

Summary
Somatic activating mutations of BRAF are the earliest and most common genetic abnormality detected in the genesis of human melanoma. However, the mechanism(s) by which activated BRAF promotes melanoma cell cycle progression and/or survival remain unclear. Here we demonstrate that expression of BIM, a pro-apoptotic member of the BCL-2 family, is inhibited by BRAF → MEK → ERK signaling in mouse and human melanocytes and in human melanoma cells. Trophic factor deprivation of melanocytes leads to elevated BIM expression. However, re-addition of trophic factors or activation of a conditional form of BRAFV600E leads to rapid inhibition of BIM expression. In both cases, inhibition of BIM expression was dependent on the activity of MEK1/2 and the proteasome. Consistent with these observations, pharmacological inhibition of BRAFV600E or MEK1/2 in human melanoma cells (using PLX4720 and CI-1040 respectively) led to a striking elevation of BIM expression. Re-activation of BRAF → MEK → ERK signaling led to phosphorylation of BIM-EL on serine 69 and its subsequent degradation. Interestingly, endogenous expression of BIM in melanoma cells was insufficient to induce apoptosis unless combined with serum deprivation. Under these circumstances, inhibition of BIM expression by RNA interference provided partial protection from apoptosis. These data suggest that regulation of BIM expression by BRAF → MEK → ERK signaling is one mechanism by which oncogenic BRAFV600E can influence the aberrant physiology of melanoma cells.
Keywords: BRAF, Bim, melanoma, apoptosis, Bcl-2
Introduction
Pigment-producing melanocytes in the epidermis give rise to malignant melanoma, the most aggressive form of skin cancer (Chin et al., 1998). The increasing incidence of this disease, combined with the paucity of successful clinical strategies to combat it, highlights the challenge of metastatic melanoma treatment in medical oncology (Gray-Schopfer et al., 2007).
Somatic activating mutations in BRAF are detected in approximately 85% of benign melanocytic nevi and 60–70% of all melanomas (Davies et al., 2002; Pollock et al., 2003). The most common BRAF mutation is a T1799A transversion, encoding BRAFV600E with constitutive protein kinase activity promoting sustained activation of the BRAF → MEK → ERK MAP kinase signaling pathway. This pathway has pleiotropic effects that promote the aberrant physiology of the melanoma cell (Pollock et al., 2003; Wan et al., 2004). Indeed, ectopic expression of BRAFV600E in p53 nullizygous zebrafish or in immortalized mouse Melan-a cells leads to melanocyte transformation (Patton et al., 2005). In addition, inhibition of BRAFV600E expression or signaling inhibits melanoma cell proliferation (Hingorani et al., 2003; Sharma et al., 2005). These data indicate that mutated BRAF is important for both melanoma initiation and maintenance and raise the important question of how sustained BRAFV600E → MEK → ERK signaling contributes to the aberrant physiology of the melanoma cell.
Melanoma cells display remarkable resistance to apoptosis, which contributes to their metastatic potential and striking resistance to chemotherapy (Gray-Schopfer et al., 2007; Soengas and Lowe, 2003). Although activated RAF protein kinases are reported to influence apoptosis in a variety of different cell types, it is unclear which, if any, of these mechanisms may be operative in melanoma cells (Baccarini, 2002; Christensen and Guldberg, 2005).
BCL-2 family proteins are essential regulators of apoptosis that contribute to the deregulation of survival pathways in cancer cells (Youle and Strasser, 2008). Pro-survival members of the family, such as BCL-2, BCL-XL and MCL-1, possess four BCL-2 homology (BH) domains. The pro-apoptotic BCL-2 proteins are further divided into two sub-families. Proteins such as BAX or BAK contain BH1–BH3 domains but lack the N-terminal BH4 domain. Proteins such as BAD, BID, BIM or PUMA lack all but the BH3 domain and are known as the ‘BH3-only’ proteins. The current model posits that BCL-2 proteins work in a hierarchical network of inhibitory interactions to regulate apoptosis. In healthy cells, the pro-apoptotic effects of BAX and BAK are restrained by the pro-survival proteins BCL-2, BCL-XL and MCL-1. However, in response to pro-apoptotic stresses, members of the BH3-only proteins are expressed or activated. BH3-only proteins inhibit the pro-survival effects of BCL-2, BCL-XL and MCL-1 thereby liberating the pro-apoptotic effects of BAX and BAK leading to cell death. Interestingly, Bcl-2 and Bim play an essential role in mouse melanocyte survival. Bcl-2−/− mice display premature de-pigmentation due to melanocyte apoptosis (Veis et al., 1993), but additional deletion of both Bim alleles prevents this defect, restoring normal pigmentation (Bouillet et al., 2001). This places Bim as having an important role in regulated melanocyte apoptosis and possibly in melanoma.
The expression and pro-apoptotic activity of BIM is regulated by several different signaling systems including the ERK, p38 and JNK MAP kinases and the PI3′-kinase → PDK → AKT pathways through transcriptional and post-transcriptional mechanisms (Cai et al., 2006; Ewings et al., 2007; Ley et al., 2005; O’Connor et al., 1998). In this study, we demonstrate that RAF → MEK → ERK signaling regulates BIM expression in mouse and human melanocytes, and also in human melanoma cells. Furthermore, MEK1/2 inhibition promotes melanoma cell apoptosis when combined with serum deprivation. This is accompanied by induced BIM expression and its mitochondrial localization. Under the same conditions, RNA interference-mediated inhibition of BIM expression provides melanoma cells with partial protection from apoptosis. These data illustrate the potential importance of this regulatory circuit in regulating apoptosis in melanoma cells expressing BRAFV600E.
Results
Trophic factor deprivation induced expression of BIM in mouse and human melanocytes
Under normal growth conditions, BIM-EL expression is weakly detectable in immortalized mouse Melan-a melanocytes (Figure 1A). However, trophic factor deprivation (TFD) of Melan-a cells leads to robust induction of BIM-EL expression reaching a maximal level by 24 h and with sustained expression up to 72 h (Figure 1A). Three major BIM isoforms exist: short (BIM-S), long (BIM-L) and extra-long (BIM-EL) (O’Connor et al., 1998). Based on electrophoretic mobility, the predominant form of BIM detected in mouse Melan-a cells was BIM-EL.
Figure 1.
