

Abstract
ARX loss-of-function mutations cause X-linked lissencephaly with ambiguous genitalia (XLAG), a severe neurological condition that results in profound brain malformations, including microcephaly, absence of corpus callosum, and impairment of the basal ganglia. Despite such dramatic defects, their nature and origin remain largely unknown. Here, we used Arx mutant mice as a model to characterize the cellular and molecular mechanisms underlying the basal ganglia alterations. In these animals, the early differentiation of this tissue appeared normal, whereas subsequent differentiation was impaired, leading to the periventricular accumulation of immature neurons in both the lateral ganglionic eminence and medial ganglionic eminence (MGE). Both tangential migration toward the cortex and striatum and radial migration to the globus pallidus and striatum were greatly reduced in the mutants, causing a periventricular accumulation of NPY+ or calretinin+ neurons in the MGE. Arx mutant neurons retained their differentiation potential in vitro but exhibited deficits in morphology and migration ability. These findings imply that cell-autonomous defects in migration underlie the neuronal localization defects. Furthermore, Arx mutants lacked a large fraction of cholinergic neurons and displayed a strong impairment of thalamocortical projections, in which major axon fiber tracts failed to traverse the basal ganglia. Altogether, these results highlight the critical functions of Arx in promoting neural migration and regulating basal ganglia differentiation in mice, consistent with the phenotype of XLAG patients.
Keywords: forebrain, interneurons, neural migration, striatum, development, GABA, thalamocortical projections, cell fate
Introduction
The Arx gene, which encodes a highly conserved homeodomain-containing transcription factor, maps to the short arm of the mammalian X chromosome (Miura et al., 1997; Campbell and Tomlinson, 1998; Melkman and Sengupta, 2005; Seufert et al., 2005; McKenzie et al., 2007). Mutations of the human ARX gene were initially characterized in patients with X-linked West syndrome, a neurological condition affecting children and young males characterized by mental retardation, infantile spasms, and hypsarrhythmia (Stromme et al., 2002a). Subsequently, ARX mutations were shown to account for additional neurological conditions, including forms of nonsyndromic X-linked mental retardation, myoclonic epilepsy, Partington disease, and severe cases of lissencephaly associated with abnormal genitalia (XLAG) (Bienvenu et al., 2002; Kitamura et al., 2002; Stromme et al., 2002a; Poirier et al., 2006). Overall, the severity of the Arx mutation correlates with the clinical prognosis (Gecz et al., 2006). Dystonia is a condition that was frequently found in different ARX-dependent disorders. Indeed, patients were described as experiencing dystonic hands movements, as well as wrist and finger contractures, accompanied by general muscle weakness (Stromme et al., 2002b; Turner et al., 2002). In other cases, stereotyped repetitive movements or tremors were reported (Turner et al., 2002). Together, these observations strongly suggest a functional alteration of the basal ganglia nuclei, whose function is to control and finely tune the activity of the motor cortex through both direct and indirect circuits connecting striatum, globus pallidus, substantia nigra, and other relay nuclei with the cerebral cortex. Accordingly, alterations of these two circuits were previously associated with impaired voluntary movements and the development of involuntary actions, such as ticks, tremors, and dystonia (Albin et al., 1989; Graybiel, 2000).
In mice, during embryonic development, the Arx ortholog exhibits an early-onset expression in the forebrain vesicle. In the ventral telencephalon, Arx is expressed in progenitor cells and neurons of the lateral and medial ganglionic eminences (LGE and MGE, respectively), primordia of the striatal and pallidal subdivision of the basal ganglia (Kitamura et al., 2002; Colombo et al., 2004; Cobos et al., 2005).
Arx mutant mice die perinatally as a result of a hypoglycemia related to pancreatic defects (Collombat et al., 2003, 2005). Arx-deficient brains were found to be dramatically altered, exhibiting poorly developed olfactory bulbs and reduced volumes of the cerebral cortex and hippocampus (Kitamura et al., 2002). Moreover, GABAergic neurons originating from the subpallium exhibited an aberrant migration toward the cerebral cortex, resulting in a decrease in the number of cortical GABAergic interneurons (Kitamura et al., 2003). Unfortunately, the perinatal death of these mice did not allow any correlation of these specific alterations with the neurological manifestations observed in humans. Overall, both the descriptions of the patient clinical conditions and the GABAergic defects characterized in Arx mutant mice suggest a functional impairment of the basal ganglia morphogenesis. Herein, we present a detailed molecular characterization of the basal ganglia development and GABAergic and cholinergic neural differentiation in Arx mutant brains.
Materials and Methods
Animals.
Arx mutant mice (Collombat et al., 2003) were maintained by backcrossing to C57BL/6 animals for at least five generations. Genotyping was performed by morphological assessment [when possible, after embryonic day 13.5 (E13.5)] and confirmed by PCR using a mixture of three primers to identify the wild-type (wt) and the mutant alleles, generating 0.25 and 0.4 kb PCR products, respectively (primer 1, CGGCTCACTACACTTGTTACCGCTTGGTCC; primer 2, AGCAGCCCTCTTCCTGGTACTGATTGCTC; and primer 3, TGCTGCAAGGCGATAAGTTGGGTAACGC) (Collombat et al., 2003). Mice were maintained at the San Raffaele Scientific Institute institutional mouse facility, and experiments were performed in accordance with experimental protocols approved by local Institutional Animal Care and Use Committees.
In situ RNA hybridization.
Unless otherwise indicated, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO). In situ RNA hybridizations on frozen sections were performed as described previously by Schaeren-Wiemers and Gerfin-Moser (1993) with the following modifications. Sections were fixed for 30 min at room temperature in 4% paraformaldehyde (PFA) in PBS and treated for 5 min with 1 μg/ml proteinase K in 1 mm EDTA and 20 mm Tris-HCl, pH 7.0. Before hybridization, they were washed twice in 2× SSC for 15 min and incubated in 0.1 m Tris/0.1 m glycine for at least 30 min. The hybridization solution (60 μl/slide) contained 50% formamide, 5× SSC, pH adjusted with citric acid to pH 6.0, 5% dextran sulfate, 2 mg/ml heparin, 100 μg/ml tRNA, and from 1:100 to 1:50 dilution of the riboprobes, and was performed overnight at 65°C under coverslips. Next, the sections were washed for 1–2 h in 0.5× SSC and 20% formamide at 65°C. Subsequently, they were treated with 10 μg/ml RNase A for 30 min at 37°C in NTE (NaCl-Tris-EDTA) and then washed for 4 h in 0.5× SSC and 20% formamide at 65°C and for 30 min in 2× SSC, and blocked for 1 h at room temperature in 1% blocking reagent (Roche Diagnostics, Basel, Switzerland) in 100 mm maleic acid, 150 mm NaCl, and 0.1% Tween 20 (MABT). A 1:5000 dilution of anti-digoxigenin–alkaline phosphatase (AP)-conjugated antibody (Roche Diagnostics) was preincubated for at least 1 h in 1% blocking reagent in MABT at 4°C. Slices were incubated with the antibody overnight at 4°C, washed for 6 h in TBST (TBS–Tween 20), washed for 30 min in 100 mm Tris-HCl, pH 9.5, 100 mm NaCl, 50 mm MgCl2, 0.1% Tween 20, and 2 mm levamisole (NTMT), and stained using centrifuged BM purple AP substrate (Roche Diagnostics) in 0.3% Tween 20 for 12–36 h at 4°C or room temperature. They were washed in NTMT and then in distillated water and mounted in Aquamount (Polysciences, Warrington, PA). The following probes were used: Dlx2 (Anderson et al., 1997), Nkx2.1, Etv1 (ER81), enkephalin (Sussel et al., 1999), EBF1 (Garel et al., 1999), Dlx5 (kindly provided by F. Guillemot), Lhx6, Lhx7, choline acetyl transferase (ChAT) (a kind gift from V. Pachnis), Lmo3 (kindly provided by T. Rabbitts), and neuregulin (a kind gift from C. Birchmeier).
Immunohistochemistry.
For immunoperoxidase staining, 10-μm-thick frozen sections were treated with 3% hydrogen peroxidase in methanol for 30 min at room temperature. Sections were rehydrated and blocked in 10% fetal calf serum and 0.5% BSA in PBS for 1 h. Primary antibodies were diluted in the same medium, applied on sections, and incubated overnight at 4°C. Then, the slices were washed and incubated with appropriate biotinylated secondary antibody (DakoCytomation, Glostrup, Denmark) for 90 min. After washing, the sections were processed with a Vectastain ABC kit (Vector Laboratories, Burlingame, CA) for 1 h at room temperature and revealed with DAB peroxidase substratum (SK-4100; Vector Laboratories). Finally, sections were dehydrated, dried, and coverslipped with Eukitt (Electron Microscopy Sciences, Hatfield, PA).
The primary antibody used were as follows: mouse anti-microtubule-associated protein 2 (Map2) (1:400; Millipore, Billerica, MA), rabbit anti-NPY (1:1000; Incstar, Stillwater, MN), rabbit anti-calretinin (1:1000; Swant, Bellinzona, Switzerland), rabbit anti-dopamine and cAMP-regulated phosphoprotein 32 (DARPP32; 1:50; Immunological Sciences, Rome, Italy), and rabbit anti-ChAT (1:500; Millipore). For immunofluorescence, secondary antibodies of the Alexa family were used (Invitrogen, Eugene, OR) in combination with the fluorescent dye Hoechst 33342 (Invitrogen). Then, slices were washed and mounted in fluorescent mounting medium (DakoCytomation).
Axonal tracing.
After overnight fixation in 4% PFA at 4°C, single crystals of the fluorescent carbocyanine dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI; Invitrogen) or 4-[4-(dihexadecyl-amino)styryl]N-methyl-pyridinium iodide (DiA; Invitro-gen) were placed in single or multiple locations in the neocortex (Metin and Godement, 1996). After 3 weeks at room temperature in 4% PFA to allow dye diffusion, the sample was embedded in 5% agarose and cut into 200-μm-thick sections on a vibratome (VT100; Leica Microsystems, Bannockburn, IL). Counterstaining was performed using Hoechst, and digital images were acquired using a digital camera (DXM 1200; Nikon, Tokyo, Japan) on a fluorescent dissection microscope (Nikon SMZ1500).
Birth-dating analysis.
Pregnant females were injected intraperitoneally at different embryonic stages (E11.5, E13.5, and E15.5) with 5-bromo-2′-deoxyuridine (BrdU) at a dose of 100 μg BrdU/g of body weight in a saline solution. Embryos were harvested at E18.5, and brains were isolated and fixed in paraformaldehyde 4% in PBS. Immunohistochemistry was performed as described with a mouse monoclonal BrdU antibody (BD Biosciences, San Jose, CA).
