

Chronic obstructive pulmonary disease (COPD) is a complex medical condition that is increasing in incidence and will be the third leading cause of death worldwide by 2030. Like cystic fibrosis (CF), one feature of COPD is small airway mucus obstruction that is associated with accelerated loss of lung function and mortality.1 Though mucus obstruction is present in the majority of patients with COPD, including those with an emphysema-dominant phenotype, it is most clinically apparent in those with chronic bronchitis.2
Cigarette smoking exerts a number of deleterious effects on airway epithelial function, including reduced cystic fibrosis transmembrane conductance regulator (CFTR) activity, enhanced mucus production, and a pronounced impairment of mucociliary clearance, resulting in a phenotype characteristic of acquired CFTR dysfunction.3 Emerging data indicate that patients with COPD without congenital CFTR mutations have reduced CFTR activity, as detected in the upper airways3, lower airways,4 sweat glands,5, 6 and intestine.5 Furthermore, CFTR dysfunction is independently associated with chronic bronchitis and dyspnea,3–6 and can persist despite smoking cessation.5, 7
The CFTR potentiator, ivacaftor, is approved for the treatment of CF patients with specific CFTR mutations.8–10 In addition to some mutant forms of CFTR, ivacaftor augments the function of wild-type CFTR protein,11 including epithelia with acquired CFTR dysfunction due to cigarette smoke exposure.3 To test whether CFTR potentiation might be helpful in COPD, we conducted a pilot study to determine the safety, efficacy, and pharmacokinetics of ivacaftor in COPD patients with chronic bronchitis. Patients were age 40–65, without significant comorbidities, and on a stable medical regimen that did not include inhibitors or inducers of cytochrome P350 3A4 (CYP450 3A4), which metabolizes ivacaftor.
Twelve active (n=4) or former smokers (n=8) with COPD (defined as post-bronchodilator FEV1/FVC ratio <0.7) and chronic bronchitis (defined as >3 months of daily productive cough for two or more consecutive years) were enrolled in a double-blind, randomized, placebo controlled pilot trial to investigate the effect of ivacaftor on safety, CFTR function and patient-reported outcomes of bronchitis symptoms after 14 days of treatment (NCT02135432). The study was approved by the Institutional Review Board of the University of Alabama at Birmingham. Patients were randomized in a 2:1 fashion (ivacaftor to placebo). Baseline characteristics were well-matched between the active and placebo arms, although mean sweat chloride for the whole group was lower than anticipated based on previous studies in a similar population, reflecting less severe CFTR dysfunction at baseline (Fig. 1A).5, 6 As expected, most patients had some evidence of emphysema on CT scan, although the means for each trial arm were relatively low. Two subjects in each arm had evidence for a minimal amount of bronchiectasis but were without clinically appreciable disease (i.e. lack of voluminous mucopurulent secretions on daily basis or history of bronchiectasis); mild bronchiectasis is detectable in a percentage of subjects with COPD and chronic bronchitis.12 All patients underwent CFTR sequencing to detect genetic mutations. Some of subjects were heterozygous for CFTR mutations (see supplemental Table 1). The subject with largest response to ivacaftor had a synonymous CFTR sequence variation of minimal significance on CFTR function.
Figure 1. Effect of Ivacaftor on CFTR function and Symptoms of Chronic Bronchitis.
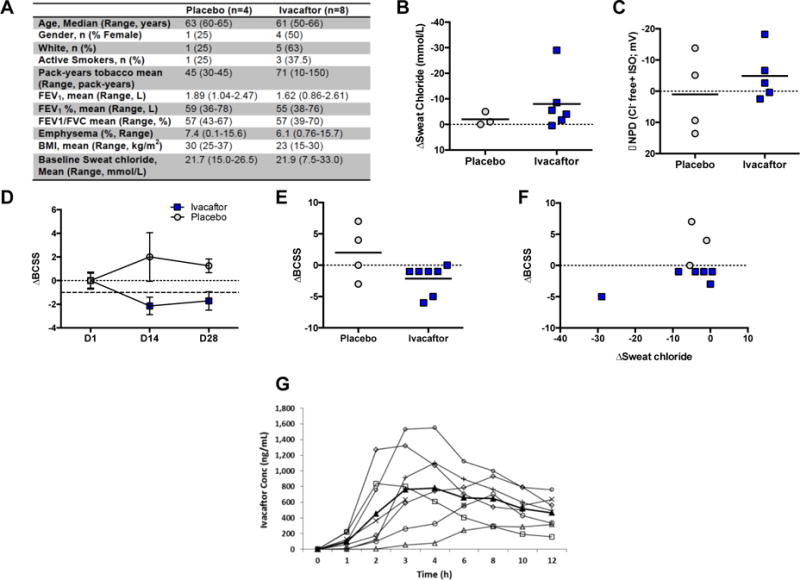
A. Baseline Characteristics of subjects enrolled in each treatment arm. BMI=body mass index. FEV1%= percent predicted forced expiratory volume in 1 second. B. Change in sweat chloride after 14 days of treatment compared to baseline. C. Change in CFTR-dependent chloride conductance (Δchloride-free plus isoproterenol) after 14 days of treatment compared to baseline. D. Change in Breathlessness, Cough, and Sputum Scale (BCSS) scores after 14 days of treatment (D14) and after 14-day washout period (D28, ±SEM). The range of BCSS is 1–12, and the MCID is 1 unit. E. Individual change in BCSS scores after 14 days of treatment compared to baseline. F. Correlation of BCSS to change in sweat chloride after 14 days of treatment. G. Pharmacokinetic profile of subjects taking ivacaftor after first dose (n=8). Each line represents an individual subject; heavy black line designates mean value. All P values are paired t-tests, within subject, per study protocol. Between subject testing was limited by small sample sizes.
