

ABSTRACT
Group A Streptococcus (GAS) is an important human pathogen responsible for both superficial infections and invasive diseases. Autoimmune sequelae may occur upon repeated infection. For this reason, development of a vaccine against GAS represents a major challenge, since certain GAS components may trigger autoimmunity. We formulated three combination vaccines containing the following: (i) streptolysin O (SLO), interleukin 8 (IL-8) protease (Streptococcus pyogenes cell envelope proteinase [SpyCEP]), group A streptococcal C5a peptidase (SCPA), arginine deiminase (ADI), and trigger factor (TF); (ii) the conserved M-protein-derived J8 peptide conjugated to ADI; and (iii) group A carbohydrate lacking the N-acetylglucosamine side chain conjugated to ADI. We compared these combination vaccines to a “gold standard” for immunogenicity, full-length M1 protein. Vaccines were adjuvanted with alum, and mice were immunized on days 0, 21, and 28. On day 42, mice were challenged via cutaneous or subcutaneous routes. High-titer antigen-specific antibody responses with bactericidal activity were detected in mouse serum samples for all vaccine candidates. In comparison with sham-immunized mice, all vaccines afforded protection against cutaneous challenge. However, only full-length M1 protein provided protection in the subcutaneous invasive disease model.
IMPORTANCE
This set of experiments demonstrates the inherent variability of mouse models for the characterization of GAS vaccine candidate protective efficacy. Such variability poses an important challenge for GAS vaccine development, as advancement of candidates to human clinical trials requires strong evidence of efficacy. This study highlights the need for an open discussion within the field regarding standardization of animal models for GAS vaccine development.
INTRODUCTION
Group A Streptococcus (GAS) causes numerous human disease manifestations from mild infections of the skin and throat to more serious and life-threatening conditions, such as necrotizing fasciitis or toxic shock syndrome, to poststreptococcal autoimmune complications, including rheumatic heart disease and poststreptococcal glomerulonephritis. Globally, more than 500,000 deaths per annum are estimated to be caused by GAS-related diseases (1), and treatment of GAS infections poses a significant economic burden (2, 3). Though an efficacious vaccine against GAS would represent the best and most cost-effective intervention to diminish disease burden, the link between GAS infection and autoimmune sequelae has hindered the development of such a vaccine. More than 30 years ago, a ban on the administration of GAS and its components into humans was enforced by the U.S. Food and Drug Administration (FDA), after children vaccinated with GAS M protein developed rheumatic fever (4). Although this ban was subsequently lifted in 2005 (5), the quest to develop an effective and safe GAS vaccine for human use is ongoing.
A recent report by the World Health Organization (WHO) lists 26 vaccine candidates that have shown promising results in preclinical studies (6). While precluded as a human vaccine candidate due to safety concerns, full-length M protein from the homologous GAS challenge strain is effective at preventing infection in a variety of animal models and is regularly used as a positive control for GAS vaccine studies (7–9). However, the lack of a standardized animal model for GAS immunization studies has prompted the use of a diverse range of mouse models for evaluation of vaccine efficacy. A few of the many variations that have been reported for GAS vaccine assessment (10, 11) include the following: infections via the intraperitoneal, cutaneous, subcutaneous, intranasal, and intramuscular routes; measurement of survival, bacterial colonization, and dissemination; evaluation of passive and active protection; and differing adjuvant formulations. This diversity in models of protection used to assess vaccine efficacy has made it difficult to compare GAS vaccine compositions.
In this study, we selected promising vaccine candidates described previously to confer protection in at least one mouse model to formulate three experimental vaccines. Homologous M1 protein was used as a positive control, and phosphate-buffered saline (PBS) was used as a negative control. All vaccine formulations were adjuvanted with aluminum hydroxide (alum). The first experimental vaccine consisted of a combination of trigger factor (TF) (7) and inactivated versions of arginine deiminase (ADI) (7, 12), streptolysin O (SLO) (8, 13), Streptococcus pyogenes cell envelope proteinase (SpyCEP) (8, 14), and group A streptococcal C5a peptidase (SCPA) (15). The second experimental vaccine consisted of the conserved M-protein-derived J8 peptide (16, 17) conjugated to ADI, while the third vaccine contained group A carbohydrate lacking the N-acetylglucosamine side chain (ΔGAC) (18) conjugated to ADI. We evaluated the immunogenicity and efficacy of vaccine candidates in mice using a superficial skin infection model and invasive disease model upon challenge with M1 GAS, allowing parallel comparison between the different experimental vaccine formulations.
RESULTS
Antibody response to experimental GAS vaccine antigens.
The immune response to each vaccine component was assessed by enzyme-linked immunosorbent assay (ELISA) following immunization with M1 protein, the five-component SLO, SpyCEP, SCPA, ADI, and TF vaccine (hereafter designated Combo#5), and the conjugate J8-ADI and ΔGAC-ADI vaccines. Both BALB/c and humanized plasminogen mice responses to all protein antigens (M1, ADI, TF, SLO, SpyCEP, and SCPA) were significantly higher in vaccinated mice than in sham-immunized control mice (Fig. 1). J8 peptide was successfully conjugated to ADI, with an average peptide-to-protein ratio of three peptide molecules per ADI molecule as determined by amino acid analysis. Immunization with this conjugate yielded both J8-specific and ADI-specific antibodies in sera of both mouse strains and with specific titers significantly higher in J8-ADI-vaccinated mice than in control PBS-immunized mice (Fig. 1). ΔGAC was also successfully conjugated to ADI, with an average glycan-to-protein ratio of 0.78 glycan molecule per ADI molecule as determined by quantification of protein and carbohydrate concentrations in the conjugate. Immunization with ΔGAC-ADI conjugate resulted in the generation of ΔGAC-specific and ADI-specific murine antibody titers significantly higher than those detected in PBS-immunized mice (Fig. 1).
FIG 1 .

Antigen-specific IgG response in serum samples from BALB/c (A) and humanized plasminogen AlbPLG1 (B) mice at day 35 (n = 10). Antigens used to coat ELISA plates are displayed. IgG titers in antigen-immunized groups (M1, Combo#5, J8-ADI, or ΔGAC-ADI) were compared to titers in PBS-immunized mice (open circles) and found to be significantly different (P < 0.0001) using a two-tailed Mann-Whitney U test. Each symbol represents the value for an individual mouse. The bars show the geometric mean titer (geomean titer) with 95% confidence interval (95% CI). α-M1, anti-M1 antibody.
Additionally, we assessed the binding of murine serum antibodies to the surfaces of live GAS, as detected by flow cytometry. Incubation of serum samples from BALB/c mice with GAS pM1.200 showed a significant shift in fluorescence in sera from vaccinated groups (solid line) compared to sera from PBS-immunized mice (shaded histogram). Serum samples from M1- and Combo#5-immunized mice showed the largest shifts in fluorescence (Fig. 2A). The sera from antigen-vaccinated humanized plasminogen mice incubated with GAS 5448 also showed a significant shift in fluorescence (solid line) compared to sera from PBS-immunized mice (shaded histogram) (Fig. 2B).
FIG 2 .

