

ABSTRACT
In the recent years, the role of actin and actin-binding proteins in gene transcription has received considerable attention. Nuclear monomeric and polymerized actin and several actin binding proteins have been detected in the mammalian cell nucleus, although their roles in transcription are just beginning to emerge. Our group recently reported that the actin-binding protein Filamin A interacts with the transcriptional coactivator MKL1 to link actin polymerization with transcriptional activity of Serum Response Factor. Here we summarize the regulation and function of MKL1, and highlight this novel mechanism of MKL1 regulation through binding to Filamin A and its implications for cell migration.
Keywords: actin, Filamin A, MKL1, MRTFA, MAL, SRF
Introduction
In many physiological and pathophysiological processes, eukaryotic cells are subject to rearrangements of the actin cytoskeleton. The transcriptional coactivator MKL1 has the exquisite property to transduce rearrangements in the actin cytoskeleton into gene expression via the transcription factor SRF.1 A communication of the state of actin rearrangements to the genome is especially important for cellular processes that involve dynamic cytoskeletal changes, such as cell motility, proliferation, contractility, adhesion, endocytosis and secretion.2,3 According to the current paradigm, the actin sensitivity of SRF is due to sequestration of MKL1 in the cytoplasm by direct binding to monomeric G-actin.4 Actin monomers occupying the N-terminal RPEL-domains block access of importins to MKL1's nuclear localization sequence, thereby impairing MKL1's nuclear entry.5 Actin polymerization in response to Rho signaling causes dissociation of the G-actin-MKL1 complex and translocation of MKL1 to the nucleus,4 where it activates the transcription of SRF target genes involved in motile, contractile and muscle-specific functions.1 It was hitherto believed that MKL1/SRF activation occurs as an indirect consequence of polymerization-induced depletion of actin monomers. Recent evidence demonstrated a direct contribution of nuclear actin polymers sufficient to promote MKL1 activation. Further support for this notion comes from the overexpression of nucleus-resident actin mutants that enhance SRF activity.6,7 However, the molecular mechanism leading to MKL1/SRF activation via nuclear actin polymerization remained unclear.8 Our group identified a new MKL1 binding partner named Filamin A (FLNA) that promotes nuclear actin polymerization and MKL1/SRF activity.9
Downregulation of SRF activity is facilitated by nuclear export of actin-bound, phosphorylated MKL1.10,11 The nuclear-cytoplasmic shuttling mechanism of MKL1 has been described most thoroughly for MKL1 in fibroblasts and muscle cells. By contrast, in mammary-, hepatocellular carcinoma and melanoma cells lacking the tumor suppressor Deleted in Liver Cancer 1 (DLC1), a constitutive nuclear localization of MKL1 is observed even in the absence of stimuli.12 MKL1 nuclear localization leads to activation of pro-tumorigenic target genes and triggers tumor cell proliferation and –migration.12 Despite the importance of MKL1 for tumor growth as illustrated by the inhibition of hepatocellular carcinoma xenograft growth upon MKL1 depletion, the modulators influencing MKL1 activity in the nucleus remain largely unclear.13,14 This question has been tackled by our lab in a recent publication in the journal Science Signaling.9
Linking actin polymerization and gene transcription
Searching for novel interaction partners involved in MKL1 activity, we identified the cytoskeletal protein Filamin A (FLNA). FLNA is an actin-binding protein that determines cell shape and locomotion by cross-linking actin filaments. FLNA binds MKL1 through a unique protein domain at aa 301–310. In functional studies on the MKL1-FLNA interaction, we obtained several pieces of evidence suggesting that FLNA stimulates MKL1/SRF activity. First, LPA-induced RhoA activation in primary human fibroblasts promoted the interaction of MKL1 and FLNA and induced SRF target genes. Second, siRNA-mediated silencing of FLNA in primary fibroblasts, mammary and hepatocellular carcinoma cells resulted in a strong reduction of target genes. Third, FLNA-induced target gene expression proved to be MKL-dependent and vice versa. Thus, FLNA binding constitutes a novel mechanism for MKL1 regulation in addition to G-actin binding.9 Elegant experiments have shown how MKL1 is regulated by the levels of free G-actin.4,10,15
Both mechanisms share one common feature, i.e. their dependency on MKL1 phosphorylation. MKL1 phosphorylation is a prerequisite for G-actin binding repressing MKL1 transcriptional activation.11 On the contrary, FLNA binding serves a positive function, because it impairs MKL1 phosphorylation.9 Released from G-actin, MKL1 can subsequently activate SRF-dependent transcription via its association with FLNA.
