

Abstract
Transient receptor potential vanilloid 1 (TRPV1) is a pronociceptive cation channel involved in persistent inflammatory and neuropathic pain. Herpes simplex virus (HSV) vector expression of TRPV1 causes cell death in the presence of capsaicin, thereby completely blocking virus replication. Here we describe a selection system for negative regulators of TRPV1 based on rescue of virus replication. HSV-based coexpression of TRPV1 and a PC12 cell-derived cDNA library identified protein phosphatase 1α (PP1α) as a negative regulator of TRPV1, mimicking the activity of “poreless” (PL), a dominant-negative mutant of TRPV1. Vectors expressing PP1α or PL reduced thermal sensitivity following virus injection into rat footpads, but failed to reduce the nocifensive responses to menthol/icilin-activated cold pain or formalin, demonstrating that the activity identified in vitro is functional in vivo with a degree of specificity. This system should prove powerful for identifying other cellular factors that can inhibit ion channel activity.
Introduction
Transient receptor potential vanilloid 1 (TRPV1) is a member of the transient receptor potential cation channel family and is activated by capsaicin, protons, and temperatures above 43°C.1,2 TRPV1 expression is upregulated during neuropathic pain, and its inhibition results in pain reduction, suggesting that it is an effective target for pain relief.3–5 Many cellular factors contribute to TRPV1 regulation. Signaling molecules such as nerve growth factor (NGF) and bradykinin initiate second messenger cascades resulting in phosphorylation and sensitization of TRPV1 (ref. 6), while calcineurin (PP3), a Ca2+-calmodulin-dependent serine/threonine protein phosphatase, causes TRPV1 desensitization.7,8 We hypothesized that there may be other proteins that can negatively affect TRPV1 activity and may thereby be applicable in gene therapy for chronic pain.
We devised an herpes simplex virus (HSV)-based system to screen a cDNA library for cellular factors that inhibit TRPV1 activity. Our library screen was based on the observations that (i) overexpression of TRPV1 from an HSV vector in the presence of capsaicin caused cell death and the absence of virus plaques and (ii) virus replication could be restored with TRPV1 antagonists or by coinfection with vectors expressing dominant-negative PL.9 This suggested that rescue of plaque formation could be used to select negative TRPV1-regulatory genes from a cellular cDNA library coexpressed with TRPV1. Using this system, we identified one candidate that ameliorated TRPV1-related nocifensive responses in animal models of pain.
Results
HSV vector development for library screening
To develop the HSV-based coexpression vector, we introduced a TRPV1 cDNA (VR1) in place of the viral thymidine kinase (tk, UL23) coding sequence in a bacterial artificial chromosome (BAC)-based HSV-1 genome (Figure 1a, top) and substituted the internal repeat (IRL-IRS; joint) region with a Gateway (GW) recombination cassette flanked by the cytomegalovirus immediate-early promoter (CMV) and bovine growth hormone (bGH) polyA region as the site for introduction of a cDNA library (T-GW). The promoters driving expression of TRPV1 (tk promoter) and the library cDNAs (CMV) were chosen to initiate transcription of library cDNAs prior to that of TRPV1. To validate this strategy, we substituted the GW core with a green fluorescent protein (GFP) cDNA. The resultant construct (T-GFP) was transfected into Vero cells and infectious virus (vT-GFP) was harvested, expanded, and expression of TRPV1 and GFP was assessed by Western blot of infected cell lysates. GFP protein was detectable as early as 3-hour post infection (hpi), while TRPV1 protein was not observed until 7 hpi (Figure 1b).
Figure 1.