Antibody binding to the surfaces of GAS. Sera from immunized BALB/c (A) and humanized plasminogen AlbPLG1 (B) mice raised against vaccine antigens (M1, Combo#5, J8-ADI, and ΔGAC-ADI) were incubated with live GAS pM1.200 and 5448, respectively. Specific binding to the bacterial surface was detected by a shift in Alexa Fluor 488 fluorescence. Representative histograms for test antigens are shown with a black solid line; histograms for PBS-immunized mice are shown as a shaded area. The populations in histograms for test antigens were statistically different (P < 0.01) from control histograms with T(X) values above the established cutoff value of T(X) = 100. T(X) values, shown on the right top corner of each panel, were determined by the probability binning algorithm in FlowJo.
Bacterial opsonization.
An indirect bactericidal assay was used to investigate the ability of heat-inactivated murine serum from vaccinated mice to enhance the killing of GAS in the presence of human blood. Serum from BALB/c mice was used to test bactericidal activity against GAS pM1.200 (Fig. 3A), while serum from humanized plasminogen mice was used with GAS 5448 (Fig. 3B). Our results are similar for both groups, with anti-M1 sera being the most opsonic, followed by anti-Combo#5 sera. Overall, all experimental vaccines were able to raise bactericidal antibodies compared to sera from sham-immunized mice.
FIG 3 .

Indirect bactericidal assay. Pooled heat-inactivated sera raised against vaccine antigens in BALB/c (A) and humanized plasminogen AlbPLG1 (B) mice were preincubated with GAS pM1.200 and 5448, respectively. Human blood tested to support the growth of both strains was then added, and following 3-h incubation at 37°C, samples were plated for CFU determination. The survival percentage was calculated using the growth of GAS in serum from PBS-immunized mice as 100%. Values are the mean survival percentage plus standard deviation (SD) (error bars) for three independent replicates. Survival percentages were compared using one-way ANOVA corrected for multiple comparisons using Dunnett’s test. Values that are significantly different from the value for the control (α PBS) are indicated by asterisks as follows: **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Murine model of skin infection.
Protection against superficial skin infection with GAS pM1.200 in immunized BALB/c mice was determined by measuring bacterial persistence in skin lesions and dissemination of GAS into the blood and spleen. Five mice per group were euthanized on days 3 and 6 postinfection, and bacterial CFU were enumerated. In skin samples, a reduction in CFU was observed in samples from vaccinated mice compared to PBS-immunized mice on day 6 postinfection; however, this difference did not reach statistical significance (Fig. 4A). GAS pM1.200 was not detected in spleen samples from M1-immunized mice from day 3 postinfection onwards, while on day 6 postinfection, Combo#5- and J8-ADI immunized mice showed significantly lower CFU in the spleen compared to PBS-immunized mice (Fig. 4B). All vaccine antigens showed significant protection against bacteremia on day 6 postinfection compared to the PBS control. Furthermore, no detectable bacteria were found in samples from M1-, Combo#5-, and ΔGAC-ADI-vaccinated mice on day 3 postinfection (Fig. 4C).
FIG 4 .
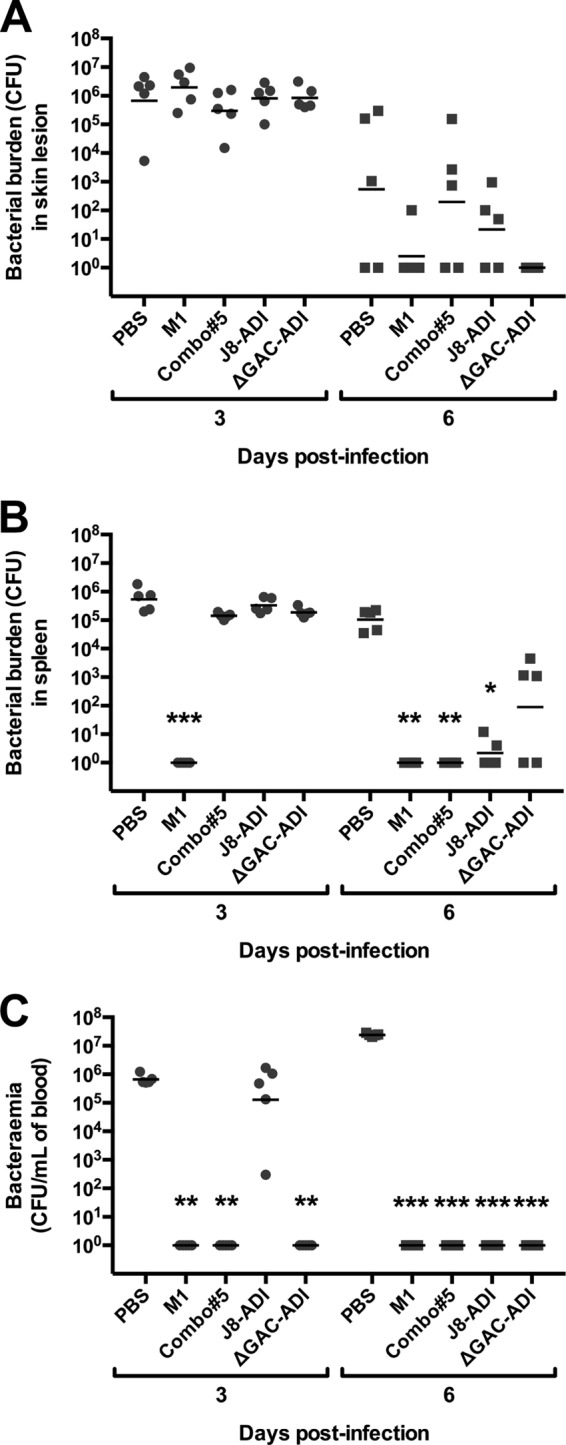
Bacterial persistence in the skin (A) and dissemination into spleen (B) and blood (C) following skin challenge of BALB/c mice (n = 10 for all groups). Mice were infected on day 42, and on days 3 and 6 postinfection, five mice per group were sacrificed to determine GAS bacterial burden. Each symbol represents the value for an individual mouse. The short lines represent geometric means of CFU. Statistical significance was determined by Kruskal-Wallis test corrected for multiple comparisons using Dunn’s test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Samples where GAS was undetected were allocated a value of 1 for graphical representation.
Murine model of invasive disease.
To assess vaccine efficacy against more severe forms of infection, an invasive mouse model of disease was employed. Following subcutaneous challenge of humanized plasminogen mice with GAS strain 5448, survival was monitored for 10 days. M1 protein was the only experimental vaccine that conferred protection against GAS strain 5448 challenge, with 100% survival (Fig. 5). Immunization with Combo#5, J8-ADI, and ΔGAC-ADI did not confer protection against lethal challenge beyond that observed in sham-immunized mice.
FIG 5 .
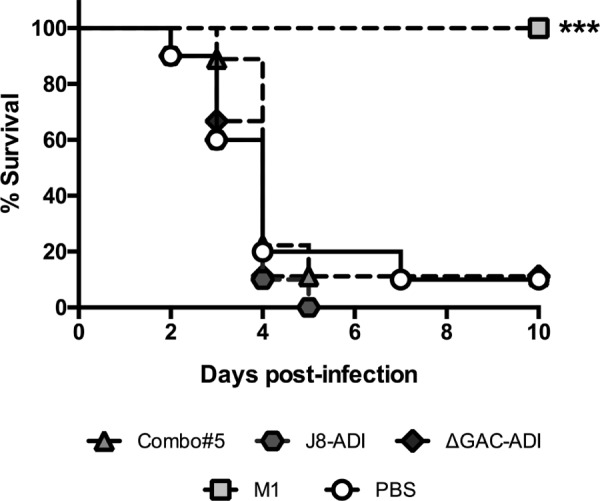
Survival of humanized plasminogen AlbPLG1 mice following subcutaneous challenge with 1.3 × 108 CFU of GAS 5448 on day 42 (n = 10 for the PBS and J8-ADI groups; n = 9 for the Combo#5 and ΔGAC-ADI groups; n = 8 for the M1 group). Curves were compared using the log rank (Mantel-Cox) test (***, P < 0.001).