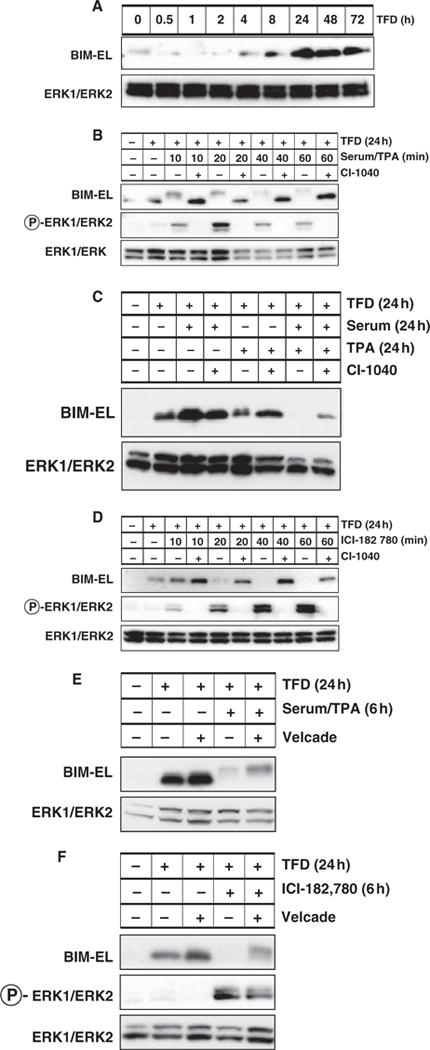
Regulation of BIM expression in mouse Melan-a melanocytes. (A) Asynchronously growing Melan-a cells were subjected to trophic factor deprivation (TFD) for up to 72 h at which time the expression of BIM-EL and ERK1/2 was assessed by Western blot analysis of cell lysates. (B) Asynchronously growing Melan-a cells were subjected to 24 h of TFD prior to addition of 2 μM CI-1040 and subsequent re-stimulation with media containing serum and TPA for 10–60 min as indicated. The phosphorylation and expression of BIM-EL and ERK1/2 were assessed by Western blotting. (C) Asynchronously growing Melan-a cells were subjected to 24 h of TFD prior to stimulation with complete media containing serum and/or TPA for 24 h in the absence or presence of 2 μM CI-1040 (as indicated). The expression of BIM-EL and ERK1/2 was assessed by Western blotting. (D) Melan-a cells engineered to express BRAFV600E:ERT1 were subjected to 24 h of TFD prior to treatment with 200 nM ICI 182,780 for 10–60 min in the absence or presence of 2 μM CI-1040 as indicated. The phosphorylation and expression of BIM-EL and ERK1/2 were assessed by Western blotting. (E) Asynchronously growing Melan-a cells were subjected to 24 h of TFD in the absence or presence of 15 nM Velcade as indicated. Cells were then treated with full media for 6 h in the absence or presence of Velcade. The phosphorylation and expression of BIM-EL and ERK1/2 were assessed by Western blotting. (F) Asynchronously growing Melan-a/BRAFV600E:ERT1 cells were subjected to 24 h of TFD in the absence or presence of 15 nM Velcade. BRAFV600E:ERT1 was then activated by addition of 200 nM ICI 182,780 in the absence or presence of Velcade for 6 h. The phosphorylation and expression of BIM-EL and ERK1/2 were assessed by Western blotting.
Re-stimulation of Melan-a cells with serum and TPA-induced rapid BIM-EL phosphorylation, as reflected by reduced mobility of the protein in SDS-PAGE, accompanied by a significant reduction in BIM-EL expression within 40–60 min (Figure 1B). Furthermore, growth factor-induced BIM-EL phosphorylation and reduced expression were inhibited by CI-1040, a specific and selective inhibitor of RAF → MEK → ERK signaling (Figure 1B) (Sebolt-Leopold et al., 1999). To test the ability of serum and/or TPA to repress BIM-EL expression, trophic factor deprived Melan-a cells were re-stimulated either with serum, TPA or both agents together. Surprisingly, neither serum nor TPA alone had a strong effect on BIM-EL expression over the course of 24 h (Figure 1C). By contrast, the combination of serum and TPA led to a striking reduction in BIM-EL expression after 24 h that was inhibited by pre-treatment of the cells with CI-1040 (Figure 1C). These data suggest that the regulation of BIM-EL expression by trophic factors stimulation requires the combined action of both serum and TPA-regulated signaling pathways. Given the effects of CI-1040, one of the major pathways is the RAF → MEK → ERK MAP kinase signaling pathway.
Expression of mutationally activated BRAFV600E promotes oncogenic transformation of Melan-a cells leading to TPA-independent cell proliferation (Wellbrock et al., 2004). To test the effects of activated BRAFV600E on BIM-EL expression, Melan-a cells expressing a conditionally active form of BRAFV600E (BRAFV600E:ERT1) were derived by retrovirus infection (Liu et al., 2007). In these cells, addition of the estrogen analog ICI-182,780 (ICI) led to rapid activation of BRAFV600E → MEK → ERK signaling (Figure 1D). Trophic factor deprived Melan-a cells expressing BRAFV600E:ERT1 were treated with ICI in the absence or presence of CI-1040. Activation of BRAFV600E → MEK → ERK signaling in Melan-a cells led to a loss of BIM-EL expression within 20–40 min, and this was potently inhibited by CI-1040 (Figure 1D). Taken together, these data indicate that the ability of oncogenic BRAFV600E to repress BIM-EL expression is dependent on MEK1/2 activity.
Phosphorylation of BIM-EL is reported to promote its destruction by the proteasome in a number of cell types (Ley et al., 2005). To test this in melanocytes, trophic factor deprived Melan-a cells were re-stimulated either with serum + TPA (Figure 1E) or by ICI-mediated activation of BRAFV600E:ERT1 (Figure 1F) in the absence or presence of Velcade, a potent proteasome inhibitor (Richardson et al., 2006). As before, TFD led to a dramatic increase in BIM-EL expression, and subsequent treatment with serum/TPA (Figure 1E) or activation BRAF → MEK → ERK (Figure 1F) led to inhibition of BIM-EL expression. The addition of Velcade led to sustained BIM-EL expression, although phosphorylation was still clearly detectable. These data indicate the requirement of proteasome activity for the suppression of BIM-EL expression in response to growth factor stimulation or BRAFV600E activation in mouse melanocytes.
Regulation of BIM-EL expression downstream of BRAF in human melanoma cells
The regulated expression of BIM-EL by oncogenic BRAFV600E in immortalized mouse Melan-a melanocytes prompted us to test whether similar mechanisms may be at work in human melanocytes and melanoma cells. As with Melan-a cells, BIM-EL levels were low under normal growth conditions in human melanocytes, were increased following 24 h of growth factor deprivation and then suppressed following growth factor replacement (Figure 2A). Again, growth factor-mediated suppression of BIM-EL expression required RAF → MEK → ERK signaling and the proteasome, as it was prevented by CI-1040 (Figure 2A) or addition of Velcade (Figure 2B). As in Melan-a cells, addition of Velcade led to accumulation of phosphorylated BIM-EL as detected by reduced mobility of the protein in SDS-PAGE (Figure 2B).
Figure 2.
