

Abstract
Understanding enzymatic Diels—Alder (DA) reactions that can form complex natural product scaffold is of considerable interest. Sch 210972 1, a potential anti-HIV fungal natural product, contains a decalin core that is proposed to form via a DA reaction. We identified the gene cluster responsible for the biosynthesis of 1 and heterologously reconstituted the biosynthetic pathway in Aspergillus nidulans to characterize the enzymes involved. Most notably, deletion of cghA resulted in a loss of stereoselective decalin core formation, yielding both an endo 1 and a diastereomeric exo adducts of the proposed DA reaction. Complementation with cghA restored the sole formation of 1. Density functional theory computation of the proposed DA reaction provided a plausible explanation of the observed pattern of product formation. Based on our study, we propose that lipocalin-like CghA is responsible for the stereoselective intramolecular [4+2] cycloaddition that forms the decalin core of 1.
Keywords: cycloaddition, decalin natural product, density functional calculations, fungal metabolite, tetramic acid
While Diels—Alder (DA) reaction, including catalytic variants of the reaction, has seen great use in chemical synthesis, the biogenic and biocatalytic DA reactions are less well characterized. However, designed proteins[1] and nucleic acids[2] are capable of catalyzing the DA reaction, suggesting that biomolecules can act as competent catalysts for the cycloaddition. The DA reaction has been established as a key transformation in biosynthesis of a growing number of natural products.[3] Despite these proposals, the identification of enzymes, Diels–Alderases (DAases), that catalyze the cycloaddition has proven challenging. In fact, only a handful of DAases have been identified based upon biochemical and structural studies, including lovastatin nonaketide synthase (LovB),[4] solanapyrone synthase,[5] SpnF involved in the biosynthesis of spinosyn A[6] and most recently VstJ from the versipelostatin biosynthetic pathway responsible for the formation of spirotetronate framework.[7] Of these examples, SpnF and VstJ are the only enzymes for which specific rate accelerations of [4+2] cycloaddition reactions have been verified experimentally. Given the limited understanding of the enzymatic DA reactions and the potential synthetic utility of (re)engineered DAases, additional studies establishing the existence and the catalytic modes of natural DAases are warranted.
We have investigated the biosynthetic pathway responsible for the formation of the tetramic acid-containing metabolite Sch 210972 1 (Figure 1) from Chaetomium globosum. This compound exhibits unique inhibitory activity against CCR-5, a cell surface receptor that allows the entry of HIV-1 into cells, and is an attractive lead compound for novel anti-HIV therapeutics.[8] Previous studies have shown that the tetramic acid moieties in related compounds, such as equisetin 2[9] (Scheme 1) and cyclopiazonic acid[10], are formed via a Dieckmann-type condensation that is catalyzed by reductive domains located at the C-termini of polyketide synthase nonribosomal peptide synthetase hybrid megaenzymes (PKS–NRPSs)[11]. Thus, we hypothesized that the framework of 1 was also biosynthesized by a hybrid PKS–NRPS. The structural similarity of 1, 2 and lovastatin 3 suggested that the decalin core of 1 may be formed by an intramolecular DA reaction, as proposed for the biocatalytic formation of the decalin cores of 2 and 3 (Figure 1B).[12] To gain further insight into the mechanism of biosynthesis of 1 and to determine if a DA reaction is indeed involved in the biosynthesis of 1, we identified the C. globosum gene cluster responsible for the biosynthesis of 1 for a detailed investigation.
Figure 1.
