviruses (Fig. 1B74,75). Second is the clade that includes Blackford virus of D. tristis, Buckhurst virus of D. obscura, and Bofa virus (also derived from the Kenyan pool,31 incorporating three unnamed fragments KP757936, KP757935, and KP757975). These viruses, along with seven transcriptome sequences from various arthropods and Muthill, Marsac, and Brandeis viruses (described above), appear to represent a major group of insect-infecting viruses that fall between the recently proposed Negeviruses76 and the plant virus family Virgaviridae.
A DNA iridescent virus in Drosophila
In D. immigrans and D. obscura, we identified more than 900 read pairs almost identical to the DNA iridescent virus of A. vulgare (Invertebrate Iridovirus 3137). Although read numbers were relatively small (around 700 high-quality read pairs in D. obscura and 250 read pairs in D. immigrans), they do not represent low-complexity sequence, they are widely distributed around the viral genome, and they suggest that viral genes were being expressed (ie, present in RNA). The longest contiguous region of coverage in D. obscura corresponded to the virus major capsid protein and displayed 98% sequence identity to A. vulgare DNA iridescent virus (KS = 0.08). These data suggest that this virus has a broad host range and represent the third DNA virus to be identified naturally infecting a drosophilid.
Small RNA data from Takaungu virus and Bofa virus
For Takaungu virus (Contigs KP757925 and KU754513) and Bofa virus (KU754515) small (19–30 nt), RNA data were available from our previous study of D. melanogaster.31 Although relatively few small RNA reads were detected from these viruses (about 200 reads from Bofa virus and about 800 reads from Takaungu virus), the small RNAs displayed the properties expected of virus-derived siRNAs in Drosophila (Supplementary File 5). Specifically, they were derived from both strands of the virus, they were distributed along the full length of the virus contigs, their size distribution peaked sharply at 21 nt (in contrast to viral siRNAs of chelicerates, hymenopterans, and nematodes that are predominantly 22 nt in length), and there was a bias against G in the 5′ position.
The distribution of virus reads across host species
To explore the distribution of viruses across hosts, we mapped high-quality reads from all libraries to new and previously reported Drosophila virus sequences (Fig. 3). We included a UK sample of D. melanogaster and a mixed drosophilid pool from Kenya and USA that were published previously.31 Overall, approximately 1% of RNA-seq reads were viral in origin, ranging from 0.02% in the D. tristis pool to 6.96% in the mixed drosophilid pool. As expected, many published Drosophila viruses were absent. These include all the Rhabdoviruses from host species not present in our collections (Rhabdoviruses from D. affinis, D. busckii, D. montana, D. sturtevanti, D. algonquin, and D. unispina32) and the Cripavirus identified in public RNA reads from D. kikkawai31 (host also absent from our collections). Absent viruses also included the five that have been previously identified only in cell culture (Drosophila X virus, American Nodavirus, D. melanogaster Birnavirus, D. melanogaster Totivirus,29 and the totivirus from public dataset SRR119746631), and also Berkeley virus (identified in reads from SRR07041631).
The number of viruses varied substantially among the metagenomic pools. Normalizing by sequence length and by the number of reads from host COI (to account for variation in total read numbers, rRNA contamination levels, and sequence lengths), we were able to detect between 4 viruses (D. tristis) and 27 viruses (D. immigrans DSN) per pool at 0.001% of COI expression. The number of detectable viruses was positively correlated with the number of flies in the single-species samples, and the strength of the relationship increased with the expression threshold for inclusion (Spearman rank correlations: at 0.001% of COI ρ = 0.86, P = 0.02; at 0.01% of COI ρ = 0.96, P = 0.0008; at 0.1% of COI ρ = 0.96, P = 0.003). For D. immigrans, the DSN library detected more viruses than the rRNA-depleted library, regardless of threshold. Note that the presence of some cross-mapping between related viruses means that the estimates of the number of viruses will tend to be slightly inflated at low thresholds.