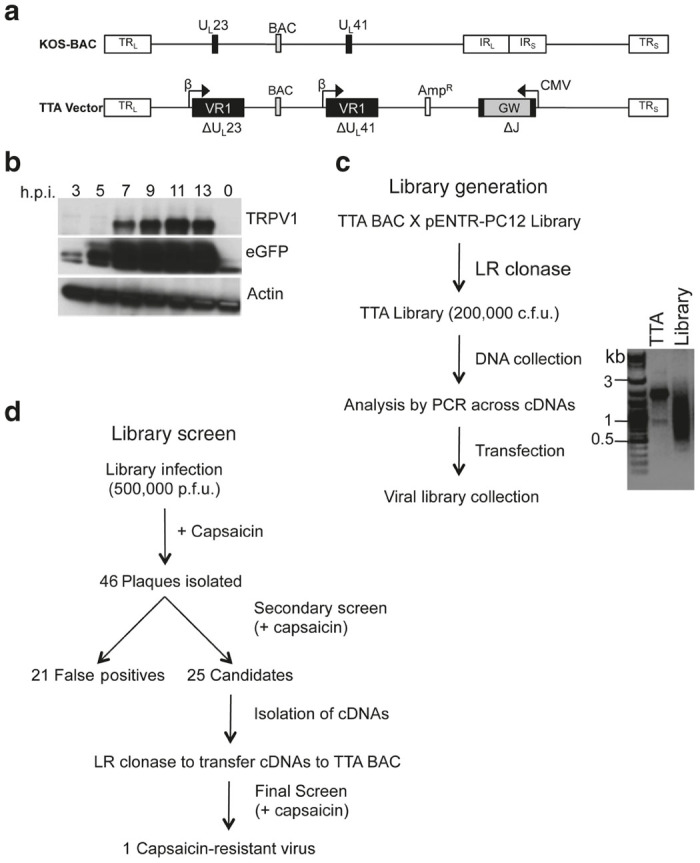
Transient receptor potential vanilloid 1 (TRPV1) vector and antagonist screen designs. (a) KOS-37 bacterial artificial chromosome (BAC) genome representation (top) with the locations of relevant genomic regions indicated. TRL, TRS, terminal repeats of the unique long (UL) and unique short (US) segment, respectively; IRL, IRS, internal repeats; BAC, loxP-flanked BAC sequences; UL23, tk gene; UL41, vhs gene. The TTA BAC genome represented underneath contained a CMV promoter–driven Gateway (GW) cassette replacing IRL and IRS (∆J); TRPV1 (VR1) cDNAs expressed from the early (β) tk promoter in place of (∆) UL23 and UL41; and an ampicillin resistance gene (AmpR) between UL55 and UL56. (b) Western blots of infected cell lysates. Vero cells were infected with the vT-GFP vector (MOI = 3) and processed at the indicated time points for immunoblotting with antibodies to TRPV1, GFP, or β-actin. (c) Generation of TTA BAC and viral libraries. The range of cDNA sizes recombined into TTA BAC was estimated by polymerase chain reaction with flanking primers and gel electrophoresis of the products (Library). TTA BAC DNA was used as control (TTA). (d) Summary of the library screen.
The ability of capsaicin to inhibit virus growth was assessed by infecting Vero cells with virus from T-GW-transfected cells (vT-GW). Plaquing was reduced but not eliminated in the presence of capsaicin (1 plaque/100 input pfu). Sequence analysis demonstrated that these plaques were the result of mutations in the TRPV1 coding sequence. Thus, we replaced the UL41 (host shut-off) gene with a second tk promoter-TRPV1 expression cassette to reduce this background. However, selection of transformed bacteria for chloramphenicol resistance, specified in the BAC region between UL37 and UL38, yielded predominantly defective genomes lacking UL42-UL22 due to recombination between the TRPV1 cDNAs at UL23 and UL41. To allow for selection of both regions of the circular BAC genome (UL24-UL40 and UL42-UL22), we inserted an ampicillin resistance gene between UL55 and UL56 (TTA construct, Figure 1a). Bacterial coselection with chloramphenicol and ampicillin eliminated the amplification of defective recombinants. vTTA virus produced 1 plaque per 10,000 input pfu on Vero cells in the presence of capsaicin, a more acceptable background level for the cDNA library screen.