DISCUSSION
In recent years, the emergence of a worldwide epidemic of invasive infections (19, 20), scarlet fever outbreaks (21, 22), and antibiotic-resistant GAS strains (23) has emphasized the urgent need for a safe and efficacious GAS vaccine for human use. A reduction in GAS carriage and superficial infection through an effective vaccine strategy would likely reduce the burden of serious GAS disease, with the additional benefit of reducing antibiotic prescription and potentially leading to reduced levels of antibiotic resistance. GAS vaccine candidates can be classified into two categories, M-protein-based vaccines and non-M-protein-based vaccines. M-protein-based vaccines target either the hypervariable or conserved domain of the M protein. Candidate vaccines targeting the hypervariable region of M protein follow a multivalent vaccine approach, where the hypervariable domain of M protein from selected serotypes are included in the formulation (24–26). Most recently, one such multivalent vaccine was formulated to include 30 serotypes (26). While cross-opsonization has been observed against some serotypes not included in the formulation (26), heterologous protection in animal models has not yet been reported. There are more than 120 GAS M serotypes reported thus far (27), which raises a concern about the emergence of serotype replacement, as observed in the use of type-specific pneumococcal vaccines in humans (28). An experimental vaccine targeting the J8 conserved domain of M protein conjugated to diphtheria toxoid has shown protection against heterologous challenge (17).
Several non-M-protein vaccine candidates have been identified and described in the literature (6, 10). Despite showing protection in animal models, with several antigens being widely conserved across serotypes, a non-M-protein vaccine candidate has yet to progress into human clinical trials (11, 29). One challenge in the development of GAS vaccines is the lack of a standardized animal model that can truly mimic human infection (29). This has resulted in the use of various models of disease for assessment of vaccine efficacy, which have been known to deliver conflicting results. Immunogenicity studies with the streptococcal serum opacity factor (SOF) showed the generation of rabbit opsonizing antibodies following immunization with SOF emulsified in complete Freund’s adjuvant, and protection in mice against intraperitoneal lethal challenge (30). However, in a subsequent study, intranasal immunization with SOF adjuvanted with cholera toxin B yielded no protection following intranasal challenge, despite the generation of antigen-specific antibodies (31). Similarly, while intranasal immunization with the fibronectin binding protein I (SfbI) provided protection in a murine intranasal challenge model (31), it failed to provide protection in a murine skin challenge model (32). Identification of GAS antigens that provide protection across different challenge models would support the evaluation of such vaccine candidates in clinical trials.
Here we selected a set of promising antigens that have shown protection in at least one murine model of infection and compared these in two different mouse challenge models (Table 1). ADI and TF are highly conserved proteins localized on the GAS surface that previously showed protection against homologous and heterologous intraperitoneal GAS challenge (7). SLO, SpyCEP, and SCPA are important virulence factors of GAS, which have also been found to be widely conserved across GAS serotypes and have shown protection in various mouse models of infection (8, 33–35). The J8 peptide of M protein, conjugated to diphtheria toxoid has been shown to provide protection in mouse models and to not elicit cross-reactive antibodies against human heart proteins (16, 17, 36). Group A carbohydrate incorporates a polyrhamnose backbone with N-acetylglucosamine (GlcNAc) side chains. GAC is found across all GAS serotypes and has been shown to elicit functional antibodies able to confer protection (37). Existing concerns regarding host cross-reactive antibodies that recognize GlcNAc (38, 39) discouraged the use of GAC as a vaccine candidate and prompted the analysis of a GAC variant lacking GlcNAc (ΔGAC) (18). ΔGAC was able to elicit antibodies that showed protection via passive immunization (18).
TABLE 1 .
Vaccine candidates used in this study
| Antigen | Function | Inactive form | Reference(s) |
|---|---|---|---|
| ADI | Arginine deiminase. | ADI D277A | 7, 12 |
| TF | Ribosome-associated chaperone. Important for protease secretion and activation. | 7, 8 | |
| SLO | Pore-forming cytolysin. Binds and damages cell membranes, resulting in lysis of the host cell. | SLO P427L W535F | 8, 13 |
| SpyCEP | Serine protease cleaves IL-8, interfering with neutrophil recruitment to the site of infection. | SpyCEP D151A S617A | 8, 14, 35 |
| SCPA | Subtilisin-like protease. Cleaves C5a, interfering with neutrophil recruitment to the site of infection. | SCPA D130A S512A | 8, 15, 33 |
| J8 | 12-amino-acid peptide from the C-repeat region of M protein. | 17, 36, 57, 68 | |
| ΔGAC | Group A carbohydrate lacking N-acetylglucosamine (GlcNAc) side chain. | 18 |
The experimental vaccines Combo#5, J8-ADI, and ΔGAC-ADI protected BALB/c mice from skin challenge, but they did not induce protection in the AlbPLG1 mouse model of invasive disease. Immunization of BALB/c and AlbPLG1 mice with M1, Combo#5, J8-ADI, and ΔGAC-ADI elicited significant antigen-specific murine antibodies against each antigen compared to PBS-immunized mice. Such antibodies were also able to bind to the surfaces of GAS and to enhance killing of GAS in the presence of human blood. The presence of antibodies that can promote opsonophagocytosis has been previously correlated with the ability to protect against infection (17, 24) and has been suggested as an immune correlate to be measured in clinical trials (40). Some studies using non-M-protein vaccine candidates have challenged the concept of opsonizing antibodies as being the main correlate of protection (41, 42). In this work, the most effective opsonizing antibody response was generated by M1 protein, which correlated with protection against GAS challenge in the AlbPLG1 invasive model. The lower opsonic activity observed for antibodies raised by J8-ADI and ΔGAC-ADI (Fig. 3) correlates with reduced detection of antibody binding to the surfaces of GAS (Fig. 2). On the other hand, a significant albeit lesser opsonizing antibody response was generated by Combo#5 compared to M1, but protection was not apparent in the invasive model of disease. To explain these observations, we speculate that the level of opsonizing antibody generated may be important but perhaps not the only immune effector mechanism necessary to prevent lethal infection. Utilization of different adjuvants that can elicit broader humoral and cellular immune responses may lead to protective efficacy in the invasive disease model. An alternate explanation for the impressive level of protection granted by M1 immunization may be due to the ability of anti-M1 antibodies to block the strong inflammatory response triggered by M protein during infection (43). Blocking of this inflammatory effect could potentially prevent vascular leakage and multiorgan failure, promoting protective efficacy. However, additional studies are required to confirm this hypothesis.