Our work demonstrates that FLNA is required for nuclear actin polymerization.9 Although the presence of nuclear actin and the structural details of the nuclear actin network are a matter of intense debate, recent studies revealed nuclear actin as a component of all 3 nuclear RNA polymerases and chromatin remodeling complexes and – beyond this - the nuclear presence of many actin associated proteins such as supervillin, cofilin, α-actinin, coronin 2A and also FLNA.16-20 Nuclear FLNA has been shown to be involved in DNA repair, regulation of nuclear shape and transcription via its association with RNA polymerase I, Smad-, FOXC1- or HIF1α transcription factors.21-27 Our finding of an interaction between MKL1 and FLNA as an F-actin binding protein in the nucleus is particularly appealing, because Baarlink and colleagues recently reported that nuclear actin is polymerized by formins in a signal-operated manner, thereby controlling MKL1/SRF transcriptional activation.8 These results raised the question how the signal for actin polymerization is transmitted to the nucleus and translated into MKL1/SRF activity. Given the actin-polymerizing and F-actin-binding properties of FLNA as novel MKL1 interaction partner we tested whether FLNA might function as a transducer of actin polymerization into MKL1/SRF activity. Indeed, FLNA was found to be required for actin polymerization to mediate MKL1/SRF activation, as shown by utilizing an actin mutant (S14C-actin) whose expression favors F-actin formation and administration of the actin polymerization enhancing cytoskeletal drug Jasplakinolide.9
Thus, our studies shed new light on the mechanism linking actin polymerization and SRF-dependent transcription. Moreover, our finding that FLNA is required for SRF activity in response to nuclear actin polymerization, triggered by a constitutive active version of mDia, lends further credence to the emerging concept of nuclear actin and actin binding proteins in transcription. This concept is consistent with a model, in which LPA stimulation promotes MKL1 nuclear translocation due to RhoA-induced stress fiber formation. FLNA then facilitates nuclear actin polymerization, thereby mediating an association between MKL1, FLNA and F-actin that is required for SRF target gene expression and counteracts the known repressive complex of MKL1 and monomeric G-actin (Fig. 1A, B).
Figure 1.