Although our sampling scheme and a small amount of species cross-contamination precludes a rigorous formal analysis of host range, some viruses do appear to be generalists and others specialists. Using the 0.01% threshold, the majority of Rhabdoviruses (including Sigmaviruses) appeared to be restricted to a single host: assuming that the apparent low level of DImmSV in D. melanogaster is due to cross-mapping, only Cherry Gardens virus (related to soybean cyst nematode associated with northern cereal mosaic virus77) was present in the two host species (D. subobscura and D. subsilvestris). In contrast, a few viruses appeared to have a broad host range: La Jolla virus (Iflavirus), Blackford virus (related to Negeviruses and the Virgaviridae), Corseley virus (related to Tombusviruses), and Pow Burn virus (Picornavirales, related to Fisavirus 1) were each present in four species at >0.01% COI, and a small number of La Jolla virus reads was detected in all pools except S. deflexa. Considering read frequencies across all viruses, members of the obscura group displayed the greatest similarity to each other (Fig. 3; D. obscura, D. subobscura, D. tristis, and D. subsilvestris), while S. deflexa was the most distinct, with six of its viruses not present in any other pool, and only two of the viruses from the other pools present in S. deflexa.
Discussion
New viruses of Drosophila
The 25 new viruses presented here bring the total number of viruses reported from the Drosophilidae to approximately 85 (see Supplementary File 1). Although it does not detract from the potential utility of the viruses we were able to identify, it should be noted that this sampling is far from comprehensive. First, more viruses are likely to have been present in these samples than we were able to detect – for example, because viral titer was too low for some viruses or (for flies other than D. immigrans) because polyA-selection biases against their discovery. Second, more virulent viruses may reduce fly movement, so that virulent viruses are underrepresented by collections from baited traps.
As for the majority of metagenomic studies, it also remains uncertain whether these viruses constitute active infections of Drosophila, or whether they are contaminants of the host surface or gut lumen, infections of an unrecognized parasite or other Drosophila-associated microflora, or fossil endogenous viral elements (EVEs) integrated into the host genome and still expressed.78 Small RNA sequencing can, in principle, be used to demonstrate that viruses do replicate within arthropod and nematode hosts and are targeted by their immune system.30,31 In addition, as hymenoptera, chelicerata, and nematodes generate predominantly 22 nt small RNAs from viruses, the presence of 21 nt virus-derived siRNAs is highly suggestive of an immune response by Drosophila. As two of the viruses reported here (Takaungu virus and Bofa virus) were identified through reanalysis of data from Webster et al.31, we were able to test whether these viruses show the expected siRNA profile. As expected, we do detect 21 nt siRNAs from both strands of these two viruses, consistent with their replication in Drosophila (Supplementary File 5). Indeed, in the earlier analysis,31 we identified an unnamed but putatively viral sequence purely on the basis of 21 nt siRNAs that can now be shown to be part of Bofa virus (GenBank accession KP757975; sufficient similarity to identify Bofa virus using blast is now provided by Buckhurst virus).
Nevertheless, in the absence of small RNA data for the other 23 putative viruses presented here, it remains possible that these virus-like sequences are EVEs,78 or infections of Drosophila-associated microflora. However, while EVEs are common in insect genomes,78 expressed EVEs are rarer, and expressed EVEs appear to be extremely rare relative to active viral infections. For example, in our previous metagenomic study of Drosophila RNA viruses, none of the 14 viruses we initially identified by RNA sequencing in D. melanogaster proved to be EVEs.31 Thus, although a minority of the sequences presented here could be recently acquired EVEs, few are likely to be as they do not appear in the genomes of closely related hosts, they are expressed, and they appear to be constrained (we detect long open reading frames).
Fifteen of the remaining 23 putative viruses in the present study are extremely closely related to known insect viruses or virus-like sequences from insect transcriptomes (Figs. 1 and 2), and/or are present at such high levels (greater than 10% of host COI in the cases of Muthill virus and Eridge virus), that it seems likely that the associated drosophilid is indeed the host. For the remaining eight, namely, Braid Burn virus, Cherry Gardens virus, Blackford virus, La Tardoire virus, Hermitage virus, Pow Burn virus, Tartou virus, and Soudat virus, conclusive demonstration of a drosophilid host must await future siRNA sequencing or experimental confirmation.
Three groups of newly discovered and currently unclassified viruses seem particularly prominent within the drosophilid samples presented here. First, near to the Sobemoviruses and Poleroviruses are a large clade of invertebrate-infecting viruses defined by Ixodes tick-associated viruses 1 and 2,79 Humaita-Tubiacanga virus,30 the Drosophila-associated Grom virus, Prestney Burn virus, Motts Mill virus, Braid Burn virus, and La Tardoire virus, and transcriptome-derived sequences predominantly from Hymenoptera and Hemiptera. Second, branching basally to the Negeviruses (and potentially between Negeviruses and Virgaviridae) are two clades including the Drosophila-associated Blackford virus, Bofa virus, Buckhurst virus, Brandeis virus, Muthill virus, and Marsac virus, along with transcriptome-derived sequences dominated by Diptera and Hymenoptera. Third, near to the Tombusviridae are the clades defined by D. citri associated C virus,73 Tartou virus and Corseley virus from the Drosophilidae, and transcripts from various invertebrates. All three groups appear to represent common and widespread infections of invertebrates that warrant taxonomic recognition.