Dissociated cell culture.
E18.5 wt and mutant mouse brains were isolated and embedded in 4% low-melt agarose to obtain 250 μm vibratome sections. Ventricular zone (VZ) and subventricular zone (SVZ) regions from the basal ganglia were dissected from wt and mutant brain slices under a dissection microscope. Tissues were treated with papain solution for 10 min at 37°C before triturating mechanically with fire-polished glass pipettes to obtain a single-cell suspension. Approximately 1 × 106 cells were plated in glass dishes coated with polylysine in 24-well plates and cultured in serum-free medium supplemented with B27. After 5 d cells, cell cultures were subjected to immunofluorescence procedure as described above with the following primary antibodies: mouse MAP2 (Sigma-Aldrich) and rabbit TuJ1 (Covance, Princeton, NJ), DARPP32 (Immunological Sciences), calbindin, (Swant) and EBF (kind gift from G. G. Consalez).
Organotypic culture and grafting experiments.
Slice cultures of embryonic mouse forebrain were prepared as described previously (Anderson et al., 1997). Briefly, mouse brains were isolated and embedded in 4% low-melt agarose (Sigma-Aldrich), and 250-μm-thick coronal sections were cut on a vibratome. The sections were then transferred to polycarbonate culture membranes (diameter, 13 mm; pore size, 8 μm; Costar, Cambridge, MA) in organ tissue dishes containing 1.5 ml of serum-containing medium (Invitrogen α-MEM with 10% fetal calf serum, glutamine, penicillin, and streptomycin). Slices were maintained for 1 h at 37°C in 5% CO2 in a standard sterile incubator. Then, medium was changed to a Neurobasal/B27 (Invitrogen). Transplantations were performed immediately after this step. Fragments of periventricular MGE were dissected out from brain slices either from green fluorescent protein (GFP) ubiquitous expressing mice or from Arx mutant animals and transplanted in homotopic positions in either mutant or wt brain slices, respectively. Grafted slices were cultured up to 72 h.
In vitro neuronal migration assays.
E14.5 mouse brain slices were obtained from isolated brain sectioned using a vibratome as described above. Small pieces or fragments of the MGE were dissected out from the slices and incubated for 1 h in 1 ml of medium with serum. Explants were then pipetted in a liquid Matrigel solution (BD Biosciences) and rapidly placed on 3.5 cm sterile culture dishes. Matrigel drops containing the explants were allowed to polymerize for ∼40 min. Serum-free medium was then added, and explants were cultured for up to 72 h in a standard CO2 incubator.
Cell counts and statistics.
Cells were counted using a Nikon Eclipse E600 fluorescent microscope connected to the DXM1200C camera and the ACT-1 Nikon software. For BrdU quantification in vivo, a minimum of four nonsequential sections taken at different levels of the basal ganglia were analyzed for both wild-type and mutant genotypes for each experiment (three independent experiments). For each section, three different fields were chosen randomly in the subpallial SVZ region. The number of DAPI-positive nuclei identifiable in a selected optical field was counted, and the fraction of labeled nuclei showing BrdU staining in the same field was assessed. A total of ∼600 DAPI-positive nuclei were analyzed for each section. Data analysis was performed blind to genotype.
Results were expressed as mean value ± SD and were tested for statistical significance by the one-tailed Student's t test for paired differences with GraphPad (San Diego, CA) Prism software.
Results
In the prenatal mouse subcortical telencephalon, Arx is normally expressed in neural progenitors and immature neurons throughout the subpallium, including in the LGE, MGE, and anterior entopeduncular area (AEP) (Miura et al., 1997; Kitamura et al., 2003; Poirier et al., 2004; Cobos et al., 2005; Colombo et al., 2004). These cells will ultimately give rise to the striatum (or caudate–putamen), globus pallidus, cholinergic nuclei, and interneurons that tangentially migrate to the cerebral cortex, olfactory bulb, and striatum (Miura et al., 1997; Marin et al., 2000; Kitamura et al., 2003; Marin and Rubenstein, 2003; Colombo et al., 2004; Poirier et al., 2004). To assess Arx function during these differentiation processes, we analyzed mice in which the LacZ gene had been inserted into the Arx locus, leading to gene inactivation (Collombat et al., 2003). Arx-deficient mice die soon after birth, probably because of a severe hypoglycemia secondary to the lack of pancreatic α-cells (Collombat et al., 2003). Here, we focused on the consequences of Arx inactivation on prenatal subcortical telencephalic morphogenesis.
Defects in striatal histogenesis in neonatal Arx mutants
Nissl staining of newborn [postnatal day 0 (P0)] mutant brain sections revealed that the dorsal striatum was relatively normal, whereas its ventral counterparts (nucleus accumbens and olfactory tubercle), as well as septum structures, appeared reduced in size (Fig. 1 A,B). Importantly, the subventricular zone of the LGE was found clearly enlarged compared with wild-type brains, suggesting an accumulation of a large number of cells (Fig. 1 A–D, dashed lines). Striatal differentiation was assessed by its expression of Ebf1, preproenkephalin RNA, and calbindin or tyrosine hydroxylase (TH) proteins. Ebf1 and preproenkephalin are normally expressed in projection neurons of the striatonigral and striatopallidal pathways, respectively (Song and Harlan, 1993; Lobo et al., 2006). Calbindin is a general marker of striatal neurons, whereas TH marks afferent fibers from the substantia nigra (Gerfen and Wilson, 1996; Jacobowitz and Abbott, 1998). In Arx mutants, enkephalin and TH expression in the dorsal striatum appeared roughly normal, whereas their expression was nearly lost in the nucleus accumbens and olfactory tubercle (Fig. 1, E–H, Ot). Although expression of Ebf1 appeared unchanged in the striatum (Fig. 1 I,J), its expression was lost in the pallidum (Fig. 1 I,J, arrowheads); the latter expression domain is associated with ventrally migrating cells that regulate the entry of thalamic axons into the striatum (López-Bendito et al., 2006). Calbindin expression was reduced in the lateral striatum (Fig. 1 K,L).
Figure 1.
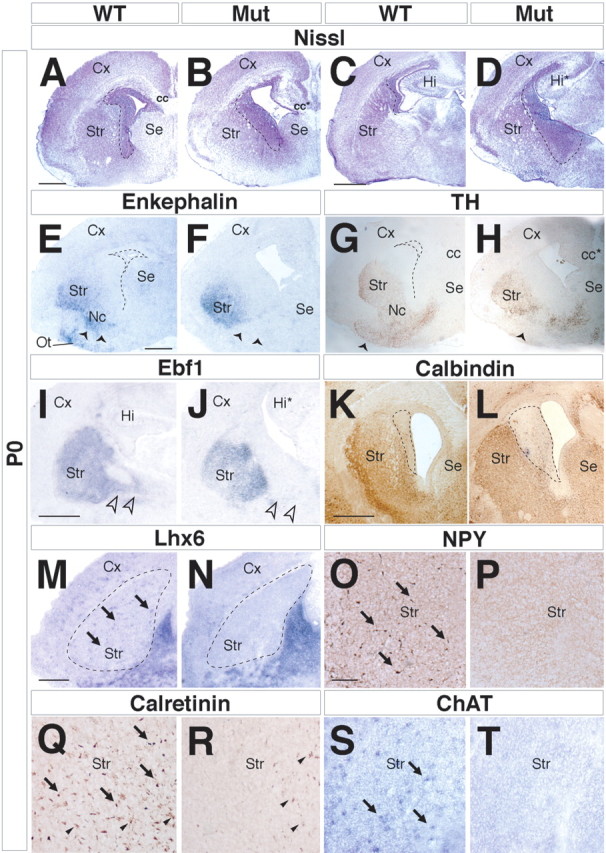
Striatal defects in Arx mutant brains. A–D, Nissl staining on P0 brain coronal sections demonstrates an abnormal, expanded, cell-dense periventricular region (outlined by dashed lines). E–L, Analysis of the mature striatal compartment on coronal sections using marker genes such as enkephalin (E, F), TH (G, H), Ebf1 (I, J), and calbindin (K, L). All of these are found to be reduced in Arx mutant brains. Furthermore, enkephalin and TH fail to label the olfactory tubercle (E, Ot) and nucleus accumbens (Nc) in Arx mutants (E–H, arrowheads). E, G, The dashed lines indicate the ventricle. I, J, Lack of Ebf1 expression domain in the MGE of Arx mutant brains (open arrowheads). K, L, The calbindin-negative area in the periventricular region of the anterior striatum is expanded in the mutant (dashed lines). M–T, Absence of GABAergic and cholinergic interneurons in Arx mutant striatum. The expression of markers of interneurons, such as Lhx6 (M, arrows), or of specific neuronal subclasses, such as NPY (O, arrows), calretinin (Q, arrows), and ChAT (S, arrows), is lacking in the mutant striatal tissue (N, P, R, T). Q, R, Arrowheads point to thalamocortical fibers positive for calretinin. cc, Corpus callosum; cc*, mutant corpus callosum; Cx, cerebral cortex; Hi, hippocampus; Hi*, mutant hippocampus; Mut, mutant; Se, septum; Str, striatum. Scale bars: A (for A, B), C (for C, D), E (for E–H), I (for I, J), K (for K, L), 200 μm; M (for M, N), 100 μm; O (for O–T), 50 μm.
The striatum contains interneurons implicated in organizing local circuits with striatal principal neurons (spiny neurons) accounting for ∼10% of the entire striatal cell population that are generated in the MGE (Marin et al., 2000; Corbin et al., 2001). Arx mutant striata were, therefore, assessed for GABAergic interneurons expressing Nkx2.1, Lhx6, NPY, or calretinin. In all instances, the mutant striatum lacked any cells positive for these markers, demonstrating a drastic reduction in the striatal GABAergic interneuron population (Fig. 1 M–R) (data not shown). These data confirmed previous findings reported by Kitamura et al. (2002). However, because Nkx2.1 is also expressed in striatal cholinergic interneurons (Marin et al., 2000), its absence in Arx mutants could reflect an additional lack of this cell population. We, then, expanded this analysis, considering also cholinergic interneuron population residing in the striatum. Although ChAT-expressing neurons were easily notable in wt striatum, no staining was detected in mutant analogous tissues, indicating a severe loss of cholinergic cells in Arx mutants (Fig. 1 S,T). Altogether, our results suggest that, in Arx mutants, the dorsal striatum, although present, is reduced in size and devoid of GABAergic and cholinergic interneurons, whereas there is a severe defect of the ventral striatum.