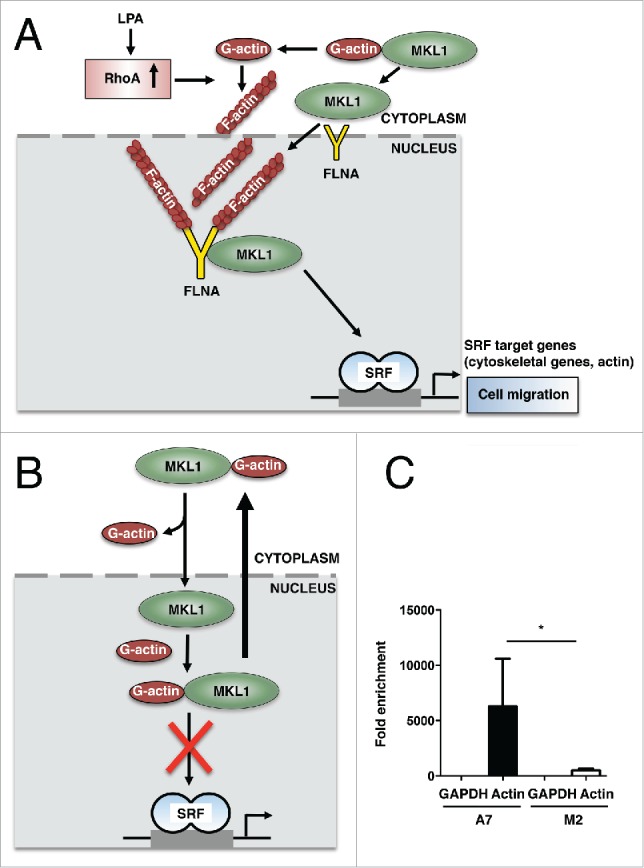
(A) Model for MKL1 regulation through binding to FLNA in stimulated fibroblasts: LPA stimulation promotes MKL1 nuclear localization due to RhoA-induced stress fiber formation in fibroblasts. FLNA facilitates nuclear actin polymerization and mediates an association between F-actin, FLNA and MKL1 that is required for SRF target gene expression. SRF target genes include cytoskeletal genes and actin itself to ensure sufficient supply of actin to meet the needs for cell motility. (B) Model for MKL1 regulation through binding to G-actin in unstimulated fibroblasts: In serum-starved fibroblasts, binding of monomeric G-actin represses SRF activation and promotes nuclear export of MKL1. (C) FLNA recruitment to the actin promoter. ChIP was performed using FLNA-expressing A7 and FLNA-deficient M2 melanoma cells and a specific antibody against FLNA. Actin and GAPDH (control) promoters were quantified by qRT-PCR from 3 independent chromatin preparations. Values are mean ± SD (n = 3), *p < 0.05.
Functional consequences on cell migration
The importance of the FLNA-MKL1 interaction is further underscored by the observation that cells expressing a MKL1 deletion mutant unable to bind FLNA displayed strongly reduced motile and invasive properties.9 While several knock-out studies of the SRF and MKL genes have revealed essential functions in cytoskeletal organization and cell migration,28-32 and many SRF target genes such as Integrin α5 and FHL1 encode migratory proteins and components of the actin cytoskeleton,33 the mechanisms how MKL1 controls motile cell functions remained enigmatic.
Our discovery of the transducer function of FLNA for actin polymerization into MKL1/SRF activity presented in the last section together with the present knowledge about actin and gene expression argue for the following 2 important functions of FLNA in cell migration:
FLNA transduces the information for actin polymerization into MKL/SRF-dependent gene expression. The communication of information about the state of actin rearrangements to the genome enables the cell to adjust the expression of MKL/SRF-dependent cytoskeletal genes accordingly. This allows the tight temporal coupling of actin dynamics required for cell motility.
FLNA promotes the expression of MKL/SRF target genes. These encode proteins that modify the actin cytoskeleton in order for cells to be able to migrate, and actin itself, as demonstrated by the direct recruitment of FLNA to the actin promoter (Fig. 1C). Interestingly, it has recently been shown that actin gene transcription itself is the key regulatory step in the control of cell migration.34 Actin protein then feeds back to the production of actin mRNA to ensure sufficient supply of actin to meet the needs of cytoskeletal alterations in the course of cell migration (Fig. 1A).
Aberrant migration leads to pathological processes including tumor formation and metastasis. It seems therefore likely that the FLNA-MKL1 complex may serve an important pathophysiological function herein. Consistent with this notion, FLNA is upregulated in hepatic, colorectal and breast carcinomas.35-37 Further support for this hypothesis comes from an elegant experimental metastasis assay in which depletion of MKL reduced cell motility.38 Furthermore, initiation of invasive cell migration was essentially abolished in the absence of MKL1 in the Drosophila ovary.39 Deeper analysis of the FLNA-MKL1 interaction will therefore help to understand how MKL1 controls metastasis.