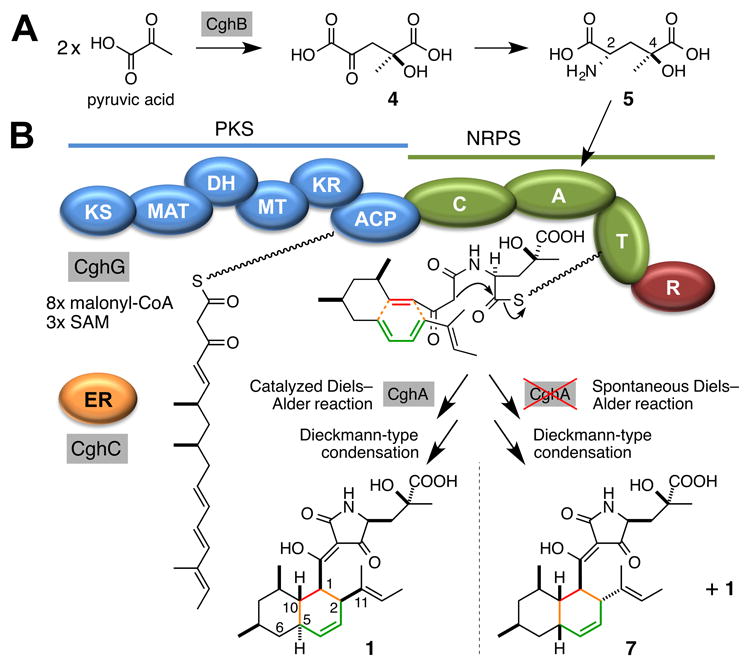
Proposed biosynthetic pathway of 1. (A) Formation of an unusual amino acid, γ-hydroxymethyl-l-glutamic acid 5, from two molecules of pyruvic acid dimerized by an aldolase CghB. (B) CghG-catalyzed synthesis of the linear substrate and a tetramic acid moiety, and the two final products that can be formed by the proposed Diels–Alder reaction. Abbreviations: KS, ketosynthase; MAT, malonyl-CoA acyltransferase; DH, dehydratase; MT, methyltransferase; KR, ketoreductase; ACP, acyl carrier protein; C, condensation; A, adenylation; T, thiolation; R, reductase; ER, enoyl reductase; SAM, S-adenosyl-l-methionine.
Scheme 1.
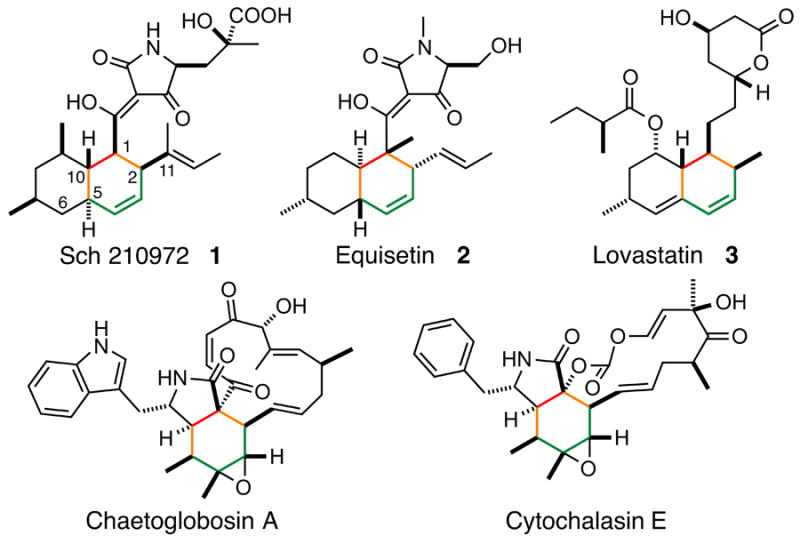
Natural products putatively biosynthesized via Diels–Alder reactions. Resulting cyclohexyl groups are shown in gray.
A BLASTP[13] search of the genome of C. globosum identified three recognizable hybrid PKS–NRPS genes: CHGG_01239, CHGG_05286 and CHGG_02374–CHGG_02378. We have recently reported that CHGG_01239 codes for the PKS–NRPS CheA responsible for the formation of chaetoglobosin-type of natural products[14] (Figure S13), and deletion of CHGG_05286 did not affect the production of 1. Therefore, the reassigned ORF CHGG_02374–CHGG_02378, which we have named cghG, was predicted and later confirmed to code for the PKS–NRPS responsible for the biosynthesis of 1 (Supporting Methods; Table S2; Figures S1–S9; S14, i vs. viii). The chemical structure of 1 was characterized using high-resolution (HR) electrospray ionization (ESI) LCMS, 1H NMR and 13C NMR. (Table S3 and Figure S16, S17), and the absolute configuration of 1 was determined crystallographically (Figure S18 and Tables S5–S12). Additionally, this class of PKS NRPS is often accompanied by a stand-alone enoyl reductase (ER) that works in trans with the PKS NRPS for reduction of polyketide backbone double bonds that is required for the formation of the final products.[11, 15] Based on homology to CheB, the ER involved in chaetoglobosin biosynthesis,[16] CghC was predicted to be the stand-alone ER for the biosynthesis of 1 (Table S2), and its disruption abolished the production of 1 (Figure S14, i vs. iv). Lastly, disruption of cghD also showed a complete loss of production of 1 (Figure S14, i vs. v), presumably because CghD is the GAL4-type transcription factor that activates the expression of the cgh genes.