After 14 days of treatment, patients receiving ivacaftor exhibited non-significant improvements in CFTR function as detected by improved sweat chloride (−8.0 ± 4.4 mmol/L ivacaftor vs. +2.0 ± 1.5 mmol/L placebo, compared to baseline, P=0.38, Fig. 1B) and nasal potential difference (−4.9 ± 3.9 mV ivacaftor group vs. +1.0 ± 6.4 mV placebo, compared to baseline, P=0.56, Fig. 1C), although these changes were not statistically significant given the small sample size. Accompanying augmentation of CFTR function, subjects in the ivacaftor treatment group demonstrated non-significant improvements in symptoms as detected by changes in the Breathlessness, Cough, and Sputum Scale (BCSS) that exceeded the minimally clinical important difference of 1 unit (mean change of −2.1 ± 0.9 ivacaftor vs. +2.0 ± 2.2 placebo, compared to baseline, p=0.11, Fig 1D, E).13 The change in sweat chloride did not correlate significantly to the change in chronic bronchitis symptoms given the small sample size (Fig. 1F), although the patient with the largest response in sweat chloride (and NPD) also had the greatest improvement in BCSS and the highest sweat chloride at baseline, reflecting more severe CFTR dysfunction that was partially reversible. There were no improvements in spirometry over this duration (−2.3 ± 5.2 FEV1% ivacaftor vs. +1.8 ± 9.3 placebo, P=0.60, compared to baseline). Pharmacokinetics revealed stable ivacaftor concentrations with an AUC that mirrored experience in CF (Fig 1G).14 One patient discontinued drug due to a COPD exacerbation; no serious adverse events were attributed to study drug. Where available, all data was analyzed for the key trial endpoints, although one subject was unable to complete each assessment at subsequent visits.
Overall, ivacaftor was safe and well tolerated in these COPD patients and exhibited reasonable absorption and metabolism. Although this pilot trial was underpowered and potentially too brief to detect definitive changes in lung function, there were improvements in CFTR activity and respiratory symptoms that did not reach statistical significance; each of these outcomes were markedly improved after relatively brief treatment period with ivacaftor in patients with CF.8, 9 Due to less severe CFTR dysfunction, prolonged therapy may be necessary to demonstrate that improvements in acquired CFTR dysfunction that will impact clinical outcomes. The mean improvement in NPD and sweat chloride indicates a substantial improvement (~20%) in wild-type CFTR activity, even though baseline CFTR activity was not diminished at the start of the study, whereas those with diminished CFTR function at baseline had the most robust response. These results set the stage for longer trials to determine the effects of CFTR potentiation on clinical outcomes in patients with chronic bronchitis and a substantial degree of acquired CFTR dysfunction at baseline that is prevalent in ~30–40% of patients with COPD.5, 6
Supplementary Material
Acknowledgments
Vertex Pharmaceuticals provided investigational product for the conduct of the investigator-initiated study but had no role in the design, conduct, or interpretation of the data. We thank Lawrence Rasmussen (UAB) for performing sweat chloride analysis. The UAB CCTS assisted with preparation of an investigator-initiated IND. CFTR sequencing was conducted by the Johns Hopkins Institute for Clinical and Translational Research (ICTR) which is funded in part by Grant Number UL1 TR 001079 from the National Center for Advancing Translational Sciences (NCATS) a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the Johns Hopkins ICTR, NCATS or NIH. We are grateful to the patients for their participation in this study.