Vaccine efficacy was evaluated using two different murine models of disease. The first model, using BALB/c mice, resembles a superficial non-life-threatening skin infection. This model requires the use of mouse-passaged GAS strains, which have been adapted to the murine host. The second model employed here is the humanized plasminogen murine model. It is well established that activation of human plasminogen by GAS streptokinase is key for systemic dissemination. GAS streptokinase has a greater affinity for human plasminogen than mouse plasminogen; therefore, mice that express human plasminogen are more susceptible to GAS dissemination with nonpassaged GAS strains (44, 45). Each model represents a different disease manifestation, and therefore, the immune mechanisms required for protection may differ for each model. However, the protection afforded by M1 immunization in this study suggests the possibility of developing a vaccine that can confer protection against several clinical manifestations.
Lack of understanding of innate and adaptive immunity following infection in humans has resulted in a lack of standardized correlates of protection (29). An alternative to address this issue is the use of nonhuman primate (NHP) models to characterize immune responses against GAS in an animal model biologically much closer to humans. Experimental GAS colonization in the upper and lower respiratory tracts of NHPs has been reported in rhesus and cynomolgus macaques (46, 47) and in baboons (48). Moreover, NHPs show clinical symptoms of pharyngitis and tonsillitis (46, 47). NHPs may thus represent a powerful tool to investigate immune markers and correlates of protection during streptococcal pharyngitis and ultimately to assess vaccine efficacy (47, 49). During the 1970s, human trials were also carried out where naive and experimentally vaccinated volunteers were infected with GAS (50–52). Controlled infection of volunteers allowed important observations about M-protein immunity and clinical symptoms associated with GAS pharyngeal infection. Following the ban enforced by the U.S. Food and Drug Administration, this type of study has not been undertaken for almost 30 years. Recently, there is increased interest in the development of a GAS human challenge model, with the aim of acquiring better understanding of human immunity to GAS and ultimately to accelerate assessment of vaccine efficacy (53).
Human clinical trials represent huge economic investments. Therefore, a comprehensive portfolio of vaccine antigen efficacy evidence needs to be available. A combination of convincing preclinical data in small-animal models, NHPs, and potentially humans may represent one pathway to progress safe and efficacious GAS vaccine candidates into human clinical trials. We observed a clear difference in the level of protection granted by the experimental vaccine candidates Combo#5, J8-ADI, and ΔGAC-ADI in two mouse models of GAS infection. These results raise important questions regarding the use of mouse models to assess the efficacy of GAS vaccines and the lack of uniformity within the field, particularly as new protective antigens continue to be discovered (54, 55). We strongly believe that correlates of protection in humans and the use of standardized animal models of protection should be openly discussed among investigators and pharmaceutical interests in order to develop the best possible GAS vaccine for safe and efficacious use in humans.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
For recombinant protein expression, Escherichia coli BL21 Star (DE3) was grown in Luria-Bertani medium (LB) with antibiotic selection as appropriate. Streptococcus pyogenes M1T1 5448 strain, an invasive clinical isolate (56), was grown in Todd-Hewitt medium supplemented with 1% (wt/vol) yeast extract (THY). S. pyogenes pM1.200, a mouse-passaged M1 streptomycin-resistant reference strain (57) was grown in THY supplemented with 1% (wt/vol) neopeptone and in the presence of streptomycin (200 µg/ml) when required.
Expression and purification of streptococcal antigens.
SpyCEP (amino acids 40 to 683, D151A S617A) sequence was cloned into the pET151/D-TOPO vector (Invitrogen) following amplification from GAS 5448 genomic DNA and QuikChange site-directed mutagenesis (Agilent Technologies). SCPA (amino acids 40 to 1039, D130A S512A) sequence was cloned into the pET151d vector by sequence- and ligation-independent cloning (SLIC) using synthetic double-stranded DNA (dsDNA) (gBlock; Integrated DNA Technologies) for the 5′ and 3′ sequences and a PCR-amplified internal sequence from SF30 genomic DNA. SLO (amino acids 1 to 571) cloned into pET-15b (58) was modified by site-directed mutagenesis to incorporate P427L and W535A mutations. ADI (amino acids 1 to 411, D277A) was previously cloned into pET151/D-TOPO (12). TF (amino acids 1 to 427) has been cloned into pET151/D-TOPO (7). The gene encoding M1 protein (amino acids 13 to 455) was cloned into pGEX-2T (GE Healthcare Life Sciences), incorporating a carboxy-terminal 6×His tag (9).
ADI, TF, SpyCEP, SLO, and SCPA antigens were expressed in E. coli BL21 Star (DE3) cells and purified by immobilized metal ion affinity chromatography (IMAC). Bacterial endotoxins were removed during IMAC by supplementing washing buffers with 0.1% (vol/vol) Triton X-114 (Sigma) (59) or by incubating IMAC-purified proteins with Pierce high-capacity endotoxin removal resin (Thermo Fisher Scientific) following the manufacturer’s protocol. To prepare antigens for ELISA, tobacco etch virus (TEV) protease was used to cleave the His tag from purified ADI, TF, SpyCEP, and SCPA; uncleaved protein and TEV were removed by IMAC. Thrombin protease (Sigma-Aldrich) was used for His tag removal from SLO, followed by size exclusion chromatography and IMAC to remove thrombin and uncleaved SLO, respectively. M1 protein was purified as described previously (9). The final protein concentration was determined using a Direct Detect infrared spectrometer (Millipore). Endotoxin levels were measured using the Pierce Limulus amebocyte lysate (LAL) chromogenic endotoxin quantitation kit (Thermo Fisher Scientific).
Peptide and carbohydrate conjugation to ADI.
J8 peptide was commercially sourced (China Peptides Co.) and conjugated to purified ADI using N-(ε-maleimidocaproyloxy)sulfosuccinimide ester (Sulfo-EMCS; Thermo Fisher Scientific) following the manufacturer’s protocol. The ratio of J8 peptide to ADI carrier protein was determined using amino acid analysis (Australian Proteome Analysis Facility) and found to average three peptide molecules per ADI molecule. This ratio translates into a dose of 5 µg of J8 and 25 µg of ADI delivered per vaccinated mouse.
Streptococcal group A carbohydrate lacking N-acetylglucosamine (GlcNAc) side chain (ΔGAC) was purified from the GAS 5448ΔgacI strain as previously reported (18). Purified ΔGAC was directly conjugated to ADI by cyanylation using 1-cyano-4-dimethylaminopyridinium tetrafluoroborate (CDAP) (Sigma) (60). Briefly, ΔGAC in lipopolysaccharide (LPS)-free water was activated by slowly adding CDAP while vortexing. After 30 s, the pH was raised to pH 8 with triethylamine. At 2.5 min, purified ADI was added, and the reaction mixture was incubated for 4 h at room temperature. The reaction was quenched with excess glycine, and the ΔGAC-ADI conjugate was further purified by size exclusion chromatography. Carbohydrate concentration in the conjugate was measured by the phenol-sulfuric acid method using rhamnose as a standard (61). Specifically, 4.6 µg of ΔGAC was found to be conjugated to 30 µg of ADI, which was the dose delivered per vaccinated mouse.
Immunization and challenge.