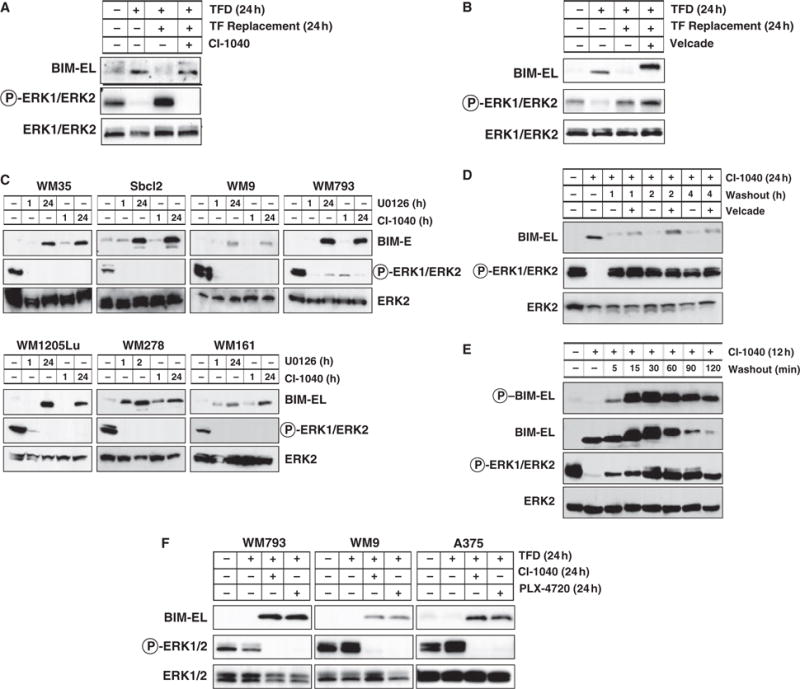
Regulation of BIM expression in human melanocytes and melanoma cells. (A, B) Asynchronously growing primary human melanocytes were subjected to 24 h of TFD. These cells were then re-stimulated with full media containing serum and growth factors for 24 h in the absence or presence of 2 μM CI-1040 or 15 nM Velcade. The phosphorylation and expression of BIM-EL and ERK1/2 were assessed by Western blotting. (C) A panel of human melanoma cells (for details of each cell line visit http://www.wistar.org/herlyn/melanoma.htm) was treated with either 10 μM U0126 or 2 μM CI-1040 to inhibit MEK1/2 for 1 or 24 h as indicated. Cell extracts were then analyzed for BIM-EL expression and phosphorylation and expression of ERK1/2. (D) Asynchronously growing WM793 cells were treated with 2 μM CI-1040 for 24 h. Cells were then cultured in the absence (washout) of 2 μM CI-1040 for 1, 2 or 4 h in the absence or presence of 15 nM Velcade as indicated. Cell extracts were analyzed by Western blotting. (E) Asynchronously growing WM793 cells were treated with 2 μM CI-1040 for 12 h. Cells were then cultured in the absence (washout) of 2 μM CI-1040 for 120 min. Cell extracts were analyzed by Western blotting for Bim-EL expression and serine 69 phosphorylation, and expression and phosphorylation of ERK1/2. (F) Human WM793, WM9 melanoma cells (all expressing BRAFV600E) were subjected to 24 h TFD in the absence or presence of either 2 μM CI-1040 or 10 μM PLX4720. Cell extracts were then analyzed for BIM-EL expression and phosphorylation and expression of ERK1/2.
Most human melanoma cells display elevated ERK1/2 activity as a consequence of expression of mutationally activated NRAS or BRAF (Gray-Schopfer et al., 2007). Consistent with this, we readily detected ERK1/2 phosphorylation accompanied by low to undetectable levels of BIM-EL expression in a panel of human melanoma cell lines (Figure 2C). Inhibition of MEK → ERK signaling with either U0126 or CI-1040, structurally unrelated MEK inhibitors, led to induced BIM-EL expression within 24 h in all cell lines tested (Figure 2C). These data indicate that the regulation of BIM-EL expression by RAF → MEK → ERK signaling observed in mouse and human melanocytes is operative in bona fide human melanoma cells.
To determine if the MEK1/2-dependent suppression of BIM-EL protein expression was dependent on the proteasome, WM793 cells were treated with CI-1040 for 24 h to induce BIM-EL expression (Figure 2D). CI-1040 was then removed and the cells cultured in full media in the absence or presence of Velcade. Following CI-1040 removal, ERK1/2 activity was restored within 60 min and was not influenced by Velcade addition (Figure 2D). ERK1/2 reactivation was accompanied by both a phosphorylation-induced mobility shift and reduced BIM-EL expression. Velcade addition had no effect on the mobility shift but prevented the suppression of BIM-EL expression that occurred following ERK1/2 reactivation (Figure 2D). Together, these data demonstrate that BIM-EL expression is regulated by RAF → MEK → ERK signaling and the proteasome in human melanoma cells.
Phosphorylation of BIM-EL on serine 69 (pS69) with the motif PASP (serine 65 in mouse) is required for its subsequent degradation by the proteasome (Ley et al., 2003). To test if BIM-EL is phosphorylated on this site we raised a mouse monoclonal antibody against a suitable phosphopeptide that spans this site (to be described in detail elsewhere). This antibody has excellent specificity and selectivity for phospho-serine 69 in endogenous mouse and human BIM-EL.
WM793 cells were treated with CI-1040 for 12 h at which time the drug was removed and the cells cultured in fresh media for 5–120 min (Figure 2E). Cell lysates were immunoblotted for expression of total BIM or for the presence of pS69 BIM-EL using appropriate antisera. As expected, MEK inhibition leads to the accumulation of hypophosphorylated BIM-EL that is not recognized by the pS69 phospho-BIM-EL-specific antibody. However, within 5 min following release from MEK inhibition, phosphorylation of BIM-EL on S69 was readily detected. Peak phosphorylation of BIM-EL was detected 15–30 min after release from MEK inhibition after which phosphorylation decreased in accordance with the overall decrease in BIM-EL expression (Figure 2E). These data are in accordance with the proposed role of S69 phosphorylation in targeting BIM-EL for destruction.
Recently, a specific pharmacological inhibitor of oncogenic BRAFV600E (PLX4720) has been reported (Tsai et al., 2008). Consequently, we tested the ability of PLX4720 to induce BIM-EL expression in WM793, WM9 and A375 melanoma cells, all of which express mutationally activated BRAFV600E. Asynchronously growing cells were subjected to trophic factor deprivation in the absence or presence of either PLX4720 or CI-1040. In all three melanoma cell lines, PLX4720 treatment led to inhibition of ERK phosphorylation with concomitant induction of BIM-EL that was comparable to that observed in response to MEK inhibition with CI-1040 (Figure 2F). These data indicate that BIM-EL is induced by pharmacological inhibition of either oncogenic BRAFV600E or its downstream targets MEK1/2.
MEK1/2 inhibition in combination with serum deprivation promotes melanoma cell apoptosis
As inhibition of RAF → MEK → ERK signaling led to increased BIM-EL expression, we tested whether MEK inhibition was sufficient to induce apoptosis. WM793 cells were treated either with CI-1040 alone, subjected to serum deprivation or a combination of both for 48 h (Figure 3A). In these cells, CI-1040 or serum deprivation alone had only modest effects on apoptosis. However, the combination of both CI-1040 and serum deprivation led to a dramatic increase in cell death (Figure 3A, B). In other melanoma cell lines, the sensitivity to CI-1040 varied with WM35 cells showing the greatest sensitivity and WM9 cells the least. However, in most cell lines, serum deprivation strongly augmented the apoptotic response to MEK1/2 inhibition as measured by Annexin V and propidium iodide staining (Figure 3B). For example, Sbcl2 cells displayed a fourfold increase of apoptotic cells in response to CI-1040, a twofold increase in response to serum deprivation and an eightfold increase in response to both challenges.
Figure 3.