Virus diversity and host range
Rapid viral discovery, facilitated by large-scale metagenomic sequencing and the serendipitous discovery of viral genomes in transcriptomic data, is revolutionizing our understanding of virus diversity. The Drosophilidae provide a clear example of this, with approximately 10 viruses reported prior to the year 2000, 11 more between 2001 and 2014, and more than 60 since 2015. Particularly striking is the frequency with which completely new, and deeply divergent, lineages of RNA viruses are being identified. Recent examples include the enormous and unexpected diversity of basally branching ssRNA viruses80 and the diversity of basal Flaviviridae,75 the Negeviruses,76 and the Phasmaviruses.81
How many invertebrate viruses are there, and when will the accelerating virus discovery curve start to saturate? Our ad hoc but intensive sampling of Drosophila suggests that such questions will require systematic estimates of the distribution of virus host ranges, the distribution of virus geographic ranges, and the distribution of virus prevalences. First, many Drosophila viruses are multihost and widely distributed. Around 10 of the 25 new viruses reported are detectable in multiple species, and we also detect previously published viruses of D. melanogaster in D. immigrans and members of the obscura group (Fig. 3). Similarly, our earlier PCR survey of D. melanogaster viruses31 detected 12 of the 16 viruses in more than a third of D. melanogaster populations, and 10 of them in at least one D. simulans population. Second, it seems likely that more closely related hosts share more viruses. This is consistent with the apparently high overlap in virus community between D. melanogaster and D. simulans31 and among the members of the obscura group, and the divergent set of viruses associated with S. deflexa (Fig. 3, but note that the D. subobscura sample was slightly contaminated by D. tristis, and the D. subsilvestris sample by D. bifasciata). It is also consistent with the absence of D. melanogaster viruses from metagenomic surveys of other invertebrate taxa (although Goose Dicistrovirus is closely related to Empeyrat virus of S. deflexa). Third, it is clear that viruses vary enormously in prevalence, such that few viruses are common and many are rare. Of the 16 viruses previously surveyed by PCR, only three ever exceeded 50% prevalence and most of them only exceeded 10% prevalence in two or three of the surveyed populations. This is consistent with the positive relationship found between sample size and virus number and suggests that many hundreds of Drosophila individuals are required to comprehensively survey a population.
Conclusions
The 25 new viruses presented here expand the catalog of recorded drosophilid-associated viruses by nearly 50% and identify several new clades of insect-associated viruses. These include a new clade related to the Iflaviruses (Kinkell virus), new clades related to the Tombusviridae (Corseley virus and Tartou virus), and new clades related to the Negeviruses and Virgaviridae (including six viruses detected in Drosophila). Nevertheless, the large number of undescribed viruses present in transcriptome datasets illustrates that, across the invertebrates as a whole, there are many more viruses and many more deeply divergent virus lineages to uncover.
We expect that the future isolation of these Drosophila-associated viruses will provide useful laboratory tools to better understand host–virus biology and host range. However, it is possible to capitalize on viral sequences to address these questions even in the absence of viable viral isolates, and new virus sequences per se are likely to prove valuable.33 In addition, given the widespread experimental use of model viruses that are not known to infect D. melanogaster in the wild, such as Flock House virus,82,83 Drosophila X virus,84,85 and Invertebrate Iridovirus 6,86 it is reassuring to know that these viruses have close relatives naturally associated with the Drosophilidae (Newington virus, Eridge virus, and A. vulgare iridescent virus in D. immigrans, respectively).
Acknowledgments
We thank the City of Edinburgh Council, Friends of the Hermitage of Braid, Keith and Sue Obbard, Sandy and Helen-May Bayne, Elizabeth Bayne, Graeme Pratt, and Neil and Jacky Longdon for permission to collect Drosophila and/or for support while making collections. We thank Fergal Waldron and Gytis Dudas for discussion, Roy Hall for encouraging us to submit an article to this special issue, and all six anonymous reviewers for their positive and helpful comments on the article.
Footnotes
ACADEMIC EDITOR: Jike Cui, Deputy Editor in Chief
PEER REVIEW: Six peer reviewers contributed to the peer review report. Reviewers’ reports totaled 2,393 words, excluding any confidential comments to the academic editor.