Five groups (n = 10 for all groups) of BALB/c mice and transgenic humanized plasminogen mice heterozygous for the human plasminogen gene (AlbPLG1) were immunized intramuscularly on days 0, 21, and 28 with 30 µg of total protein adjuvanted with alum (Alhydrogel [2%]; Brenntag) at a 1:1 ratio (50-µl immunization dose)/mouse. The negative-control group received PBS in alum as a sham vaccine. Serum samples were taken before immunization and on day 35. On day 42, immunized mice were challenged with M1 GAS. BALB/c mice were infected cutaneously with 1 × 106 CFU of GAS pM1.200 as previously described (62). At days 3 and 6 postinfection, five mice per group were euthanized to obtain skin, blood, and spleen samples for CFU quantification. Two AlbPLG1 mice from the M1 group and one mouse from the Combo#5 group (Combo#5 is the five-component SLO, SpyCEP, SCPA, ADI, and TF vaccine) and the ΔGAC-ADI group were lost prior to challenge (e.g., did not recover from anesthesia) and were excluded from survival analysis. Humanized plasminogen mice (n = 10 for the PBS and J8-ADI groups; n = 9 for the Combo#5 and ΔGAC-ADI groups; n = 8 for the M1 group) were infected subcutaneously with 1.3 × 108 CFU of GAS strain 5448, and survival was monitored for 10 days (63).
ELISA.
Individual protein antigens (His tags removed) at 5 µg/ml in carbonate coating buffer (50 mM Na2CO3-NaHCO3, pH 9.6), were adsorbed to Titertek polyvinyl chloride (PVC) microplates (M.P. Biomedicals) using 100 µl per well overnight at 4°C. Plates were blocked using 5% (wt/vol) skim milk in phosphate-buffered saline (PBS) containing 0.05% (vol/vol) Tween 20 (90 min, 37°C) and incubated with mouse sera (90 min, 37°C). Antigen-specific mouse antibodies were detected with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG antibody (Thermo Fisher Scientific) and SIGMAFAST o-phenylenediamine dihydrochloride (OPD) (Sigma-Aldrich) as an HRP substrate with absorbance measured at 450 nm. Endpoint titers were determined as the highest dilution of serum for which the absorbance was 3 standard deviations above the mean optical density of blank wells.
Purified ΔGAC was activated with 15 mM NaIO4 in sodium acetate buffer (0.1 M sodium acetate [pH 5.5]) for 30 min at room temperature. The reaction was stopped with ethylene glycol, and activated ΔGAC was dialyzed against sodium acetate buffer. Costar carbohydrate binding plates (Corning) were coated with 100 µl of activated GAC (10 µg/ml) in sodium acetate buffer for 1 h at room temperature. Plates were blocked with 1% (wt/vol) bovine serum albumin (BSA) in 50 mM Tris (pH 8.2) for 30 min at room temperature. Mouse serum samples in PBS supplemented with 10% (vol/vol) goat serum were added and incubated at 37°C for 90 min. Detection of ΔGAC-specific mouse antibodies and determination of endpoint titers was done as described above.
Detection of antibody binding to the GAS surface by flow cytometry.
GAS strains were grown to mid-logarithmic phase (optical density at 600 nm [OD600] of 0.6), washed in PBS, and blocked using nonspecific human IgG (200 μg/ml; Merck Millipore) in PBS with 3% (wt/wt) BSA (3% BSA/PBS) (1 h, 4°C). Bacterial cells were washed and resuspended in PBS. A volume of 0.3 ml of bacterial suspension in PBS (OD600 of 0.6) was incubated overnight at 4°C in 100 µl of pooled mouse sera and then diluted 1:50 in 0.3% BSA/PBS (wt/vol). Pellets were washed in PBS and resuspended in 100 µl of a 1:200 dilution of goat anti-mouse IgG (H+L) conjugated to Alexa Fluor 488 (Thermo Fisher Scientific) in 0.3% BSA/PBS (wt/vol). Cells were washed with PBS and fixed in 1.5% paraformaldehyde/PBS (wt/vol). A total of 50,000 events were analyzed using a BD Accuri C6 flow cytometer (BD Biosciences), and further analysis was undertaken using FlowJo software (Tree Star Inc.).
Bacterial opsonization assay.
The indirect bactericidal activity of mouse antibodies was measured as previously described with minor changes (9). Briefly, GAS strains were grown until early logarithmic phase (OD600 of 0.4). Pooled heat-inactivated serum from immunized BALB/c mice was incubated for 20 min at room temperature with GAS pM1.200 diluted in PBS to a 1 × 10−4 dilution. Likewise, pooled heat-inactivated serum from immunized humanized plasminogen AlbPLG1 mice was incubated with GAS 5448 diluted in PBS to a 5 × 10−5 dilution. Fresh human blood from a volunteer, previously tested to support the growth of both strains, was added, and samples were incubated (3 h, 37°C) with end-to-end rotation before being plated out in triplicate for CFU determination. Opsonic activity of the immune sera (percent reduction in mean CFU) was calculated as follows: (1 − CFU in the presence of vaccine immune sera/mean CFU in the presence of sham-immunized mouse sera) × 100. Three independent replicates were performed for each treatment.
Statistical analysis.
Differences in antigen-specific endpoint titers were analyzed using the two-tailed Mann-Whitney U test with P of <0.05 considered statistically significant (GraphPad Prism 6). Flow cytometry data were analyzed using the probability binning algorithm in FlowJo 10.1 (Tree Star Inc.), a cutoff value of T(X) of 100 was empirically determined, and samples having T(X) of >100 were considered significant (P < 0.01) (99% confidence) (64, 65). [T(X) is a statistic metric developed to provide an indication of the probability that two populations differ from each other by using the probability binning algorithm. The higher the value of T(X), the less alike the populations are.] Survival times in the indirect bactericidal assay were compared using one-way analysis of variance (ANOVA) corrected for multiple comparisons using Dunnett’s test, with P < 0.05 considered statistically significant (GraphPad Prism 6). Differences in bacterial persistence and dissemination were analyzed using the Kruskal-Wallis test corrected for multiple comparisons using Dunn’s test (GraphPad Prism 6). Murine survival curves were analyzed using the Mantel-Cox log rank test with P < 0.05 considered statistically significant (GraphPad Prism 6).
Ethics approvals.
All animal procedures were conducted according to the Australian Code for the Care and Use of Animals for Scientific Purposes (66). Procedures using BALB/c mice were approved by the Griffith University Animal Ethics Committee, and procedures using humanized plasminogen AlbPLG1 mice were approved by the University of Queensland Animal Ethics Committee. Human blood donation for use in indirect bactericidal assays was conducted in accordance with the National Statement on Ethical Conduct in Human Research (67), complied with the regulations governing experimentation on humans, and was approved by the University of Queensland Medical Research Ethics Committee.
ACKNOWLEDGMENTS
Amino acid analysis was carried out by the Australian Proteome Analysis Facility (APAF Ltd.).
This work was supported by funding from the National Health and Medical Research Council (NHMRC) of Australia, U.S. National Institutes of Health (NIH), and the Queensland Emory Development Alliance.
M.F.G. and M.R.B. hold the patent for J8. V.N. holds the patent for ΔGAC. M.J.W., A.H., and J.C. hold the patent for ADI and TF. All other authors declare no conflict of interest.
Footnotes
Citation Rivera-Hernandez T, Pandey M, Henningham A, Cole J, Choudhury B, Cork AJ, Gillen CM, Ghaffar KA, West NP, Silvestri G, Good MF, Moyle PM, Toth I, Nizet V, Batzloff MR, Walker MJ. 2016. Differing efficacies of lead group A streptococcal vaccine candidates and full-length M protein in cutaneous and invasive disease models. mBio 7(3):e00618-16. doi:10.1128/mBio.00618-16.