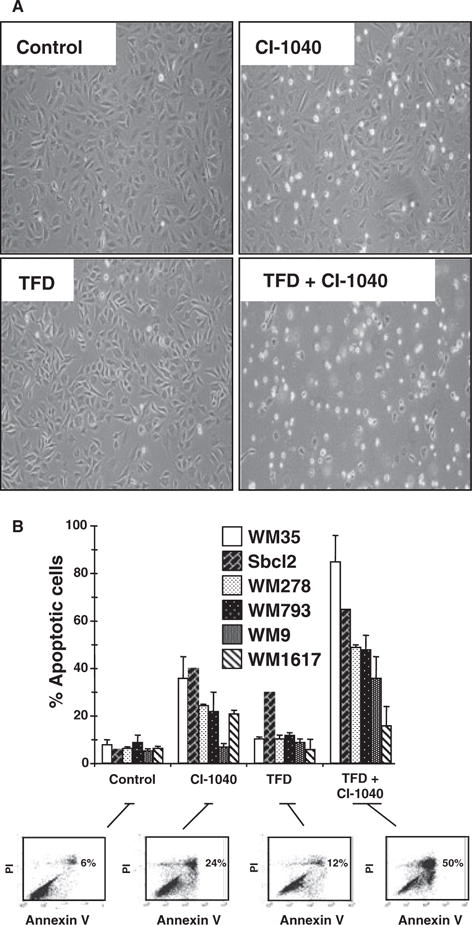
Regulation of apoptosis in human melanoma cells. (A) WM793 cells were treated with 2 μM CI-1040 in the absence or presence of serum for 48 h as indicated. Cells were photographed under phase contrast using a Nikon TMS microscope (magnification 100×). (B) A panel of human melanoma cell lines was subjected to 48 h of TFD in the absence or presence of 2 μM CI-1040 as indicated. Cells were harvested and their viability assessed by Annexin V/propidium iodide co-staining followed by analysis by flow cytometry. Results shown for each cell line are the mean ± SD of at least three individual experiments. Flow cytometry data are shown for one representative experiment for the WM278 cell line.
If the cell death induced by CI-1040 treatment/serum deprivation of WM793 melanoma cells was due to apoptosis, the cells should be protected by ectopic expression of BCL-2. Hence, we generated WM793 cells overexpressing BCL-2. Addition of CI-1040 had no effect on BCL-2 overexpression or BIM-EL induction (Figure 4A). However, BCL-2 overexpression clearly inhibited WM793 cell death in response to the combination of CI-1040 and serum deprivation suggesting that the cells are dying as a consequence of apoptosis (Figure 4B, C).
Figure 4.
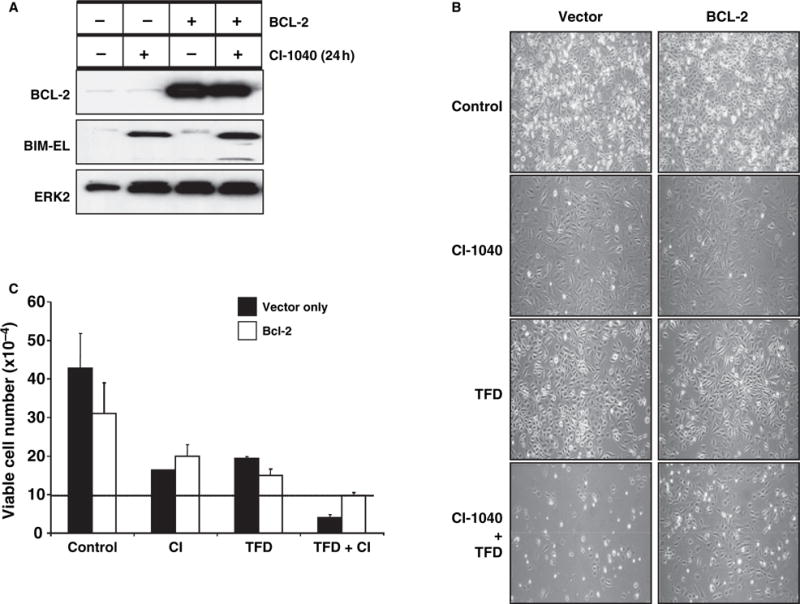
BCL-2 overexpression protects WM793 cells from apoptosis induced by CI-1040 combined with serum deprivation. (A) WM793 cells engineered to overexpress BCL-2 were treated with 2 μM CI-1040 for 48 h. The expression of BCL-2, BIM-EL and ERK2 was analyzed by Western blot analysis of cell extracts. (B) Vector control and BCL-2 overexpressing WM793 cells were plated at 105 cells per well and then subjected to TFD in the absence or presence of 2 μM CI-1040 for 48 h. Cells were photographed under phase-contrast microscopy (magnification 100×). (C) Vector control (black bars) and BCL-2 overexpressing (white bars) WM793 cells were plated at 105 cells per well (line represents plating density). Cells were then subjected to TFD in the absence or presence of 2 μM CI-1040 for 48 h. Cells were harvested and viable cells were determined by Trypan blue exclusion. Values represent mean ± standard deviation.
Effect of MEK inhibition plus serum deprivation on BIM-EL mitochondrial localization
Cytochrome c and Smac/DIABLO are mitochondrial proteins released into the cytosol from the inter-mitochondrial membrane space following initiation of apoptosis (Saelens et al., 2004). In asynchronously growing WM793 cells, both cytochrome c and Smac/DIABLO are only weakly detected in the cytosolic fraction of the cell (Figure 5A). In response to treatment with either CI-1040 or serum deprivation alone, we observed a modest accumulation of cytoplasmic cytochrome c and Smac/DIABLO, while the combination of CI-1040 and serum deprivation led to a striking accumulation of these proteins in the cytoplasm (Figure 5A). Consistent with this, accumulation of the 17 kDa active form of caspase 3 was also cooperatively induced by the combination of MEK inhibition and serum deprivation (Figure 5B). However, the pan-caspase inhibitor Z-VAD-FMK had no effect on BIM-EL expression levels (data not shown) suggesting that caspases themselves were not regulating BIM-EL expression. These biochemical data are consistent with the biological results above suggesting that MEK inhibition cooperates with serum deprivation to promote apoptosis in melanoma cells.
Figure 5.
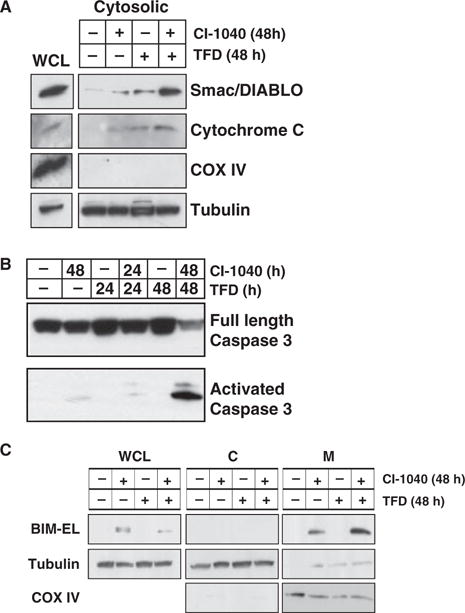
Effect of MEK inhibition and trophic factor deprivation on markers of apoptosis. (A) WM793 cells were treated with 2 μM CI-1040 in the absence or presence of serum for 48 h as indicated at which time cells were harvested. Whole cell and cytosolic lysates (cytosolic) were analyzed by Western blot analysis for the presence of Smac/DIABLO, cytochrome c, the inner mitochondrial marker COX IV and the cytosolic marker β-tubulin. (B) WM793 cells were treated with 2 μM CI-1040 in the absence or presence of serum for 24 or 48 h as indicated. The presence of full length pro-caspase 3 and activated caspase 3 was assessed by Western blot analysis of cell lysates. (C) WM793 cells were treated with 2 μM CI-1040 in the absence or presence of serum for 48 h at which time extracts were prepared. WCL, cytosolic (C) and mitochondrial fractions (M) were prepared as described in Materials and Methods. The presence of BIM-EL, tubulin (cytosolic marker) and COX IV (mitochondrial marker) in each cell fraction was assessed by Western blot analysis of cell extracts.
