

Abstract
Immature glutamatergic synapses in cultured neurons contain high-release probability (Pr) presynaptic sites coupled to postsynaptic sites bearing GluN2B-containing NMDA receptors (NMDARs), which mature into low-Pr, GluN2B-deficient synapses. Whether this coordinated maturation of high-Pr, GluN2B+ synapses to low-Pr, GluN2B-deficient synapses actually occurs in vivo, and if so, what factors regulate it and what role it might play in long-term synapse function and plasticity are unknown. We report that loss of the integrin-regulated Abl2/Arg kinase in vivo yields a subpopulation of “immature” high-Pr, GluN2B+ hippocampal synapses that are maintained throughout late postnatal development and early adulthood. These high-Pr, GluN2B+ synapses are evident in arg−/− animals as early as postnatal day 21 (P21), a time that precedes any observable defects in synapse or dendritic spine number or structure in arg−/− mice. Using focal glutamate uncaging at individual synapses, we find only a subpopulation of arg−/− spines exhibits increased GluN2B-mediated responses at P21. As arg−/− mice age, these synapses increase in proportion, and their associated spines enlarge. These changes coincide with an overall loss of spines and synapses in the Arg-deficient mice. We also demonstrate that, although LTP and LTD are normal in P21 arg−/− slices, both forms of plasticity are significantly altered by P42. These data demonstrate that the integrin-regulated Arg kinase coordinates the maturation of presynaptic and postsynaptic compartments in a subset of hippocampal synapses in vivo, and this coordination is critical for NMDAR-dependent long-term synaptic stability and plasticity.
SIGNIFICANCE STATEMENT Synapses mature in vitro from high-release probability (Pr) GluN2B+ to low-Pr, GluN2B−, but it is unknown why this happens or whether it occurs in vivo. High-Pr, GluN2B+ synapses persist into early adulthood in Arg-deficient mice in vivo and have elevated NMDA receptor currents and increased structural plasticity. The persistence of these high-Pr, GluN2B+ synapses is associated with a net synapse loss and significant disruption of normal synaptic plasticity by early adulthood. Together, these observations suggest that the maturation of high-Pr, GluN2B+ synapses to predominantly low-Pr, GluN2B− synapses may be essential to preserving a larger dynamic range for plasticity while ensuring that connectivity is distributed among a greater number of synapses for optimal circuit function.
Keywords: hippocampus, integrin–Arg signaling, maturation, NMDA receptor, synaptic plasticity, synaptic stability
Introduction
The remarkable synaptic plasticity observed at excitatory synapses is driven by NMDA receptors (NMDARs) that along with AMPA receptors (AMPARs) are responsible for excitatory glutamatergic transmission. NMDAR heterotetramers are composed of obligatory GluN1 subunits as well as a variable configuration of GluN2A-D and GluN3A-B subunits (Cull-Candy et al., 2001; Paoletti et al., 2013). Differences in NMDAR composition throughout development regulate synapse stability and plasticity. NMDARs in early-developing synapses are enriched for GluN2B subunits, but GluN2A subunit incorporation increases as synapses mature (Carmignoto and Vicini, 1992; Hestrin, 1992; Williams et al., 1993). Consequently, these maturing synapses exhibit faster NMDAR decay times. This transition correlates closely with reduced activity-dependent synaptic plasticity and a reduction or loss of critical periods (Carmignoto and Vicini, 1992; Hestrin, 1992; Williams et al., 1993).
Functional changes also occur on the presynaptic side in maturing synapses. At hippocampal synapses formed between CA3 axons and CA1 pyramidal neurons (CA3–CA1 synapses), neurotransmitter release probability (Pr) is high in early postnatal synapses but decreases with age (Muller et al., 1989; Bolshakov and Siegelbaum, 1995). Using single hippocampal neuron microisland cultures that form autaptic synapses, Chavis and Westbrook (2001) provided evidence that the maturation of presynaptic and postsynaptic compartments is tightly coordinated. They found that synapses with high-Pr presynaptic terminals apposed to postsynaptic compartments with GluN2B-containing NMDARs mature in an activity- and integrin-dependent manner into synapses containing low-Pr terminals coupled to GluN2B-deficient synapses. It is not known whether the maturation from high-Pr, GluN2B+ synapses to low-Pr, GluN2B− synapses observed in this model system actually occurs in vivo, and if so, what factors might regulate it or what role it might play in long-term synapse function and plasticity.
Integrins are transmembrane heterodimeric (αβ) adhesion receptors that both mediate cell–cell and cell–extracellular matrix interactions, and initiate kinase signaling cascades that modulate neighboring transmembrane proteins, including NMDARs (Lin et al., 2003; Bernard-Trifilo et al., 2005; Shi and Ethell, 2006). Integrin α3β1 adhesion receptor signaling through the Abl2/Arg nonreceptor tyrosine kinase (Arg) is critical for dendrite arbor and synapse stability. Neurons lacking integrin α3β1 or Arg develop normally through postnatal day 21 (P21), but then exhibit widespread dendrite, dendritic spine, and synapse loss by P42 (Sfakianos et al., 2007; Gourley et al., 2012; Kerrisk et al., 2013). Integrin–Arg signaling stabilizes dendrite arbors by activating the Rho GTPase inhibitor p190RhoGAP to attenuate RhoA signaling (Sfakianos et al., 2007; Gourley et al., 2012; Kerrisk et al., 2013). Inhibition of RhoA signaling rescues the dendrite arbor loss resulting from reduced integrin–Arg signaling, but these stabilized dendrites still exhibit dendritic spine and synapse loss (Sfakianos et al., 2007; Lin et al., 2013). Thus, integrin–Arg signaling must act via a distinct mechanism to influence dendritic spine and synapse stability.
We provide evidence here that Arg is required for the coordinated maturation of presynaptic release probability and postsynaptic NMDAR composition at hippocampal synapses in vivo. Loss of Arg function yields a subpopulation of “immature” high-Pr, GluN2B+ synapses, which increases from P21 through early adulthood. NMDAR-mediated currents are larger at these synapses at P21, a time that precedes observable defects in synapse or dendritic spine number or structure in arg−/− mice. Using focal glutamate uncaging at individual synapses, we find that only a subpopulation of spines exhibits increased GluN2B-mediated responses at P21. The proportion of these spines increases through P42, coinciding temporally with overall synapse loss. We also demonstrate that although long-term potentiation (LTP) and long-term depression (LTD) are normal in P21 arg−/− slices, both forms of plasticity are significantly altered by P42. Together, these data strongly suggest that the maturation of presynaptic release probability and postsynaptic NMDAR composition is coordinated by the integrin-regulated Arg kinase at a subset of hippocampal synapses in vivo, and that the disruption of this coordination leads to synapse loss and altered synaptic plasticity.
Materials and Methods
Animals and reagents.
arg−/− mice on a mixed 129Sv/J × C57BL/6 background were previously described (Koleske et al., 1998), and genotypes were determined by PCR. To control for potential genetic background and sex differences, we used male littermates at P31 and P42 (ranges, P30–32, P42–P45) and littermates of both sexes at P10 (range, P9–P10), P15 (range, P14–P15), and P21 (range, P20–P22) for all experiments. Mice were segregated by sex and held to no more than five per cage. Animals were on a 12 h light/dark cycle with food and water available ad libitum. All procedures were approved by the Yale University Animal Care and Use Committee. In some experiments, one or more of the following drugs were added to the artificial CSF (ACSF): 10 μm bicuculline methiodide (BMI), 50 μm aminophosphonovalerate (APV), 20 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 10 μm 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX), 20 μm NMDA, 5 μm AMPA, 3 μm ifenprodil (Ifen), 10 μm SU6656 (SU), 1 μm tetrodotoxin (TTX), 5 μm MK801, 10 μm Alexa Fluor-594, 300 μm Fluo-5F, 2.5 mm MNI-caged-l-glutamate (MNI-Glutamate, Tocris Bioscience), and 50 μm 3,5-dihydroxyphenylglycine (DHPG; Tocris Bioscience). All chemicals were purchased from Sigma-Aldrich, unless otherwise noted.
Hippocampal slice preparation.
Hippocampal slices were prepared as previously described with slight modifications (Moresco et al., 2003). Briefly, after rapid removal under deep Nembutal anesthesia, brains were cooled in ice-cold sucrose-containing ACSF as follows (in mm): 2.5 KCl, 7 MgSO4 · 7H2O, 1.25 NaH2PO4, 0.5 CaCl2 · 2H2O, 28 NaHCO3, 7 dextrose, and 205 sucrose saturated with 95% O2-5% CO2 at 290–320 mOsmol/L, pH 7.4. Transverse hippocampal slices (350 μm) were cut at 4°C on a vibratome (VT 1000S, Leica). Slices were transferred to a humidified, oxygenated environment at 33.5°C for at least 30 min before recording. During recording, slices were perfused at 1 ml/min with ACSF as follows (in mm): 118 NaCl, 3 KCl, 1 MgSO4 · 7H2O, 1.25 NaH2PO4, 2 CaCl2 · 2H2O, 26 NaHCO3, 10 dextrose, 0.4 ascorbic acid, 4 Na-lactic acid, and 2 Na-pyruvic acid.
Electrophysiological recordings.
Whole-cell recordings were obtained from pyramidal neurons in the hippocampal CA1 region with a patch-clamp amplifier (Axopatch 700B; Molecular Devices) under visual control using differential interference contrast and infrared optics via a water-immersion objective (NIR Apo 40×; BX51, Nikon) at room temperature. Evoked excitatory postsynaptic currents (EPSCs) were induced by repetitive 0.05 Hz stimulations delivered by a bipolar tungsten-stimulating electrode placed in the Schaffer collaterals (SCs) and were recorded in pyramidal neurons ∼100 μm from the stimulating electrode. Stimulus intensities were adjusted to achieve 50–60% of maximal responses. Recording electrodes made from 1.5 mm glass capillaries were pulled on a Flaming-Brown micropipette puller (P-96, Sutter Instrument) and filled with an intracellular solution containing the following (in mm): 137 cesium gluconate, 10 NaCl, 0.2 EGTA, 4 Na2ATP, 0.3 Na3GTP, 5 Na2-phosphocreatine, 10 HEPES, and 5 QX-314 chloride adjusted to 300 mOsmol/L and pH 7.4. With this solution, the recording electrodes exhibited a resistance of 2–6 MΩ. Signals were low-pass filtered at 2 kHz and digitized at 5 kHz with a digitizer (Digidata 1440, Molecular Devices). To ensure that EPSCs were comparable in different slices, we computed the input–output relations of the evoked EPSCs in CA1. The input is given by the amplitude of the Schaffer collateral stimulus that precedes the postsynaptic response, whereas the output is given by the initial amplitude of the EPSC. The input–output relations were indistinguishable between wild-type (WT) and arg−/− mice slices.