Concluding remarks
The discovery of the essential role of FLNA for MKL1/SRF transcriptional activity and cell motility highlights the importance of studying the molecular mechanisms of the MKL1-FLNA interaction and their role in pathophysiology. Regarding the molecular mechanisms, the crucial question arises whether MKL1 binding to FLNA and FLNA interaction with F-actin occur simultaneously. Another pressing issue is whether the actin-binding activity of FLNA is required for the expression of SRF target genes. These questions will be challenging tasks to tackle in the future.
Disclosure of potential conflicts of interest
No potential conflict of interest was disclosed.
Acknowledgments
We thank laboratory members for discussions.
Funding
Our work is supported by the DFG (MU 2737/2–1 and 2–2, TRR 152).
References
- [1].Olson EN, Nordheim A. Nat Rev Mol Cell Biol 2010; 11:353; PMID:20414257; http://dx.doi.org/ 10.1038/nrm2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chen H, Bernstein BW, Bamburg JR. Trends Biochem Sci 2000; 25:19; PMID:10637608; http://dx.doi.org/ 10.1016/S0968-0004(99)01511-X [DOI] [PubMed] [Google Scholar]
- [3].Carlier MF, Le C Clainche, Wiesner S, Pantaloni D. Bioessays 25:336; PMID:12655641; http://dx.doi.org/ 10.1002/bies.10257 [DOI] [PubMed] [Google Scholar]
- [4].Miralles F, Posern G, Zaromytidou AI, Treisman R. Cell 2003; 113:329; PMID:12732141; http://dx.doi.org/ 10.1016/S0092-8674(03)00278-2 [DOI] [PubMed] [Google Scholar]
- [5].Pawlowski R, Rajakyla EK, Vartiainen MK, Treisman R. EMBO J 2010; 29:3448; PMID:20818336; http://dx.doi.org/ 10.1038/emboj.2010.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kokai E, et al.. Histochem Cell Biol 2014; 141:123; PMID:24091797; http://dx.doi.org/ 10.1007/s00418-013-1151-4 [DOI] [PubMed] [Google Scholar]
- [7].Stern S, et al.. J Neurosci 2009; 29:4512; PMID:19357276; http://dx.doi.org/ 10.1523/JNEUROSCI.0333-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Baarlink C, Wang H, Grosse R. Science 2013; 340:864; PMID:23558171; http://dx.doi.org/ 10.1126/science.1235038 [DOI] [PubMed] [Google Scholar]
- [9].Kircher P, et al.. Sci Signal 2015; 8:ra112; PMID:26554816; http://dx.doi.org/ 10.1126/scisignal.aad2959 [DOI] [PubMed] [Google Scholar]
- [10].Vartiainen MK, Guettler S, Larijani B, Treisman R. Science 2007; 316:1749.; PMID:17588931; http://dx.doi.org/ 10.1126/science.1141084 [DOI] [PubMed] [Google Scholar]
- [11].Muehlich S, et al.. Mol Cell Biol 2008; 28:6302; PMID:18694962; http://dx.doi.org/ 10.1128/MCB.00427-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Muehlich S, et al.. Oncogene 2012; 31:3913; PMID:22139079; http://dx.doi.org/ 10.1038/onc.2011.560 [DOI] [PubMed] [Google Scholar]
- [13].Hampl V, et al.. EMBO Mol Med 2013; 5:1367; PMID:23853104; http://dx.doi.org/ 10.1002/emmm.201202406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Muehlich S, Gudermann T. Aging (Albany NY) 2013; 5:639; PMID:24057649; http://dx.doi.org/ 10.18632/aging.100601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sotiropoulos A, Gineitis D, Copeland J, Treisman R. Cell 1999; 98:159; PMID:10428028; http://dx.doi.org/ 10.1016/S0092-8674(00)81011-9 [DOI] [PubMed] [Google Scholar]
- [16].Skarp KP, Vartiainen MK. Cytoskeleton (Hoboken) 2010; 67:487; PMID:20593452 [DOI] [PubMed] [Google Scholar]