A library PP1α cDNA rescues virus growth in the presence of capsaicin
We recombined a cDNA library derived from capsaicin-responsive PC12 cells10–12 into the GW locus of the TTA BAC and converted bulk recombinants into infectious virus, as outlined in Figure 1c. BAC cDNA insert sizes ranged from roughly 400 bp to 3 kb (Figure 1c). Approximately 500,000 pfu of virus were screened in the presence of 1-µM capsaicin (Figure 1d). We isolated 46 candidate plaques, amplified the resultant viruses on Vero cells, and retested each for capsaicin-resistant plaque formation; approximately 50% of the isolated plaques retained a capsaicin-resistant phenotype. Insert cDNAs were polymerase chain reaction (PCR) amplified, introduced into a fresh TTA BAC backbone, and recombinant viruses were retested for capsaicin resistance. One candidate consistently demonstrated the ability to rescue virus replication in the presence of capsaicin. Sequencing identified the insert as full-length protein phosphatase 1α (PP1α) cDNA.
We compared the growth of vTTA-PP1α on Vero cells at 48 hpi (multiplicity of infection (MOI) = 0.01) with that of a negative control vector (vTTA) and a positive control vector (vTTA-PL) containing “poreless” (PL), a dominant negative mutant of TRPV1.13 vTTA produced no detectable infectious progeny in the presence of capsaicin, while vTTA-PP1α replicated with an efficiency similar to vTTA-PL, both yielding approximately 1 × 103 pfu/ml compared with 0.5 to 1 × 106 pfu/ml produced in the absence of capsaicin (Figure 2a). The yields in the absence of capsaicin were comparable between the three viruses, indicating that PP1α did not universally improve virus yield.
Figure 2.

Herpes simplex virus vector-mediated protein phosphatase 1α (PP1α) or poreless (PL) expression in vitro and in vivo reduces transient receptor potential vanilloid 1 (TRPV1) channel activity. (a) Growth of vTTA, vTTA-PP1α, and vTTA-PL viruses on capsaicin-treated or untreated Vero cells. Cells were infected at an MOI of 0.01 and virus yields were determined at 48 hpi. Bars represent the mean ± SD of duplicate experiments; no virus was detected in TTA virus-infected, capsaicin-treated cells. (b) Effect of virus-mediated PP1α expression on calcium influx in response to capsaicin. Neurons isolated from DRGs of fetal rats were infected with DBAC-PP1α, DBAC-PL, or DBAC-GFP, and the increase in intracellular calcium (mean increase ± SD in the F340/F380 ratio) after capsaicin treatment is shown (*P < 0.001; Student’s t-test). (c–e) Male Sprague-Dawley rats were injected subcutaneously into the right hind paw with 1 × 108 pfu of DBAC-GFP, DBAC-PP1α, or DBAC-PL virus. (c) Thermal latency responses were measured using a Hargreaves apparatus and plotted for each group as the mean ± SD of the response times (in seconds) for uninjected left (white) and injected right (black) hind paws. (*P < 0.001, **P < 0.0001). (d–e) Thermal allodynia was assessed using a dynamic hot plate in the (d) absence or (e) presence of capsaicin, and the results were plotted as the mean ± SD of the number of responses by each animal per group. Student’s t-test was used to compare the control DBAC-GFP group with DBAC-PL and DBAC-PP1α (*P < 0.05 and **P < 0.01). BAC, bacterial artificial chromosome.
PP1α inhibits capsaicin-induced calcium influx in vitro
To express PP1α in the absence of viral replication, we chose an HSV BAC vector, DBAC-GW, which is deleted for the essential ICP4 IE gene and has the essential IE gene ICP27 replaced with a GW cassette between the CMV promoter and a polyA region.12 We recombined PP1α, PL, and GFP cDNAs with the GW cassette and produced infectious viruses in Vero cells engineered to complement the essential genes in trans.14 We compared capsaicin-induced calcium influx using Fura-2, a ratiometric Ca2+ indicator, in isolated rat fetal dorsal root ganglion (DRG) neurons exposed to the three vectors. Figure 2b shows the average increase in the Fura-2 fluorescence ratio (F340/F380) following capsaicin application. A depolarization (50 mM K+)-induced increase in fluorescence ratio was used as a positive control. We observed a significant decrease in the magnitude of the capsaicin-induced Ca2+ transient in neurons exposed to PP1α or PL expression vectors compared with the GFP vector (P < 0.01).