The AMPAR-mediated component of EPSCs (AMPAR-EPSCs) was recorded in ACSF containing APV in neurons voltage clamped at −70 mV. NMDAR-EPSCs were recorded in ACSF containing CNQX in neurons voltage clamped at negative (−40 mV) as well as at positive (+40 mV) potentials. NMDAR-EPSCs were measured at 40 ms after the stimulus artifact, a time point at which AMPAR currents have decayed (Choi et al., 2000). Some experiments included the GluN2B-specific antagonist ifenprodil (3 μm). Intrinsic neuronal membrane properties and excitability were measured in the presence of BMI, APV, and CNQX to block synaptic inputs via GABAA receptor, NMDAR, and AMPAR, respectively.
In the paired-pulse ratio (PPR) experiments, the paired-pulse stimulation (interpulse intervals, 75 ms) was delivered via a bipolar tungsten stimulating electrode placed in CA3 Schaffer collateral axons in the hippocampus. The second EPSC (EPSC2), which was evoked by the second of the paired-pulse stimulations, was divided by EPSC1 to obtain a PPR (Zucker and Regehr, 2002).
To examine the rate of amplitude decay at −70 mV, we applied 11 pulses at 40 Hz every 6 s. Stimulus intensity was adjusted to obtain an ∼100 pA response on the first pulse. The time constant of synaptic depression was obtained by fitting the following single exponential equation: I(t) = (I0 − PL) exp(−t/τd) + PL, where I(t) is the normalized EPSC amplitude when time is t, τd is the rate constant of the synaptic depression (Schneggenburger et al., 2002), PL is the normalized steady-state amplitude and I0 + PL = 1.
To measure the probability of transmitter release, the kinetics of MK801 blockade were calculated from the rate of exponential decay of the amplitude of the evoked NMDAR-EPSCs (Hessler et al., 1993; Rosenmund et al., 1993; Huang and Stevens, 1997). MK801 (5 μm) was added to the bath, and responses were evoked every 20 s until the NMDAR-mediated response was abolished. The time constant of MK801 blockade (τblock) was calculated via the following equation: I(t) = I0 exp(−t/τblock), where I(t) is the normalized amplitude of NMDAR-EPSC when time is t, and I0 is the amplitude of the first pulse.
To describe the decay kinetics of NMDAR-EPSCs, a nonlinear regression in pClamp version 10.3 software (Molecular Devices) was used to fit double exponentials to decay curves of NMDAR-EPSCs via the following equation: I(t) = Ifast exp(−t/τfast) + Islow exp(−t/τslow), where I(t) is the amplitude of NMDAR-EPSCs when time is t, Ifast and Islow are the peak amplitudes of the fast and slow components, respectively, and τfast and τslow are their respective time constants. The weighted time constant (τweighted) was used for quantification purposes and was calculated as τweighted = τfast * (Ifast/(Ifast + Islow)) + τslow * (Islow/(Ifast + Islow)).
For AMPAR-mediated miniature EPSC (mEPSC) recordings, TTX was added to the external solution. To record NMDAR-mediated mEPSCs, neurons were held in voltage clamp at −40 mV in the presence of TTX, BMI, and CNQX in ACSF with low Mg2+ (0.1 mm) and high Ca2+ (3.8 mm) to partially remove the Mg2+ block. The amplitude, frequency, and decay time of miniature events were detected using MiniAnalysis software (Synaptosoft). The events were further inspected visually to exclude overlapped events and events on a noisy baseline.
Theta-burst stimulation (TBS; five trains of four pulses at 100 Hz, 200 ms intervals; repeated four times with intervals of 10 s) was used to induce LTP. Low-frequency stimulation (LFS; 900 stimuli at 1 Hz) or DHPG (50 μm) was used to induce LTD. In these experiments, BMI was always present. The series resistance (Rs) was monitored during recording. Cells were excluded from the analysis if Rs deviated >20% or if Rs exceeded 20 MΩ. Data were collected with pClamp version 10.3 software and analyzed using Clampfit version 10.3.
Two-photon glutamate uncaging.
NMDAR-mediated responses in single dendritic spines were obtained using two-photon laser uncaging of glutamate. Glass electrodes (3.2–3.8 MΩ) were filled with an internal solution containing the following (in mm): 135 CsMeSO3, 10 HEPES, 4 MgCl2, 4 Na2ATP, 0.4 NaGTP, and 10 sodium creatine phosphate, adjusted to pH 7.3 with KOH and used to voltage clamp CA1 pyramidal neurons at −70 mV. Slices were bathed in ACSF containing 0 mm extracellular Mg2+, TTX, and the AMPAR antagonist NBQX. Neurons were filled with red-fluorescent Alexa Fluor-594 for at least 15 min before imaging to visualize cell morphology. Two-photon imaging was accomplished with a custom-built microscope essentially as previously described (Chiu et al., 2013). Fluorophores were excited by a pulsed titanium-sapphire laser tuned to 840 nm. A second titanium-sapphire laser tuned to 720 nm was used to photo-uncage glutamate for focal stimulation of single dendritic spines. Laser power was calibrated for each spine by directing the uncaging spot to the middle of the spine head and adjusting the uncaging power to achieve 50% photobleaching of the Alexa Fluor-594 dye filling the spine. MNI-Glutamate was bath applied. Stimulating spines on the secondary or tertiary apical dendrites (within 150 μm from the soma) evoked uncaging EPSCs. Synaptic glutamate receptors were stimulated by uncaging MNI-Glutamate with brief two-photon laser flashes at four to five separate locations around a single spine head to find a “hot spot,” the place of the largest response, considered the synaptic area. To selectively stimulate receptors on the targeted spine head membrane, we chose spines that were well separated from their parent dendrite and from other spines. We collected six to eight uncaging EPSC trials (20 s between stimuli) from each hot spot location, and amplitudes were averaged.
Tissue collection and subcellular fractionation.
In all cases, hippocampi were dissected under deep Nembutal anesthesia. Wild-type and arg−/− littermate hippocampi were homogenized in 2× SDS sample buffer (50 mm Tris, pH 6.8, 4% SDS, and 10% glycerol) containing protease (10 μg/ml leupeptin, 20 μg/ml aprotinin, 10 μg/ml pepstatin A, 10 μg/ml chymostatin, 1 mm benzamidine, and 1 mm PMSF) and phosphatase (1 mm NaVO4, 2 mm NaF) inhibitors, boiled for 10 min, and centrifuged at 18,000 × g for 10 min to remove the remaining insoluble material.
To isolate a synapse-enriched fraction to measure GluN2B levels and phosphorylation, hippocampi were dissected from WT and arg−/− mice, snap frozen in liquid nitrogen, and stored at −80°C until processing. All fractionation steps took place at 4°C, all centrifugation steps were completed in a tabletop centrifuge, and all buffers contained protease inhibitors (see above) and 1× HALT Phosphatase Inhibitor Cocktail (Pierce). Hippocampi from individual mice were homogenized in ice-cold homogenization buffer (0.32 m sucrose, 10 mm HEPES, pH 7.4, and 1 mm EDTA) and centrifuged twice at 900 × g for 10 min and once for 6 min to remove unhomogenized material and nuclei. The supernatant was centrifuged at 12,000 × g for 20 min to separate the cytosolic and crude synaptosomal membrane fractions. The synaptosomal membrane was washed twice by resuspending the pellet in wash buffer (4 mm HEPES, pH 7.4, and 1 mm EDTA) and centrifuging at 12,000 × g for 20 min, then resuspended in homogenization buffer containing 0.5% Triton X-100, rotated for 15 min, then centrifuged at 12,000 × g for 20 min. The supernatant from this spin (TxS) is enriched for Triton-soluble proteins that are not associated with postsynaptic densities, while the pellet (TxP) is enriched in synaptic proteins associated with the postsynaptic density. The TxP was solubilized for 1 h in homogenization buffer containing 1% Triton X-100, 1% SDS, and 1% sodium deoxycholate, then centrifuged at 10,000 × g for 15 min to remove any remaining insoluble material, and the supernatant was used as the synapse-enriched fraction. All fractions were snap frozen in liquid nitrogen and stored at 80°C until use.
Immunoblotting.
Protein concentrations were determined by BCA assay (Pierce). Equal amounts of protein (4–30 μg) were completely solubilized by boiling for 10 min with 1× SDS sample buffer (plus 2% β-mercaptoethanol), resolved by SDS-PAGE, and transferred to nitrocellulose membranes. The samples were immunoblotted with antibodies to GluA1, GluA2/3, GluA4, actin (Millipore), GluN1 (Cell Signaling Technology), GluN2A (BD Biosciences), GluN2B (NeuroMab), pY1472 GluN2B (PhosphoSolutions), or HSP70 (Santa Cruz Biotechnology), and were detected using chemiluminescence. Bands on scanned films were quantified using Quantity One software (Bio-Rad) to determine relative changes between WT and arg−/− mice.
Statistical analysis.
Analyses were performed in small groups with ongoing power analysis to estimate the number of replicates needed. Our tests are specified as two sided. Variance was similar between the groups. Data were excluded if they were >3 SDs from the mean. Wild-type littermate controls were used for comparison with the mutant mice. While experiments were not blinded to genotype, the same criteria were applied to all experiments within an experimental group.
EPSCs were calculated from stimulus–response curves obtained from at least four slices from four animals in each experimental group. For whole-cell patch recordings, the amplitudes of Schaffer collateral-evoked EPSCs were calculated around the peak response and were averages of 10 EPSCs. Evoked EPSCs were interleaved with a 20 s intertrial interval. Analysis of mEPSCs was performed using Mini Analysis Software (Synaptosoft). Uncaging data were acquired using National Instruments data acquisition boards and custom software written in MATLAB (MathWorks). Off-line analysis was performed using custom routines written in MATLAB and Igor Pro (Wavemetrics), as described previously (Chiu et al., 2013).