One unique structural feature of 1 is the tetramic acid motif proposed to be derived from a non-proteinogenic amino acid, γ-hydroxymethyl-l-glutamic acid 5 via a Dieckmann-type condensation catalyzed by the reductase domain of CghG (Figure 1B) like in the case of the biosynthesis of 2.[17] Because the cgh gene cluster possessed an aldolase-coding cghB, we proposed CghB to catalyze the aldol condensation of two molecules of pyruvic acid to yield the intermediate 4, which can then be stereoselectively transaminated to form 5 (Figure 1A). To test this hypothesis, we prepared 5 and its diastereomers chemoenzymatically[18] and used them in feeding experiments with an engineered C. globosum strain, CGKW14[19] (Supporting Methods). After confirming the loss of production of 1 by ΔcghB/CGKW14 (Figure S14 i vs. iii), we fed 5 to this strain to observe the recovery of the production of 1 (Figure 2). Amination of 4 is likely performed by a transaminase in the cell. On the other hand, feeding of a diastereomer of 5, (2S,4R)-4-hydroxy-4-methylglutamic acid 6 (Supporting Methods) failed to restore the production of 1 in ΔcghB/CGKW14 (Figure S15). These results suggest that CghB is responsible for supplying 5 to CghG to complete the biosynthesis of 1, and also revealed the stereospecificity of the adenylation domain of the NRPS segment of CghG.
Figure 2.
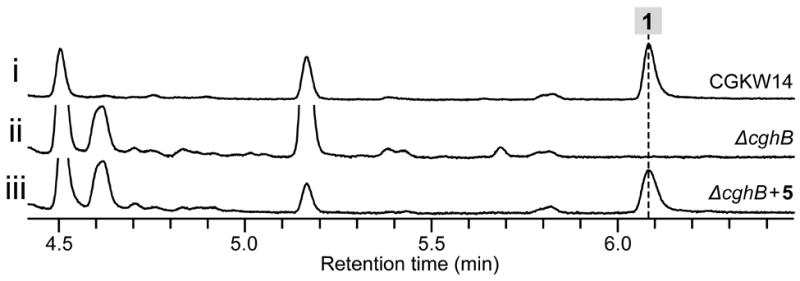
Confirmation of the activity of CghB by bioconversion of synthetic substrate 5 to 1 in C. globosum. HPLC traces of extracts of the culture of (i) wild-type control CGKW14, (ii) negative control of ΔcghB/CGKW14 without fed 5 and (iii) ΔcghB/CGKW14 with fed 5.
The biosynthetic gene clusters for all of the compounds except 3 in Scheme 1 contain a gene encoding hypothetical proteins that are homologous to one another (Figure S13). For 1, this gene corresponds to cghA (Table S2). While CcsF, the homolog of CghA in the cytochalasin E biosynthetic pathway, was suggested to play a possible role in the formation of the isoindolone core by catalyzing a DA reaction,[15b] none of those cghA-like genes has been examined experimentally until now. When cghA was disrupted, 1 was still formed, albeit in a reduced quantity (Figure S14, i vs. ii) but also resulted in the formation of a second compound, 7 (Figure 3, ii). The wild type CGKW14 strain did not form 7 (Figure 3, i). Detailed characterizations by HR-ESI-LCMS, 1H NMR and 13C NMR (Table S4 and Figure S19–S24) and X-ray crystallography (Figure S18), 7 was determined to be a diastereomer of 1 where 1 and 7 correspond to the endo and exo adduct of the proposed decalin-forming [4+2] cycloaddition reaction, respectively. The transition states leading to these cycloadducts are shown in Scheme 2.
Figure 3.