FUNDING: This work was funded by a Wellcome Trust Research Career Development Fellowship (WT085064) to DJO. BL was supported by grants from the UK Natural Environment Research Council (NE/L004232/1) and the European Research Council (281668, Drosophila Infection). SHL was supported by a Natural Environment Research Council Doctoral Training Grant (NERC DG NE/J500021/1). Work in DJO’s laboratory is partly supported by a Wellcome Trust strategic award to the Centre for Immunity, Infection and Evolution (WT095831). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: DJO, BL. Performed the experiments: DJO, BL, SHL, CLW. Analyzed the data: DJO. Wrote the first draft of the article: DJO. Contributed to the writing of the article: DJO, BL, SHL, CLW. All authors reviewed and approved of the final manuscript.
Supplementary Materials
Supplementary File 1. Viruses of the Drosophilidae. A comprehensive list of all Drosophila viruses reported to date (excluding retroelements) is provided as an xlsx file. Recorded data include the virus name, its Baltimore classification, the drosophilid hosts in which it has been detected (excluding experimental infections), its year of discovery, its approximate classification, reference GenBank accession identifiers, and citation for its discovery.
Supplementary File 2. Raw metagenomic assemblies. Compressed fasta files containing the transcriptome assemblies generated for this study [note that as mixed-species (metagenomic) assemblies these cannot be submitted to the NCBI Transcriptome Shotgun Assembly database].
Supplementary File 3. Alignments used for phylogenetic inference. Compressed fasta-format protein alignments used to infer phylogenetic trees.
Supplementary File 4. Phylogenetic trees. Mid-point rooted maximum-likelihood trees, with percentage support marked on nodes for which tree inference method identified less than 100% support (recorded as SH|bootstrap) and NCBI accession identifiers for the sequences used to infer the phylogeny.
Supplementary File 5. Small RNAs (19–30 nt) that map to Takaungu virus and Bofa virus from D. melanogaster. Bar charts (left) show the size distribution of small RNAs mapping to the positive-sense (above x-axis) and negative-sense (below x-axis) viral strands, and their base composition at the 5′ position (red, U; yellow, G; blue, C; green, A). Bar charts (right) show the distribution of 19–30 nt reads along the length of the virus contig (blue bars represent reads mapping to the positive strand and red bars represent reads mapping to the negative strand).
REFERENCES
- 1.Huszart T, Imler JL. Drosophila viruses and the study of antiviral host-defense. Adv Virus Res. 2008;72:227–65. doi: 10.1016/S0065-3527(08)00406-5. [DOI] [PubMed] [Google Scholar]
- 2.Xu J, Cherry S. Viruses and antiviral immunity in Drosophila. Dev Comp Immunol. 2014;42(1):67–84. doi: 10.1016/j.dci.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bronkhorst AW, van Rij RP. The long and short of antiviral defense: small RNA-based immunity in insects. Curr Opin Virol. 2014;7(0):19–28. doi: 10.1016/j.coviro.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 4.Chtarbanova S, Imler JL. Innate antiviral immunity in Drosophila. Virologie. 2011;15(5):296–306. doi: 10.1684/15-5.2011.17800. [DOI] [PubMed] [Google Scholar]
- 5.Dostert C, Jouanguy E, Irving P, et al. The Jak-STAT signaling pathway is required but not sufficient for the antiviral response of Drosophila. Nat Immunol. 2005;6(9):946–53. doi: 10.1038/ni1237. [DOI] [PubMed] [Google Scholar]
- 6.van Rij RP, Saleh M-C, Berry B, et al. The RNA silencing endonuclease Argonaute 2 mediates specific antiviral immunity in Drosophila melanogaster. Genes Dev. 2006;20(21):2985–95. doi: 10.1101/gad.1482006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teixeira L, Ferreira A, Ashburner M. The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol. 2008;6(12):2753–63. doi: 10.1371/journal.pbio.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costa A, Jan E, Sarnow P, Schneider D. The Imd pathway is involved in antiviral immune responses in Drosophila. PLoS One. 2009;4(10):e7436. doi: 10.1371/journal.pone.0007436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamoto M, Moy Ryan H, Xu J, et al. Virus recognition by Toll-7 activates antiviral autophagy in Drosophila. Immunity. 2012;36(4):658–67. doi: 10.1016/j.immuni.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kemp C, Mueller S, Goto A, et al. Broad RNA interference-mediated antiviral immunity and virus-specific inducible responses in Drosophila. J Immunol. 2013;190(2):650–8. doi: 10.4049/jimmunol.1102486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powell JR. Progress and Prospects in Evolutionary Biology: The Drosophila Model. New York, NY: Oxford University Press; 1997. [Google Scholar]