For gamma mixture analysis of uncaging-evoked EPSCs (uEPSCs), uEPSC amplitudes from WT and arg−/− slices were fit using an expectation maximization algorithm (mixtools package in R) to a mixture of gamma distributions containing either one or two components. The theoretical distribution of the log likelihood ratio under the null hypothesis that the data from each condition are gamma distributed with one component is asymptotically χ2 with df = 2. The data were assumed to fit a two-component gamma model if p < 0.05.
To measure the spine head area, z-stacks of each image were collapsed by maximum intensity projection and noise was suppressed by Gaussian filtering in ImageJ. We used an empirically determined cutoff that was the same for every image to threshold the images. Under manual supervision and blinded to sample identity, spines in each image were segmented by watershed transform, spines were identified, and the head area in the x–y-plane was quantified. To obtain quantitative estimates of the association between amplitude and area by genotype group, we performed multivariate linear least-squares regression of the NMDAR-uEPSC amplitude–genotype interaction in the spine head area, while controlling for age and genotype. Although amplitude alone is a significant predictor of spine head area, we wished to obtain stratified estimates of its relationship with spine head area by genotype. Regression results are expressed as estimates with SE, and associated p values obtained from the respective t statistic.
For immunoblots, WT and littermate arg−/− lysates were run simultaneously, and each experimental sample was normalized to its WT littermate. For all experiments, Student's t test (two-tailed, unpaired), and one-way, two-way, or repeated-measures ANOVA followed by post hoc Student–Newman–Keuls test were used to identify significant differences. In all cases, data are presented as the mean ± SEM, and p < 0.05 was considered statistically significant. Throughout the figures, representative traces of evoked EPSCs are the average of 10 recordings; representative NMDA-induced, AMPA-induced, and mEPSC traces are from individual recordings; and uEPSC traces are the average of all recordings.
Results
Release probability is increased at late postnatal arg−/− hippocampal synapses
Early postnatal hippocampal synapses exhibit a higher Pr values that is reduced by P15 (Muller et al., 1989; Bolshakov and Siegelbaum, 1995). Paired-pulse facilitation (PPF) is absent or even negative at hippocampal synapses at P5–P6 because of the high initial Pr (Zucker and Regehr, 2002; Collin et al., 2005), but it manifests by P15 in concert with the conversion of these synapses to lower Pr. Our previous studies using extracellular field recordings indicated that PPF was reduced in arg−/− hippocampal CA3–CA1 synapses (Moresco et al., 2003), suggesting a possible alteration in Pr. To further explore this, we used whole-cell recordings of CA3 SC-evoked EPSCs in hippocampal CA1 pyramidal neurons to measure the PPR in these mice. While PPR did not differ between WT and arg−/− slices at P10 and P15 (Fig. 1A; unpaired t test: P10: p = 0.63; P15: p = 0.35), PPR was significantly reduced in arg−/− slices relative to WT at P21, P31, and P42 (Fig. 1A; unpaired t test: P21: p = 0.01; P31: p = 0.005; P42: p = 0.01). Notably, reduced PPR was observed at P21 (Fig. 1A), before detectable dendrite, spine, or synapse loss is observed in arg−/− CA1 neurons (Sfakianos et al., 2007; Gourley et al., 2012). We next monitored the rate of synaptic response decay in CA1 neurons in WT and arg−/− slices at P31 following bursts of 40 Hz stimulation to CA3-SC axons. The rate of synaptic depression was greatly increased in arg−/− slices relative to WT, as evidenced by a shorter depression time decay constant (τd; Fig. 1B; two-way ANOVA: WT: τd = 19.94 ± 3.94 stimuli; arg−/−: τd = 9.16 ± 1.77 stimuli; p = 0.03; F(1,66) = 30.3). These results are consistent with a higher Pr in arg−/− mice, leading to increasingly restricted neurotransmitter availability for subsequent release (Schneggenburger et al., 2002; Zucker and Regehr, 2002).
Figure 1.

Pr is increased at synapses in arg−/− hippocampal slices. A, arg−/− hippocampal synapses have decreased PPR compared with WT synapses. Top, Representative traces of EPSCs from P10 and P42 WT and arg−/− hippocampal CA1 neurons at −70 mV holding potential evoked by paired-pulse stimulation of hippocampal CA3 neurons at 70 ms interstimulus intervals. Bottom, Summary graph of PPRs recorded from WT and arg−/− mice at P10, P15, P21, P31, and P42. Throughout the figures, data represent the mean ± SEMs, and the values in each column are the number of cells used. *p < 0.05, **p < 0.01 vs age-matched WT; #p < 0.05 vs P10 WT. B, Loss of Arg increases the rate of synaptic depression. Top, Representative traces of synaptic responses recorded at −70 mV in hippocampal slices from WT and arg−/− mice at P31 in response to a brief 40 Hz stimulation train (11 pulses). Bottom, Summary plot of the normalized amplitude of synaptic responses in WT (open circles) and arg−/− (closed circles) mice. Responses were normalized to the first pulse. *p < 0.05 vs WT. C, NMDAR-EPSC blockade by MK801 is faster in hippocampal neurons from arg−/− mice than from WT mice. Graph, NMDAR-EPSC amplitude in WT (open circles) and arg−/− (closed circles) slices in response to 20 s interval stimuli for 25 min. Amplitudes were normalized to the first stimulus. Inset, Representative traces of NMDAR-EPSCs after MK801 treatment in WT (points 1, 3 and 5) and arg−/− slices (points 2, 4, and 6). Points 1 and 2, points 3 and 4, and points 5 and 6 are from fifth, 30th, and 55th stimulus, respectively. *p < 0.05 vs WT. D, E, Membrane potential (Vm; D) and input resistance (Rin; E) in CA1 neurons from P42 hippocampal slices are not altered by the loss of Arg. F, Neuron excitability is not altered by the loss of Arg function. Spikes were evoked by injection of a 2.0 nA, 600 ms depolarizing current in CA1 neurons from P42 WT and arg−/− mice. Excitability was measured as the mean number of spikes fired in response to increasing depolarizing current steps in CA1 neurons from P42 WT (open circles) and arg−/− (closed circles) hippocampal slices. G, CA1 pyramidal cell EPSCs evoked by a range of SC stimulation intensities do not differ between WT (open circles) and arg−/− (closed circles) hippocampal slices. The input–output relationship of evoked EPSCs is indistinguishable between WT and arg−/− slices, suggesting that SC-evoked EPSCs are comparable across hippocampal slices. EPSCs were recorded in the presence of BMI.
We also took advantage of the pharmacological properties of MK801, an irreversible open-channel NMDAR blocker, to monitor Pr in WT and arg−/− mice (Fig. 1C). MK801 block relates directly to Pr: the higher the Pr, the faster the rate of block by MK801 (Hessler et al., 1993; Rosenmund et al., 1993; Huang and Stevens, 1997). Consistent with the reduced PPF and increased synaptic depression rate, we found that MK801 yielded a faster inhibition rate in P31 arg−/− slices than in WT slices (Fig. 1C; two-way ANOVA: WT: τblock = 26.46 ± 3.93 stimuli; arg−/−: τblock = 15.56 ± 1.97 stimuli; p = 0.03; F(1,760) = 347.5). Collectively, these results indicate that Pr is increased at CA3–CA1 synapses in arg−/− hippocampus. To rule out the possibility that age-dependent changes in dendrite structure and spines previously reported in arg−/− mice impact basic neuronal physiology, we also measured the intrinsic membrane properties and excitability of CA1 pyramidal neurons in hippocampal slices from WT and arg−/− mice at P42. CA1 pyramidal neurons in arg−/− slices did not exhibit differences in resting membrane potential (Fig. 1D; unpaired t test: p = 0.25), input resistance (Fig. 1E; unpaired t test: p = 0.28), or spike frequency in response to increasing depolarizing current steps (Fig. 1F; unpaired t test: p = 0.80) compared with WT slices. In addition, the input–output relationship of SC-evoked EPSCs was indistinguishable between WT and arg−/− slices, suggesting that SC-evoked EPSCs are comparable across hippocampal slices (Fig. 1G; unpaired t test: p = 0.89).
NMDAR amplitudes and decay kinetics are altered in arg−/− mice
To test whether the increased Pr at hippocampal synapses in arg−/− mice is accompanied by postsynaptic changes, we next measured SC-evoked NMDAR-mediated and AMPAR-mediated EPSCs in the same neurons by voltage clamping at +40 and −70 mV, respectively. The NMDAR/AMPAR ratio was increased at P21, P31, and P42 in arg−/− slices compared with WT littermates (Fig. 2A; unpaired t test: P21: p = 0.02; P31: p = 0.03; P42: p = 0.02). Strikingly, the increased NMDAR/AMPAR ratio was observed at P21, a time that precedes dendrite, dendritic spine, and synapse loss in arg−/− CA1 neurons (Sfakianos et al., 2007; Gourley et al., 2012). Given that neuronal excitability and input–output relations are unaffected by loss of Arg (Fig. 1D–G), this robust increase in evoked NMDAR/AMPAR ratio strongly suggests that loss of Arg function differentially affects synaptic NMDAR-mediated versus AMPAR-mediated transmission.
Figure 2.

Arg-deficient slices exhibit unaffected AMPAR function and larger NMDAR-mediated currents. A, arg−/− CA1 neurons have an increased NMDAR/AMPAR-EPSC ratio. Top, Representative traces of NMDAR-EPSCs (recorded at +40 mV holding potential) and AMPAR-EPSCs (recorded at −70 mV holding potential) recorded in the same WT and arg−/− CA1 neurons at P21, P31, and P42. Bottom, Histogram represents the average ratios of evoked NMDAR-EPSCs to AMPAR-EPSCs in the same WT and arg−/− CA1 neurons at each age. *p < 0.05 vs age-matched WT. B, Loss of Arg does not affect AMPA-induced currents. Left, Representative traces of AMPA-induced currents recorded in WT and arg−/− CA1 neurons at P42 after 60 s of AMPA bath application in the presence of TTX and NMDAR antagonist APV. Right, Bar graph represents the average amplitude of AMPA-induced currents in WT and arg−/− CA1 neurons at P21 and P42. C, Cell surface NMDAR levels are increased in arg−/− mice. Top, Representative traces of NMDA-induced currents recorded in WT and arg−/− CA1 neurons after 60 s of NMDA bath application in low-Mg2+ ACSF with TTX and AMPAR antagonist CNQX. Bottom, Bar graph represents the average amplitude of NMDA-induced currents in WT and arg−/− CA1 neurons at P21, P31, and P42. *p < 0.05 vs WT.