PP1α reduces thermal sensitivity in vivo
We next asked whether PP1α overexpression affects paw withdrawal latency (PWL) in vivo. We injected the DBAC-PP1α, -PL, and -GFP vectors into the right footpad of male Sprague-Dawley rats and measured PWL at 0 and 7 days post inoculation (dpi). At 0 dpi, the response was comparable in all animals (data not shown). At 7 dpi, no difference in PWL was observed between the infected and uninfected paws for animals injected with the DBAC-GFP vector (Figure 2c). In contrast, rats infected with the PP1α or PL vector showed increases of 1.7- or 1.8-fold, respectively, in the time to withdrawal, demonstrating that these animals experienced reduced basal thermal sensitivity. A qualitatively similar effect has been reported using the selective TRPV1 agonist resiniferatoxin to ablate TRPV1-containing C fibers,15 in studies with HSV vectors expressing PL16 or dominant-negative PKCϵ,17 and in TRPV1−/− mice18 or mice treated with the selective TRPV1 antagonist JNJ-17203212 (ref. 19). However, other studies have observed robust reductions in PWL typically only after sensitization by TRPV1 agonist or inflammatory insult,20–22 raising the possibility that the reduced thermal sensitivity we see is mediated by factors other than TRPV1.
To further examine the effect of PP1α on heat sensitivity, we observed vector-inoculated animals for alterations in thermal allodynia using a dynamic hot plate where temperature increased from 30 to 45°C over a 15-minute period. DBAC-GFP-inoculated animals showed significant increases in pain-related behaviors at temperatures of 42°C or higher (Figure 2d). In contrast, animals infected with DBAC-PL or DBAC-PP1α exhibited substantially reduced pain-related responses at all temperatures. Application of capsaicin to the plantar surface of the paw previously inoculated with DBAC-GFP caused thermal allodynia, characterized by both a lower temperature at which pain behavior was induced, 32°C, and an increase in the number of pain-related responses observed in the sensitive range (32°C–45°C) (Figure 2e). However, in animals infected with DBAC-PL or DBAC-PP1α, there was no capsaicin-induced increase in the number of pain-related responses observed at any temperature tested (Figure 2e). These data demonstrated the ability of exogenously expressed PP1α to reduce thermal sensitivity. The robust reduction in capsaicin-enhanced nociceptive responses suggests that TRPV1 channel activity was inhibited, while the reduction in responses seen in the absence of capsaicin may indicate an effect on other thermal TRPs.
PP1α does not alter cold allodynia- or formalin-induced nociceptive behavior
We next tested the effect of PP1α on cold allodynia. Noxious cold responses are reportedly transmitted by both TRPM8 and TRPA1.23,24 TRPM8 is also activated and sensitized by menthol and icilin. In the absence of agonists, rats were largely unresponsive to cold temperatures down to 4°C, regardless of the vector with which they had been inoculated (Figure 3a). However, treatment of injected animals with icilin (Figure 3b) or menthol (Figure 3c) prior to exposure to cold triggered pain-related behaviors at temperatures below 10°C. No significant difference was observed between the three different vector groups, suggesting that PP1α overexpression does not modulate TRPM8 activity. As seen with the TRPM8 agonists, no significant difference was observed between the three different vector groups when the TRPA1 agonists cinnamaldehyde (CA) and AITC were applied prior to exposure to cold, suggesting that PP1α overexpression does not modulate TRPA1 cold-related activity (data not shown).
Figure 3.

Herpes simplex virus vector-mediated PP1α or poreless (PL) expression did not significantly alter cold- or formalin-induced pain responses. Male Sprague-Dawley rats injected subcutaneously into the right rear hindpaw with DBAC-GFP, DBAC-PP1α, or DBAC-PL virus (n = 4/group). (a) Animals were placed on a dynamic cold plate where temperature decreased from 20 to 4°C over a 15-minute period. The total number of pain-related behaviors (licking, paw withdrawals) observed for the injected paw during each degree interval are plotted as the mean ± SD. (b) The injected paws were treated with icilin, and cold allodynia was assessed as in a. (c) The injected paws were treated with menthol, and responses were assessed as in a. (d) Quantitation of nocifensive behavior in the formalin footpad test. A weighted pain score was plotted against time and compared between control and treatment groups using one-way analysis of variance. No significant differences were noted between the PP1α, PL, and GFP vector-injected animals in any of the above assays. BAC, bacterial artificial chromosome.