To determine if BIM-EL localizes to mitochondria, WM793 cells were treated with CI-1040 for 48 h in the absence or presence of serum, at which time mitochondrial fractions were prepared. In asynchronously growing or serum deprived WM793 cells, we detected no BIM-EL expression either in whole cell lysates (WCL) or in fractions enriched for mitochondria (M, Figure 5C). MEK inhibition, either in the absence or presence of serum, led to robust BIM-EL induction that was readily detected in the mitochondrial fraction. Despite reports that BIM-EL associates with microtubules (Day et al., 2004), we did not detect any BIM-EL protein in the cytoplasmic fraction in these cells (Figure 5C). These data clearly implicate BIM-EL in the regulation of the mitochondrial checkpoint that leads to caspase activation and subsequent apoptosis.
RNAi-mediated inhibition of BIM-EL expression confers partial protection against apoptosis
To determine the importance of BIM-EL in the apoptotic response of WM793 cells to combined MEK inhibition and serum deprivation, we utilized a retrovirus vector expressing a short hairpin RNA (shRNA) designed to inhibit expression of all BIM isoforms. Two stable WM793 derived clones were derived (4.3 and 4.5) that displayed an c. 60–70% reduction of BIM-EL expression following MEK1/2 inhibition by CI-1040 (Figure 6A). A control clone (4.1) derived by infection with the empty retrovirus vector, displayed normal induction of BIM-EL in response to CI-1040 (Figure 6A). All cells were treated with CI-1040 in the absence of serum for either 24 or 48 h before apoptosis was assessed by staining cells with Annexin V and propidium iodide (Figure 6B). As expected, expression of the anti-BIM shRNA had no effect on basal levels of apoptosis in these cells. However, cells with reduced expression of BIM-EL displayed a partial protection against apoptosis following 48 h of MEK inhibition and serum deprivation. These data suggest that induced expression of BIM-EL serves as one mechanism of several by which RAF → MEK → ERK signaling regulates apoptosis in melanoma cells.
Figure 6.
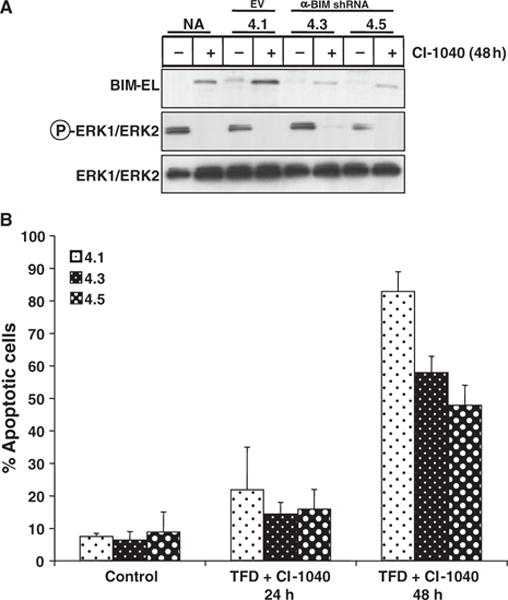
Inhibition of BIM-EL expression by RNA interference provides WM793 protection from apoptosis. (A) Parental WM793 cells (NA) or WM793 cells carrying an empty vector control (4.1), or two different WM793 clones expressing an shRNA designed to inhibit BIM expression (4.3 and 4.5) were treated with 2 μM CI-1040 for 24 h. The effects of CI-1040 and the anti-BIM shRNA on the phosphorylation of ERK1/2 or the expression of BIM-EL was assessed by Western blot analysis of cell extracts. (B) Cells described in (A) were treated with 2 μM CI-1040 in the absence of serum for 24 or 48 h as indicated. The presence of apoptotic cells was assessed by co-staining with Annexin V and propidium iodide followed by flow cytometry as described in Materials and Methods. Results shown are representative of three independent experiments.
Discussion
The striking prevalence of BRAF mutations in human melanoma suggests that BRAF → MEK → ERK signaling plays an essential role in the malignant transformation of melanocytes. Here we demonstrate that in non-transformed mouse and human melanocytes and in human melanoma cells, BIM-EL expression is under the concerted control of RAF → MEK → ERK signaling and the proteasome. In separate studies, we have also observed regulation of BIM mRNA levels through RAF → MEK → ERK signaling, consistent with previous observations (Figure S1). These data suggest that the regulation of BIM expression may be regulated through direct effects on both mRNA and protein abundance, as has observed previously in other cell types (Ley et al., 2005; O’Connor et al., 1998). Consistent with this, we detected rapid phosphorylation of endogenous BIM-EL in WM793 cells released from MEK inhibition using a new phosphospecific monoclonal antibody that detects phoshpho-serine 69 in human BIM-EL.
Induced expression of BIM-EL in WM793 human melanoma cells did not lead to a dramatic induction of apoptosis unless combined with serum deprivation. As serum deprivation had no effect on the overall level of induced BIM-EL expression in melanoma cells, the nature of the cooperation must be mediated through effects on other apoptotic regulators. Importantly however, both BCL-2 overexpression and RNAi-mediated inhibition of BIM-EL expression was sufficient to afford WM793 cells a measure of protection from apoptosis in response to MEK inhibition and serum deprivation. Interestingly, we observed that trophic factor deprivation suppressed the expression of the pro-survival BCL-2 and MCL-1 proteins when combined with MEK inhibition (Figure S2), suggesting that pathways parallel to RAF → MEK → ERK signaling cooperate in regulating key mediators of apoptosis.
Protection of WM793 melanoma cells from apoptosis through suppression of BIM-EL expression is consistent with reports from other experimental systems that utilize either epithelial or hematopoietic cells or indeed cancer cells derived from a variety of solid tumors (Collins et al., 2005; Marani et al., 2004; Ramjaun et al., 2007; Reginato et al., 2003). In addition, it is perhaps not surprising that inhibition of BIM expression offers only partial protection against apoptosis, as RAF → MEK → ERK signaling is reported to control apoptosis by multiple mechanisms. For example, RAF activation leads to release of autocrine EGF family growth factors that can suppress anoikis through activation of the EGF receptor (McCarthy et al., 1995; Schulze et al., 2001). Moreover, RAF activation can also promote expression of MDM2 that in turn inhibits p53-dependent apoptosis (Ries et al., 2000). It is also reported that ERK-mediated phosphorylation of caspase 9 inhibits its activation by APAF1, an observation that may explain observations that BRAF can inhibit apoptosis downstream of cytochrome c release (Allan et al., 2003; Erhardt et al., 1999). Finally, in melanoma cells, RAF → MEK → ERK signaling regulates the expression of β3-integrin, which in conjunction with its heterodimerization partner αV-integrin, is a known regulator of anoikis (Woods et al., 2001).