To determine whether altered NMDAR- or AMPAR-mediated transmission caused the increased NMDAR/AMPAR ratio, we bath applied AMPA for 60 s to activate surface AMPARs and measured AMPA-evoked currents. AMPA-induced currents were not different in arg−/− slices compared with WT slices at P21 or P42 (Fig. 2B; unpaired t test: P21: p = 0.88; P42: p = 0.70). We also recorded NMDAR-mediated currents in WT and arg−/− slices by applying NMDA for 60 s in low Mg2+ ACSF. NMDA-induced currents were significantly increased in arg−/− slices relative to WT at P21, P31, and P42 (Fig. 2C; unpaired t test: P21: p = 0.04; P31: p = 0.03; P42: p = 0.01). In fact, we observed a 30% age-dependent increase in NMDAR-evoked currents in arg−/− mice between P21 and P42. Together with the increased NMDAR/AMPAR ratio, these results demonstrate that loss of Arg selectively increases NMDAR function while leaving AMPAR-mediated responses unchanged.
To ascertain whether synaptic AMPAR function is specifically modified in arg−/− mice, we recorded AMPAR-mediated mEPSCs (AMPAR-mEPSCs) in both WT and arg−/− mice at P21 and P31. AMPAR-mEPSC amplitudes were similar in WT and arg−/− mice at both P21 and P31 (Fig. 3Ai,Aii; unpaired t test: P21: p = 0.58; P31: p = 0.33). AMPAR-mEPSC frequency, which is thought to reflect both the number of release sites and Pr (Kerchner and Nicoll, 2008), was not different in arg−/− mice relative to WT mice at P21 (Fig. 3Aiii; unpaired t test: p = 0.36), but was significantly reduced in arg−/− mice at P31 (Fig. 3Aiii; unpaired t test: p = 0.04). This reduced mEPSC frequency is consistent with the previously documented loss of CA1 spine and synapse density in arg−/− mice as early as P26 (Sfakianos et al., 2007; Gourley et al., 2012). To characterize whether the persistence of high-Pr, GluN2B+ synapses was also specifically associated with changes in synaptic NMDAR responses in arg−/− mice, we also examined NMDAR-mEPSCs at P21 and P31. A trend toward increased NMDAR-mEPSC amplitudes was observed at P21 in arg−/− slices, and NMDAR-mEPSC amplitudes were significantly increased by P31 (Fig. 3Bi,Bii; unpaired t test: P21: p = 0.06; P31: p < 0.0001), consistent with increased synaptic NMDAR function. NMDAR-mEPSC frequency did not differ between WT and arg−/− mice at P21, but it was significantly reduced in arg−/− slices compared with WT slices at P31 (Fig. 3Bi,Biii; unpaired t test: P21: p = 0.82; P31: p = 0.03), again consistent with the time course of synapse loss in these mice.
Figure 3.

arg−/− CA1 neurons have enhanced synaptic NMDAR function. A, Frequency of AMPAR-mEPSCs is decreased in arg−/− mice at P31. Ai, Representative AMPAR-mEPSCs recorded at a −70 mV holding potential in the presence of TTX from WT and arg−/− CA1 neurons at P21 and P31. Aii, AMPAR-mEPSC amplitude is similar in CA1 neurons from arg−/− hippocampal slices compared with WT neurons at both P21 and P31. Aiii, AMPAR-mEPSC frequency trends toward an increase at P21 and is decreased at P31 in arg−/− CA1 neurons compared with WT. *p < 0.05 vs WT. B, NMDAR-mEPSCs are altered in arg−/− mice. Bi, Representative NMDAR-mEPSCs recorded at −40 mV holding potential in the presence of CNQX from WT and arg−/− CA1 neurons at P21 and P31. Bii, NMDAR-mEPSC amplitude is increased in CA1 neurons from arg−/− hippocampal slices compared with WT at P31. **p < 0.01 vs WT. Biii, NMDAR-mEPSC frequency is decreased in arg−/− CA1 neurons compared with WT neurons at P31. *p < 0.05 vs WT; #p < 0.05 vs P21 arg−/−.
Decay times of synaptic NMDAR-mediated responses are increased at arg−/− synapses
NMDAR-mediated EPSCs in early postnatal synapses have long decay times that transition in an activity-dependent manner to shorter decay times (Carmignoto and Vicini, 1992; Hestrin, 1992). We noted that, in addition to their increased amplitudes in arg−/− mice, the shape of NMDAR-mediated response curves was also changed. As measured by τweighted, NMDAR-EPSC decay kinetics were noticeably slower in P31 arg−/− slices than WT slices (Fig. 4Ai; unpaired t test: p = 0.01). The NMDAR-EPSC decay phases were well fit by the sum of two exponentials (τfast and τslow). The τslow was selectively increased in arg−/− neurons (Fig. 4Aii; unpaired t test: p = 0.01), while τfast was not significantly different between arg−/− and WT neurons (Fig. 4Aiii; unpaired t test: p = 0.07), indicating that the changes in τweighted mainly occurred because of a change in τslow. Together, these data indicate that arg−/− hippocampal synapses exhibit larger NMDAR-mediated EPSCs with slower decay kinetics.
Figure 4.

arg−/− CA1 neurons have longer NMDAR-EPSC decay time constants. A, Loss of Arg increases the NMDAR-EPSC decay time constant. Ai, Left, Representative traces of CA1 neuron NMDAR-EPSCs recorded at +40 mV holding potential from WT and arg−/− hippocampal slices. Right, Bar graph shows the average weighted decay time constant of NMDAR-EPSCs from WT and arg−/− CA1 neurons. Aii, Aiii, Bar graph shows the average slow component (Aii) and fast component (Aiii) of the decay time constant of NMDAR-EPSCs recorded in WT and arg−/− CA1 neurons. **p < 0.01 vs WT. B, Loss of Arg increases the proportion of NMDAR-mEPSCs with slow decay times. Bi, Representative NMDAR-mEPSC recordings from WT and arg−/− hippocampal neurons at P31. WT events, n = 22,196 from 37 neurons; arg−/− events, n = 23,367 from 45 neurons. Bii, Histogram shows NMDAR-mEPSC event decay times were shifted toward longer decay times in arg−/− (red) compared with WT (blue) mice. Biii, Biv, Decay times of NMDAR-mEPSC events in WT (Biii) and arg−/− (Biv) mice were well fit by a two-component Gaussian distribution (green, faster decay time constant; red, slower decay time constant). The proportion of NMDAR-mEPSCs with slower decay times was larger in arg−/− mice and smaller in WT mice.
We also analyzed the decay times of individual NMDAR-mEPSC events at P31. These events were aligned by their onset and fitted to a single exponential function. NMDAR-mEPSC decay times were differently distributed in slices from WT versus arg−/− mice (two-sample Kolmogorov–Smirnov test: p < 0.01; Fig. 4Bi,Bii). The individual events in the WT slices were well fit as a mixture of the following two Gaussian distributions: one fast-decaying and one slow-decaying population. The overall distribution in arg−/− slices was more heterogeneous, but a slow-decaying population predominated in arg−/− slices (Fig. 4Biv), while the fast-decaying population predominated in WT slices (Fig. 4Biii; WT: 56% fast, 44% slow; arg−/−: 32% fast, 68% slow). Together, these data indicate that the persistence of immature-like synapses in arg−/− mice is associated with increased synaptic NMDAR response amplitude and decay time.
Enhanced NMDAR postsynaptic responses in arg−/− mice are mediated by GluN2B-containing NMDARs apposed to high-Pr presynaptic terminals
Longer NMDAR decay times in early-developing synapses are mediated by GluN2B subunits, which are sensitive to the GluN2B-selective antagonist ifenprodil (Carmignoto and Vicini, 1992; Hestrin, 1992; Williams et al., 1993). The finding that NMDAR-EPSC decay times are slower in arg−/− slices (Fig. 4) suggests that GluN2B-containing NMDARs (GluN2B-NMDARs) may contribute more substantially to NMDAR currents in these mutants. Indeed, the GluN2B-NMDAR-specific inhibitor ifenprodil partly inhibited NMDAR-EPSCs in both WT and arg−/− slices, but had a significantly greater inhibitory effect on arg−/− slices at all ages. Ifenprodil normalized NMDAR-EPSCs in arg−/− slices, making them similar in magnitude to those observed in age-matched ifenprodil-treated WT slices [Fig. 5A: unpaired t test; WT + Ifen: p = 0.02 (Fig. 5Ai); p = 0.001 (Fig. 5Aii); p < 0.0001 (Fig. 5Aiii); arg−/−: p = 0.04 (Fig. 5Ai); p = 0.006 (Fig. 5Aii); p = 0.007 (Fig. 5Aiii); arg−/− + Ifen: p = 0.001 (Fig. 5Ai); p < 0.0001 (Fig. 5Aii); p = 0.0004 (Fig. 5Aiii)], indicating that the increased NMDAR-EPSCs in arg−/− mice are mediated by GluN2B-containing receptors. Proportionally more of the NMDAR current was inhibited by ifenprodil in both WT and arg−/− slices as the mice aged, and at all ages, the ifenprodil-sensitive fraction of the NMDAR-EPSC was greater in arg−/− slices than in age-matched WT slices (Fig. 5B). These observations indicate that NMDARs containing GluN2B subunits are the major component of the increased NMDAR-EPSCs observed in arg−/− mice.
Figure 5.