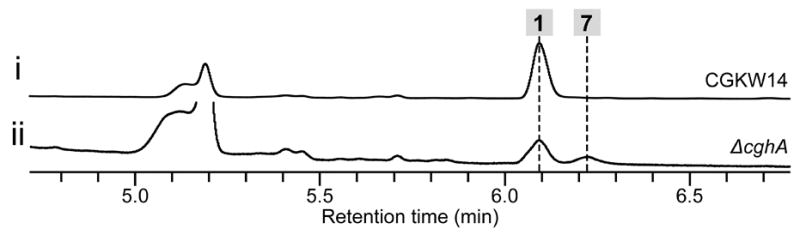
HPLC analysis of the metabolites from cghA strain of C. globosum. HPLC traces of extracts of the culture of (i) wild-type control CGKW14 and (ii) deletion mutant ΔcghA/CGKW14. The yield of 1 and 7 from the ΔcghA/CGKW14 culture were 2 and 1 mg/L, respectively, whereas 60 mg/L of 1 was isolated from the wild-type reference CGKW14 culture.
Scheme 2.
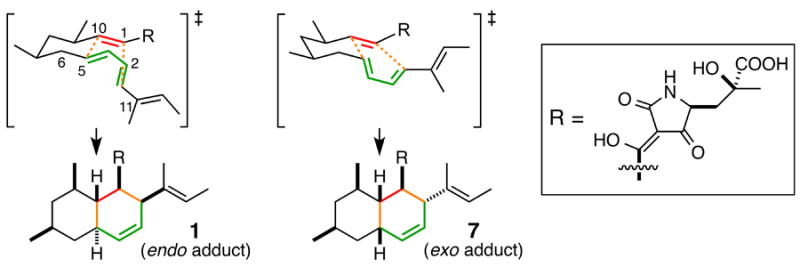
The endo and exo transition state of the [4+2] cycloaddition, leading to the formation of 1 and 7, respectively.
To confirm the involvement of CghA in dictating the course of the stereoselective intramolecular [4+2] cycloaddition reaction, we conducted a complementation experiment in which a functional copy of the cghA gene was reintroduced to the ΔcghA/CGKW14 strain. We chose to integrate cghA into the chaetoviridin biosynthetic PKS gene cazM[20] so that the integration event would be indicated by the loss of formation of chaetoviridin A 8 and B 9.[19] A control experiment verified that disruption of cazM would not affect the production of 1 (Figure 4, i vs. ii). Comparison of the extract from the cghA-knockout strain and the cghA-complemented strain showed that the biosynthesis of 7 was no longer observed and exclusive production of 1 was restored with cghA (Figure 4, iii vs. iv), indicating CghA is directly responsible for ensuring the stereoselective formation of 1 and suppressing the nonenzymatic formation of the alternative stereoisomer, 7. Furthermore, the Sch 210972 biosynthetic pathway was reconstituted in a heterologous host to eliminate any interfering effects in C. globosum that can obscure our findings (see SI for details). As expected, introduction of cghA, cghB, cghC and cghG to Aspergillus nidulans A1145 resulted in the formation of 1 (Figure 5 i vs. ii), while exclusion of cghA led to the formation of 1 and 7 (Figure 5 iii). These results clearly indicate that CghA selectively promotes a single stereochemical pathway among the decalin-forming intramolecular [4+2] cycloaddition reaction pathways to yield 1 exclusively.
Figure 4.
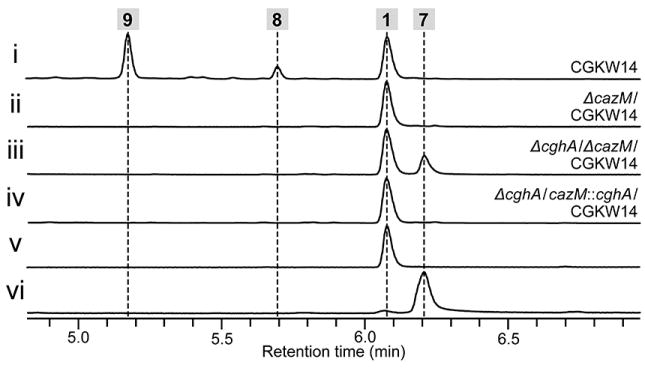
Confirmation of the role of CghA in the selective formation of 1 by complementing the cghA-disruption mutant with cghA in C. globosum. HPLC traces of extracts of the culture of (i) wild-type control CGKW14, (ii) ΔcazM/CGKW14 as a control showing the loss of production of 8 and 9, (iii) ΔcghA/ ΔcazM/CGKW14 and (iv) ΔcghA/cazM∷cghA/CGKW14. Authentic reference of (v) 1 and (vi) 7.
Figure 5.