Globus pallidus formation is impaired in Arx mutant mice
Arx expression is normally more prominent in the SVZ of the MGE than in the LGE (Cobos et al., 2005) and is maintained in differentiating neurons of the pallidum, including the globus pallidus (GP), bed nucleus of stria terminalis, and a more superficial region called the ventral pallidum. Nissl staining of P0 mutant brains suggests a dramatic expansion of the SVZ of the MGE and a fragmentation of the pallidal nuclei (Fig. 2 A,B). Immunohistochemical analysis confirms that the pallidum has major deficits. The GP normally receives TH+ collaterals of nigrostriatal fibers (Fig. 2 C) (Lindvall and Bjorklund, 1979; Cossette et al., 1999; Fuchs and Hauber, 2003); in Arx mutants, TH+ fibers were not identified in this location (Fig. 2 D). Along the same line, the glutamic acid decarboxylase 67 (Gad67) expression, normally marking GABAergic neurons, was decreased in the mutant pallidum (Fig. 2 E,F). Calretinin, which is a calcium-binding protein detected in a subset of pallidal interneurons (Cooper and Stanford, 2002) and in a subpopulation of thalamocortical axons, normally labels the region of the GP but failed to delineate this structure in Arx mutants (Fig. 2 G,H).
Figure 2.
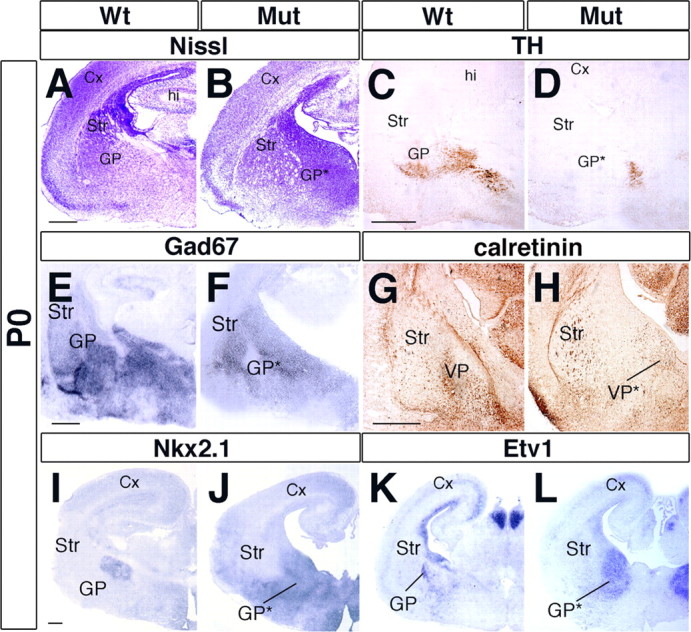
Impairment of GP formation in Arx mutant brains. A, B, P0 Coronal sections stained for Nissl unravel drastic alterations in the cytoarchitecture of the ventral telencephalon and a loss of the GP in the Arx mutants. C, D, TH+ dopaminergic afferent fibers to the GP are lacking in Arx mutants. E, F, GAD67 staining failed to identify the basal nuclei in Arx mutants found in control tissue. G, H, Calretinin typically labels GP interneurons and allows the visualization of the entire GP. In mutant tissue, such a structure is lacking (H). I–L, Molecular markers of GP neurons, such as Nkx2.1 (I, J) and Etv1 (K, L), are not detected in the putative GP location in mutant tissue but appear to be ectopically expressed in a medial–ventral location. Cx, Cerebral cortex; hi, hippocampus; GP*, mutant globus pallidus; Mut, mutant; Str, striatum; VP, ventral pallidum; VP*, mutant ventral pallidum. Scale bars: A (for A, B), C (for C, D), I (for I–L), 200 μm; E (for E, F), G (for G, H), 150 μm.
Other GP molecular features were present but lacked a normal distribution. Nkx2.1 expression was greatly expanded, extending from the progenitor zone to the pial surface, rather than labeling a distinct GP nucleus (Fig. 2 I,J). Etv1 (ER81) showed a similar expanded expression (Fig. 2 K,L). Thus, at P0, albeit the key molecular characteristics of the MGE territory appeared correctly specified (Nkx2.1 and Etv1), its histogenesis into pallidal nuclei was altered.
Arx mutant neurons exhibit a time-dependent arrest of migration and differentiation
We next investigated the degree of differentiation of the cells present in the expanded SVZ of the E18.5 mutant subpallium (Fig. 3 A,B). These exhibited a reduced expression of MAP2, which is a marker of differentiated and mature neurons (Chun and Shatz, 1989; Johnson and Jope, 1992) (Fig. 3 C,D), and failed later on to express additional postmitotic markers (enkephalin and calbindin) (Fig. 1 E,F,K,L). Ebf1 expression was also found altered despite a residual detection in the SVZ of the prenatal LGE (Fig. 3 E,F). On the contrary, they accumulated Dlx2 transcripts, which usually mark cells within the ventricular and subventricular zones of the subpallium (Anderson et al., 1997; Eisenstat et al., 1999) (Fig. 3 G,H). This suggested that the cells present in the expanded SVZ may be at a less mature state than those of control tissues. A BrdU pulse labeling performed for 1 h at E18.5 indicated that the SVZ of the controls and mutants had a similar density of S-phase cells (13 ± 3 and 15 ± 3% for wt and mutants, respectively; 2500 neurons counted for each genotype; three independent experiments; p > 0.05) (Fig. 3 I,J), suggesting that the expanded SVZ had normal mitotic properties.
Figure 3.
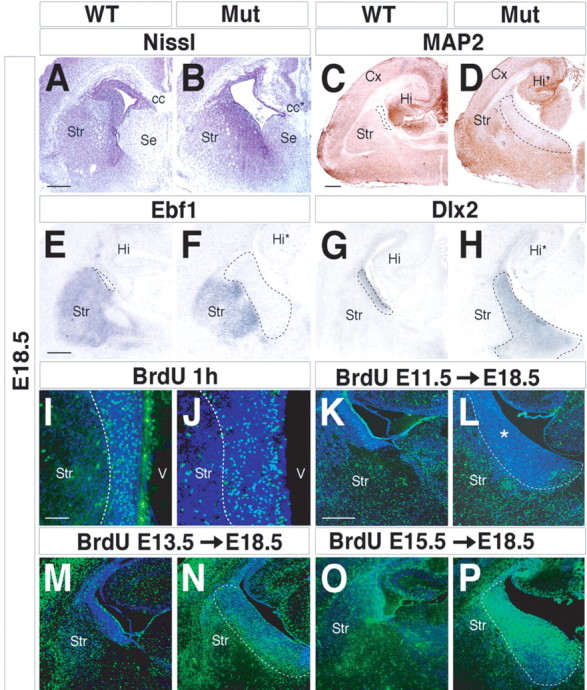
Characterization of the abnormal periventricular area found in Arx mutants. A, B, E18.5 coronal sections stained for Nissl outline the cytoarchitectural defects of the Arx mutant striatum. Marker genes for mature striatal neurons, such as MAP2 (C, D) and Ebf1 (E, F), are not detected in the mutant periventricular area (dashed lines). G, H, Dlx2 transcripts identify the enlarged mutant periventricular region. I, J, Labeling of proliferating cells located within the periventricular area. An expanded area of BrdU-positive cells was observed in mutants (I, J, dashed lines), although without any modification in the number of proliferating cells. K–P, Birth-dating analysis of basal ganglia neurons during development. Pregnant females received a single BrdU injection at E11.5 (K, L), E13.5 (M, N), and E15.5 (O, P), and the location of labeled cells was examined at E18.5. K, L, A single BrdU pulse at E11.5 marks cells distributed throughout the mature compartment both in controls and Arx mutants. *, Expanded mutant SVZ. M–P, Mutant cells labeled at E13.5 and E15.5 are progressively accumulated within the periventricular area (dashed lines) and fail to migrate into the striatal mantle zone. cc, Corpus callosum; cc*, mutant corpus callosum; Cx, cerebral cortex; Hi, hippocampus; Hi*, mutant hippocampus; Mut, mutant; Se, septum; Str, striatum; V, ventricle. Scale bars: A (for A, B), E (for E–H), K (for K–P), 150 μm; C (for C, D), 200 μm; I (for I, J), 100 μm.
BrdU pulse-chase analyses at E18.5 of embryos that received BrdU at E11.5, E13.5, and E15.5 provided insights into the timing of the accumulation of cells in the mutant SVZ. We found that E11.5 BrdU-labeled cells were not retained in the E18.5 mutant SVZ, whereas they were detectable within the striatum and the pallidum, indicating that early-born neurons were able to migrate out of the mutant's progenitor zones (Fig. 3 K,L). In contrast, E13.5 and E15.5 BrdU-labeled cells accumulated in the mutant SVZ, consistent with a failure in their ability to migrate out of the SVZ (Fig. 3 M–P). E13.5-labeled BrdU cells were located both in the periventricular area and in the striatum in a ratio of 2:1 (2200 neurons counted; three independent experiments). By E15.5, 75 ± 11% of the total BrdU-positive cells remained in the periventricular area, whereas 25 ± 4% were detected in the differentiated regions (2500 neurons counted; three independent experiments). These findings suggest that Arx mutants exhibit a progressive impairment of cells to migrate from the proliferative areas to the mature compartments. Beginning at ∼E12.5, some LGE cells begin to accumulate in the periventricular area, whereas at E15.5, most of them remained confined to this domain. Similar results were found for the MGE/pallidum, although molecular features of this region were more severely altered by the Arx mutation.
Analysis of prenatal subcortical patterning and differentiation
To elucidate the mechanisms underlying the neonatal histological and molecular defects provoked by the Arx deficiency, we examined the expression of transcription factors known for their involvement in regulating the patterning and differentiation of the progenitor region. The expression of Mash1 (bHLH) and Vax1 (homeobox) in Arx mutants appeared normal at E12.5; these genes are required for multiple steps in subpallial development (Casarosa et al., 1999; Yun et al., 2002; Taglialatela et al., 2004) (Fig. 4 A,B) (data not shown). The expression domain of Dlx2 was expanded, particularly in the MGE/pallidal domain at E12.5, E14.5, and E18.5 (Figs. 3 G,H, 4 C,D,M,N). Dlx2 acts, together with Dlx1, to promote differentiation throughout the subpallium (Anderson et al., 1997; Yun et al., 2002). A similar phenotype was also observed for the expression of the Nkx2.1 and Lhx6 homeobox genes (Fig. 4 E–H,K,L). Nkx2.1 specifies the MGE progenitor cell identity (Sussel et al., 1999; Marin and Rubenstein, 2003), whereas Lhx6 is implicated in the differentiation of MGE-derived cells (Grigoriou et al., 1998; Marin et al., 2000; Alifragis et al., 2004). Thus, although the regional expression of these genes was not altered, suggesting that regional specification is not altered, their expression ectopically expands into the mantle zone, suggesting that these neurons may have differentiation defects. This hypothesis is further supported by the reduction in MAP2 expression exhibited in particular by the Arx mutant LGE region starting from E14.5 (Fig. 4 O,P, dashed lines).