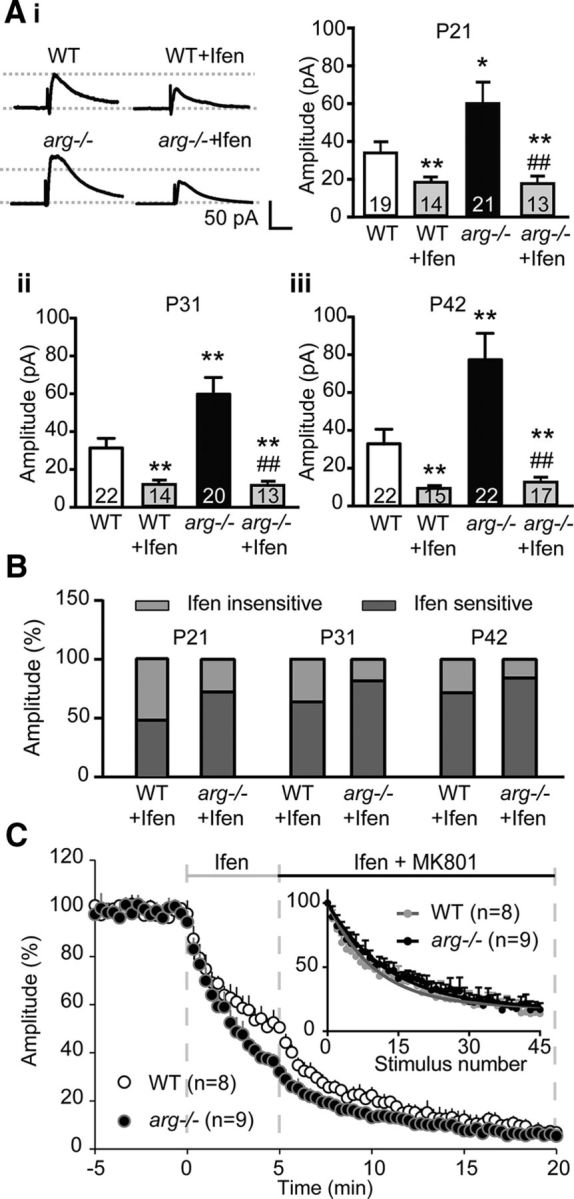
GluN2B-mediated NMDAR-EPSCs are increased in arg−/− hippocampal slices. A, Loss of Arg increases GluN2B-containing NMDAR-EPSC amplitude. Ai, Left, Representative traces of NMDAR-EPSCs recorded from CA1 hippocampal neurons with or without the GluN2B antagonist Ifenprodil (Ifen) from WT and arg−/− hippocampal slices at P42. Ai–Aiii, Loss of Arg increases NMDAR-EPSCs compared with WT. Ifenprodil partially blocks both WT and arg−/− NMDAR-EPSCs at P21 (Ai), P31 (Aii), and P42 (Aiii). Note that ifenprodil treatment blocks the increased NMDAR-EPSCs in arg−/− neurons to levels comparable to those of WT + Ifen. *p < 0.05, **p < 0.01 vs WT; ##p < 0.01 vs arg−/−. B, Ifenprodil blocks a larger portion of the NMDAR-EPSC in arg−/− neurons. Histogram represents the percentage of the NMDAR-EPSC that is sensitive and insensitive to ifenprodil. C, Block of GluN2B-containing NMDARs silences high-Pr synapses in arg−/− neurons. Five minutes of ifenprodil treatment reduces NMDAR-EPSC amplitude in CA1 neurons from arg−/− slices faster than in WT neurons. The addition of MK801 after 5 min further inhibits NMDAR-EPSCs, but at a similar rate in WT and arg−/−. Inset, When NMDAR-EPSC amplitudes are renormalized to the time of MK801 addition, it is evident that the rate of MK801 inhibition of NMDAR-EPSC amplitude in WT (gray circles) and arg−/− (black circles) neurons is not different. The stimulus interval is 20 s.
Previous work on autaptic synapses formed in culture revealed that high-Pr terminals are apposed to postsynaptic compartments bearing GluN2B-containing NMDARs (Chavis and Westbrook, 2001). If these were occurring in vivo, blocking GluN2B-containing NMDARs should “silence” the high-Pr terminals, and the remaining synapses should be ifenprodil resistant with low Pr and thus exhibit slow inhibition by MK801. We recorded at least 5 min of stable baseline recordings of SC-evoked NMDAR-EPSCs, and then added ifenprodil for 5 min to block GluN2B-containing NMDARs in both WT and arg−/− slices at P31. Ifenprodil reduced NMDAR-EPSCs by 67% in arg−/− neurons, but only by 47% in WT neurons (Fig. 5C), which is consistent with the increased GluN2B-mediated component in arg−/− neurons. The MK801 inhibition rate was much slower in arg−/− neurons in the presence of ifenprodil than in its absence (compare Fig. 5C and Fig. 1C). In fact, when ifenprodil was present, the MK801 inhibition rate in NMDAR-EPSCs was not significantly different between arg−/− and WT mice (two-way ANOVA: WT: τblock = 18.43 ± 2.11 stimuli; arg−/−: τblock = 20.97 ± 1.62 stimuli; p = 0.36). This is most clearly observed when the WT and arg−/− plots are renormalized to the beginning of MK801 treatment (Fig. 5C, inset). These data are consistent with an increased population of high-Pr, GluN2B+ synapses in arg−/− mice hippocampi.
Loss of Arg does not affect GluN2B tyrosine 1472 phosphorylation
NMDAR and AMPAR subunit levels were not different at the whole-cell level at P21 (data not shown) or P42 (Fig. 6A; two-way ANOVA: interaction: p = 0.27, F(5,94) = 1.3; main effect of protein: p = 0.27, F(5,94) = 1.3; main effect of genotype: p = 0.9434, F(1,94) = 0.005), suggesting that the loss of Arg function does not grossly impact glutamate receptor subunit expression. Src family kinase-mediated GluN2B phosphorylation at tyrosine (Y) 1472 promotes GluN2B surface localization and increases NMDAR-EPSCs (Salter and Kalia, 2004; Prybylowski et al., 2005). We hypothesized that the loss of Arg might somehow trigger increased Src-mediated Y1472 phosphorylation, resulting in increased surface GluN2B. However, we found that Y1472 phosphorylation was not significantly different in the synapse-enriched fractions of P31 WT and arg−/− hippocampi (Fig. 6B; two-way ANOVA: interaction: p = 0.27, F(4,65) = 1.3; main effect of protein: p = 0.27, F(4,65) = 1.3; main effect of genotype: p = 0.60, F(1,65) = 0.3). GluN1, GluN2B, and GluA1 levels in the synaptic fraction of WT and arg−/− hippocampi were also not different (Fig. 6B). Further, treating arg−/− slices with the Src family kinase inhibitor SU6656 did not reduce the enhanced arg−/− NMDAR-EPSCs to WT levels [Fig. 6C; unpaired t test: WT + SU: p = 0.02 (P21); p = 0.01 (P31); p = 0.02 (P42); arg−/−: p = 0.04 (P21); p = 0.008 (P31); p = 0.01 (P42); arg−/− + SU: p = 0.56 (P21); p = 0.007 (P31); p = 0.01 (P42)] as ifenprodil does (Fig. 3A). Together, these data indicate that the increased NMDAR function in arg−/− slices does not result from increased Src-mediated GluN2B Y1472 phosphorylation.
Figure 6.

Src-mediated GluN2B tyrosine phosphorylation does not underlie the increased NMDAR-EPSCs in arg−/− mice. A, Whole-cell hippocampal levels of NMDAR and AMPAR subunits are not changed at P42 in arg−/− mice. Top, Representative Western blots, with HSP70 as a loading control. Bottom, Quantification normalized to HSP70. Protein levels are normalized to WT for each protein combination. B, Tyrosine phosphorylation of Y1472 in the synapse-enriched fraction of the hippocampus is not changed by loss of Arg at P31. In addition, the levels of GluN1, GluN2B, and GluA1 in the synapse-enriched fraction are not changed. Left, Representative Western blots. PSD95 levels are shown as a loading control. Right, Graphed data represent the signal of the first protein normalized to the second, and protein levels are normalized to WT for each protein combination. C, While the Src inhibitor SU6656 (SU) partially blocks NMDAR-EPSCs in both WT and arg−/− neurons at P31 and P42, NMDAR-EPSCs are still increased in arg−/− slices with SU6656 relative to WT slices with SU6656, which differs from the normalization of NMDAR-EPSCs following ifenprodil treatment (Fig. 3). Top, Representative traces of NMDAR-EPSCs at +40 mV in the presence of CNQX from P42 WT and arg−/− mice with or without SU6656. Bottom, Current amplitudes were normalized to the average value calculated from WT at the same age. *p < 0.05, **p < 0.01 vs WT; #p < 0.05 vs arg −/−; ∧p < 0.05 vs WT + SU6656.
A subset of synapses in arg−/− neurons exhibits exuberant NMDAR-mediated currents
We next used glutamate uncaging-evoked EPSCs to measure NMDAR-mediated responses at single dendritic spines. Two-photon uncaging released glutamate in a sufficiently small volume to excite receptors on individual dendritic spines, as described previously (Chiu et al., 2013). Glutamate uncaging evoked NMDAR-uEPSCs that were on average significantly increased in arg−/− neurons at P31 (Fig. 7Aiii) and P42 [Fig. 7Ai and Aiv; unpaired t test: WT + Ifen: p < 0.0001 (Fig. 7Aii); p < 0.0001 (Fig. 7Aiii); p < 0.0001 (Fig. 7Aiv); arg−/−: p = 0.93 (Fig. 7Aii); p = 0.01 (Fig. 7Aiii); p = 0.004 (Fig. 7Aiv); arg−/− + Ifen: p = 0.004 (Fig. 7Aii); p < 0.0001 (Fig. 7Aiii); p < 0.0001 (Fig. 7Aiv)] compared with those in WT mice. Consistent with previous results (Fig. 5), ifenprodil normalized the NMDAR-uEPSCs in arg−/− neurons to the same level as in WT neurons.
Figure 7.