- 12.Longdon B, Hadfield JD, Day JP, et al. The causes and consequences of changes in virulence following pathogen host shifts. PLoS Pathog. 2015;11(3):e1004728. doi: 10.1371/journal.ppat.1004728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Longdon B, Hadfield JD, Webster CL, Obbard DJ, Jiggins FM. Host phylogeny determines viral persistence and replication in novel hosts. PLoS Pathog. 2011;7(9):e1002260. doi: 10.1371/journal.ppat.1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brun G, Plus N. The viruses of Drosophila. In: Ashburner M, Wright TRF, editors. The Genetics and Biology of Drosophila. New York, NY: Academic Press; 1980. pp. 625–702. [Google Scholar]
- 15.Berkalof A, Breglian JC, Ohanessi A. Mise en evidence de virions dans de drosophiles infetees par le virus hereditare Sigma. C R Acad Sci Hebd Seances Acad Sci. 1965;260(22):5956–9. [PubMed] [Google Scholar]
- 16.L’Héritier PH, Teissier G. Une anomalie physiologique héréditaire chez la Drosophile. C R Acad Sci Paris. 1937;205:1099. [Google Scholar]
- 17.L’Heritier PH. Sensitivity to CO2 in Drosophila – a review. Heredity. 1948;2(3):325–48. doi: 10.1038/hdy.1948.20. [DOI] [PubMed] [Google Scholar]
- 18.Plus N, Duthoit JL. Un nouveau virus de Drosophila melanogaster, le virus P. C R. Acad Sci Hebd Seances Acad Sci D. 1969;268(18):2313. [PubMed] [Google Scholar]
- 19.Jousset FX, Plus N, Croizier G, Thomas M. Existence chez Drosophila de deux groupes de Picornavirus de propriétés sérologiques et biologiques différentes. C R Acad Sci Hebd Seances Acad Sci D. 1972;275(25):3043. [PubMed] [Google Scholar]
- 20.Plus N, Croizier G, Jousset FX, David J. Picornaviruses of laboratory and wild Drosophila melanogaster – geographical distribution and serotypic composition. Ann Microbiol (Paris) 1975;A126(1):107–17. [PubMed] [Google Scholar]
- 21.Plus N, Croizier G, Duthoit JL, David J, Anxolabehere D, Periquet G. Découverte, chex la Drosophile, de virus appartenant à trois nouveaux groupes. C R Acad Sci Hebd Seances Acad Sci D. 1975;280(12):1501. [PubMed] [Google Scholar]
- 22.Teninges D, Ohanessian A, Richardmolard C, Contamine D. Isolation and biological properties of Drosophila X virus. J Gen Virol. 1979;42(FEB):241–54. [Google Scholar]
- 23.Teninges D, Ohanessian A, Richard-Molard C, Contamine D. Contamination and persistent infection of Drosophila cell lines by reovirus type particles. In Vitro. 1979;15(6):425–8. [Google Scholar]
- 24.Haars R, Zentgraf H, Gateff E, Bautz FA. Evidence for endogenous reovirus-like particles in a tissue culture cell line from Drosophila melanogaster. Virology. 1980;101(1):124–30. doi: 10.1016/0042-6822(80)90489-4. [DOI] [PubMed] [Google Scholar]
- 25.Pasyukova EG, Mukha DV. Reovirus-like double stranded RNA fraction in a Drosophila melanogaster line containing individual second chromosome from natural population. In: Connell CJ, Ralsto DP, editors. Insect Viruses: Detection, Characterization and Roles. New York City, NY: Nova Science Publishers Inc; 2009. pp. 157–64. [Google Scholar]
- 26.Jousset FX. Virus extracted from Drosophila immigrans inducing a CO2 sensitivity syndrom in males of Drosophila melanogaster. C R Acad Sci Hebd Seances Acad Sci D. 1970;271(13):1141. [PubMed] [Google Scholar]
- 27.Louis C, Lopez-Ferber M, Comendador M, Plus N, Kuhl G, Baker S. Drosophila S virus, a hereditary reolike virus, probable agent of the morphological S character in Drosophila simulans. J Virol. 1988;62(4):1266–70. doi: 10.1128/jvi.62.4.1266-1270.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Habayeb MS, Ekengren SK, Hultmark D. Nora virus, a persistent virus in Drosophila, defines a new picorna-like virus family. J Gen Virol. 2006;87:3045–51. doi: 10.1099/vir.0.81997-0. [DOI] [PubMed] [Google Scholar]