In addition to its reported involvement in the sensation of noxious cold, TRPA1 has a clear role in sensation of noxious chemicals such as formalin.23,24 Thus, we tested the effect of our vectors on TRPA1 activity by observation of nociceptive behavior induced by intradermal formalin.25 We observed a characteristic response in control GFP vector-injected animals, i.e., an immediate pain response followed by a period of normal behavior, an increasing response and then a decreasing response over a period of approximately 60 minutes (Figure 3d).26 PP1α and PL vector-injected animals responded in a similar fashion, suggesting that neither vector influenced TRPA1 channel activity in response to noxious chemicals.
Discussion
Afferent nociceptors consist of heterogeneous functional types, responsive to different noxious stimuli. HSV-based gene therapy vectors that block the activation of individual nociceptors may be an attractive strategy for treating specific types of pain. Here we searched for genes that inhibit TRPV1 signaling and may function to reduce TRPV1-mediated pain. We developed a unique library screening methodology based on the differential replication of TRPV1-expressing viruses in the presence and absence of the TRPV1 agonist capsaicin. By introducing a library of cDNAs into the TRPV1 vector, we could rescue virus growth in the presence of capsaicin and identified the gene encoding PP1α as the responsible library component.
PP1α is a member of the serine/threonine phosphatase family known to regulate a wide variety of biological processes through the dephosphorylation of various substrate targets.27 This diverse portfolio of activities is regulated by numerous PP1α binding partners that affect PP1α subcellular localization, determine PP1α substrates, and ultimately control PP1α function.28 TRPV1 activity is regulated by many cellular signals, existing at a point of convergence of several second messenger cascades, with phosphorylation sensitizing and dephosphorylation desensitizing TRPV1 activation.1 While a key role for calcineurin (PP3/PP2B) in regulating the activity of TRPV1 through direct dephosphorylation of the channel itself has been clearly demonstrated,8,29 previous studies have not excluded PP1α as a regulator of TRPV1 activity.8,30 We show here that vector-expressed PP1α reduced capsaicin-induced calcium influx in DRG neurons in vitro. Moreover, vector-mediated PP1α expression reduced thermal sensitivity but not TRPM8 or TRPA1 agonist-enhanced sensitivity to cold, or formalin sensitivity mediated by TRPA1, consistent with a degree of specificity in vivo for channels underlying responses to heat, including TRPV1. There remains the possibility that HSV vector-mediated overexpression of PP1α may modify the heat-activated responses of other thermoTRPs,1,24 such as TRPM3, TRPM4, TRPM5, TRPV3, or TRPV4, that were not directly assessed. Desensitization of one or more of the thermoTRP channels, either as a result of dephosphorylation directly by PP1α or as part of a PP1α-regulated signaling cascade, may have contributed to the reduced thermal sensitivity we observed.
Application of PP1α expression to pain gene therapy could be adapted to minimize potential off-target effects on other TRP channels or target proteins by the use of transductional or transcriptional targeting to limit vector distribution or expression to only those cells expressing TRPV1. Moreover, PP1α could be used to treat pain arising from specific tissues or organs, such as the urinary bladder,31 where local application of the PP1α-expressing vector would further limit its action to the specific painful area and provide long-term therapeutic gene expression. This targeted approach to chronic pain therapy offers a benefit over many analgesic treatments that are administered systemically and can generate widespread side effects.
A variety of TRP channels, as well as ligand-gated and voltage-gated ion channels, are involved in either the detection of pain or propagation of the pain signal. Our ability to utilize TRPV1 as a target to identify a gene that inhibits pain-related thermal channels in vivo suggests that our library screening methodology should be generally useful to identify negative regulators of other channels whose activation causes cell death.32–34
Materials and Methods
Cell culture
U2OS and Vero cells (ATCC, Manassas, VA) and Vero-based ICP4/ICP27-complementing 7B cells14 were grown in 5% CO2 at 37°C in Dulbecco’s Modified Eagle’s medium (DMEM) (Lonza, Walkersville, MD) with 5–10% (v/v) fetal bovine serum (FBS; Sigma-Aldrich, Saint Louis, MO), 100 units/ml penicillin, and 100 μg/ml streptomycin (ThermoFisher Scientific, Pittsburgh, PA).