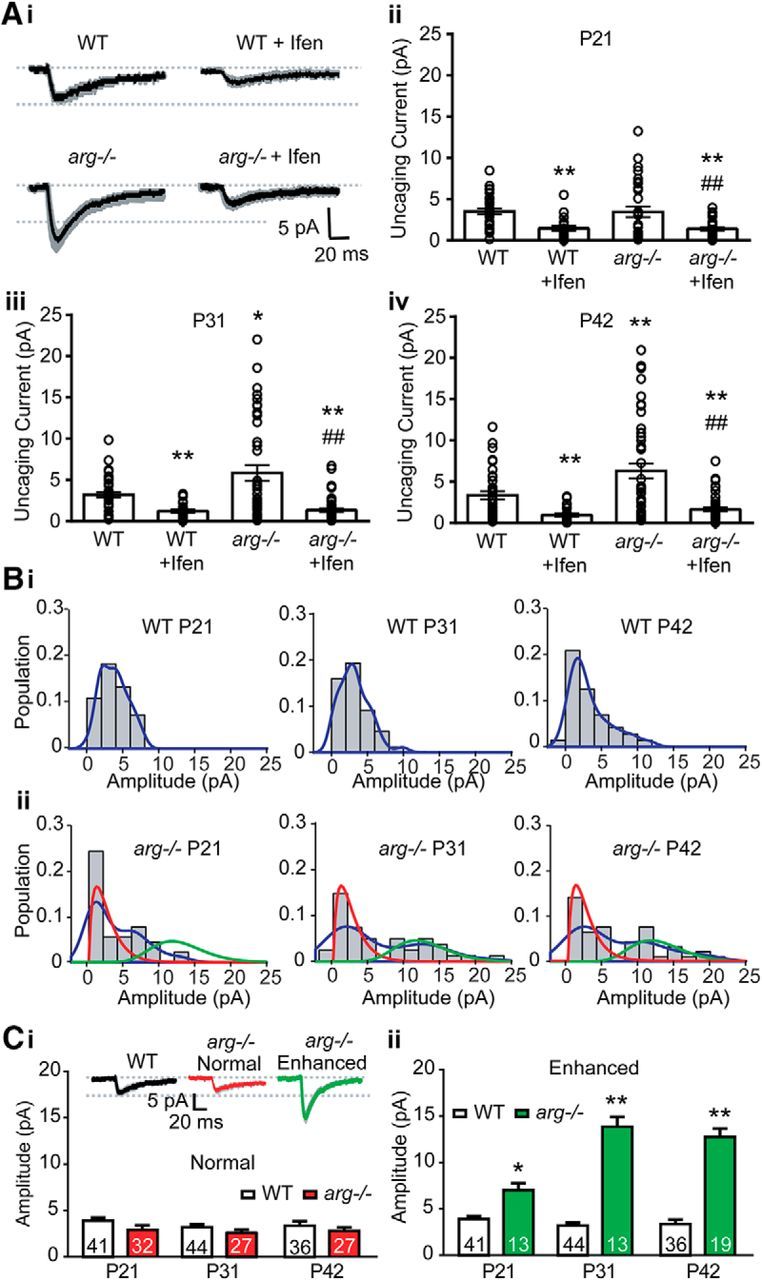
Loss of Arg increases GluN2B-mediated glutamate uncaging-induced NMDAR-EPSCs in a subpopulation of dendritic spines. A, Loss of Arg increases GluN2B-mediated NMDAR-uEPSCs. Ai, Average traces of NMDAR-uEPSCs recorded from CA1 neurons in WT and arg−/− hippocampal slices at P42 without or with ifenprodil (Ifen) ± SEM (shaded areas). Aii–Aiv, The average NMDAR-uEPSC is increased in arg−/− CA1 neurons at P31 (Aiii; n = 40–44) and P42 (Aiv, n = 36–46) compared with WT neurons, but not at P21 (Aii, n = 40–45). Histograms show the average NMDAR-uEPSC amplitude, and open circles represent individual uEPSCs from single spines. Ifenprodil blocks the increased NMDAR-uEPSCs in arg−/− neurons to similar levels as WT + Ifen. *p < 0.05, **p < 0.01 vs WT; ##p < 0.01 vs arg−/−. B, NMDAR-uEPSC amplitudes from single arg−/− spines fall into two subpopulations. Bi–Bii, Histograms of the probability density (population of the spines) for given NMDAR-uEPSCs amplitudes from WT (Bi) and arg−/− (Bii) slices at P21, P31, and P42. NMDAR-uEPSC amplitudes from WT spines are best fit as a single population. By contrast, NMDAR-uEPSCs from arg−/− spines are best fit as a mixture of two distributions, one with an average amplitude similar to WT and one with an enhanced amplitude compared with WT. Fits were calculated as gamma mixtures (red, smaller amplitude; green, larger amplitude) in arg−/− slices. The empirical probability density is outlined in blue. C, NMDAR-uEPSC amplitudes are increased as early as P21 in one subpopulation of spines in arg−/− hippocampal slices. Ci, Inset, Traces of NMDAR-uEPSCs from the single WT population and the arg−/− normal and enhanced populations defined in B. Histogram shows that the average NMDAR-uEPSC amplitude of arg−/− spines from the normal population (Bii, red traces) is not different from the WT population at any age. Cii, The average NMDAR-uEPSC from the enhanced population of arg−/− spines (Bii, green traces) is greater than WT at all three ages, and the difference increases between P21 and P31. *p < 0.05, **p < 0.01 vs WT.
Since the distribution of NMDAR-uEPSC responses was substantially positively skewed in arg−/− spines compared with WT spines, we tested whether the single-spine responses contained subpopulations by modeling each dataset as a single or mixture of two gamma distributions. Comparing the fits of each model, we determined whether the data were more likely to be generated by two subpopulations or a single population. NMDAR-uEPSCs from WT slices were better fit as a single population (p value for mixture of two vs one at P21 = 0.24, at P31 = 0.15, at P42 = 0.35; Fig. 7Bi), while the responses from arg−/− slices were better modeled as two subpopulations (at P21, p = 0.004; at P31, p = 0.002; at P42, p = 0.006; Fig. 7Bii). NMDAR-uEPSCs in arg−/− slices were assigned to a subpopulation by maximum-likelihood estimation, using best-fit gamma mixture parameters. The average NMDAR-uEPSC from the first subpopulation of spines in arg−/− slices did not differ from the mean of the single WT distribution (Fig. 7Bii, red traces, Ci; unpaired t test: P21: p = 0.05; P31: p = 0.06; P42: p = 0.06), while the average NMDAR-uEPSC from the second population was significantly increased in arg−/− neurons compared with WT at all ages (Fig. 7Bii, green traces, Cii; unpaired t test: P21: p = 0.03; P31: p < 0.0001; P42: p < 0.0001). Twenty-nine percent of spines (13 of 45 spines) in arg−/− mice had enhanced NMDAR-uEPSCs at P21, and this proportion increased in arg−/− mice with age (13 of 40 spines, 33% at P31; 19 of 46 spines, 41% at P42). NMDAR-uEPSC amplitude in the “enhanced” population also increased between P21 and P31 (Fig. 7Cii; unpaired t test: p < 0.0001, t = 5.069), suggesting that both the proportion and the magnitude of enhanced NMDAR-uEPSCs are increased in arg−/− mice with age. These results demonstrate that the increase in NMDAR-EPSCs due to loss of Arg occurs heterogeneously in a subpopulation of spines, and this population is already detectable by P21, before spine and synapse loss are observed (Sfakianos et al., 2007; Gourley et al., 2012).
Spine head enlargement correlates with elevated NMDAR-mediated responses at arg−/− synapses
We determined the relationship between spine head morphology and increased NMDAR-uEPSCs in WT and arg−/− neurons (Fig. 8A). The average maximum intensity projection spine head area was significantly increased at P31 and P42 specifically in the subpopulation of arg−/− spines with enhanced NMDAR-uEPSCs (Fig. 8B; unpaired t test: P21 enhanced: p = 0.14; P31 enhanced: p = 0.009; P42 enhanced: p = 0.007). By contrast, arg−/− spines that had WT-like NMDAR-uEPSCs (Fig. 8B; unpaired t test: P21 normal: p = 0.19; P31 normal: p = 0.82; P42 normal: p = 0.33) had spine head areas that were indistinguishable from WT spines at each age. This result suggests that the increased NMDAR-uEPSCs might correlate directly with changes in spine head size in arg−/− neurons. We performed a linear regression analysis by regressing spine head area on NMDAR-uEPSC amplitude for each genotype, while controlling for age and genotype as covariates. Spine head area and NMDAR-uEPSC were significantly associated in arg−/− slices (estimate coefficient = 0.036 pA/μm2, SE = 0.008, p < 0.01) but not in WT slices (estimate coefficient = 0.007 pA/μm2, SE = 0.019, p = 0.71; Fig. 8C,D; n ≥ 121 spines from 24 slices in each genotype), indicating that spines with a larger head area have larger NMDAR-uEPSCs, but this relationship only held in arg−/− neurons.
Figure 8.

Spine head area correlates with NMDAR-uEPSC amplitude only in arg−/− CA1 neurons. A, Neurons were filled with Alexa Fluor-594 to visualize cell morphology. Spine head area (red arrowhead indicates measured spine) was measured in WT (Ai) and arg−/− (Aii) neurons. For each genotype, the left image is a raw two-photon capture, and the right image is a watershed transform used for head area quantitation. B, Average spine head area was unchanged at all ages between WT and the previously defined subpopulation of arg−/− spines that had normal NMDAR-uEPSCs (Fig. 7). In the previously defined subpopulation of arg−/− spines that had enhanced NMDAR-uEPSCs, the average spine head area was significantly increased at P31 and P42. **p < 0.01 vs WT. C, Scatter plot showing the relationship between spine head area and NMDAR-uEPSC amplitude in each spine from WT mice (blue circles) or arg−/− mice (red circles). D, Table of linear regression of NMDAR-uEPSC amplitude–genotype interaction on spine head area, with age and genotype as covariates. Estimates are given as regression coefficients (SEs). The baseline regression model is as follows: Genotypearg−/−, AgeP21. GenotypeWT, AgeP31, and AgeP42 are dummy variables representing the estimate for difference in mean from the baseline model. The coefficients for Genotypearg−/−:Amplitude and GenotypeWT:Amplitude are estimates of the slope of amplitude vs area for each genotype in units of pA/μm2. p Values are calculated from the appropriate t statistic for each regression coefficient. *p < 0.05; **p < 0.01.