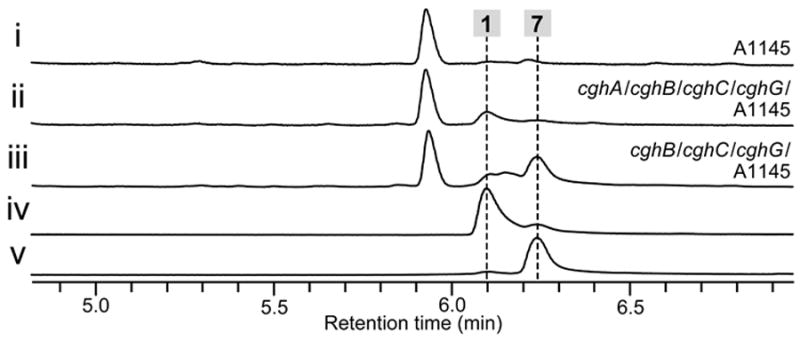
Reconstitution of the biosynthesis of 1 in Aspergillus nidulans A1145 and loss of stereoselective product formation upon elimination of cghA. HPLC traces of metabolic extracts from the culture of (i) wild-type A. nidulans A1145 control, (ii) A. nidulans harboring cghA, cghB, cghC and cghG and (iii) A. nidulans harboring cghB, cghC and cghG. Authentic reference of (iv) 1 and (v) 7.
Lastly, to quantitate the stereoselectivity of the cycloaddition, we computed the DA reaction of model substrates 10 and 11 shown in Figure 6A and S25 using the M06-2X density functional and 6-31+G(d,p) basis with the SMD implicit solvation model. Computations of the reaction with both substrates led to similar conclusions. Therefore, the discussion here will focus on the computations of the DA reaction of a truncated substrate 10. Details regarding density functional theory computations of the cycloaddition of 10 and 11 and our computational methods are presented in the Supporting Methods. The activation free energies and the transition structures for the formation of the four stereoisomeric cycloadduct of 10 are shown in Figure 6B. The barriers for the formation of these stereoadducts ranged from 18 to 27 kcal/mol. TS10endo1 and TS10exo1, the lowest energy transition structures, led to cycloadducts 12a and 12b, which are analogous in terms of stereochemistry to the adducts 1 and 7 formed by the cghA strain. The dominant adduct formed in the ΔcghA strain was 1 with 7 being a less prevalent product (Figure 3 ii), and our computations reproduced this selectivity trend correctly. The transition state for the formation of 12a (TS10endo1) is 1.3 kcal/mol lower in energy than the transition state leading to 12b (TS10exo1), suggesting approximately a 10:1 preference for the formation of 12a. TS10exo1 is destabilized by a 1,3-diaxial interaction involving the methylene group of the diene. Similar, but more severe 1,3-diaxial interactions destabilize the other endo and exo transition states (TS10exo2 and TS10endo2), causing them to be more than 4 kcal/mol higher in energy than TS10endo1.
Figure 6.
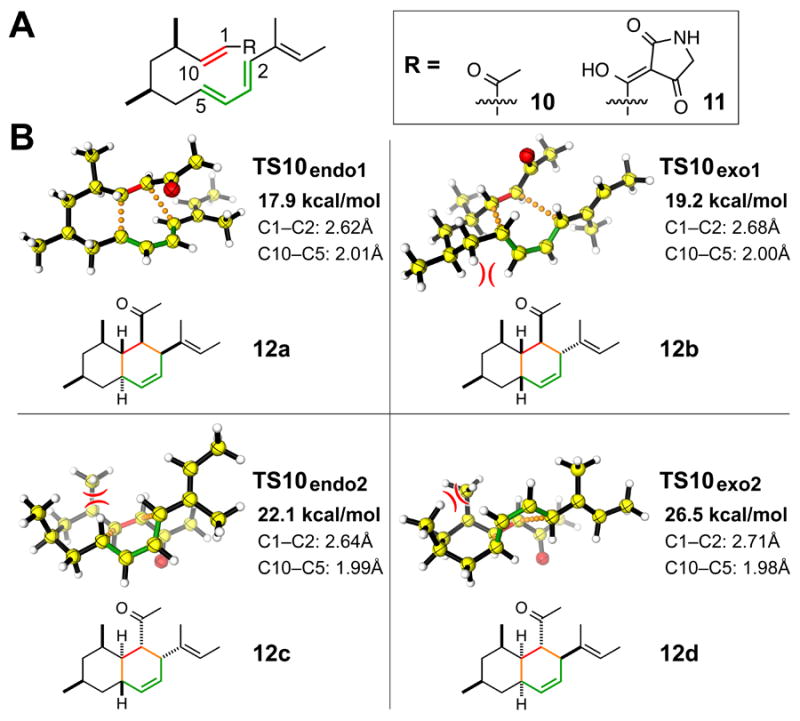
Computational studies of the proposed Diels–Alder reaction involved in the biosynthesis of Sch 210972. (A) Truncated substrates examined using density functional theory. (B) Computed transition structures of the intramolecular Diels–Alder reaction of 10. The activation free energy (bold) and th e forming bond lengths are given. Unfavorable 1,3-diaxial interactions are indicated by opposing arcs.