Library screen
All infections to test capsaicin resistance were performed in the presence of 1 μmol/l capsaicin (Sigma-Aldrich) diluted in Dulbecco’s Modified Eagle’s medium (DMEM) + 5% FBS. Media were changed daily to provide fresh capsaicin. The viral library was titered on Vero cells in the absence of capsaicin. Based on this titer, 500,000 pfu of virus were screened on Vero cells in the presence of capsaicin. Cells were infected for 2 hours at 0.01 pfu/cell and overlaid with fresh medium containing 1% (w/v) methylcellulose and capsaicin. 46 plaque-forming viruses were harvested by scraping and sonicating infected cells, amplified on Vero cells and retested by infecting duplicate wells of Vero cells +/− capsaicin. Plaques were visualized by brightfield microscopy and crystal violet staining. Viruses that formed plaques under both conditions were considered capsaicin resistant. DNA was isolated from those viruses and insert cDNAs were PCR amplified and introduced into a fresh TTA BAC backbone by LR clonase II reaction. Viruses produced by transfection of the resulting BAC DNAs were retested for capsaicin resistance.
cDNA amplification from viral DNA
Viral DNA was isolated from plaques growing on Vero cells in 24-well plates by proteinase K digestion (100 mmol/l Tris-Cl pH8, 10-mmol/l ethylenediamine tetraacetic acid (EDTA), 100 mmol/l NaCl, 10 μg/ml proteinase K, 0.5% SDS), phenol:chloroform extraction, and ethanol precipitation. DNA was resuspended in 25-μl TE buffer, and PCR was performed in two rounds with nested primers in the CMV promoter and bGH polyA (round 1: CMV-F1 and bGH-R2 and round 2: CMV-F3 and bGH-R1; Table 1). Phusion DNA polymerase (New England Biolabs, Ipswich, MA) was used in reactions with 2-μl template DNA, 0.4 μmol/l of each primer, 0.2 mmol/l of each dNTP in 1× GC buffer (New England Biolabs) at the following cycler settings: 98°C for 30 seconds; 30 cycles of 98°C for 10 seconds, 68°C for 10 seconds, 72°C for 1 minute; and 72°C for 10 minute. 1 μl of the 50-μl first-round PCR reaction was used for the second round, with the same PCR conditions. PCR products were resolved on a 1% agarose gel, visualized by ethidium bromide staining, and isolated with the Qiaquick gel extraction kit.
Table 1. PCR primers.
| Name | Sequence |
|---|---|
| 3′Joint/CMV | CCGCCCCCCGCCGCCCGGGC CCGCCCCCGGGGCCGGCG CGGAGTCGGGCACGATG TACGGGCCAGATATACGCG |
| 5′Joint/bGHpA | GCCTTTTATAACCCCG GGGGTCATTCCCAACGATCACA TGCAATCTAACTGGCTAT CCCCAGCATGCCTGCTATTGTC |
| CS_UL23F | GTGGCGTGAAACTCCC GCACTCTTCG GCCAGCGCCTGTAGAAGC GCGTATAGGGCCTGGTGATCAT GGCGGGATCG |
| VR1_UL23R | TTATTCGGCT CATCGCGCGGGT TCCTTCCGGTA TTGTCTCCTT CCGTGTTTCAGAA GAACTCGTCAA GAAGGCG |
| VR1_UL23F | GTGGCGTGAAA CTCCCGCACTCTTCGGC CAGCGCCTGTAGA AGCGCGTATGGAACA ACGGGCTAGCTTAGAC |
| VR1_UL23R | TTATTCGGCTC ATCGCGCGGGTTCCT TCCGGTATTGTCT CCTTCCGTCTTT TATTTCTCCCCT GGGACCATGG |
| CS_UL41F | GCAGGGTGCTGC ACCAATCCGCGTGGAG TTGGGCCATCGA AATTATAAAGAG GGCCTGGTGATCA TGGCGGGATCG |
| CS_UL41R | ATTGGTGGGTCGTT TGTTCGGGGACAAGCGCGCTC GTCTGACGTTTG GGCTCA GAAGAACTC GTCAAGAAGGCG |
| VR1_UL41F | GCAGGGTGCTGCACCA ATCCGCGTGGAGTTGGG CCATCGAAATTATAAAGAG AGTTCTATGATGACACA AACCCCG |
| VR1_UL41R | ATTGGTGGGT CGTTTCTTCGGGGACAA GCGCGCTCGTCTGA CGTTTGGGCTTATT TCTCCCCT GGGACCATGG |
| UL55/56_AmpF | TAAAGAGC CGTAACCCAACCAA ACCAGGCGTGGTGT GAGTTTGTGGACC CTGGAACGAAAA CTCACGTTAAGG |