REFERENCES
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 2.Pfoh E, Wessels MR, Goldmann D, Lee GM. 2008. Burden and economic cost of group A streptococcal pharyngitis. Pediatrics 121:229–234. doi: 10.1542/peds.2007-0484. [DOI] [PubMed] [Google Scholar]
- 3.Hughes GJ, Van Hoek AJ, Sriskandan S, Lamagni TL. 2015. The cost of hospital care for management of invasive group A streptococcal infections in England. Epidemiol Infect 143:1719–1730. doi: 10.1017/S0950268814002489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Massell BF, Honikman LH, Amezcua J. 1969. Rheumatic fever following streptococcal vaccination: report of three cases. JAMA 207:1115–1119. doi: 10.1001/jama.207.6.1115. [DOI] [PubMed] [Google Scholar]
- 5.Federal Register 2005. Revocation of status of specific products; group A Streptococcus. Direct final rule. Fed Regist 70:72197–72199. [PubMed] [Google Scholar]
- 6.World Health Organization 2015. Status of vaccine research and development of vaccines for Streptococcus pyogenes. World Health Organization, Geneva, Switzerland: http://www.who.int/immunization/research/meetings_workshops/GroupAStrep_VaccineRD_Sept2014.pdf. [Google Scholar]
- 7.Henningham A, Chiarot E, Gillen CM, Cole JN, Rohde M, Fulde M, Ramachandran V, Cork AJ, Hartas J, Magor G, Djordjevic SP, Cordwell SJ, Kobe B, Sriprakash KS, Nizet V, Chhatwal GS, Margarit IYR, Batzloff MR, Walker MJ. 2012. Conserved anchorless surface proteins as group A streptococcal vaccine candidates. J Mol Med 90:1197–1207. doi: 10.1007/s00109-012-0897-9. [DOI] [PubMed] [Google Scholar]
- 8.Bensi G, Mora M, Tuscano G, Biagini M, Chiarot E, Bombaci M, Capo S, Falugi F, Manetti AGO, Donato P, Swennen E, Gallotta M, Garibaldi M, Pinto V, Chiappini N, Musser JM, Janulczyk R, Mariani M, Scarselli M, Telford JL, Grifantini R, Norais N, Margarit I, Grandi G. 2012. Multi high-throughput approach for highly selective identification of vaccine candidates: the group A Streptococcus case. Mol Cell Proteomics 11:M111.015693. doi: 10.1074/mcp.M111.015693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanderson-Smith M, Batzloff M, Sriprakash KS, Dowton M, Ranson M, Walker MJ. 2006. Divergence in the plasminogen-binding group A streptococcal M protein family: functional conservation of binding site and potential role for immune selection of variants. J Biol Chem 281:3217–3226. doi: 10.1074/jbc.M508758200. [DOI] [PubMed] [Google Scholar]
- 10.Steer AC, Batzloff MR, Mulholland K, Carapetis JR. 2009. Group A streptococcal vaccines: facts versus fantasy. Curr Opin Infect Dis 22:544–552. doi: 10.1097/QCO.0b013e328332bbfe. [DOI] [PubMed] [Google Scholar]
- 11.Pandey M, Batzloff MR, Good MF. 2012. Vaccination against rheumatic heart disease: a review of current research strategies and challenges. Curr Infect Dis Rep 14:381–390. doi: 10.1007/s11908-012-0263-7. [DOI] [PubMed] [Google Scholar]
- 12.Henningham A, Ericsson DJ, Langer K, Casey LW, Jovcevski B, Chhatwal GS, Aquilina JA, Batzloff MR, Kobe B, Walker MJ. 2013. Structure-informed design of an enzymatically inactive vaccine component for group A Streptococcus. mBio 4:e00509-13. doi: 10.1128/mBio.00509-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiarot E, Faralla C, Chiappini N, Tuscano G, Falugi F, Gambellini G, Taddei A, Capo S, Cartocci E, Veggi D, Corrado A, Mangiavacchi S, Tavarini S, Scarselli M, Janulczyk R, Grandi G, Margarit I, Bensi G. 2013. Targeted amino acid substitutions impair streptolysin O toxicity and group A Streptococcus virulence. mBio 4:e00387-12. doi: 10.1128/mBio.00387-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zingaretti C, Falugi F, Nardi-Dei V, Pietrocola G, Mariani M, Liberatori S, Gallotta M, Tontini M, Tani C, Speziale P, Grandi G, Margarit I. 2010. Streptococcus pyogenes SpyCEP: a chemokine-inactivating protease with unique structural and biochemical features. FASEB J 24:2839–2848. doi: 10.1096/fj.09-145631. [DOI] [PubMed] [Google Scholar]
- 15.Cleary PP, Matsuka YV, Huynh T, Lam H, Olmsted SB. 2004. Immunization with C5a peptidase from either group A or B streptococci enhances clearance of group A streptococci from intranasally infected mice. Vaccine 22:4332–4341. doi: 10.1016/j.vaccine.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 16.Hayman WA, Brandt ER, Relf WA, Cooper J, Saul A, Good MF. 1997. Mapping the minimal murine T cell and B cell epitopes within a peptide vaccine candidate from the conserved region of the M protein of group A Streptococcus. Int Immunol 9:1723–1733. doi: 10.1093/intimm/9.11.1723. [DOI] [PubMed] [Google Scholar]
- 17.Batzloff MR, Hayman WA, Davies MR, Zeng M, Pruksakorn S, Brandt ER, Good MF. 2003. Protection against group A Streptococcus by immunization with J8-diphtheria toxoid: contribution of J8- and diphtheria toxoid-specific antibodies to protection. J Infect Dis 187:1598–1608. doi: 10.1086/374800. [DOI] [PubMed] [Google Scholar]
- 18.Van Sorge NM, Cole JN, Kuipers K, Henningham A, Aziz RK, Kasirer-Friede A, Lin L, Berends ETM, Davies MR, Dougan G, Zhang F, Dahesh S, Shaw L, Gin J, Cunningham M, Merriman JA, Hütter J, Lepenies B, Rooijakkers SHM, Malley R, Walker MJ, Shattil SJ, Schlievert PM, Choudhury B, Nizet V. 2014. The classical Lancefield antigen of group A Streptococcus is a virulence determinant with implications for vaccine design. Cell Host Microbe 15:729–740. doi: 10.1016/j.chom.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cleary PP, Kaplan EL, Handley JP, Wlazlo A, Kim MH, Hauser AR, Schlievert PM. 1992. Clonal basis for resurgence of serious Streptococcus pyogenes disease in the 1980s. Lancet 339:518–521. doi: 10.1016/0140-6736(92)90339-5. [DOI] [PubMed] [Google Scholar]
- 20.Cole JN, Barnett TC, Nizet V, Walker MJ. 2011. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol 9:724–736. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 21.Davies MR, Holden MT, Coupland P, Chen JHK, Venturini C, Barnett TC, Zakour NLB, Tse H, Dougan G, Yuen KY, Walker MJ. 2015. Emergence of scarlet fever Streptococcus pyogenes emm12 clones in Hong Kong is associated with toxin acquisition and multidrug resistance. Nat Genet 47:84–87. doi: 10.1038/ng.3147. [DOI] [PubMed] [Google Scholar]
- 22.Ben Zakour NL, Davies MR, You Y, Chen JHK, Forde BM, Stanton-Cook M, Yang R, Cui Y, Barnett TC, Venturini C, Ong CLY, Tse H, Dougan G, Zhang J, Yuen KY, Beatson SA, Walker MJ. 2015. Transfer of scarlet fever-associated elements into the group A Streptococcus M1T1 clone. Sci Rep 5:15877. doi: 10.1038/srep15877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beatson SA, Walker MJ. 2014. Tracking antibiotic resistance. Science 345:1454–1455. doi: 10.1126/science.1260471. [DOI] [PubMed] [Google Scholar]
- 24.Hu MC, Walls MA, Stroop SD, Reddish MA, Beall B, Dale JB. 2002. Immunogenicity of a 26-valent group A streptococcal vaccine. Infect Immun 70:2171–2177. doi: 10.1128/IAI.70.4.2171-2177.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McNeil SA, Halperin SA, Langley JM, Smith B, Warren A, Sharratt GP, Baxendale DM, Reddish MA, Hu MC, Stroop SD, Linden J, Fries LF, Vink PE, Dale JB. 2005. Safety and immunogenicity of 26-valent group A Streptococcus vaccine in healthy adult volunteers. Clin Infect Dis 41:1114–1122. doi: 10.1086/444458. [DOI] [PubMed] [Google Scholar]
- 26.Dale JB, Penfound TA, Chiang EY, Walton WJ. 2011. New 30-valent M protein-based vaccine evokes cross-opsonic antibodies against non-vaccine serotypes of group A streptococci. Vaccine 29:8175–8178. doi: 10.1016/j.vaccine.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Centers for Disease Control and Prevention. Streptococcus Laboratory, Centers for Disease Control and Prevention. http://www2a.cdc.gov/ncidod/biotech/strepblast.asp. Accessed 2 March 2016.