The high frequency of RAS and BRAF mutations in human cancer has led to considerable effort being expended to identify pharmacologic inhibitors of RAF → MEK → ERK signaling such as BAY43-9006 (Sorafenib), PD0325901 and AZD6244 (Thompson and Lyons, 2005). In melanoma cells, pharmacological or genetic inhibition of BRAF inhibits cell proliferation and survival of melanoma cells in vitro (Gray-Schopfer et al., 2007). In other studies, inhibition of MEK inhibits the proliferation of pancreatic cancer cells in vitro (Gysin et al., 2005). However, data presented here suggest that melanoma cells might be largely refractory to apoptosis induced by a therapeutic that targets BRAF or MEK. Consistent with this, little if any response was observed in melanoma patients treated with BAY43-9006 as a monotherapy (Gray-Schopfer et al., 2007). However, it is possible that inhibition of BRAF → MEK → ERK signaling might predispose melanoma cells to killing by conventional chemotherapeutic agents. Furthermore, in melanoma, the combination of both MEK inhibition with the addition of a BH3 mimetic compound has been shown more effective than MEK inhibition alone (Verhaegen et al., 2006), suggesting that multiple drugs targeting BCL-2 proteins may be used in successful therapy (Mohammad et al., 2008). Additional efficacy may be obtained by the use of high doses of agents such as PLX4720 that selectively target the activity of BRAFV600E as the side effects of such agents may be minimized by their apparent specificity for melanoma cells expressing mutated BRAF (Tsai et al., 2008).
Although these studies emphasize the potential importance of BIM downstream of BRAF → MEK → ERK signaling in the regulation of melanoma cell apoptosis, they do not preclude the possibility that other signaling pathways contribute to the dysregulation of apoptosis in melanoma cells. As mutation or silencing of PTEN is frequently observed in BRAFV600E expressing melanoma cells, it is possible that PI3′-lipid signaling contributes to the regulation of the aberrant physiology of the melanoma cell (Goel et al., 2006; Tsao et al., 2004). Indeed, the AKT target BAD has recently been implicated in the prevention of melanoma cell anoikis (Boisvert-Adamo and Aplin, 2008).
At the initiation of this research, the role of BRAF → MEK → ERK signaling in melanoma cell survival was unclear. We have demonstrated that BIM-EL is a relevant downstream target of this pathway, and that its suppression by oncogenic BRAF is one mechanism by which melanoma cells evade apoptosis. These findings have been supported by two recent studies. Inhibition of MEK has been shown to lead to the induction of PUMA, BIM, and MCL-1 in a panel of melanoma cells resulting in a modest increase in apoptosis (Wang et al., 2007). Consistent with this, our findings also demonstrated an increase in MCL-1 expression following MEK inhibition (Figure S2). Interestingly, the induction of PUMA following MEK inhibition may explain why RNAi-mediated suppression of BIM-EL expression gave melanoma cells only partial protection from apoptosis. BIM and BAD have also been implicated in the protection of melanoma cells from anoikis in the presence of oncogenic BRAF (Boisvert-Adamo and Aplin, 2008). In this study, both BIM and BAD were shown to be regulated at the transcriptional and post-transcriptional level downstream of BRAF → MEK → ERK signaling. Again, this study demonstrated that suppression of BIM expression alone was insufficient to protect melanoma cells from death, but that this protein played a role in melanoma cell survival. Taken together with our data, it is likely that BIM is a mediator of melanocyte and melanoma cell apoptosis in response to a range of apoptogenic insults. Moreover, even if BIM plays only a minor role in regulating apoptosis in melanoma cells, it may serve as a useful surrogate biomarker of the response of cancer cells to therapeutics targeted against the BRAF → MEK → ERK pathway, or even in evaluating the stage of disease progression for a patient (Dai et al., 2008).
Materials and methods
Cell culture and treatments
Melanoma cell lines were the kind gift of Dr. M. Herlyn (Wistar Institute, Philadelphia, PA, USA) and were cultured in DME-H16 media with 3 mg/ml glucose, and 0.584 mg/ml L-glutamine, 0.11 mg/ml Na pyruvate and 3.7 mg/ml NaHCO3, supplemented with 10% (v/v) fetal calf serum, 5 μg/ml of insulin, L-glutamine and penicillin (100 units/ml) and streptomycin (100 mg/ml). Trophic factor-deficient media consisted of DME-H16 supplemented only with L-glutamine, penicillin and streptomycin at the concentrations described above. Mouse Melan-a cells were obtained from Prof. D. Bennett (St. George’s Hospital, London, UK). Cells were cultured in RPMI 1640 media with fetal calf serum, penicillin, streptomycin, L-glutamine (as described above) and 200 nM TPA, tetradecanoyl phorbol-13-acetate (Sigma-Aldrich, St Louis, MO, USA). Trophic factor-deficient media consisted of RPMI 1640 media with penicillin, streptomycin and L-glutamine only. Human melanocytes expressing an anti-INK4A shRNA to promote their extended lifespan (Dr P. Gupta, Whitehead Institute, Cambridge, MA, USA) were cultured in Hams F-12 media supplemented with 15 μg/ml bovine pituitary extract, 5 μg/ml insulin, 0.5 μg/ml hydrocortisone, 0.5%(v/v) fetal calf serum, 200 ng/ml TPA, 5 ng/ml bFGF, penicillin, streptomycin and L-glutamine. Trophic factor-deficient media consisted of Hams F-12 media supplemented with penicillin, streptomycin and L-glutamine only.
Stock solutions of U0126 (Calbiochem, Gibbstown, NJ, USA), CI-1040 (Prof. Sir P. Cohen, University of Dundee), PLX4720 (Gideon Bollag, Plexxicon) and Z-VAD-FMK (BD BioSciences, San Jose, CA, USA) were prepared in DMSO. ICI 182,780 (Faslodex) (Dr Keith Wakeling, AstraZeneca, Alderley Edge, UK) was dissolved in ethanol. Velcade/PS341 (Dr Mark Rolfe, Millenium Pharmaceuticals Inc., Cambridge, MA, USA) was dissolved in water.
Retroviral infections and generation of stable cell lines
Retroviral vectors encoding a fusion protein consisting of full-length human BRAFV600E linked to the T1 form of the human estrogen receptor hormone-binding domain were generated by standard techniques (Liu et al., 2007; Morgenstern and Land, 1990a,b). Retrovirus stocks were generated by transient transfection of Phenix-E cells as described previously (Pear et al., 1993).
WM793 melanoma cells stably overexpressing human BCL-2 were generated by infection with an amphotropic retroviral stock obtained by transfection of pBabe-BCL-2-hygro into Phenix-A cells (Pear et al., 1993). The effect of BCL-2 expression on cell viability was assessed by Trypan blue staining.
RNA interference techniques to inhibit BIM expression
Human WM793 melanoma cells with reduced BIM expression were derived by infection with a pSuperRetro retrovirus (OligoEngine) expressing anti-BIM shRNA sequences complementary to human BIM. The shRNA sequence used to generate the hairpin was GATCCCCGACCACCCACGAATGGTTATTCAAGA-GATAACCATTCGTGGGTGGTCTTTTTGGAAA, with the regions targeted to BIM in bold. Puromycin-resistant clones were exposed to CI-1040 for 24 h before harvesting and analyzing cells lysates by Western blotting. Cell death was assessed by co-staining with Annexin V and propidium iodide as described below.