Figure 4.

Analysis of the expression of ventral telencephalic marker genes during the morphogenesis of the basal ganglia. A–H, Analysis at E12.5 of the different ganglionic eminence domains, including the proliferative zone (Nkx2.1 and Mash1), the SVZ (Dlx2, Nkx2.1, and Lhx6), and the immature neuronal compartment (Dlx2 and Lhx6). A, B, The extension of the striatal ventricular zone is unaltered in Arx mutants as evidenced by Mash1 expression domain analysis. C–H, Dlx2- (C, D), Nkx2.1- (E, F), and Lhx6 (G, H)-positive neuronal areas are drastically enlarged compared with controls, suggesting a block of neuronal differentiation. I, J, E14.5 coronal sections of brains stained for Nissl. K–N, Lhx6 (K, L) and Dlx2 (M, N) expression domains are enlarged in the ganglionic eminences of E14.5 mutant brains. O, P, The MAP2+ region of the mature ventral telencephalon (red) is decreased in Arx mutant brains compared with age-matched controls. This result is evident in the LGE (dashed lines). aH, Anterior hypothalamus; Cge, caudal ganglionic eminence; Cx, Cerebral cortex; dt, dorsal thalamus; Mut, mutant; Str, striatum. Scale bars: A–H (in A), 400 μm; I–N (in I), 300 μm; O, P (in O), 250 μm.
Next, we assessed the consequences of the Arx deficiency on the expression of Lhx7 (Lhx8) and Gbx1, two homeobox genes present in the SVZ and mantle of the MGE. Lhx7 (Lhx8) function is required for the differentiation of a subset of subpallial cholinergic neurons (Zhao et al., 2003; Mori et al., 2004; Fragkouli et al., 2005), whereas Gbx1 expression is associated with the differentiation of these neurons (Asbreuk et al., 2002; Zhao et al., 2003). Arx expression largely overlaps with those of Lhx7 and Gbx1 in the SVZ and mantle regions of the MGE (data not shown). In Arx mutant E12.5 brains, the expression of Gbx1 appeared normal, whereas Lhx7 transcripts were not detectable in the MGE and pallidal mantle (Fig. 5 A–D). Lhx7 expression was increased in the SVZ of the ventral MGE (Fig. 5 D, arrowheads), whereas its expression was reduced in the mantle zone, being maintained only in a small ventral domain (Fig. 5 D, arrows). By E14.5, Gbx1 expression was greatly reduced, demonstrating that Arx is required to maintain its expression (Fig. 5 E,F,I,J). At this stage, Lhx7 expression remained weak, except in progenitor and mantle regions related to the AEP and preoptic area (POA), including part of the septum (Fig. 5 G,H,K,L).
Figure 5.

Analysis of cholinergic neuronal commitment and differentiation in Arx mutant brains. A, B, Gbx1+ cholinergic neurons are detected in the MGE mantle zone in Arx-null brains, indicating an early cholinergic commitment, at E12.5. C, D, A large number of mutant cholinergic neurons fail to express Lhx7. Only neurons in the SVZ (D, arrowheads) and preoptic area (D, arrows) activate Lhx7 expression. E, F, I, J, Gbx1 expression fades in E14.5 Arx mutant tissue, indicating a loss of cholinergic neurons (arrows). G, H, K, L, Lhx7 expression remains largely undetectable in E14.5 mutant brains. Only a few cells located in the SVZ (H, L, arrowheads) or in the rostral mantle zone (H, arrows) are Lhx7 positive. Cge, Caudal ganglionic eminence; Cx, cerebral cortex; Mut, mutant; Se, septum. Scale bars: A–D (in A), 300 μm; E–L (in E), 400 μm.
At P0, the expression of both Gbx1 and Lhx7 was mostly undetectable (Fig. 6 A–D,G–J). Because both genes are implicated in cholinergic neuron differentiation, the expression of choline acetyltransferase was examined and found dramatically decreased in Arx mutants (Fig. 6 E,F,K,L). These findings demonstrate that Arx is essential for differentiation of cholinergic neurons, probably through promoting expression of Lhx7 and Gbx1.
Figure 6.

Severe reduction of cholinergic neuron numbers in P0 Arx mutant brains. A–D, G–J, Gbx1 (A, B, G, H) and Lhx7 (C, D, I, J) specifically label forebrain cholinergic neurons. Both genes are typically expressed in most cholinergic ventral centers in controls, whereas, in Arx mutant brains, their expression is dramatically reduced. E, F, K, L, ChAT RNA in situ hybridization fails to outline principal basal cholinergic nuclei [i.e., the magnocellular preoptic nucleus (mcpo) in rostral sections and the bed nucleus (nb) in more caudal brain regions]. bst, Bed nucleus of stria terminalis; Cx, cerebral cortex; db, diagonal band; hi, hippocampus; Mut, mutant; Se, septum; si, substantia innominata; Str, striatum; vp, ventral pallidum. Scale bars: A (for A–D), G (for G–J), 300 μm; E (for E, F, K, L), 100 μm.
Failure of interneuron migration from the basal telencephalon to the LGE and cortex in Arx mutants
The expression domain of Lhx6 in the subcortical telencephalon (basal telencephalon) includes several progenitor regions: MGE, AEP, and POA. The MGE, and probably the AEP and POA, generates neuronal populations that tangentially migrate to reach the cerebral cortex, striatum, and other telencephalic areas (Marin and Rubenstein, 2001). Alterations in the genesis of cortical GABAergic interneurons were previously reported in Arx mutants (Kitamura et al., 2003). We detected a severe reduction of Lhx6+ cells migrating from the MGE to the cerebral cortex and the striatum at E18.5 (Fig. 7 A,B, arrowheads). Similarly, the Arx mutant cerebral cortex appeared largely devoid of GABAergic neurons (GAD67+ and calbindin+) that originate from the MGE and caudal ganglionic eminence (Fig. 7 C–F). Only a few Lhx6+ and/or GAD67+ mutant neurons were detected in the cerebral cortex; these were confined to its periventricular zone (12 ± 3% Lhx6+ and 15 ± 3% GAD67+ neurons were detected in the mutant compared with control cortices at E18.5; 600 neurons were counted from two independent experiments; p < 0.001) (Fig. 7 B,D, arrowheads).
Figure 7.
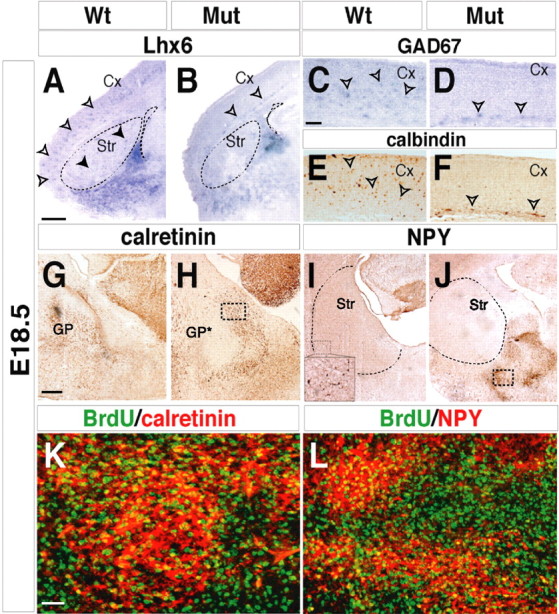
Arx deficiency induces an impairment of radial and tangential migration from the MGE. A–D, Lhx6 (A, open arrowheads), Gad67 (C, open arrowheads), and calbindin (E, open arrowheads) normally mark GABAergic neurons that have migrated from the MGE in the E18.5 cerebral cortex. In Arx mutant, the cerebral cortex appears largely devoid of Gad67+/Lhx6+/calbindin+ interneurons (B, D). Few interneurons are detected in ectopic periventricular locations in mutant cortices (B, D, F, open arrowheads). The mature striatum is reduced in Arx mutant brains compared with controls (A, B, dashed lines) and is devoid of interneurons (A, filled arrowheads). G, H, Mutant calretinin+ cells are accumulated in the MGE area, instead of being localized in the GP and striatal structures (G). I, J, NPY+ striatal interneurons (I and inset) are ectopically accumulated in the MGE area but undetectable in the striatum (dashed lines) in mutants (J). K, L, Several NPY+ and calretinin+ cells (yellow) in the periventricular region of E18.5 mutant brains are labeled by a single E13.5 BrdU pulse, indicating that they correspond to postmitotic cells that left the cell cycle at that specific stage. K, L, Higher magnifications of MGE areas outlined in the boxed regions in H and J, respectively. Cx, Cerebral cortex; GP*, mutant globus pallidus; Mut, mutant; Str, Striatum. Scale bars: A, B (in A), 200 μm; C–F (in C), 100 μm; G–J (in G), 150 μm; K, L (in K), 25 μm.
Evidence suggesting that the reduced number of cortical interneurons results from a migration defect comes from analysis of calretinin and NPY expression. Calretinin+ interneurons are normally detected throughout the mature striatum and GP in wild-type tissue. In Arx mutants, calretinin+ cells accumulated within the periventricular zone (Fig. 7 G,H). Similarly, mutant NPY+ cells were detected in an overlapping area, unlike in control brains, in which NPY+ cells constituted a large class of striatal and cortical interneurons (Fig. 7 I,J). Using BrdU birth dating, we assessed the stage when the ectopically placed calretinin+ and NPY+ neurons became postmitotic. It is known that striatal interneurons originate from the MGE field after stage E11.5 (Marin et al., 2000). Therefore, single BrdU pulses at E11.5, E13.5, and E15.5 were performed, and the tissues were analyzed at E18.5. Similar to the wt conditions, BrdU+ cells labeled by injections performed at late stages (E13.5 and E15.5) were costained with calretinin or NPY (Fig. 7 K,L) (data not shown). In contrast, BrdU/calretinin or BrdU/NPY double-positive cells were never detected after E11.5 BrdU injections (data not shown). Thus, in Arx-deficient animals, calretinin+ and NPY+ neurons are found ectopically localized, although their pattern of BrdU incorporation is similar to normal, peaking between E13.5 and E15.5 during embryonic development. Together, these results strongly suggest that Arx mutant subpallial neurons failed to tangentially migrate to the LGE and cortex and accumulated as periventricular ectopia.