- 28.Weinberger DM, Malley R, Lipsitch M. 2011. Serotype replacement in disease after pneumococcal vaccination. Lancet 378:1962–1973. doi: 10.1016/S0140-6736(10)62225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dale JB, Fischetti VA, Carapetis JR, Steer AC, Sow S, Kumar R, Mayosi BM, Rubin FA, Mulholland K, Hombach JM, Schödel F, Henao-Restrepo AM. 2013. Group A streptococcal vaccines: paving a path for accelerated development. Vaccine 31(Suppl 2):B216–B222. doi: 10.1016/j.vaccine.2012.09.045. [DOI] [PubMed] [Google Scholar]
- 30.Courtney HS, Hasty DL, Dale JB. 2003. Serum opacity factor (SOF) of Streptococcus pyogenes evokes antibodies that opsonize homologous and heterologous sof-positive serotypes of group A streptococci. Infect Immun 71:5097–5103. doi: 10.1128/IAI.71.9.5097-5103.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schulze K, Medina E, Guzmán CA. 2006. Intranasal immunization with serum opacity factor (SOF) of Streptococcus pyogenes fails to protect mice against lethal mucosal challenge with a heterologous strain. Vaccine 24:1446–1450. doi: 10.1016/j.vaccine.2005.06.039. [DOI] [PubMed] [Google Scholar]
- 32.McArthur J, Medina E, Mueller A, Chin J, Currie BJ, Sriprakash KS, Talay SR, Chhatwal GS, Walker MJ. 2004. Intranasal vaccination with streptococcal fibronectin binding protein Sfb1 fails to prevent growth and dissemination of Streptococcus pyogenes in a murine skin infection model. Infect Immun 72:7342–7345. doi: 10.1128/IAI.72.12.7342-7345.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji Y, Carlson B, Kondagunta A, Cleary PP. 1997. Intranasal immunization with C5a peptidase prevents nasopharyngeal colonization of mice by the group A Streptococcus. Infect Immun 65:2080–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park HS, Cleary PP. 2005. Active and passive intranasal immunizations with streptococcal surface protein C5a peptidase prevent infection of murine nasal mucosa-associated lymphoid tissue, a functional homologue of human tonsils. Infect Immun 73:7878–7886. doi: 10.1128/IAI.73.12.7878-7886.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turner CE, Kurupati P, Wiles S, Edwards RJ, Sriskandan S. 2009. Impact of immunization against SpyCEP during invasive disease with two streptococcal species: Streptococcus pyogenes and Streptococcus equi. Vaccine 27:4923–4929. doi: 10.1016/j.vaccine.2009.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pandey M, Batzloff MR, Good MF. 2009. Mechanism of protection induced by group A Streptococcus vaccine candidate J8-DT: contribution of B and T-cells towards protection. PLoS One 4:e5147. doi: 10.1371/journal.pone.0005147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabharwal H, Michon F, Nelson D, Dong W, Fuchs K, Manjarrez RC, Sarkar A, Uitz C, Viteri-Jackson A, Suarez RS, Blake M, Zabriskie JB. 2006. Group A Streptococcus (GAS) carbohydrate as an immunogen for protection against GAS infection. J Infect Dis 193:129–135. doi: 10.1086/498618. [DOI] [PubMed] [Google Scholar]
- 38.Goldstein I, Rebeyrotte P, Parlebas J, Halpern B. 1968. Isolation from heart valves of glycopeptides which share immunological properties with Streptococcus haemolyticus group A polysaccharides. Nature 219:866–868. doi: 10.1038/219866a0. [DOI] [PubMed] [Google Scholar]
- 39.Kirvan CA, Swedo SE, Heuser JS, Cunningham MW. 2003. Mimicry and autoantibody-mediated neuronal cell signaling in Sydenham chorea. Nat Med 9:914–920. doi: 10.1038/nm892. [DOI] [PubMed] [Google Scholar]
- 40.Dale JB, Penfound T, Chiang EY, Long V, Shulman ST, Beall B. 2005. Multivalent group A streptococcal vaccine elicits bactericidal antibodies against variant M subtypes. Clin Diagn Lab Immunol 12:833–836. doi: 10.1128/CDLI.12.7.833-836.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu M, Zhu H, Zhang J, Lei B. 2007. Active and passive immunizations with the streptococcal esterase Sse protect mice against subcutaneous infection with group A streptococci. Infect Immun 75:3651–3657. doi: 10.1128/IAI.00038-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan X, Wang X, Li N, Cui H, Hou B, Gao B, Cleary PP, Wang B. 2014. Sortase A induces Th17-mediated and antibody-independent immunity to heterologous serotypes of group A streptococci. PLoS One 9:e107638. doi: 10.1371/journal.pone.0107638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herwald H, Cramer H, Mörgelin M, Russell W, Sollenberg U, Norrby-Teglund A, Flodgaard H, Lindbom L, Björck L. 2004. M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell 116:367–379. doi: 10.1016/S0092-8674(04)00057-1. [DOI] [PubMed] [Google Scholar]
- 44.Sun H, Ringdahl U, Homeister JW, Fay WP, Engleberg NC, Yang AY, Rozek LS, Wang X, Sjöbring U, Ginsburg D. 2004. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 305:1283–1286. doi: 10.1126/science.1101245. [DOI] [PubMed] [Google Scholar]
- 45.Cole JN, McArthur JD, McKay FC, Sanderson-Smith ML, Cork AJ, Ranson M, Rohde M, Itzek A, Sun H, Ginsburg D, Kotb M, Nizet V, Chhatwal GS, Walker MJ. 2006. Trigger for group A streptococcal M1T1 invasive disease. FASEB J 20:1745–1747. doi: 10.1096/fj.06-5804fje. [DOI] [PubMed] [Google Scholar]
- 46.Virtaneva K, Porcella SF, Graham MR, Ireland RM, Johnson CA, Ricklefs SM, Babar I, Parkins LD, Romero RA, Corn GJ, Gardner DJ, Bailey JR, Parnell MJ, Musser JM. 2005. Longitudinal analysis of the group A streptococcus transcriptome in experimental pharyngitis in cynomolgus macaques. Proc Natl Acad Sci U S A 102:9014–9019. doi: 10.1073/pnas.0503671102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skinner JM, Caro-Aguilar IC, Payne AM, Indrawati L, Fontenot J, Heinrichs JH. 2011. Comparison of rhesus and cynomolgus macaques in a Streptococcus pyogenes infection model for vaccine evaluation. Microb Pathog 50:39–47. doi: 10.1016/j.micpath.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 48.Ashbaugh CD, Moser TJ, Shearer MH, White GL, Kennedy RC, Wessels MR. 2000. Bacterial determinants of persistent throat colonization and the associated immune response in a primate model of human group A streptococcal pharyngeal infection. Cell Microbiol 2:283–292. doi: 10.1046/j.1462-5822.2000.00050.x. [DOI] [PubMed] [Google Scholar]