Cell fractionation
Sub-confluent WM793 cells were treated with CI-1040 for 48 h in the presence or absence of serum. 5 × 107 adherent or non-adherent cells were harvested, washed in phosphate-buffered saline and collected. Mitochondrial and cytosolic fractions were prepared using a mitochondrial/cytosolic fractionation kit (Alexis Biochemicals, San Diego, CA, USA) according to the manufacturer’s instructions.
Immunoblot analysis
Whole cell lysates were prepared using radio-immunoprecipitation assay (RIPA) lysis buffer as described previously (Samuels et al., 1993). Protein concentrations were estimated using Coomassie protein concentration reagent (BioRad, Hercules, CA, USA) according to the manufacturer’s instructions, and equal amounts of protein were analyzed by SDS-polyacrylamide gels electrophoresis (PAGE), followed by transfer to polyvinylidene difluoride membranes (PVDF; Millipore, Billeria, MA, USA), and Western blot analysis using standard techniques. Primary antibodies were obtained from the following sources: α-phosphorylated pT202/pY204 ERK1/2 and total ERK1/2 (Cell Signaling Technology, Davis, MA, USA), α-caspase 3, α-BIM (Calbiochem), α-BCL-2 (Santa Cruz Antibodies, Santa Cruz, CA, USA), α-cytochrome C, α-cytochrome C Oxidase IV (Invitrogen, Carlsbad, CA, USA), α-Smac/DIABLO (BD Pharmingen), α-MCL-1 (Invitrogen) and α-β-tubulin (Sigma). The phospho-serine 69 specific anti-BIM-EL monoclonal antibody was generated in collaboration with BD BioSciences and details of its characterization will be published elsewhere (Cagnol, S. and McMahon, M., unpublished data).
Assay for apoptosis
Apoptotic cells were quantitatively identified using Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, Pharmingen) according to the manufacturer’s instructions. Harvested cells were washed twice in cold PBS then suspended in binding buffer at a concentration of 106 cells/ml. 105 cells were stained in 100 μl with FITC-conjugated Annexin V and propidium iodide for 15 min at room temperature in the dark. Cells were analyzed within an hour using a Becton Dickinson FACscan.
Supplementary Material
Fig. S1 Regulation of BIM expression though mRNA abundance in human melanoma cells. Total RNA was extracted from the WM35 (RGP), WM793 (VGP) and WM9 (Met) melanoma cell lines following treatment with 2 μM CI-1040 (in DMSO) for 24 h. mRNA expression was assessed by real-time PCR as described in Materials and Methods. Expression data are shown for the genes BIM and MCL-1. Graphs indicate the abundance of mRNA levels relative to those of cells treated with DMSO alone (a value arbitrarily set to 1) for duplicate samples expressed as mean ± SEM.
Fig. S2 Regulation of BCL-2 family protein expression by trophic factor deprivation or MEK inhibition in WM793 cells. Asynchronously growing WM793 cells were treated with either 2 μM CI-1040 alone, subjected to TFD, or both 2 μM CI-1040 and TFD combined for 24 or 48 h as indicated. Cells were lysed and lysates analysed by immunoblotting with antibodies which recognize BIM, MCL-1, BCL-2 and ERK1/ERK2 total proteins or phosphorylated ERK1/ERK2.
Acknowledgments
We wish to thank several individuals for the provision of key materials and reagents used in these experiments: Alan Wakeling and Gary Nunn (Astra Zeneca) for ICI 182,780; Mark Rolfe (Millenium Pharmaceuticals) for Velcade; Sir Philip Cohen and Robert Macintosh (University of Dundee) for CI-1040, Gideon Bollag (Plexxicon) for PLX4720; Dorothy Bennett (St George’s Hospital Medical School) for Melan-a cells; Meenhard Herlyn (Wistar Institute) for human melanoma cells and for ongoing advice, support and guidance; Wendy Fantl (Chiron Corporation) for full length human BRAF cDNAs and Efthalia Chronopoulous and Roberto Campos (BD BioSciences) for the phospho-serine 69 specific BIM-EL mAb. We thank all the members of the McMahon Laboratory, Gerard Evan, David Stokoe (Genentech Inc.), Simon Cook (Babraham Institute), Joan Brugge & Tobias Schmelzle (Harvard Medical School) and Maria Soengas (CNIO, Madrid) for advice, suggestions and support. This research was supported by grants to M.M. from the NIH (CA108972), the Stewart Foundation, the Charlotte Geyer Foundation and the Cancer Research Coordinating Committee of the University of California. G.R.T. was supported by a UNCF/Merck post-doctoral fellowship and S.A.M. was supported by a post-doctoral fellowship from The Sass Foundation for Medical Research, New York.
Footnotes
Supporting information
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol. 2003;5:647–654. doi: 10.1038/ncb1005. [DOI] [PubMed] [Google Scholar]
- Baccarini M. An old kinase on a new path: Raf and apoptosis. Cell Death Differ. 2002;9:783–785. doi: 10.1038/sj.cdd.4401070. [DOI] [PubMed] [Google Scholar]
- Boisvert-Adamo K, Aplin AE. Mutant B-RAF mediates resistance to anoikis via Bad and Bim. Oncogene. 2008;27:3301–3312. doi: 10.1038/sj.onc.1211003. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Cory S, Zhang LC, Strasser A, Adams JM. Degenerative disorders caused by Bcl-2 deficiency prevented by loss of its BH3-only antagonist Bim. Dev Cell. 2001;1:645–653. doi: 10.1016/s1534-5807(01)00083-1. [DOI] [PubMed] [Google Scholar]
- Cai B, Chang SH, Becker EB, Bonni A, Xai Z. p38 MAP kinase mediates apoptosis through phosphorylation of BIM-EL at Ser65. J Biol Chem. 2006;281:25215–25222. doi: 10.1074/jbc.M512627200. [DOI] [PubMed] [Google Scholar]
- Chin L, Merlino G, DePinho RA. Malignant melanoma: modern black plague and genetic black box. Genes Dev. 1998;12:3467–3481. doi: 10.1101/gad.12.22.3467. [DOI] [PubMed] [Google Scholar]
- Christensen C, Guldberg P. Growth factors rescue cutaneous melanoma cells from apoptosis induced by knockdown of mutated (V 600 E) B-RAF. Oncogene. 2005;24:6292–6302. doi: 10.1038/sj.onc.1208758. [DOI] [PubMed] [Google Scholar]
- Collins NL, Reginato MJ, Paulus JK, Sgroi DC, Labaer J, Brugge JS. G1/S cell cycle arrest provides anoikis resistance through ERK-mediated Bim suppression. Mol Cell Biol. 2005;25:5282–5291. doi: 10.1128/MCB.25.12.5282-5291.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DL, Wang Y, Liu M, Martinka M, Li G. Bim expression is reduced in human cutaneous melanomas. J Invest Dermatol. 2008;128:403–407. doi: 10.1038/sj.jid.5700989. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Day CL, Puthalakath H, Skea G, Strasser A, Barsukov I, Lian LY, Huang DC, Hinds MG. Localization of dynein light chains 1 and 2 and their pro-apoptotic ligands. Biochem J. 2004;377:597–605. doi: 10.1042/BJ20031251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt P, Schremser EJ, Cooper GM. B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol Cell Biol. 1999;19:5308–5315. doi: 10.1128/mcb.19.8.5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewings KE, Wiggins CM, Cook SJ. Bim and the pro-survival Bcl-2 proteins; opposites attract, ERK repels. Cell Cycle. 2007;6:2236–2240. doi: 10.4161/cc.6.18.4728. [DOI] [PubMed] [Google Scholar]