Sequence analysis of CghA and its homologs by FFAS03[21] identified a calycin-like β-barrel protein NE1406[22] as the closest structural homolog of CghA. Another family of calycin-like β-barrel proteins called dirigent proteins is known to sequester and orient phenoxy radicals to direct stereospecific formation of dilignols, precursors to lignin and lignans.[23] In a similar fashion, CghA-like proteins may also bind specific conformations of their straight-chain polyketide substrates to promote the [4+2] cycloaddition reaction and to control the stereoselectivity of the reaction. Computations indicate that catalysis by substrate preorganization could accelerate the cycloaddition by as much a 1,000-fold at 30 °C, as the reactive conformer for the formation of 12a is 4 kcal/mol higher in energy than the global minimum conformer. Hydrogen bonding of the enone carbonyl in the active site of CghA could also activate the precursor and further accelerate the cycloaddition. While detailed in vitro characterization of the biosynthesis of 3[4] and the recent identification of SpnF[6] and VstJ[7] indicate strongly the existence of natural enzymes that catalyze DA reactions, little remains known about how exactly these enzymes catalyze asymmetric DA reactions. Efforts to further study the CghA-catalyzed intramolecular [4+2] cycloaddition reaction are currently underway to understand the possible involvement of DA-catalyzing enzymes in natural product biosynthesis.
Supplementary Material
Acknowledgments
We would like to thank financial support from Japan Society for the Promotion of Science “Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented” (No. G2604), Amano Enzyme Foundation and Nagase Science and Technology Foundation Japan (K.W.). K.N.H acknowledges the financial support of the NSF (CHE-1059084) and the NIH (1R01GM097200). A.P. thanks the Chemistry Biology Training Grant Program (T32 GM 008496) for their support and the University of California, Los Angeles for funding. We thank UCLA’s Hoffman2 cluster and NSF’s Extreme Science and Engineering Discovery Environment (TG-CHE040013N) for computing time on the Gordon and Stampede cluster. Y.T. is supported by NIH (1DP1GM106413).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.20xxxxxxx.
References
- 1.a) Xu Y, Yamamoto N, Janda KD. Bioorg Med Chem. 2004;12:5247–5268. doi: 10.1016/j.bmc.2004.03.077. [DOI] [PubMed] [Google Scholar]; b) Preiswerk N, Beck T, Schulz JD, Milovnik P, Mayer C, Siegel JB, Baker D, Hilvert D. Proc Natl Acad Sci USA. 2014;111:8013–8018. doi: 10.1073/pnas.1401073111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaschke A, Seelig B. Curr Opin Chem Biol. 2000;4:257–262. doi: 10.1016/s1367-5931(00)00086-7. [DOI] [PubMed] [Google Scholar]
- 3.Oikawa H. In: Comprehensive Natural Products II. Chap 8. Liu H-W, Mander L, editors. VIII. Elsevier; Oxford: 2010. pp. 277–314. [Google Scholar]
- 4.Auclair K, Sutherland A, Kennedy J, Witter DJ, Van den Heever JP, Hutchinson CR, Vederas JC. J Am Chem Soc. 2000;122:11519–11520. [Google Scholar]
- 5.Kasahara K, Miyamoto T, Fujimoto T, Oguri H, Tokiwano T, Oikawa H, Ebizuka Y, Fujii I. Chembiochem. 2010;11:1245–1252. doi: 10.1002/cbic.201000173. [DOI] [PubMed] [Google Scholar]