| UL55/56_AmpR | TAAAAGTATCACG GTCCATACTGGCC TGTCGCGTTGTCT CTGAGGGCTTTGAC GTCAGGTGGCAC TTTTCGGG |
| CMV-F1 | CGCAAATGGGCGGTAGGCGTG |
| CMV-F3 | GAGAACCCACTGCTTACTGGCTTA |
| bGH-R2 | GACACCTACTCAGACAATGCGATGC |
| bGH-R1 | TAGAAGGCACAGTCGAGG |
Italicized sequences indicate homology arms.
Western blotting
Vero cells were infected with vT-GFP vector (MOI = 3), and infected cell lysates were prepared by incubating the cells in 1× RIPA lysis buffer (Millipore, Billerica, MA) with protease inhibitors (cOmplete, Mini; Roche, Indianapolis, IN) for 30 minutes on ice. Lysates were boiled for 5 minutes and resolved on a sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel at 100 V. Proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore). The membrane was blocked with 5% nonfat dry milk/TBS-T and probed overnight at 4°C with 1:500 diluted primary antibody followed by incubation for 1 hour at room temperature (RT) with HRP-conjugated secondary antibodies (Sigma-Aldrich) diluted to 1:50,000 in 5% nonfat dry milk/TBS-T. After washing with 1× TBS-T, proteins were visualized with ECL reagents (ThermoFisher).
Calcium imaging
Dissociated fetal rat primary DRG neurons were plated on poly-d-lysine-coated coverslips in 24-well dishes.35 Cover slips with populations of DRG cells were exposed to DBAC-PP1α, DBAC-PL, or DBAC-GFP (4 × 104 pfu/coverslip; three coverslips per virus), and cells were studied at 3 hpi. GFP expression on control coverslips indicated high efficiency of infection and expression from the CMV promoter at this time point (data not shown). DRG cells were loaded with Fura-2AM (2 μmol/l; Molecular Probes, Grand Island, NY) for 30 minutes at 37°C. Cover slips were then placed on an epifluorescence microscope and continuously superfused (3–4 ml/minute) with hanks’ balanced salt solution. Fura-2 was excited alternately with ultraviolet light at 340 and 380 nm, and the fluorescence emission was detected at 510 nm using a computer-controlled monochromator. Image pairs were acquired every 1–30 seconds using illumination periods between 20 and 50 ms. Wavelength selection, timing of excitation, and the acquisition of images were controlled using the program C-Imaging (Compix, Cranberry Township, PA) running on a personal computer. For each coverslip, baseline ratios were measured for 2 minutes (values between 0.2 and 0.3). Cells were then stimulated with 0.5-μmol/l capsaicin for 10 seconds, and after return to baseline, these were treated with 80-mmol/l KCl for 10 seconds. Only neurons showing both a KCl and a capsaicin response were chosen for analysis. For each neuron, the change in intracellular calcium after capsaicin treatment was calculated as the peak response minus the average baseline value. The results were plotted as the mean value (±SD) for all KCl responsive neurons (PP1α, n = 29; PL, n = 19; GFP, n = 19).
Animal studies
All housing and experimental manipulations of animals were performed in BSL-2 facilities approved by the Association for the Accreditation of Laboratory Animal Care (AALAC). All protocols were in agreement with procedures approved by the University of Pittsburgh Institutional Animal Care and Use Committee (IACUC) in accordance with the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain (IASP).