NMDAR-dependent long-term potentiation is occluded, and long-term depression is enhanced by early adulthood in arg−/− mice
Extensive work shows a strong correlation between hippocampal long-term synaptic plasticity and morphological changes in dendritic spines (Engert and Bonhoeffer, 1999; Harvey and Svoboda, 2007; Brigman et al., 2010; Hayama et al., 2013). LTP and LTD of excitatory synaptic transmission are important models for studying the cellular mechanisms of circuit reorganization and are proposed cellular substrates of learning and memory (Hayashi-Takagi et al., 2015). We therefore tested whether the disrupted synaptic maturation and structural changes observed in arg−/− mice were associated with changes in NMDAR-dependent long-term plasticity. We measured TBS-induced LTP and low-frequency stimulation-induced LTD at −70 mV in low Mg2+ ACSF in WT and arg−/− slices (Fig. 9). Both LTP and LTD were successfully induced in WT and arg−/− slices from P21 mice, and there were no significant differences between the normalized amplitudes measured in WT and arg−/− mice at P21 in either LTP (Fig. 9A; two-way ANOVA: p = 1.05, F(1,4338) = 58.92) or LTD (Fig. 9B; two-way ANOVA: p = 0.07, F(1,4116) = 3.4). At P42, loss of Arg partially occluded LTP induction and greatly attenuated LTP maintenance, with the evoked response dropping back to baseline levels by 60 min after TBS (Fig. 9C; two-way ANOVA: p < 0.0001, F(1,4151) = 368.5). By contrast, LTD was significantly enhanced in slices from arg−/− mice when compared with control littermates at P42 (Fig. 9D; two-way ANOVA: p < 0.0001, F(1,3528) = 541.5). A distinct form of LTD that depends on activation of metabotropic glutamate receptors (mGluRs) can also be induced at SC–CA1 synapses using the selective group 1 mGluR agonist DHPG (Oliet et al., 1997). After brief application of DHPG for 5 min, we observed a rapid and long-lasting depression of AMPAR-mediated currents that was not different in WT and arg−/− P42 slices, indicating that mGluR-induced LTD is not affected in arg−/− mice (Fig. 9E; two-way ANOVA: p = 0.07, F(1,4338) = 3.4). Therefore, the persistence of immature high-Pr, GluN2B+ synapses along with overall synapse loss in arg−/− mice is associated with altered circuit remodeling and a shift in NMDAR-mediated synaptic plasticity toward reduced enhancement and exaggerated depression.
Figure 9.

arg−/− slices exhibit age-dependent alterations in NMDAR-dependent LTP and LTD. A, LTP was recorded in hippocampal slices from WT and arg−/− mice at P21. The magnitude of LTP in arg−/− slices did not differ at any time after TBS at P21. EPSCs were recorded in 20 s intervals at −70 mV for at least 100 min. LTP induction by TBS is indicated by the arrow. Insets show averages of two EPSCs before and after TBS in WT (points 1 and 2) and arg−/− (points 3 and 4) neurons. B, The magnitude of LTD recorded from WT and arg−/− hippocampal slices at P21 was not different. Horizontal bar indicates LTD induction by LFS. Insets show averages of two EPSCs before and after LFS in WT (points 1 and 2) and arg−/− (points 3 and 4) neurons. C, arg−/− neurons at P42 had reduced LTP stability. Insets show averages of two EPSCs before and after TBS in WT (points 1 and 2) and arg−/− (points 3 and 4) neurons. n ≥ 9 slices (four animals). **p < 0.01 vs WT. D, LTD magnitude was significantly enhanced after LFS in arg−/− slices at P42. Insets show the averages of two EPSCs before and after LFS in WT (points 1 and 2) and arg−/− (points 3 and 4) neurons. n ≥ 10 slices (five animals). **p < 0.01 vs WT. E, The magnitude of mGluR-LTD from WT and arg−/− slices at P42 was not different. Horizontal bar indicates DHPG application. Insets show the averages of two EPSCs before and after DHPG in WT (points 1 and 2) and arg−/− (points 3 and 4) neurons. n = 10 slices (five animals).
Discussion
Coordinated maturation of presynaptic and postsynaptic function is critical for synapse stability and synaptic plasticity
Previous studies of autaptic hippocampal synapses in culture demonstrated that synapses mature from a high-Pr, GluN2B+ state to a low-Pr, GluN2B− state. We report here that the loss of Arg function allows a subpopulation of high-Pr, GluN2B+ synapses to persist into adolescence and early adulthood. These immature-like synapses have larger NMDAR-mediated currents, and their spines enlarge between P21 and P31, a developmental window during which significant SC–CA1 synapse loss is observed in arg−/− mice (Sfakianos et al., 2007; Gourley et al., 2012). The proportion of GluN2B-containing synapses increases during the same time interval that net synapse and dendritic spine numbers are decreasing (Sfakianos et al., 2007; Gourley et al., 2012). These observations collectively suggest that enhanced NMDAR-mediated currents at a subset of synapses may drive dendritic spine loss at other synapses. Individual dendritic segments on mature CA1 neurons maintain a homeostatic balance of excitatory inputs: plasticity-related spine loss is accompanied by an increase in synapse size at remaining inputs (Bourne and Harris, 2011; Oh et al., 2015). We speculate that the activity-dependent enhancement of some synapses in arg−/− mice may trigger homeostatic loss of smaller, weaker synapses, precipitating a vicious cycle that leads to the spine destabilization and synapse loss observed in these mice (Fig. 10). In normal development, this propensity may allow immature high-Pr, GluN2B+ synapses to compete in an activity-dependent manner for limited synaptic building materials.
Figure 10.

Model for the role of integrin–Arg signaling in the coordinated maturation of presynaptic and postsynaptic function. A, A subpopulation of high-Pr, GluN2B+ synapses persists into adolescence and early adulthood in arg−/− mice. The enhanced NMDAR function at this subset of synapses may drive homeostatic dendritic spine loss at other synapses. The loss of these synapses may free up limiting synaptic materials, increasing the spine size at high-Pr, GluN2B+ synapses in arg−/− mice. B, Integrin–Arg signaling normally reduces GluN2B-dependent NMDAR currents and reduces presynaptic Pr. Arg may inhibit GluN2B-containing NMDAR function by regulating receptor trafficking pathways. Disruption of this coordination in presynaptic and postsynaptic properties leads to synapse loss and disruption of normal synaptic plasticity.
Arg acts in the postsynaptic compartment to regulate NMDAR function and spine size and stability
Sparse knockdown of Arg in cultured hippocampal neurons leads to dendritic spine destabilization, implying that Arg has a direct, postsynaptic role in controlling synaptic signaling properties (Lin et al., 2013). Dendritic spines are enriched in actin filament networks, which support spine structure and power changes in spine shape, and act as a scaffold to organize the postsynaptic signaling apparatus (Cingolani and Goda, 2008; Hotulainen and Hoogenraad, 2010; Koleske, 2013). Abl family kinases, including Arg, are master regulators of actin structure in diverse biological contexts (Bradley and Koleske, 2009), suggesting that Arg regulates dendritic spine size and stability through direct regulation of the actin cytoskeleton. Arg interacts with actin and the Arp2/3 complex activator cortactin to promote actin branch nucleation and stabilize actin filaments (MacGrath and Koleske, 2012; Courtemanche et al., 2015), and Arg knockdown in cultured hippocampal neurons causes cortactin depletion from spines coincident with spine loss. Tethering cortactin in dendritic spines rescues this spine loss, suggesting that cortactin depletion is a key trigger for spine loss in arg−/− spines (Lin et al., 2013). Interestingly, elevated NMDAR activity can disrupt cortactin localization to spines and cause spine destabilization (Hering and Sheng, 2003; Iki et al., 2005), suggesting that an NMDAR–Arg–cortactin axis is a critical determinant of spine stabilization.
We do not understand what predisposes only a subset of spines to exhibit altered structure and physiological properties in Arg-deficient mice. We detect Arg-RFP in every dendritic spine following overexpression in cultured neurons using fluorescence microscopy (Lin et al., 2013), but only in 30–35% of spines in hippocampal tissue using electron microscopy (EM; Moresco et al., 2003). It has been difficult to resolve whether the more limited distribution in EM studies reflects the true distribution of Arg or a failure to detect antigen more widely (Koleske et al., 1998; Moresco et al., 2003), leaving open the question of whether Arg preferentially localizes to some synapses and not others. It is possible that the dysfunction of a combination of potential Arg-interacting proteins may contribute to the heterogeneous effects that loss of Arg function has on single-spine NMDAR responses and structural plasticity.
A subset of spines becomes enlarged in arg−/− mice by P31 (Fig. 8B), a time that coincides with the first significant synapse and dendritic spine loss in these mice (Sfakianos et al., 2007; Gourley et al., 2012). It is possible that Arg normally restricts spine enlargement and loss of Arg relieves this brake on spine size. Mechanistically, Arg binding to actin filaments can promote actin filament severing by cofilin (Courtemanche et al., 2015), suggesting that Arg may restrict spine volume by limiting the size or complexity of the actin cytoskeleton of the spine. Alternatively, recent studies (Govindarajan et al., 2011; Makino and Malinow, 2011; Rochefort and Konnerth, 2012) suggest that the size and plasticity of spines clustered along a dendritic segment may be coordinately regulated via their shared use of a limited pool of plasticity-related proteins. According to this model, the enlargement of some spines in arg−/− mice may be fueled by the release of these limiting spine materials from the lost spines (Fig. 10).
How might Arg regulate presynaptic release probability?
Immunoelectron microscopic studies have revealed that, in addition to its localization to dendritic spines, Arg also localizes to the presynaptic compartment (Moresco et al., 2003). Actin has been proposed to play diverse presynaptic roles at synapses: actin polymerization inhibitors can reduce Pr (Kuromi and Kidokoro, 1998; Kim and Lisman, 1999) or increase presynaptic release (Morales et al., 2000), while stabilizing actin with phalloidin blocks synaptic transmission (Bernstein and Bamburg, 1989). We speculate that loss of Arg function may disrupt presynaptic actin in a way that promotes high Pr (Fig. 10B). However, it is also possible that alterations in the postsynaptic compartment of arg−/− synapses signal across the synapse to influence neurotransmitter release properties at presynaptic terminals (Davis and Murphey, 1993; Scanziani et al., 1998).
Immature and mature synapses likely differentially impact synaptic plasticity and network function
LTP is accompanied by new spine formation or a shift from thin to mushroom spines, whereas LTD results in spine shrinkage and synapse retraction (Okamoto et al., 2004; Segal, 2005; Bourne and Harris, 2008). Loss of Arg function leads to a state where high-Pr, GluN2B+ synapses persist and increase proportionally, while overall synapse numbers decline. This state may make it difficult for Arg-deficient neurons to add or enlarge synapses to achieve or maintain LTP, but may enhance the propensity to lose synapses, leading to a greater LTD induction. These age-dependent alterations in synaptic plasticity in arg−/− mice likely contribute to the progressive defects in learning, memory, and behavioral flexibility that emerge by early adulthood in these mice (Sfakianos et al., 2007; Gourley et al., 2009; Gourley et al., 2012). We propose that the maturation of high-Pr, GluN2B+ synapses to predominantly low-Pr, GluN2B− synapses in WT mice may be essential to preserve a larger dynamic range for plasticity while also ensuring that connectivity is distributed among a much larger number of synapses for optimal circuit integration and function.