- 29.Wu Q, Luo Y, Lu R, et al. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc Natl Acad Sci U S A. 2010;107(4):1606–11. doi: 10.1073/pnas.0911353107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguiar Eric Roberto Guimarães R, Olmo RP, Paro S, et al. Sequence-independent characterization of viruses based on the pattern of viral small RNAs produced by the host. Nucleic Acids Res. 2015 Jul 27;43(13):6191–206. doi: 10.1093/nar/gkv587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webster CL, Waldron FM, Robertson S, et al. The discovery, distribution, and evolution of viruses associated with Drosophila melanogaster. PLoS Biol. 2015;13(7):e1002210. doi: 10.1371/journal.pbio.1002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Longdon B, Murray GGR, Palmer WJ, et al. The evolution, diversity, and host associations of rhabdoviruses. Virus Evolution. 2015;1(1):1–12. doi: 10.1093/ve/vev014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Mierlo JT, Overheul GJ, Obadia B, et al. Novel Drosophila viruses encode host-specific suppressors of RNAi. PLoS Pathog. 2014;10(7):e1004256. doi: 10.1371/journal.ppat.1004256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Longdon B, Obbard DJ, Jiggins FM. Sigma viruses from three species of Drosophila form a major new clade in the rhabdovirus phylogeny. Proc Biol Sci. 2010;277(1678):35–44. doi: 10.1098/rspb.2009.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Longdon B, Wilfert L, Osei-Poku J, Cagney H, Obbard DJ, Jiggins FM. Host-switching by a vertically transmitted rhabdovirus in Drosophila. Biol Lett. 2011;7(5):747–50. doi: 10.1098/rsbl.2011.0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Unckless RL. A DNA virus of Drosophila. PLoS One. 2011;6(10):e26564. doi: 10.1371/journal.pone.0026564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piégu B, Guizard S, Yeping T, et al. Genome sequence of a crustacean iridovirus, IIV31, isolated from the pill bug Armadillidium vulgare. J Gen Virol. 2014;95(7):1585–90. doi: 10.1099/vir.0.066076-0. [DOI] [PubMed] [Google Scholar]
- 38.Joshi NA, Fass JN. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files. 2011. Available at: https://github.com/najoshi/sickle.
- 39.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–2. [Google Scholar]
- 40.Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benson DA, Cavanaugh M, Clark K, et al. GenBank. Nucleic Acids Res. 2013;41(D1):D36–42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10(1):421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brister JR, Ako-adjei D, Bao Y, Blinkova O. NCBI viral genomes resource. Nucleic Acids Res. 2015;43(D1):D571-7. doi: 10.1093/nar/gku1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Notredame C, Higgins DG, Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment1. J Mol Biol. 2000;302(1):205–17. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 46.Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 47.Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics. 2002;18(3):452–64. doi: 10.1093/bioinformatics/18.3.452. [DOI] [PubMed] [Google Scholar]
- 48.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9(4):286–98. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 50.Morgenstern B. DIALIGN 2: improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics. 1999;15(3):211–8. doi: 10.1093/bioinformatics/15.3.211. [DOI] [PubMed] [Google Scholar]
- 51.Pei J, Sadreyev R, Grishin NV. PCMA: fast and accurate multiple sequence alignment based on profile consistency. Bioinformatics. 2003;19(3):427–8. doi: 10.1093/bioinformatics/btg008. [DOI] [PubMed] [Google Scholar]
- 52.Do CB, Mahabhashyam MSP, Brudno M, Batzoglou S. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15(2):330–40. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 54.Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25(7):1307–20. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 55.Anisimova M, Gil M, Dufayard J-F, Dessimoz C, Gascuel O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst Biol. 2011;60(5):685–99. doi: 10.1093/sysbio/syr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chung HK, Kordyban S, Cameron L, Dobos P. Sequence analysis of the bicistronic Drosophila X virus genome segment A and its encoded polypeptides. Virology. 1996;225(2):359–68. doi: 10.1006/viro.1996.0610. [DOI] [PubMed] [Google Scholar]
- 57.Plus N. Endogenous viruses of Drosophila melanogaster cell lines: their frequency, identification and origin. In Vitro. 1978;14(12):1015–21. [Google Scholar]