- 49.Rivera-Hernandez T, Carnathan DG, Moyle PM, Toth I, West NP, Young PR, Silvestri G, Walker MJ. 2014. The contribution of non-human primate models to the development of human vaccines. Discov Med 18:313–322. [PMC free article] [PubMed] [Google Scholar]
- 50.Fox EN, Waldman RH, Wittner MK, Mauceri AA, Dorfman A. 1973. Protective study with a group A streptococcal M protein vaccine. I. Infectivity challenge of human volunteers. J Clin Invest 52:1885–1892. doi: 10.1172/JCI107372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Polly SM, Waldman RH, High P, Wittner MK, Dorfman A. 1975. Protective studies with a group A streptococcal M protein vaccine. II. Challenge of volunteers after local immunization in the upper respiratory tract. J Infect Dis 131:217–224. doi: 10.1093/infdis/131.3.217. [DOI] [PubMed] [Google Scholar]
- 52.D’Alessandri R, Plotkin G, Kluge RM, Wittner MK, Fox EN, Dorfman A, Waldman RH. 1978. Protective studies with group A streptococcal M protein vaccine. III. Challenge of volunteers after systemic or intranasal immunization with type 3 or type 12 group A Streptococcus. J Infect Dis 138:712–718. doi: 10.1093/infdis/138.6.712. [DOI] [PubMed] [Google Scholar]
- 53.Tsoi SK, Smeesters PR, Frost HRC, Licciardi P, Steer AC. 2015. Correlates of protection for M protein-based vaccines against group A Streptococcus. J Immunol Res 2015:167089. doi: 10.1155/2015/167089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reglinski M, Lynskey NN, Choi YJ, Edwards RJ, Sriskandan S. 2016. Development of a multicomponent vaccine for Streptococcus pyogenes based on the antigenic targets of IVIG. J Infect 72:450–459. doi: 10.1016/j.jinf.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mortensen R, Nissen TN, Fredslund S, Rosenkrands I, Christensen JP, Andersen P, Dietrich J. 2016. Identifying protective Streptococcus pyogenes vaccine antigens recognized by both B and T cells in human adults and children. Sci Rep 6:22030. doi: 10.1038/srep22030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kansal RG, Nizet V, Jeng A, Chuang W-J, Kotb M. 2003. Selective modulation of superantigen-induced responses by streptococcal cysteine protease. J Infect Dis 187:398–407. doi: 10.1086/368022. [DOI] [PubMed] [Google Scholar]
- 57.Batzloff MR, Yan H, Davies MR, Hartas J, Lowell GH, White G, Burt DS, Leanderson T, Good MF. 2005. Toward the development of an antidisease, transmission-blocking intranasal vaccine for group A Streptococcus. J Infect Dis 192:1450–1455. doi: 10.1086/466528. [DOI] [PubMed] [Google Scholar]
- 58.Timmer AM, Timmer JC, Pence MA, Hsu LC, Ghochani M, Frey TG, Karin M, Salvesen GS, Nizet V. 2009. Streptolysin O promotes group A Streptococcus immune evasion by accelerated macrophage apoptosis. J Biol Chem 284:862–871. doi: 10.1074/jbc.M804632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zimmerman T, Petit Frère C, Satzger M, Raba M, Weisbach M, Döhn K, Popp A, Donzeau M. 2006. Simultaneous metal chelate affinity purification and endotoxin clearance of recombinant antibody fragments. J Immunol Methods 314:67–73. doi: 10.1016/j.jim.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 60.Shafer DE, Toll B, Schuman RF, Nelson BL, Mond JJ, Lees A. 2000. Activation of soluble polysaccharides with 1-cyano-4-dimethylaminopyridinium tetrafluoroborate (CDAP) for use in protein-polysaccharide conjugate vaccines and immunological reagents. II. Selective crosslinking of proteins to CDAP-activated polysaccharides. Vaccine 18:1273–1281. doi: 10.1016/S0264-410X(99)00370-9. [DOI] [PubMed] [Google Scholar]
- 61.Masuko T, Minami A, Iwasaki N, Majima T, Nishimura S-I, Lee YC. 2005. Carbohydrate analysis by a phenol–sulfuric acid method in microplate format. Anal Biochem 339:69–72. doi: 10.1016/j.ab.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 62.Pandey M, Langshaw E, Hartas J, Lam A, Batzloff MR, Good MF. 2015. A synthetic M protein peptide synergizes with a CXC chemokine protease to induce vaccine-mediated protection against virulent streptococcal pyoderma and bacteremia. J Immunol 194:5915–5925. doi: 10.4049/jimmunol.1500157. [DOI] [PubMed] [Google Scholar]
- 63.Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG, Simpson AJ, Buchanan JT, Chhatwal GS, Kotb M, Nizet V. 2007. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med 13:981–985. doi: 10.1038/nm1612. [DOI] [PubMed] [Google Scholar]
- 64.Cox C, Reeder JE, Robinson RD, Suppes SB, Wheeless LL. 1988. Comparison of frequency distributions in flow cytometry. Cytometry 9:291–298. doi: 10.1002/cyto.990090404. [DOI] [PubMed] [Google Scholar]
- 65.Roederer M, Moore W, Treister A, Hardy RR, Herzenberg LA. 2001. Probability binning comparison: a metric for quantitating multivariate distribution differences. Cytometry 45:47–55. doi:. [DOI] [PubMed] [Google Scholar]
- 66.National Health and Medical Research Council 2013. Australian code for the care and use of animals for scientific purposes, 8th ed. National Health and Medical Research Council, Canberra, Australia. [Google Scholar]
- 67.National Health and Medical Research Council 2015. National Statement on ethical conduct in human research 2007 (updated May 2015). National Health and Medical Research Council, Canberra, Australia. [Google Scholar]
- 68.Pandey M, Wykes MN, Hartas J, Good MF, Batzloff MR. 2013. Long-term antibody memory induced by synthetic peptide vaccination is protective against Streptococcus pyogenes infection and is independent of memory T cell help. J Immunol 190:2692–2701. doi: 10.4049/jimmunol.1202333. [DOI] [PMC free article] [PubMed] [Google Scholar]