- [17].Samwer M, et al.. EMBO J 2013; 32:1886; PMID:23727888http://dx.doi.org/ 10.1038/emboj.2013.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dopie J, et al.. J Cell Sci 128:2388; PMID:26021350; http://dx.doi.org/ 10.1242/jcs.169441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kumeta M, Yoshimura SH, Harata M, Takeyasu K. J Cell Sci 2010; 123:1020; PMID:20197409; http://dx.doi.org/ 10.1242/jcs.059568 [DOI] [PubMed] [Google Scholar]
- [20].Huang W, et al.. Nature 2011; 470:414; PMID:21331046; http://dx.doi.org/ 10.1038/nature09703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yuan Y, Shen Z. J Biol Chem 2001; 276:48318.; PMID:11602572 [DOI] [PubMed] [Google Scholar]
- [22].Yue J, et al.. Cancer Res 2009; 69:7978; PMID:19808958; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Deng W, et al.. Proc Natl Acad Sci U S A 2012; 109:1524; PMID:22307607; http://dx.doi.org/ 10.1073/pnas.1107879109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gay O, et al.. Proc Natl Acad Sci U S A 2011; 108:11464; PMID:21709252; http://dx.doi.org/ 10.1073/pnas.1104211108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sasaki A, Masuda Y, Ohta Y, Ikeda K, Watanabe K. J Biol Chem 2001; 276:17871; PMID:11278410; http://dx.doi.org/ 10.1074/jbc.M008422200 [DOI] [PubMed] [Google Scholar]
- [26].Berry FB, O'Neill MA, Coca-Prados M, Walter MA. Mol Cell Biol 2005; 25:1415; PMID:15684392; http://dx.doi.org/ 10.1128/MCB.25.4.1415-1424.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zheng X, et al.. Proc Natl Acad Sci U S A 2014; 111:2560; PMID:24550283; http://dx.doi.org/ 10.1073/pnas.1320815111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schratt G, et al.. J Cell Biol 2002; 156:737; PMID:11839767; http://dx.doi.org/ 10.1083/jcb.200106008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Alberti S, et al.. Proc Natl Acad Sci U S A 2005; 102:6148; PMID:15837932; http://dx.doi.org/ 10.1073/pnas.0501191102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stritt C, et al.. Nat Neurosci 2009; 12:418; PMID:19270689; http://dx.doi.org/ 10.1038/nn.2280 [DOI] [PubMed] [Google Scholar]
- [31].Mokalled MH, Johnson A, Kim Y, Oh J, Olson EN. Development 2010; 137:2365; PMID:20534669; http://dx.doi.org/ 10.1242/dev.047605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Costello P, et al.. Blood 2015; 125:1244; PMID:25573994; http://dx.doi.org/ 10.1182/blood-2014-08-595603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Leitner L, et al.. J Cell Sci 2011; 124:4318; PMID:22223881; http://dx.doi.org/ 10.1242/jcs.092791 [DOI] [PubMed] [Google Scholar]
- [34].Salvany L, Muller J, Guccione E, Rorth P. Genes Dev 2014; 28:1048; PMID:24831700; http://dx.doi.org/ 10.1101/gad.237743.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Guedj N, et al.. J Hepatol 2009; 51:93; PMID:19446907; http://dx.doi.org/ 10.1016/j.jhep.2009.03.017 [DOI] [PubMed] [Google Scholar]
- [36].Notterman DA, Alon U, Sierk AJ, Levine AJ. Cancer Res 2001; 61:3124; PMID:11306497 [PubMed] [Google Scholar]
- [37].Tian HM, et al.. Oncol Lett 2013; 6:681; PMID:24137390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. 2009. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol.; 11(3):257-68. http://dx.doi.org/10.1038/ncb1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Somogyi K, Rorth P. Dev Cell 2004; 7:85; PMID:15239956; http://dx.doi.org/ 10.1016/j.devcel.2004.05.020 [DOI] [PubMed] [Google Scholar]