- 58.Duff MO, Olson S, Wei X, et al. Genome-wide identification of zero nucleotide recursive splicing in Drosophila. Nature. 2015;521(7552):376–9. doi: 10.1038/nature14475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodriguez J, Menet Jerome S, Rosbash M. Nascent-seq indicates widespread cotranscriptional RNA editing in Drosophila. Mol Cell. 2012;47(1):27–37. doi: 10.1016/j.molcel.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dasgupta R, Sgro JY. Nucleotide sequences of three Nodavirus RNA2’s: the messengers for their coat protein precursors. Nucleic Acids Res. 1989;17(18):7525–6. doi: 10.1093/nar/17.18.7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dacheux L, Cervantes-Gonzalez M, Guigon G, et al. A preliminary study of viral metagenomics of French bat species in contact with humans: identification of new mammalian viruses. PLoS One. 2014;9(1):e87194. doi: 10.1371/journal.pone.0087194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sim SB, Calla B, Hall B, DeRego T, Geib SM. Reconstructing a comprehensive transcriptome assembly of a white-pupal translocated strain of the pest fruit fly Bactrocera cucurbitae. GigaScience. 2015;4(1):1–5. doi: 10.1186/s13742-015-0053-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Calla B, Hall B, Hou S, Geib SM. A genomic perspective to assessing quality of mass-reared SIT flies used in Mediterranean fruit fly (Ceratitis capitata) eradication in California. BMC Genomics. 2014;15(1):1–19. doi: 10.1186/1471-2164-15-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Greninger AL, Jerome KR. Draft genome of goose dicistrovirus. Genome Announc. 2016 Mar-Apr;4(2):e00068-16. doi: 10.1128/genomeA.00068-16. Published online March 3, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reinhardt JA, Brand CL, Paczolt KA, Johns PM, Baker RH, Wilkinson GS. Meiotic drive impacts expression and evolution of X-linked genes in stalk-eyed flies. PLoS Genet. 2014;10(5):e1004362. doi: 10.1371/journal.pgen.1004362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Geib S, Calla B, Hall B, Hou S, Manoukis N. Characterizing the developmental transcriptome of the oriental fruit fly, Bactrocera dorsalis (Diptera: Tephritidae) through comparative genomic analysis with Drosophila melanogaster utilizing modENCODE datasets. BMC Genomics. 2014;15:942. doi: 10.1186/1471-2164-15-942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tan Q-Q, Zhu L, Li Y, et al. A de novo transcriptome and valid reference genes for quantitative real-time PCR in Colaphellus bowringi. PLoS One. 2015;10(2):e0118693. doi: 10.1371/journal.pone.0118693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stafford-Banks CA, Rotenberg D, Johnson BR, Whitfield AE, Ullman DE. Analysis of the salivary gland transcriptome of Frankliniella occidentalis. PLoS One. 2014;9(4):e94447. doi: 10.1371/journal.pone.0094447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clarke TH, Garb JE, Hayashi CY, et al. Multi-tissue transcriptomics of the black widow spider reveals expansions, co-options, and functional processes of the silk gland gene toolkit. BMC Genomics. 2014;15(1):1–17. doi: 10.1186/1471-2164-15-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Signor S, Seher T, Kopp A. Genomic resources for multiple species in the Drosophila ananassae species group. Fly. 2013;7(1):47–56. doi: 10.4161/fly.22353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hull JJ, Chaney K, Geib SM, et al. Transcriptome-based identification of ABC transporters in the western tarnished plant bug Lygus hesperus. PLoS One. 2014;9(11):e113046. doi: 10.1371/journal.pone.0113046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scully ED, Hoover K, Carlson JE, Tien M, Geib SM. Midgut transcriptome profiling of Anoplophora glabripennis, a lignocellulose degrading cerambycid beetle. BMC Genomics. 2013;14(1):1–26. doi: 10.1186/1471-2164-14-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nouri S, Salem N, Nigg JC, Falk BW. A diverse array of new viral sequences identified in worldwide populations of the Asian citrus psyllid (Diaphorina citri) using viral metagenomics. J Virol. 2015 doi: 10.1128/JVI.02793-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kobayashi K, Atsumi G, Iwadate Y, et al. Gentian Kobu-sho-associated virus: a tentative, novel double-stranded RNA virus that is relevant to gentian Kobu-sho syndrome. J Gen Plant Pathol. 2012;79(1):56–63. [Google Scholar]
- 75.Shi M, Lin X-D, Vasilakis N, et al. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related viruses. J Virol. 2016;90(2):659–69. doi: 10.1128/JVI.02036-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vasilakis N, Forrester NL, Palacios G, et al. Negevirus: a proposed new taxon of insect-specific viruses with wide geographic distribution. J Virol. 2013;87(5):2475–88. doi: 10.1128/JVI.00776-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bekal S, Domier LL, Niblack TL, Lambert KN. Discovery and initial analysis of novel viral genomes in the soybean cyst nematode. J Gen Virol. 2011;92(8):1870–9. doi: 10.1099/vir.0.030585-0. [DOI] [PubMed] [Google Scholar]