This research is sponsored by the NIH (R34 HL127166 to SMR., R01 HL105487 to SMR., P30 DK072482 to SMR., and 5UL1 RR025777) and the Cystic Fibrosis Foundation (CLANCY09Y0 to GMS. and R464-CF to SMR.). SVR is supported by American Lung Association (RG-305752) and the Flight Attendants Medical Research Association (YFA130008).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
GMS reports grants from NIH and Cystic Fibrosis Foundation during the conduct of the study. HH, BL, and GR report grants from NIH and Cystic Fibrosis Foundation during the conduct of the study. EPA reports grants from NIH and Cystic Fibrosis Foundation, during the conduct of the study. MTD reports personal fees and other from AstraZeneca, personal fees and other from Boehringer Ingelheim, personal fees and other from Boston Scientific, personal fees and other from GlaxoSmithKline, personal fees from Ikaria, Skyepharma, grants from NIH, US Department of Defense, and American Heart Association, and other funding from Aeris, Otsuka, Pearl, Pfizer, PneumRx, Pulmonx, and Yungjin, outside the submitted work. SMR reports grants from NIH and Cystic Fibrosis Foundation during the conduct of the study. Vertex pharmaceuticals provided study drug through an investigator initiated study program. UAB received grants from Vertex Pharmaceuticals, Novartis, and Nivalis Pharmaceuticals outside the submitted work.
Author Contributions:
GMS, MTD, and SMR contributed to design of the study. GMS, HH, BL, SVR, GR, EPA, MTD, and SMR contributed to study execution. GMS, BL, EPA, MTD, and SMR conducted data analysis. GMS, MTD, and SMR prepared the manuscript. MTD and SMR supervised the project. All authors approved the final manuscript prior to submission.
References
- 1.Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233–47. doi: 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elbehairy AF, Raghavan N, Cheng S, et al. Physiologic characterization of the chronic bronchitis phenotype in GOLD grade IB COPD. Chest. 2015;147(5):1235–45. doi: 10.1378/chest.14-1491. [DOI] [PubMed] [Google Scholar]
- 3.Sloane PA, Shastry S, Wilhelm A, et al. A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS One. 2012;7(6):e39809. doi: 10.1371/journal.pone.0039809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dransfield MT, Wilhelm AM, Flanagan B, et al. Acquired Cystic Fibrosis Transmembrane Conductance Regulator Dysfunction in the Lower Airways in COPD. Chest. 2013;144(2):498–506. doi: 10.1378/chest.13-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raju SV, Jackson PL, Courville CA, et al. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am J Respir Crit Care Med. 2013;188(11):1321–30. doi: 10.1164/rccm.201304-0733OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Courville CA, Tidwell S, Liu B, Accurso FJ, Dransfield MT, Rowe SM. Acquired defects in CFTR-dependent Beta-adrenergic sweat secretion in chronic obstructive pulmonary disease. Respir Res. 2014;15(1):25. doi: 10.1186/1465-9921-15-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Courville CA, Raju SV, Liu B, Accurso FJ, Dransfield MT, Rowe SM. Recovery of Acquired Cystic Fibrosis Transmembrane Conductance Regulator Dysfunction after Smoking Cessation. American journal of respiratory and critical care medicine. 2015;192(12):1521–4. doi: 10.1164/rccm.201502-0396LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rowe SM, Heltshe SL, Gonska T, et al. Clinical Mechanism of the CFTR Potentiator Ivacaftor in G551D-Mediated Cystic Fibrosis. Am J Respir Crit Care Med. 2014 doi: 10.1164/rccm.201404-0703OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. The New England journal of medicine. 2011;365(18):1663–72. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moss RB, Flume PA, Elborn JS, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med. 2015;3(7):524–33. doi: 10.1016/S2213-2600(15)00201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106(44):18825–30. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez-Garcia MA, Soler-Cataluna JJ, Donat Sanz Y, et al. Factors associated with bronchiectasis in patients with COPD. Chest. 2011;140(5):1130–7. doi: 10.1378/chest.10-1758. [DOI] [PubMed] [Google Scholar]
- 13.Leidy NK, Rennard SI, Schmier J, Jones MK, Goldman M. The breathlessness, cough, and sputum scale: the development of empirically based guidelines for interpretation. Chest. 2003;124(6):2182–91. doi: 10.1378/chest.124.6.2182. [DOI] [PubMed] [Google Scholar]
- 14.KalydecoTM prescribing information. Vertex Pharmaceuticals, Inc.; Jan, 2012. 2015 http://www.kalydeco.com (accessed January 6, 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.