Arx mutant LGE SVZ cells are able to differentiate in vitro
In Arx mutants, many immature neurons produced after E11.5 failed to migrate out of the subpallial germinal layers. In the SVZ of the LGE, these cells express βIII-tubulin but only weakly express MAP2 (Fig. 3 C,D) and are negative for Ebf1 (Fig. 3 E,F), enkephalin (Fig. 1 E,F), calbindin (Fig. 1 K,L), DARPP32, and dopamine 2 receptor (data not shown). Thus, they appear to be blocked in their ability to differentiate. We tested whether these cells could overcome this impairment in vitro. We compared in vitro differentiation of Arx mutant and wild-type cells from dissected E18.5 LGE and MGE subventricular zone (Fig. 8 A, dashed lines). Unlike the in vitro situation, the dissociated SVZ cells expressed a general neural marker (MAP2) (Fig. 8 C,F) and striatal markers (DARPP32, calbindin, and EBF) (Fig. 8 G–L) (n = 4). This suggests that Arx mutant SVZ cells of the LGE are capable of differentiation but that local factors in the SVZ impede their maturation. Furthermore, it suggests that their block in migration out of the SVZ may be the major phenotype.
Figure 8.
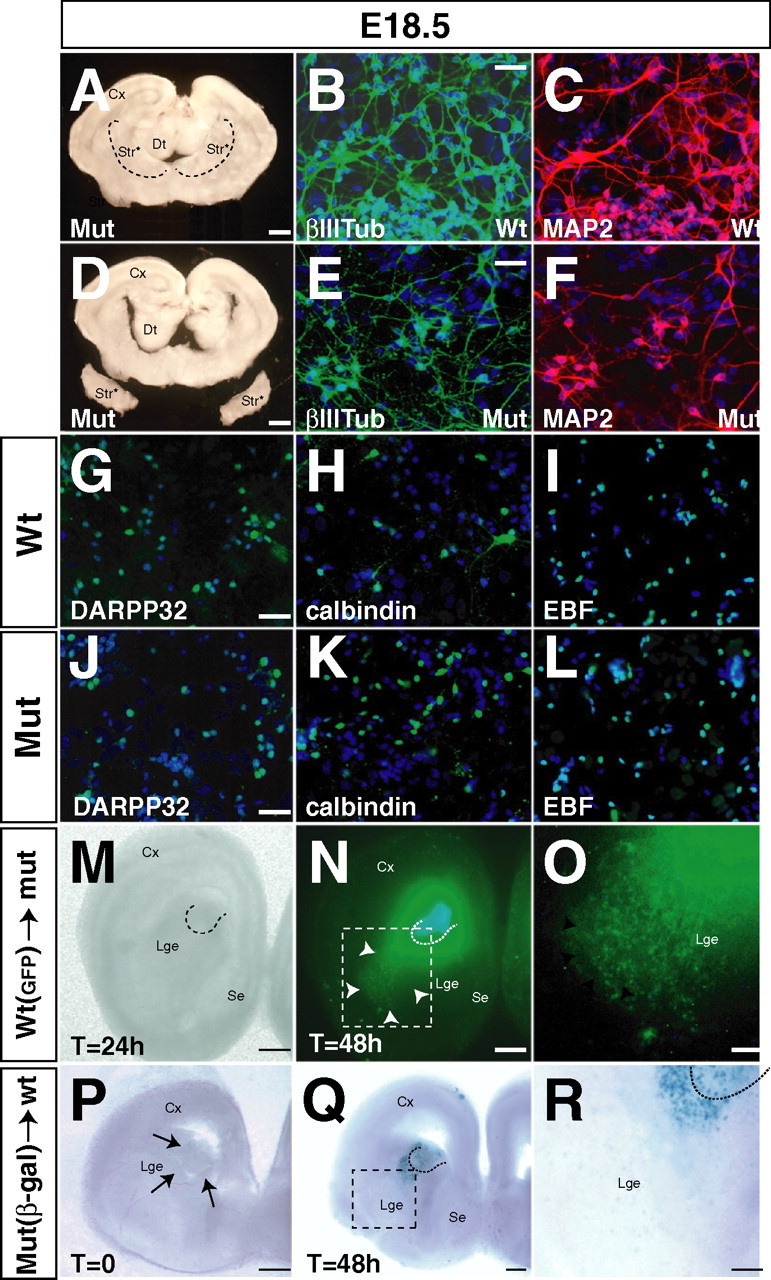
In vitro differentiation and ex vivo transplantations of Arx mutant cells. A, D, Surgical procedure to isolate E18.5 SVZ mutant tissue for in vitro cell cultures. A, Dashed lines surround the area that was isolated as source of tissue for cell culture experiments. B, C, E, F, Analysis of wt (B, C) and mutant (E, F) differentiated neural cultures derived from ventral forebrain. Mutant neurons express βIII-tubulin (βIIITub) and MAP2 and display normal axonal–dendrite morphologies. G–L, Analysis of differentiation in mature striatal neurons using DARPP32 (G, J), calbindin (H, K), and pan-EBF (I, L) antibodies. Mutant and wt cells exhibit a normal labeling for all three striatal markers. M–O, Transplantation of wt GFP-labeled LGE explant into homotopic sites in mutant brain slices cultured in vitro. M, Bright view of brain slice transplanted after 24 h. N, Analysis of cell migration of wt GFP cells. Arrowheads indicate a robust migration of the cells far away from the graft reaching the marginal areas of the mantle zone. Dashed lines indicate the site of graft. O, Higher magnification of the boxed area in N. P–R, Transplantation of mutant LacZ-labeled cells in wt brain slices cultured in vitro. P, Site of graft in the periventricular striatal region. Arrows indicate the grafted tissue. Q, Identification of the grafted mutant neurons by X-gal staining after 48 h of in vitro culture. Dashed lines indicate the site of graft. R, Higher magnification of the boxed area in Q. Mutant cells fail to migrate far away from the graft and remain localized in the graft area. Dashed lines surround the grafted tissue. β-gal, β-Galactosidase; Cx, cerebral cortex; Dt, dorsal thalamus; Mut, mutant; Se, septum; Str, striatum; Str*, mutant striatum. Scale bars: A, D, 200 μm; B (for B, C), E (for E, F), G (for G–I), J (for J–L), 20 μm; M–R, 200 μm.
Arx mutant ventral telencephalic SVZ cells have a block in migration activity
To analyze cell migration, we performed isochronic and homotopic transplantations of WT cells (GFP+) into Arx mutant slices (n = 4) and Arx mutant cells (LacZ+) into wt slices (n = 5). In the first case, small fragments of E14.5 wt LGE subventricular zone were grafted in the corresponding location of wt E14.5 telencephalic slices. After 48 h, GFP-positive cells were detected outwards from the graft and even located at a far distance in the periphery of the LGE mantle zone (Fig. 8 N, arrowheads). Conversely, in the second case, pieces of Arx mutant LGE were grafted into analogous locations in wt brain slices. After 48 h, slices were harvested and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) stained. Labeled cells were always found nearby the graft, failing to migrate away from the transplantation site (Fig. 8 Q,R). Thus, these results clearly suggested a cell-autonomous defect of Arx mutant cells preventing their normal migration and proper targeting of their final position. Similar results were obtained transplanting fragments of E14.5 mutant MGE into analogous locations of wt brain tissue (data not shown).
To further test this deficiency, explants of E14.5 wt and mutant MGE were isolated, embedded in Matrigel, and grew for up to 3 d in vitro. Matrigel matrix (BD Biosciences) provides a good substrate for neuronal migration and, because of its transparency, allows a direct monitoring of cell migration. To validate our approach, static analysis and video-recordings were used to follow the migration of individual wt neurons (n = 42). In all cases, the analyzed neurons showed the following features: (1) extension of a leading process, (2) translocation of the nucleus (nucleokinesis), and (3) retraction of the trailing part of the cell (data not shown). We therefore used this method to assay for defects in Arx mutant cells. Cells in wt explants showed robust migration away from the explant after 48 h (Fig. 9 A), whereas Arx mutant explants showed a severe reduction in migration (Fig. 9 B). In both genotypes, approximately the same number of neurons extended neurites away from the explant (Fig. 9 D,E). Quantitative analysis demonstrated that the Arx mutants exhibited a >70% reduction in mean migration distance from the edge of the explant (distance for wt cells = 1000 ± 250 μm; n = 50 for each genotype; p < 0.01). Morphological defects were noted comparing wt and mutant neurons in vitro. In fact, the mutant leading processes exhibited an increase in length and branching. In particular, 10 ± 2% of wt migrating cells had more than two branches (Fig. 9 H), whereas 32 ± 4% of Arx mutant migrating cells had more than two branches (Fig. 9 G,I) (p < 0.05), and inside this group, 18 ± 2% of mutant cells exhibited exuberant branching of the leading process and protrusion from cell soma, counting four or more secondary protrusions (Fig. 9 G), whereas only 2 ± 0.5% of wt cells exhibited a similar phenotype (Fig. 9 I) (80 neurons analyzed for both genotypes; p < 0.05). Twenty-three percent of mutant cells had leading processes that were two to four times the length of those in wt cells (48 ± 12 μm; n = 16 wt and 21 mutant cells) (Fig. 9 J,K). This excessive extension was associated with severe thinning and excessive branching (Fig. 9 J–M). These observations revealed cell-intrinsic morphological defects that may account for the altered migration of Arx mutant neurons.
Figure 9.

Reduced migration and altered morphology of Arx mutant neurons. A–D, In vitro migration assay in a three-dimensional Matrigel matrix. A, C, Wt neurons show a robust outward migration from the MGE explants after 48 h of culture. C, Wt neurons exhibit a well developed leading process either unipolar (arrows) or branched (arrowhead) in direction of migration. B, D, Mutant neurons fail to migrate out from the MGE explant and remain localized near the graft after 48 h. D, Mutant cells display leading processes in a correct outward orientation but fail to show active and robust migration. E–L, Aberrant morphology of Arx migrating neurons in vitro. Pattern of abnormalities found in Arx mutant neurons. E–H, A fraction of mutant neurons (F, H) display an increase in leading process branching compared with wt cells (E, G). Arrows indicate the branching points. I–L, A significant fraction of mutant cells (J, L) display a long leading process from two to four times longer than wt cells (I, K). K, L, Positive staining with βIII-tubulin (βIIItubul) antibody ascertains the neuronal origin of the mutant abnormal cells analyzed in the migration assays. Arrowheads indicate branching points of the leading process. Arrows point to the overall length of the neurons. Mut, Mutant. Scale bars: A, B, 200 μm; C, D, 50 μm; E (for E–J), K (for K, L), 20 μm.