- 6.Kim HJ, Ruszczycky MW, Choi SH, Liu YN, Liu HW. Nature. 2011;473:109–112. doi: 10.1038/nature09981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hashimoto T, Hashimoto J, Teruya K, Hirano T, Shin-Ya K, Ikeda H, Liu HW, Nishiyama M, Kuzuyama T. J Am Chem Soc. 2015;137:572–575. doi: 10.1021/ja510711x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang SW, Mierzwa R, Terracciano J, Patel M, Gullo V, Wagner N, Baroudy B, Puar M, Chan TM, McPhail AT, Chu M. J Nat Prod. 2006;69:1025–1028. doi: 10.1021/np060121y. [DOI] [PubMed] [Google Scholar]
- 9.Kakule TB, Sardar D, Lin Z, Schmidt EW. ACS Chem Biol. 2013;8:1549–1557. doi: 10.1021/cb400159f. [DOI] [PubMed] [Google Scholar]
- 10.Liu X, Walsh CT. Biochemistry. 2009;48:8746–8757. doi: 10.1021/bi901123r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boettger D, Hertweck C. Chembiochem. 2013;14:28–42. doi: 10.1002/cbic.201200624. [DOI] [PubMed] [Google Scholar]
- 12.Campbell CD, Vederas JC. Biopolymers. 2010;93:755–763. doi: 10.1002/bip.21428. [DOI] [PubMed] [Google Scholar]
- 13.Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. Nucleic Acids Res. 2008;36:W5–9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishiuchi K, Nakazawa T, Yagishita F, Mino T, Noguchi H, Hotta K, Watanabe K. J Am Chem Soc. 2013;135:7371–7377. doi: 10.1021/ja402828w. [DOI] [PubMed] [Google Scholar]
- 15.a) Ma SM, Li JW, Choi JW, Zhou H, Lee KK, Moorthie VA, Xie X, Kealey JT, Da Silva NA, Vederas JC, Tang Y. Science. 2009;326:589–592. doi: 10.1126/science.1175602. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qiao K, Chooi YH, Tang Y. Metab Eng. 2011;13:723–732. doi: 10.1016/j.ymben.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schümann J, Hertweck C. J Am Chem Soc. 2007;129:9564–9565. doi: 10.1021/ja072884t. [DOI] [PubMed] [Google Scholar]
- 17.Sims JW, Schmidt EW. J Am Chem Soc. 2008;130:11149–11155. doi: 10.1021/ja803078z. [DOI] [PubMed] [Google Scholar]
- 18.Hélaine V, Rossi J, Gefflaut T, Alaux S, Bolte J. Adv Synth Catal. 2001;343:692–697. [Google Scholar]
- 19.Nakazawa T, Ishiuchi K, Sato M, Tsunematsu Y, Sugimoto S, Gotanda Y, Noguchi H, Hotta K, Watanabe K. J Am Chem Soc. 2013;135:13446–13455. doi: 10.1021/ja405128k. [DOI] [PubMed] [Google Scholar]
- 20.Winter JM, Sato M, Sugimoto S, Chiou G, Garg NK, Tang Y, Watanabe K. J Am Chem Soc. 2012;134:17900–17903. doi: 10.1021/ja3090498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaroszewski L, Li Z, Cai XH, Weber C, Godzik A. Nucleic Acids Res. 2011;39:W38–44. doi: 10.1093/nar/gkr441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu HJ, Bakolitsa C, Skerra A, Lomize A, Carlton D, Miller MD, Krishna SS, Abdubek P, Astakhova T, Axelrod HL, Clayton T, Deller MC, Duan L, Feuerhelm J, Grant JC, Grzechnik SK, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kozbial P, Kumar A, Marciano D, McMullan D, Morse AT, Nigoghossian E, Okach L, Paulsen J, Reyes R, Rife CL, van den Bedem H, Weekes D, Xu Q, Hodgson KO, Wooley J, Elsliger MA, Deacon AM, Godzik A, Lesley SA, Wilson IA. Acta Crystallogr F. 2010;66:1153–1159. doi: 10.1107/S1744309109037749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Davin LB, Wang HB, Crowell AL, Bedgar DL, Martin DM, Sarkanen S, Lewis NG. Science. 1997;275:362–366. doi: 10.1126/science.275.5298.362. [DOI] [PubMed] [Google Scholar]; b) Pickel B, Pfannstiel J, Steudle A, Lehmann A, Gerken U, Pleiss J, Schaller A. FEBS J. 2012;279:1980–1993. doi: 10.1111/j.1742-4658.2012.08580.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.