- 78.Katzourakis A, Gifford RJ. Endogenous viral elements in animal genomes. PLoS Genet. 2010;6(11):e1001191. doi: 10.1371/journal.pgen.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tokarz R, Williams SH, Sameroff S, Leon MS, Jain K, Lipkin WI. Virome analysis of Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis ticks reveals novel highly divergent vertebrate and invertebrate viruses. J Virol. 2014;88(19):11480–92. doi: 10.1128/JVI.01858-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li C-X, Shi M, Tian J-H, et al. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife. 2015;4:e05378. doi: 10.7554/eLife.05378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ballinger MJ, Bruenn JA, Hay J, Czechowski D, Taylor DJ. Discovery and evolution of bunyavirids in arctic phantom midges and ancient bunyavirid-like sequences in insect genomes. J Virol. 2014;88(16):8783–94. doi: 10.1128/JVI.00531-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Han YH, Luo YJ, Wu QF, et al. RNA-based immunity terminates viral infection in adult Drosophila in the absence of viral suppression of RNA interference: characterization of viral small interfering RNA populations in wild-type and mutant flies. J Virol. 2011;85(24):13153–63. doi: 10.1128/JVI.05518-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Flynt A, Liu N, Martin R, Lai EC. Dicing of viral replication intermediates during silencing of latent Drosophila viruses. Proc Natl Acad Sci U S A. 2009;106(13):5270–5. doi: 10.1073/pnas.0813412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zambon RA, Vakharia VN, Wu LP. RNAi is an antiviral immune response against a dsRNA virus in Drosophila melanogaster. Cell Microbiol. 2006;8(5):880–9. doi: 10.1111/j.1462-5822.2006.00688.x. [DOI] [PubMed] [Google Scholar]
- 85.van Cleef KWR, van Mierlo JT, Miesen P, et al. Mosquito and Drosophila entomobirnaviruses suppress dsRNA- and siRNA-induced RNAi. Nucleic Acids Res. 2014;42(13):8732–44. doi: 10.1093/nar/gku528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bronkhorst AW, van Cleef KWR, Venselaar H, van Rij RP. A dsRNA-binding protein of a complex invertebrate DNA virus suppresses the Drosophila RNAi response. Nucleic Acids Res. 2014;42(19):12237–48. doi: 10.1093/nar/gku910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li L, Victoria JG, Wang C, et al. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol. 2010;84(14):6955–65. doi: 10.1128/JVI.00501-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File 1. Viruses of the Drosophilidae. A comprehensive list of all Drosophila viruses reported to date (excluding retroelements) is provided as an xlsx file. Recorded data include the virus name, its Baltimore classification, the drosophilid hosts in which it has been detected (excluding experimental infections), its year of discovery, its approximate classification, reference GenBank accession identifiers, and citation for its discovery.
Supplementary File 2. Raw metagenomic assemblies. Compressed fasta files containing the transcriptome assemblies generated for this study [note that as mixed-species (metagenomic) assemblies these cannot be submitted to the NCBI Transcriptome Shotgun Assembly database].
Supplementary File 3. Alignments used for phylogenetic inference. Compressed fasta-format protein alignments used to infer phylogenetic trees.
Supplementary File 4. Phylogenetic trees. Mid-point rooted maximum-likelihood trees, with percentage support marked on nodes for which tree inference method identified less than 100% support (recorded as SH|bootstrap) and NCBI accession identifiers for the sequences used to infer the phylogeny.
Supplementary File 5. Small RNAs (19–30 nt) that map to Takaungu virus and Bofa virus from D. melanogaster. Bar charts (left) show the size distribution of small RNAs mapping to the positive-sense (above x-axis) and negative-sense (below x-axis) viral strands, and their base composition at the 5′ position (red, U; yellow, G; blue, C; green, A). Bar charts (right) show the distribution of 19–30 nt reads along the length of the virus contig (blue bars represent reads mapping to the positive strand and red bars represent reads mapping to the negative strand).