Aberrant topography of thalamocortical projections in mutant Arx brains
Corticothalamic and thalamocortical projections allow the connection of a given thalamic nucleus to a specific neocortical region to enable the processing of sensory and motor information (O'Leary et al., 1994; López-Bendito and Molnár, 2003; O'Leary and Nakagawa, 2002; Garel and Rubenstein, 2004; Vanderhaeghen and Polleux, 2004). Throughout development, both cortical and thalamic projections extend through the subcortical telencephalon, in which they encounter each other (Metin and Godement, 1996; Auladell et al., 2000). There is evidence that interactions between these axons and between local structures/signals are essential for the growth and topography of these axon pathways (López-Bendito and Molnár, 2003; Garel and Rubenstein, 2004; Vanderhaeghen and Polleux, 2004). Recently, it was established that neuregulin1+ LGE-derived cells migrate into the MGE and thereby provide a signal that enables the entry of thalamic axons into an otherwise nonpermissive territory (López-Bendito et al., 2006). Given the fact that Arx mutants exhibit alterations in the MGE and LGE, we examined the development of cortical and thalamic projections. DiI injections in the cortex of E14.5 embryos normally label corticofugal projections descending through the internal capsule (IC) toward their extracortical target nuclei (Fig. 10 A). Mutant cortical fibers failed to migrate into the MGE, being unable to cross the LGE/MGE border, and instead turned ventrally toward the marginal edge (Fig. 10 F, arrowheads). The topography of E18.5 descending corticofugal projections was examined by introducing DiI and DiA crystals in the occipital or parietal neocortex of Arx mutants (n = 5) and controls (n = 5) (Fig. 10 B,G, insets). Somatosensory cortical neurons typically direct their axons toward the thalamic ventral basal complex, whereas visual cortical neurons connect to the dorsal lateral geniculate nucleus (dLGN) (Fig. 10 B–D). Both tracts grow through the striatum and GP within the IC (Fig. 10 B–D). In E18.5 Arx mutants, these projections were highly disorganized and misrouted (Fig. 10 G–I). The tracts were widely dispersed in the basal ganglia (Fig. 10 G,H, arrowheads); in the striatum, the fibers were misrouted to a more superficial position, some fibers entering the external capsule (Fig. 10 G,H, open arrowheads). Axons reached the thalamus in only 3 of 5 mutants; numerous axons failed to reach their targets and remained in the basal telencephalon (Fig. 10 G,H, arrows), Once in the thalamus, axons from the mutant occipital cortex ectopically entered more medial and more dorsal areas (Fig. 10 J, arrowheads) compared with controls (Fig. 10 E). Likewise, somatosensory cortical efferents ectopically entered the dLGN (Fig. 10 J). Hence, it appears that the majority of the cortical efferents were routed to the cerebral peduncle and, unlike the proper segregation found in wild type, they were then largely intermixed (Fig. 10 E,J, arrows). Next, we analyzed the origins of such defects throughout embryo morphogenesis. Hematoxylin–eosin staining provided useful information about the cytoarchitectural alterations in Arx mutants. In control brains, the internal capsule is noticeable because of its cell-free nature in the subcortical telencephalon (Fig. 10 K, arrowheads). In mutants, this area was not detectable, whereas a cell-free domain was noted near the marginal edge of the ventral telencephalon, at the junction between diencephalon and telencephalon (Fig. 10 P, arrowheads). To verify that these areas corresponded to domains accumulating disorganized fibers, thalamic axons were traced at E14.5 by placing DiI crystals in the dorsal thalamus. Rostrally, mutant thalamocortical projections were found confined to a more superficial position with respect to control fibers and failed to reach the cortical plate (Fig. 10 L,Q). At more caudal levels, a large amount of thalamic axons were unable to grow from the dorsal thalamus (Fig. 10 M,R). Some fibers reached the diencephalic–telencephalic junction but were misrouted toward the hypothalamus, failing to enter into the basal ganglia (Fig. 8 R, arrowheads). Similar results were obtained by labeling thalamocortical projections with the cell adhesion molecule L1 at E16.5 (Fukuda et al., 1997) (Fig. 10 N,O,S,T). Altogether, these data show a severe impairment of thalamocortical connections, which mostly fail to extend into the correct pathway (i.e., striatal IC) and miss reaching the correct target (i.e., specific dorsal thalamic nuclei as targets of corticothalamic fibers) (Fig. 10 U,V).
Figure 10.

Alterations of cortical and thalamic axons pathfinding in Arx mutant brains. A, F, At E14.5, DiI-labeled corticofugal axons normally migrate through the IC (A), whereas mutant fibers do not cross the LGE/MGE boundary and are directed toward the ventral marginal edge (F, filled arrowheads). Some mutant axons are located in the external capsule (F, open arrowhead). B–E, G–J, At E18.5, corticofugal axons were labeled using DiI and DiA crystals placed in the occipital and parietal cortex (B, G, insets, arrows). Cortical neural projections appear highly disorganized. Some tracts migrate inside the external capsule (G, H, open arrowheads), whereas others extend to the ventral striatum or near the ventral marginal edge (G, H, arrowheads). The few fibers entering the dorsal thalamus fail to specifically reach the proper nuclei (J, open arrowheads). Bundles of axons coming from different cortical areas are highly intermixed and not separated as in control brains (E, J, arrows). K, P, Hematoxylin/eosin staining of brain sections outlines axonal fibers in the IC in controls but not in mutant brains, in which they are found in more a ventral position (P, arrowheads). L, M, Q, R, Coronal sections of E14.5 brain in which thalamocortical axons were labeled using DiI crystals placed in the dorsal thalamus. In control brains (L, M), axons travel into the basal ganglia through the IC, reaching the cerebral cortex (L, arrowheads). In mutants, many thalamic fibers remain blocked into the dorsal thalamus or misrouted toward the hypothalamus (R, arrowheads). The few thalamic axons that entered into the basal ganglia are extended in an abnormal ventral position (Q) compared with wild-type fibers (L). N, O, S, T, Labeling of thalamocortical projections using immunohistochemistry targeted against L1. L1-positive thalamocortical fibers behave as described previously for DiI-labeled axons. T, Open arrowheads indicate thalamic axons that failed to exit the dorsal thalamus, whereas filled arrowheads label thalamic fibers ectopically directed into the hypothalamus. U, V, Schematic representation of the phenotypes observed in Arx mutant brains. Few thalamocortical axons (red) enter into the basal ganglia of Arx mutant brains and do not organize into the IC. Other axonal tracts remain confined to the dorsal thalamus or are misrouted toward the hypothalamus. Corticofugal projections (green) are largely disorganized in the striatum, in which some tracts are misoriented or ectopically directed toward the ventral marginal edge. Asterisk in V labels the ectopic periventricular area described in the Arx mutant brains (dashed line). Cx, Cerebral cortex; DT, dorsal thalamus; dTh dorsal thalamus; ET, epithalamus; HT, hypothalamus; LGN, lateral geniculate nucleus; LGN*, mutant lateral geniculate nucleus; Mut, mutant; Str, striatum; VB, thalamic ventral basal complex; VB*, mutant thalamic ventral basal complex; VT, ventral thalamus. Scale bars: A (for A, F), K (for K, P), L (for L, M, Q, R), N (for N, O, S, T), 200 μm; B (for B–E, G–J), 400 μm.
Discussion
Comparing brain malformations in Arx mutant mice and XLAG patients
Herein, we describe the phenotypic alteration of the basal ganglia in Arx mutant mice. A related phenotype may arise in humans having a null Arx mutation (i.e., in patients with the XLAG syndrome). In fact, most XLAG patients exhibit malformed and reduced basal ganglia structures with cavitations, indistinct borders between the structures, and fragmentation (Kato and Dobyns, 2003; Uyanik et al., 2003; Kato et al., 2004). In mice, the loss of Arx function in the cerebral cortex leads to lissencephaly (smooth cortex) with absence of corpus callosum (Kitamura et al., 2003). However, in previous publications, it was unclear whether Arx mouse mutants display defects in the histogenesis of the subcortical telencephalon, including the striatum, globus pallidus, and nucleus basalis. In this work, we describe abnormalities in the striatum and pallidum, which are consistent with magnetic resonance imaging descriptions of the analogous human structures in XLAG patients (Kato et al., 2004). Thus, the forebrain defects observed in Arx mutant mice appear to faithfully reflect the phenotype of XLAG and related disorders.
An arrest in GABAergic differentiation and migration in the Arx mutant ventral telencephalon
Subsequent to the cell cycle withdrawal, neural progenitors migrate out of the germinal layer and follow a radial or tangential route to reach their final position in the mantle zone. Cell migration is greatly decreased in the embryonic basal ganglia of Arx mutants. This results in the ectopic accumulation of immature neurons in the SVZ. In the LGE, these cells lack the expression of markers of the striatal neurons, such as calbindin, Ebf1, and enkephalin, suggesting an impairment in neuronal maturation. The MGE also showed an accumulation of unmigrated neurons, many of which were immature, based on low MAP2 expression. However, some of the ectopic neurons expressed markers associated with MGE-derived mature neurons, such as NPY and ER81.
An arrest in neuronal differentiation may account for the defects observed in mutants, including the arrest in migration. To test this hypothesis, we dissociated mutant SVZ cells and differentiated them in vitro. Interestingly, mutant LGE cells differentiated into neurons expressing striatal markers. However, migration was not restored in any of our in vitro analyses. Hence, two hypotheses, which are not mutually exclusive, may explain our observations: (1) a non-cell-autonomous mechanism may contribute to the arrest in cell maturation in vivo, at least in the LGE; and (2) there is an intrinsic defect in cell migration in both the LGE and MGE.
Like Arx mutants, Dlx1/2 mutants show ectopic collection of unmigrated cells in the LGE and MGE. Furthermore, Arx expression is reduced in Dlx1/2 mutants (Cobos et al., 2005). However, unlike in Arx mutants, the Dlx1/2−/− ectopia express high levels of MAP2 (Anderson et al., 1997) (I. Cobos, O. Borello, and J. L. R. Rubenstein, unpublished results), suggesting that these neurons are more differentiated than the Arx mutant cells. Thus, Dlx1 and Dlx2 appear to repress MAP2 expression. Arx mutants express increased Dlx2; therefore, the block of MAP2 expression may be through increased Dlx2 expression.