Footnotes
This work was supported by American Heart Association Postdoctoral Fellowship Grant 16POST27250302 (X.X.), and by National Institutes of Health Grants R01-NS-089662-01 (A.J.K. and J. Grutzendler), R21-NS-089439-01 (A.J.K. and T. Pollard), R01-MH-099045-03 (M.J.H.), F31-MH-105043-01 (A.D.L.), and T32-GM-7223-39 (A.D.L.). We thank Gyorgy Lur and Chiayu Chiu for advice on the two-photon uncaging experiments, and James Howe and members of the Koleske and Higley laboratories for helpful discussions.
The authors declare no competing financial interests.
References
- Bernard-Trifilo JA, Kramár EA, Torp R, Lin CY, Pineda EA, Lynch G, Gall CM. Integrin signaling cascades are operational in adult hippocampal synapses and modulate NMDA receptor physiology. J Neurochem. 2005;93:834–849. doi: 10.1111/j.1471-4159.2005.03062.x. [DOI] [PubMed] [Google Scholar]
- Bernstein BW, Bamburg JR. Cycling of actin assembly in synaptosomes and neurotransmitter release. Neuron. 1989;3:257–265. doi: 10.1016/0896-6273(89)90039-1. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Regulation of hippocampal transmitter release during development and long-term potentiation. Science. 1995;269:1730–1734. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Coordination of size and number of excitatory and inhibitory synapses results in a balanced structural plasticity along mature hippocampal CA1 dendrites during LTP. Hippocampus. 2011;21:354–373. doi: 10.1002/hipo.20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley WD, Koleske AJ. Regulation of cell migration and morphogenesis by Abl-family kinases: emerging mechanisms and physiological contexts. J Cell Sci. 2009;122:3441–3454. doi: 10.1242/jcs.039859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigman JL, Wright T, Talani G, Prasad-Mulcare S, Jinde S, Seabold GK, Mathur P, Davis MI, Bock R, Gustin RM, Colbran RJ, Alvarez VA, Nakazawa K, Delpire E, Lovinger DM, Holmes A. Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J Neurosci. 2010;30:4590–4600. doi: 10.1523/JNEUROSCI.0640-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignoto G, Vicini S. Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science. 1992;258:1007–1011. doi: 10.1126/science.1279803. [DOI] [PubMed] [Google Scholar]
- Chavis P, Westbrook G. Integrins mediate functional pre- and postsynaptic maturation at a hippocampal synapse. Nature. 2001;411:317–321. doi: 10.1038/35077101. [DOI] [PubMed] [Google Scholar]
- Chiu CQ, Lur G, Morse TM, Carnevale NT, Ellis-Davies GC, Higley MJ. Compartmentalization of GABAergic inhibition by dendritic spines. Science. 2013;340:759–762. doi: 10.1126/science.1234274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Klingauf J, Tsien RW. Postfusional regulation of cleft glutamate concentration during LTP at “silent synapses.”. Nat Neurosci. 2000;3:330–336. doi: 10.1038/73895. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Collin T, Marty A, Llano I. Presynaptic calcium stores and synaptic transmission. Curr Opin Neurobiol. 2005;15:275–281. doi: 10.1016/j.conb.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Courtemanche N, Gifford SM, Simpson MA, Pollard TD, Koleske AJ. Abl2/Abl-related gene stabilizes actin filaments, stimulates actin branching by actin-related protein 2/3 complex, and promotes actin filament severing by cofilin. J Biol Chem. 2015;290:4038–4046. doi: 10.1074/jbc.M114.608117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–335. doi: 10.1016/S0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- Davis GW, Murphey RK. A role for postsynaptic neurons in determining presynaptic release properties in the cricket CNS: evidence for retrograde control of facilitation. J Neurosci. 1993;13:3827–3838. doi: 10.1523/JNEUROSCI.13-09-03827.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Gourley SL, Koleske AJ, Taylor JR. Loss of dendrite stabilization by the Abl-related gene (Arg) kinase regulates behavioral flexibility and sensitivity to cocaine. Proc Natl Acad Sci U S A. 2009;106:16859–16864. doi: 10.1073/pnas.0902286106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley SL, Olevska A, Warren MS, Taylor JR, Koleske AJ. Arg kinase regulates prefrontal dendritic spine refinement and cocaine-induced plasticity. J Neurosci. 2012;32:2314–2323. doi: 10.1523/JNEUROSCI.2730-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan A, Israely I, Huang SY, Tonegawa S. The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron. 2011;69:132–146. doi: 10.1016/j.neuron.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450:1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayama T, Noguchi J, Watanabe S, Takahashi N, Hayashi-Takagi A, Ellis-Davies GC, Matsuzaki M, Kasai H. GABA promotes the competitive selection of dendritic spines by controlling local Ca2+ signaling. Nat Neurosci. 2013;16:1409–1416. doi: 10.1038/nn.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Yagishita S, Nakamura M, Shirai F, Wu YI, Loshbaugh AL, Kuhlman B, Hahn KM, Kasai H. Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature. 2015;525:333–338. doi: 10.1038/nature15257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Sheng M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci. 2003;23:11759–11769. doi: 10.1523/JNEUROSCI.23-37-11759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R. The probability of transmitter release at a mammalian central synapse. Nature. 1993;366:569–572. doi: 10.1038/366569a0. [DOI] [PubMed] [Google Scholar]
- Hestrin S. Developmental regulation of NMDA receptor-mediated synaptic currents at a central synapse. Nature. 1992;357:686–689. doi: 10.1038/357686a0. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Hoogenraad CC. Actin in dendritic spines: connecting dynamics to function. J Cell Biol. 2010;189:619–629. doi: 10.1083/jcb.201003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EP, Stevens CF. Estimating the distribution of synaptic reliabilities. J Neurophysiol. 1997;78:2870–2880. doi: 10.1152/jn.1997.78.6.2870. [DOI] [PubMed] [Google Scholar]
- Iki J, Inoue A, Bito H, Okabe S. Bi-directional regulation of postsynaptic cortactin distribution by BDNF and NMDA receptor activity. Eur J Neurosci. 2005;22:2985–2994. doi: 10.1111/j.1460-9568.2005.04510.x. [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerrisk ME, Greer CA, Koleske AJ. Integrin α3 is required for late postnatal stability of dendrite arbors, dendritic spines and synapses, and mouse behavior. J Neurosci. 2013;33:6742–6752. doi: 10.1523/JNEUROSCI.0528-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Lisman JE. A role of actin filament in synaptic transmission and long-term potentiation. J Neurosci. 1999;19:4314–4324. doi: 10.1523/JNEUROSCI.19-11-04314.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske AJ. Molecular mechanisms of dendrite stability. Nat Rev Neurosci. 2013;14:536–550. doi: 10.1038/nrn3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske AJ, Gifford AM, Scott ML, Nee M, Bronson RT, Miczek KA, Baltimore D. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21:1259–1272. doi: 10.1016/S0896-6273(00)80646-7. [DOI] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y. Two distinct pools of synaptic vesicles in single presynaptic boutons in a temperature-sensitive Drosophila mutant, shibire. Neuron. 1998;20:917–925. doi: 10.1016/S0896-6273(00)80473-0. [DOI] [PubMed] [Google Scholar]
- Lin B, Arai AC, Lynch G, Gall CM. Integrins regulate NMDA receptor-mediated synaptic currents. J Neurophysiol. 2003;89:2874–2878. doi: 10.1152/jn.00783.2002. [DOI] [PubMed] [Google Scholar]
- Lin YC, Yeckel MF, Koleske AJ. Abl2/Arg controls dendritic spine and dendrite arbor stability via distinct cytoskeletal control pathways. J Neurosci. 2013;33:1846–1857. doi: 10.1523/JNEUROSCI.4284-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGrath SM, Koleske AJ. Arg/Abl2 modulates the affinity and stoichiometry of binding of cortactin to F-actin. Biochemistry. 2012;51:6644–6653. doi: 10.1021/bi300722t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron. 2011;72:1001–1011. doi: 10.1016/j.neuron.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Colicos MA, Goda Y. Actin-dependent regulation of neurotransmitter release at central synapses. Neuron. 2000;27:539–550. doi: 10.1016/S0896-6273(00)00064-7. [DOI] [PubMed] [Google Scholar]
- Moresco EM, Scheetz AJ, Bornmann WG, Koleske AJ, Fitzsimonds RM. Abl family nonreceptor tyrosine kinases modulate short-term synaptic plasticity. J Neurophysiol. 2003;89:1678–1687. doi: 10.1152/jn.00892.2002. [DOI] [PubMed] [Google Scholar]
- Muller D, Oliver M, Lynch G. Developmental changes in synaptic properties in hippocampus of neonatal rats. Brain Res Dev Brain Res. 1989;49:105–114. doi: 10.1016/0165-3806(89)90063-1. [DOI] [PubMed] [Google Scholar]
- Oh WC, Parajuli LK, Zito K. Heterosynaptic structural plasticity on local dendritic segments of hippocampal CA1 neurons. Cell Rep. 2015;10:162–169. doi: 10.1016/j.celrep.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/S0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochefort NL, Konnerth A. Dendritic spines: from structure to in vivo function. EMBO Rep. 2012;13:699–708. doi: 10.1038/embor.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Gähwiler BH, Charpak S. Target cell-specific modulation of transmitter release at terminals from a single axon. Proc Natl Acad Sci U S A. 1998;95:12004–12009. doi: 10.1073/pnas.95.20.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T, Neher E. Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci. 2002;25:206–212. doi: 10.1016/S0166-2236(02)02139-2. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Sfakianos MK, Eisman A, Gourley SL, Bradley WD, Scheetz AJ, Settleman J, Taylor JR, Greer CA, Williamson A, Koleske AJ. Inhibition of rho via arg and p190RhoGAP in the postnatal mouse hippocampus regulates dendritic spine maturation, synapse and dendrite stability, and behavior. J Neurosci. 2007;27:10982–10992. doi: 10.1523/JNEUROSCI.0793-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Ethell IM. Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II-mediated actin reorganization. J Neurosci. 2006;26:1813–1822. doi: 10.1523/JNEUROSCI.4091-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Russell SL, Shen YM, Molinoff PB. Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron. 1993;10:267–278. doi: 10.1016/0896-6273(93)90317-K. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]