- Goel VK, Lazar AJ, Warneke CL, Redston MS, Haluska FG. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol. 2006;126:154–160. doi: 10.1038/sj.jid.5700026. [DOI] [PubMed] [Google Scholar]
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–857. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- Gysin S, Lee SH, Dean NM, McMahon M. Pharmacologic inhibition of RAF → MEK → ERK signaling elicits pancreatic cancer cell cycle arrest through induced expression of p27Kip1. Cancer Res. 2005;65:4870–4880. doi: 10.1158/0008-5472.CAN-04-2848. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003;63:5198–5202. [PubMed] [Google Scholar]
- Ley RK, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein. Bim J Biol Chem. 2003;278:18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- Ley R, Ewings KE, Hadfield K, Cook SJ. Regulatory phosphorylaiton of Bim: sorting out the ERK from the JNK. Cell Death Diff. 2005;12:1008–1014. doi: 10.1038/sj.cdd.4401688. [DOI] [PubMed] [Google Scholar]
- Liu J, Suresh Kumar KG, Yu D, Molton SA, McMahon M, Herlyn M, Thomas-Tikhonenko A, Fuchs SY. Oncogenic BRAF regulates beta-Trcp expression and NF-kappaB activity in human melanoma cells. Oncogene. 2007;26:1954–1958. doi: 10.1038/sj.onc.1209994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marani M, Hancock D, Lopes R, Tenev T, Downward J, Lemoine NR. Role of Bim in the survival pathway induced by Raf in epithelial cells. Oncogene. 2004;23:2431–2441. doi: 10.1038/sj.onc.1207364. [DOI] [PubMed] [Google Scholar]
- McCarthy SA, Samuels ML, Pritchard CA, Abraham JA, McMahon M. Rapid induction of heparin-binding epidermal growth factor/diphtheria toxin receptor expression by Raf and Ras oncogenes. Genes Dev. 1995;9:1953–1964. doi: 10.1101/gad.9.16.1953. [DOI] [PubMed] [Google Scholar]
- Mohammad R, Giri A, Goustin AS. Small-molecule inhibitors of Bcl-2 family proteins as therapeutic agents in cancer. Recent Patents Anticancer Drug Discov. 2008;3:20–30. doi: 10.2174/157489208783478676. [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990a;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern JP, Land H. A series of mammalian expression vectors and characterisation of their expression of a reporter gene in stably and transiently transfected cells. Nucleic Acids Res. 1990b;18:1068. doi: 10.1093/nar/18.4.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton EE, Widlund HR, Kutok JL, et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr Biol. 2005;15:249–254. doi: 10.1016/j.cub.2005.01.031. [DOI] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- Ramjaun AR, Tomlinson S, Eddaoudi A, Downward J. Upregulation of two BH3-only proteins, Bmf and Bim, during TGFbeta-induced apoptosis. Oncogene. 2007;26:970–981. doi: 10.1038/sj.onc.1209852. [DOI] [PubMed] [Google Scholar]
- Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, Muthuswamy SK, Brugge JS. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat Cell Biol. 2003;5:733–740. doi: 10.1038/ncb1026. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Mitsiades C, Hideshima T, Anderson KC. Bortezomib: proteasome inhibition as an effective anticancer therapy. Annu Rev Med. 2006;57:33–47. doi: 10.1146/annurev.med.57.042905.122625. [DOI] [PubMed] [Google Scholar]
- Ries S, Biederer C, Woods D, Shifman O, Shirasawa S, Sasazuki T, McMahon M, Oren M, McCormick F. Opposing effects of Ras on p53: transcriptional activation of mdm2 and induction of p19ARF. Cell. 2000;103:321–330. doi: 10.1016/s0092-8674(00)00123-9. [DOI] [PubMed] [Google Scholar]
- Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P. Toxic proteins released from mitochondria in cell death. Oncogene. 2004;23:2861–2874. doi: 10.1038/sj.onc.1207523. [DOI] [PubMed] [Google Scholar]
- Samuels ML, Weber MJ, Bishop JM, McMahon M. Conditional transformation of cells and rapid activation of the mitogen-activated protein kinase cascade by an estradiol-dependent human raf-1 protein kinase. Mol Cell Biol. 1993;13:6241–6252. doi: 10.1128/mcb.13.10.6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze AK, Lehmann K, Jefferies HB, McMahon M, Downward J. Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev. 2001;15:981–994. doi: 10.1101/gad.191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebolt-Leopold JS, Dudley DT, Herrera R, et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res. 2005;65:2412–2421. doi: 10.1158/0008-5472.CAN-04-2423. [DOI] [PubMed] [Google Scholar]
- Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene. 2003;22:3138–3151. doi: 10.1038/sj.onc.1206454. [DOI] [PubMed] [Google Scholar]
- Thompson N, Lyons J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr Opin Pharmacol. 2005;5:350–356. doi: 10.1016/j.coph.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004;122:337–341. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- Verhaegen M, Bauer JA, Martin de la Vega C, et al. A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res. 2006;66:11348–11359. doi: 10.1158/0008-5472.CAN-06-1748. [DOI] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- Wang YF, Jiang CC, Kieja KA, Gillespie S, Zhang XD, Hersey P. Apoptosis induction in human melanoma cells by inhibition of MEK is caspase-independent and mediated by the Bcl-2 family members PUMA, Bim and Mcl-1. Clin Cancer Res. 2007;13:4934–4942. doi: 10.1158/1078-0432.CCR-07-0665. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Ogilvie L, Hedley D, Karasarides M, Martin J, Niculescu-Duvaz D, Springer CJ, Marais R. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004;64:2338–2342. doi: 10.1158/0008-5472.can-03-3433. [DOI] [PubMed] [Google Scholar]
- Woods D, Cherwinski H, Venetsanakos E, Bhat A, Gysin S, Humbert M, Bray PF, Saylor VL, McMahon M. Induction of beta3-integrin gene expression by sustained activation of the Ras-regulated Raf-MEK-extracellular signal-regulated kinase signaling pathway. Mol Cell Biol. 2001;21:3192–3205. doi: 10.1128/MCB.21.9.3192-3205.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Regulation of BIM expression though mRNA abundance in human melanoma cells. Total RNA was extracted from the WM35 (RGP), WM793 (VGP) and WM9 (Met) melanoma cell lines following treatment with 2 μM CI-1040 (in DMSO) for 24 h. mRNA expression was assessed by real-time PCR as described in Materials and Methods. Expression data are shown for the genes BIM and MCL-1. Graphs indicate the abundance of mRNA levels relative to those of cells treated with DMSO alone (a value arbitrarily set to 1) for duplicate samples expressed as mean ± SEM.
Fig. S2 Regulation of BCL-2 family protein expression by trophic factor deprivation or MEK inhibition in WM793 cells. Asynchronously growing WM793 cells were treated with either 2 μM CI-1040 alone, subjected to TFD, or both 2 μM CI-1040 and TFD combined for 24 or 48 h as indicated. Cells were lysed and lysates analysed by immunoblotting with antibodies which recognize BIM, MCL-1, BCL-2 and ERK1/ERK2 total proteins or phosphorylated ERK1/ERK2.