Dlx1/2 mutants exhibit increased expression of Mash1 and Delta1 and show evidence of increased Notch signaling (i.e., increased Hes5 expression) (Yun et al., 2002). Thus, we analyzed in Arx mutants expression of signaling molecules belonging to the Notch pathway, including Notch1 and its downstream targets Hes1, Hes5, and CoupTF1. However, we did not observe any differences between wt and mutant tissues (M. Bianchi and V. Broccoli, unpublished results). Thus, although Dlx1/2 and Arx mutant mice exhibit similar defects in migration of the basal ganglia cells, some of the underlying mechanisms may be divergent.
We found that Arx mutant cells displayed a strong impairment in migration ability. This was confirmed through the in vitro analysis of cell migration using MGE explant cultures embedded in a Matrigel matrix. This approach allowed to easily follow neuronal migration in a three-dimensional manner, without forcing cells to adhere to a unique plan of migration. In these conditions, mutant cells displayed a severe reduction in migration ability, suggesting intrinsic defects in migration capabilities. These results suggest that the altered development of basal ganglia in Arx mutants may be related to a cell-autonomous impairment of the ability of Arx mutant neurons to migrate out of the periventricular space, thereby explaining the large ectopia observed in these animals. These cells remain located nearby the germinal layer and, therefore, subjected to the influence of diffusible molecules of the Notch or FGF signaling that may prevent their maturation. This may explain why mutant cells in the SVZ striatal compartment failed to express markers specific for mature neurons. Furthermore, the impaired movement activity may explain the time-dependent arrest in radial migration noticed in developing basal ganglia. Indeed, at early embryonic stages (E11.5–E12.5), mutant neurons may cover short distances more efficiently than at later stages (from E13.5 onward). Altogether, our findings strongly suggest that a cell-autonomous impairment of neuronal migration accounts for numerous aspects of basal ganglia malformations in Arx mutants. However, additional investigations, possibly using conditional mouse mutants, would be required to definitively rule out the existence of cell-nonautonomous defects in these animals.
Arx mutation impairs cholinergic differentiation in the ventral forebrain
The expression of Arx in the basal telencephalon was previously correlated with GABAergic neural differentiation (Kitamura et al., 2003; Cobos et al., 2005). Indeed, Arx is expressed in GAD67-positive neurons, and its inactivation impairs the differentiation of cortical GABAergic interneurons. Interestingly, the ventral telencephalon is also the source of most of the forebrain cholinergic neurons. In particular, cholinergic cells are thought to be derived from MGE and/or AEP progenitors and to differentiate early during embryonic development (Marin et al., 2000). There are two main types of cholinergic neurons: interneurons of the striatum and projection neurons of the basal telencephalon. Importantly, the Arx deficiency led to a dramatic decrease in both populations. Such alterations are probably not caused by an impairment of the early cholinergic commitment or a misspecification, because at E12.5, the expression of Gbx1, a marker for these neurons, was detected in the MGE mantle zone. However, most of these neurons lacked expression of Lhx7, a transcription factor required for their differentiation (Zhao et al., 2003; Mori et al., 2004; Fragkouli et al., 2005; Manabe et al., 2005). These findings raise the hypothesis that, in Arx mutants, cholinergic differentiation, although initiated, fail to progress because of the absence of at least one essential cell-fate-determining gene, most likely Lhx7. Lhx7 expression requires Nkx2.1 function. In Nkx2.1 mutants, Lhx7 is not activated (Sussel et al., 1999), whereas Nkx2.1 ectopic expression induces Lhx7 (Bachy and Retaux, 2006). However, Nkx2.1 appears to be able to induce Lhx7 only in the ganglionic eminences (Arx-positive territory), because its ectopic expression in the cortex fails to activate Lhx7 (Bachy and Retaux, 2006). Therefore, this suggests that Arx may allow the Nkx2.1-dependent activation of Lhx7 (Fig. 9 D). Additional studies would be required to strengthen this hypothesis and verify any interaction among these transcription factors.
Altered connectivity between cortex and thalamus in Arx mutants
Thalamocortical and corticothalamic projections develop in mice between E13 and E18, connecting specific thalamic nuclei with different cortical areas and establishing topographically ordered representations (Garel and Rubenstein, 2004). Both types of connections migrate through intermediate regions that function as “guideposts” to orientate axonal navigation (López-Bendito and Molnár, 2003; Vanderhaeghen and Polleux, 2004). In particular, López-Bendito et al. (2006) have recently described a population of LGE cells that, when migrating through the MGE, open a corridor through which thalamic axons can enter, and, thereby, avoid repulsive cues originating from the MGE. The MGE of Arx mutants lacks both Ebf1+ and Lhx6−/Nkx2.1− territories, indicating a loss of LGE corridor cells. This may contribute to the extensive defects of the corticothalamic pathway observed in the ventral telencephalon (López-Bendito et al., 2006). Two other structures have been shown to strongly influence corticothalamic axon guidance: the pallial/subpallial boundary (PSPB) and the diencephalon/telencephalon boundary (DTB) (O'Leary et al., 1994; Molnár and Butler, 2002). The PSPB domain appeared morphologically normal in Arx mutants, although a more focused molecular analysis of this region remains to be performed. Conversely, a strong impairment was detected in thalamocortical projections in entering the telencephalon through the DTB area. Some of the fibers leaving the dorsal thalamus were misoriented toward the hypothalamus. This may be caused by a severe reduction of the ventral thalamus and lack of eminentia thalami, an area connecting the diencephalon to the caudoventral telencephalon (Kitamura et al., 2003) (E. Colombo and Broccoli, unpublished data). Both regions represent important relay centers that may influence axonal pathfinding. Thus, the described altered thalamocortical projections may be the consequence of different morphological alterations in distinct brain structures acting on axonal navigation. However, the detailed mechanisms involved remain to be assessed.
In summary, we provide evidence that Arx plays a crucial role in promoting migration of ventral forebrain neurons both in radial and tangential orientations. This primary defect impacts the ability of these cells to correctly mature and differentiate in the basal ganglia structures. Accordingly, Arx inactivation leads to a general disorganization of the basal ganglia as detected in mice and humans. Furthermore, we identified a hitherto unsuspected function of Arx in regulating cholinergic neural differentiation. This defect may be initiated by a failure in activating the expression of Lhx7. Finally we described drastic alterations of the wiring system connecting the cortex to the thalamus in Arx mutant brain, occurring possibly as a secondary effect of the forebrain malformations. These findings open new avenues to evaluate whether Arx-deficient patients exhibit similar impairment and how this may impact the pathophysiology of such a disease.
Footnotes
This work was supported by Telethon Grant GP04141 (V.B.), the Fondo per gli Investimenti della Ricerca di Base project (Ministero dell'Università e della Ricerca, RBNE015242-RBLA03AF28; VB), the Max-Planck Society (P.C., A.M.), the Dr. H. Storz and Alte Leipziger Foundation (A.M.), National Institutes of Health Beta Cell Biology Consortium Grant U19DK072495-01 (P.C., A.M.), and Nina Ireland and National Institute of Mental Health Grants R01 MH49428-01 and K05 MH065670 (J.L.R.). We are thankful to Drs. F. Guillemot, V. Pachnis, T. Rabbitts, K. Campbell, R. Kageyama, C. Birchmaier, and G. Consalez for the gifts of probes and antisera. We are grateful to Drs. A. Mallamaci, L. Muzio, A. Fairen, and V. Pachnis for stimulating discussions and helpful suggestions.
References
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Alifragis P, Liapi A, Parnavelas JG. Lhx6 regulates the migration of cortical interneurons from the ventral telencephalon but does not specify their GABA phenotype. J Neurosci. 2004;24:5643–5648. doi: 10.1523/JNEUROSCI.1245-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Qiu M, Bulfone A, Eisenstat DD, Meneses J, Pedersen R, Rubenstein JL. Mutations of the homeobox genes Dlx-1 and Dlx-2 disrupt the striatal subventricular zone and differentiation of late born striatal neurons. Neuron. 1997;19:27–37. doi: 10.1016/s0896-6273(00)80345-1. [DOI] [PubMed] [Google Scholar]
- Asbreuk CHY, van Schaick SA, Cox JJ, Kromkamp M, Smidt MP, Burbach JPH. The homeobox genes Lhx7 and Gbx1 are expressed in the basal forebrain cholinergic system. Neuroscience. 2002;109:287–298. doi: 10.1016/s0306-4522(01)00466-3. [DOI] [PubMed] [Google Scholar]
- Bachy I, Retaux S. GABAergic specification in the basal forebrain is controlled by the LIM-hd factor Lhx7. Dev Biol. 2006;291:218–226. doi: 10.1016/j.ydbio.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Poirier K, Friocourt G, Bahi N, Beaumont D, Fauchereau F, Ben Jeema L, Zemni R, Vinet MC, Francis F, Couvert P, Gomot M, Moraine C, van Bokhoven H, Kalscheuer V, Frints S, Gecz J, Ohzaki K, Chaabouni H, Fryns JP, et al. ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet. 2002;11:981–991. doi: 10.1093/hmg/11.8.981. [DOI] [PubMed] [Google Scholar]
- Campbell G, Tomlinson A. The roles of the homeobox genes aristaless and Distal-less in patterning the legs and wings of Drosophila . Development. 1998;125:4483–4493. doi: 10.1242/dev.125.22.4483. [DOI] [PubMed] [Google Scholar]
- Casarosa S, Fode C, Guillemot F. Mash1 regulates neurogenesis in the ventral telencephalon. Development. 1999;126:525–534. doi: 10.1242/dev.126.3.525. [DOI] [PubMed] [Google Scholar]
- Cobos I, Broccoli V, Rubenstein JL. The vertebrate ortholog of Aristaless is regulated by Dlx genes in the developing forebrain. J Comp Neurol. 2005;483:292–303. doi: 10.1002/cne.20405. [DOI] [PubMed] [Google Scholar]
- Collombat P, Mansouri A, Hecksher-Sorensen J, Serup P, Krull J, Gradwohl G, Gruss P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003;17:2591–2603. doi: 10.1101/gad.269003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo E, Galli R, Cossu G, Gecz J, Broccoli V. Mouse orthologue of ARX, a gene mutated in several X-linked forms of mental retardation and epilepsy, is a marker of adult neural stem cells and forebrain GABAergic neurons. Dev Dyn. 2004;231:631–639. doi: 10.1002/dvdy.20164. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, Stanford IM. Calbindin D-28k positive projection neurons and calretinin positive interneurons of the rat globus pallidus. Brain Res. 2002;929:243–251. doi: 10.1016/s0006-8993(01)03263-2. [DOI] [PubMed] [Google Scholar]
- Corbin JG, Nery S, Fishell G. Telencephalic cells take a tangent: non-radial migration in the mammalian forebrain. Nat Neurosci. 2001;4:1177–1182. doi: 10.1038/nn749. [DOI] [PubMed] [Google Scholar]
- Cossette M, Levesque M, Parent A. Extrastriatal dopaminergic innervation of human basal ganglia. Neurosci Res. 1999;34:51–54. doi: 10.1016/s0168-0102(99)00029-2. [DOI] [PubMed] [Google Scholar]
- Fragkouli A, Hearn C, Errington M, Cooke S, Grigoriou M, Bliss T, Stylianopoulou F, Pachnis V. Loss of forebrain cholinergic neurons and impairment in spatial learning and memory in LHX7-deficient mice. Eur J Neurosci. 2005;21:2923–2938. doi: 10.1111/j.1460-9568.2005.04141.x. [DOI] [PubMed] [Google Scholar]
- Fuchs H, Hauber W. Dopaminergic innervation of the rat globus pallidus characterized by microdialysis and immunohistochemistry. Exp Brain Res. 2003;154:66–75. doi: 10.1007/s00221-003-1638-7. [DOI] [PubMed] [Google Scholar]
- Garel S, Rubenstein JL. Intermediate targets in formation of topographic projections: inputs from the thalamocortical system. Trends Neurosci. 2004;27:533–539. doi: 10.1016/j.tins.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Garel S, Marin F, Grosschedl R, Charnay P. Ebf1 controls early cell differentiation in the embryonic striatum. Development. 1999;126:5285–5294. doi: 10.1242/dev.126.23.5285. [DOI] [PubMed] [Google Scholar]
- Gecz J, Cloosterman D, Partington M. ARX: a gene for all seasons. Curr Opin Genet Dev. 2006;16:308–316. doi: 10.1016/j.gde.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Gerfen C, Wilson CJ. The basal ganglia. In: Swanson LW, Björklund A, Hokfelt T, editors. Handbook of chemical neuroanatomy. Vol 12. New York: Elsevier; 1996. pp. 371–468. Integrated systems of the CNS, Pt III, Chap II. [Google Scholar]
- Graybiel AM. The basal ganglia. Curr Biol. 2000;10:R509–R511. doi: 10.1016/s0960-9822(00)00593-5. [DOI] [PubMed] [Google Scholar]
- Grigoriou M, Tucker AS, Sharpe PT, Pachnis V. Expression and regulation of Lhx6 and Lhx7, a novel subfamily of LIM homeodomain encoding genes, suggests a role in mammalian head development. Development. 1998;125:2063–2074. doi: 10.1242/dev.125.11.2063. [DOI] [PubMed] [Google Scholar]
- Jacobowitz DM, Abbott LC. Boca Raton, FL: CRC; 1998. Chemoarchitectonic atlas of the developing mouse brain. [Google Scholar]
- Kato M, Dobyns WB. Lissencephaly and the molecular basis of neuronal migration. Hum Mol Genet. 2003;12:R89–R96. doi: 10.1093/hmg/ddg086. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, Omichi K, Suzuki R, Kato-Fukui Y, Kamiirisa K, Matsuo M, Kamijo S, Kasahara M, Yoshioka H, Ogata T, Fukuda T, Kondo I, Kato M, Dobyns WB, Yokoyama M, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32:359–369. doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Bjorklund A. Dopaminergic innervation of the globus pallidus by collaterals from the nigrostriatal pathway. Brain Res. 1979;172:169–173. doi: 10.1016/0006-8993(79)90907-7. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat Neurosci. 2006;9:443–452. doi: 10.1038/nn1654. [DOI] [PubMed] [Google Scholar]
- López-Bendito G, Molnár Z. Thalamocortical development: how are we going to get there? Nat Rev Neurosci. 2003;4:276–289. doi: 10.1038/nrn1075. [DOI] [PubMed] [Google Scholar]
- López-Bendito G, Cautinat A, Sánchez JA, Bielle F, Flames N, Garratt AN, Talmage DA, Role LW, Charnay P, Marín O, Garel S. Tangential neuronal migration controls axon guidance: a role for neuregulin-1 in thalamocortical axon navigation. Cell. 2006;125:127–142. doi: 10.1016/j.cell.2006.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe T, Tatsumi K, Inoue M, Matsuyoshi H, Makinodan M, Yokoyama S, Wanaka A. L3/Lhx8 is involved in the determination of cholinergic or GABAergic cell fate. J Neurochem. 2005;94:723–730. doi: 10.1111/j.1471-4159.2005.03261.x. [DOI] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2:780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. J Neurosci. 2000;20:6063–6076. doi: 10.1523/JNEUROSCI.20-16-06063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie O, Ponte I, Mangelsdorf M, Finnis M, Stifani S, Gécz J, Broccoli V. Mutations in the aristaless-related homeobox gene ARX affect its transcription repression activity. 2007 doi: 10.1016/j.neuroscience.2007.01.038. Submitted. [DOI] [PubMed] [Google Scholar]
- Melkman T, Sengupta P. Regulation of chemosensory and GABAergic motor neuron development by the C. elegans Aristaless/Arx homolog alr-1. Development. 2005;132:1935–1949. doi: 10.1242/dev.01788. [DOI] [PubMed] [Google Scholar]
- Miura H, Yanazawa M, Kato K, Kitamura K. Expression of a novel aristaless related homeobox gene “Arx” in the vertebrate telencephalon, diencephalon and floor plate. Mech Dev. 1997;65:99–109. doi: 10.1016/s0925-4773(97)00062-2. [DOI] [PubMed] [Google Scholar]
- O'Leary DD, Nakagawa Y. Patterning centers, regulatory genes and extrinsic mechanisms controlling arealization of the neocortex. Curr Opin Neurobiol. 2002;12:14–25. doi: 10.1016/s0959-4388(02)00285-4. [DOI] [PubMed] [Google Scholar]
- O'Leary DD, Schlaggar BL, Tuttle R. Specification of neocortical areas and thalamocortical connections. Annu Rev Neurosci. 1994;17:419–439. doi: 10.1146/annurev.ne.17.030194.002223. [DOI] [PubMed] [Google Scholar]
- Poirier K, Van Esch H, Friocourt G, Saillour Y, Bahi N, Backer S, Souil E, Castelnau-Ptakhine L, Beldjord C, Francis F, Bienvenu T, Chelly J. Neuroanatomical distribution of ARX in brain and its localisation in GABAergic neurons. Brain Res Mol Brain Res. 2004;122:35–46. doi: 10.1016/j.molbrainres.2003.11.021. [DOI] [PubMed] [Google Scholar]
- Poirier K, Lacombe D, Gilbert-Dussardier B, Raynaud M, Desportes V, de Brouwer AP, Moraine C, Fryns JP, Ropers HH, Beldjord C, Chelly J, Bienvenu T. Screening of ARX in mental retardation families: consequences for the strategy of molecular diagnosis. Neurogenetics. 2006;7:39–46. doi: 10.1007/s10048-005-0014-0. [DOI] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- Seufert DW, Prescott NL, El-Hodiri HM. Xenopus aristaless-related homeobox (xARX) gene product functions as both a transcriptional activator and repressor in forebrain development. Dev Dyn. 2005;232:313–324. doi: 10.1002/dvdy.20234. [DOI] [PubMed] [Google Scholar]
- Song DD, Harlan RE. Ontogeny of the proenkephalin system in the rat corpus striatum: its relationship to dopaminergic innervation and transient compartmental expression. Neuroscience. 1993;52:883–909. doi: 10.1016/0306-4522(93)90536-o. [DOI] [PubMed] [Google Scholar]
- Stromme P, Mangelsdorf ME, Shaw MA, Lower KM, Lewis SM, Bruyere H, Lutcherath V, Gedeon AK, Wallace RH, Scheffer IE, Turner G, Partington M, Frints SG, Fryns JP, Sutherland GR, Mulley JC, Gecz J. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet. 2002a;30:441–445. doi: 10.1038/ng862. [DOI] [PubMed] [Google Scholar]
- Stromme P, Mangelsdorf ME, Scheffer IE, Gecz J. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev. 2002b;24:266–268. doi: 10.1016/s0387-7604(02)00079-7. [DOI] [PubMed] [Google Scholar]
- Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- Taglialatela P, Soria JM, Caironi V, Moiana A, Bertuzzi S. Compromised generation of GABAergic interneurons in the brains of Vax1−/− mice. Development. 2004;131:4239–4249. doi: 10.1242/dev.01299. [DOI] [PubMed] [Google Scholar]
- Turner G, Partington M, Kerr B, Mangelsdorf M, Gecz J. Variable expression of mental retardation, autism, seizures, and dystonic hand movements in two families with an identical ARX gene mutation. Am J Med Genet. 2002;112:405–411. doi: 10.1002/ajmg.10714. [DOI] [PubMed] [Google Scholar]
- Uyanik G, Aigner L, Martin P, Gross C, Neumann D, Marschner-Schafer H, Hehr U, Winkler J. ARX mutations in X-linked lissencephaly with abnormal genitalia. Neurology. 2003;61:232–235. doi: 10.1212/01.wnl.0000079371.19562.ba. [DOI] [PubMed] [Google Scholar]
- Vanderhaeghen P, Polleux F. Developmental mechanisms patterning thalamocortical projections: intrinsic, extrinsic and in between. Trends Neurosci. 2004;27:384–391. doi: 10.1016/j.tins.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Yun K, Fischman S, Johnson J, Hrabe de Angelis M, Weinmaster G, Rubenstein JL. Modulation of the notch signaling by Mash1 and Dlx1/2 regulates sequential specification and differentiation of progenitor cell types in the subcortical telencephalon. Development. 2002;129:5029–5040. doi: 10.1242/dev.129.21.5029. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Marin O, Hermesz E, Powell A, Flames N, Palkovits M, Rubenstein JL, Westphal H. The LIM-homeobox gene Lhx8 is required for the development of many cholinergic neurons in the mouse forebrain. Proc Natl Acad Sci USA. 2003;100:9005–9010. doi: 10.1073/pnas.1537759100. [DOI] [PMC free article] [PubMed] [Google Scholar]