Virus injections.
Male Sprague-Dawley rats (200–250 g) (Jackson Labs, Bar Harbor, ME) were injected subcutaneously into the right hindpaw with 1 × 108 pfu of DBAC-GFP (n = 4), DBAC-PP1α (n = 4), or DBAC-PL (n = 4). The animals were subjected to the following series of behavioral tests, and all responses were recorded by a blinded observer.
Thermal latency response.
At 7 dpi, each rat was placed in a plexiglass enclosure on a glass surface maintained at 30°C (Hargreaves apparatus, IITC, Woodland Hills, CA). After a 15-minute accommodation period, a light beam was focused on the midplantar area of each hindpaw, and the amount of time to paw withdrawal from the heat source was measured. Three determinations per hindpaw were used for each animal to determine its thermal latency response. Light beam intensity was chosen such that the average paw withdrawal times for the contralateral paws and the DBAC-GFP-injected ipsilateral paws ranged from 7 to 10 seconds.
Capsaicin-induced thermal allodynia.
At 14 dpi, animals were placed on a dynamic hot plate (IITC) where temperature increased from 30°C to 45°C over a 15-minute period. During each degree interval, the total number of pain-related behaviors (licking, paw withdrawal) was counted for the injected paw and the average number of nocifensive responses per animal was plotted over the temperature range (mean ± SD). At 15 dpi, 100 μl of capsaicin (10 mmol/l in 50% ethanol) was applied to the plantar surface of the right hindpaw and after 15 minutes, pain behaviors were scored on the heat ramp.
Cold-induced pain.
At 16 dpi, animals were placed on a dynamic cold plate where temperature decreased from 20°C to 4°C over a 15-minute period. During each degree interval, the total number of pain-related behaviors (licking, paw withdrawal) for the injected paw was counted. At 14 dpi, the virus-injected hindpaw was injected subcutaneously with 50 µl of 10-mmol/l AITC (Sigma) or 50 µl of 20% trans-cinnamaldehyde (CA; Sigma).36 At 15-minute post injection, behavior was observed on the cold ramp. At 17 dpi, the injected hindpaw was treated with 50 µl of 100-µmol/l icilin (Tocris, Avonmouth, Bristol, UK) and behavior was again observed on a cold ramp. At 18 dpi, the injected hindpaw was treated with 50 µl of 100-µmol/l menthol (Sigma-Aldrich) and behavior was observed 15 minutes later on a cold ramp. For each test, the average number of responses per animal (mean ± SD) was plotted over the temperature range.
Formalin footpad test.
At 18 dpi, a dilute solution of formalin (2.5%) was injected subcutaneously in the plantar aspect of the virus-injected paw, and the rats were placed in a 48 × 27 × 20 cm plastic box positioned over a mirror tilted at a 45° angle. Beginning 30 seconds after the injection of formalin, and once every 10 minutes thereafter, nocifensive behaviors were recorded for 3 minutes. A weighted pain score was derived based on the number of times the animal exhibited a specific behavior during the 3-minute observation period (0 = plantar surface of paw flat against surface; 1 = paw cupped with only toes touching surface; 2 = paw lifting from surface; 3 = complete paw withdrawal with licking). The weighted pain score was plotted against time and compared between control and treatment groups using one-way analysis of variance.
GW recombination
Reactions were performed using LR clonase II (ThermoFisher Scientific). Individual cDNAs were cloned between the attL recombination sites of plasmid pENTR1A (ThermoFisher Scientific). BAC DNAs containing an attR-flanked GW cassette were maintained in HerpesHogs (HH8) bacteria.12 Reactions were performed with 200 ng of BAC DNA, 100 ng of pENTR1A plasmid DNA, and 2 μl of enzyme for 1 hour at RT and were inactivated with Proteinase K. DNA was spotted on 0.025-μm MF-Membrane Filters (EMD Millipore, Darmstadt, Germany), dialyzed for 1 hour against 0.5× TE, and introduced into electrocompetent DH10B-T1 Phage Resistant Escherichia coli (ThermoFisher Scientific) by electroporation (1650 V, 25 μF, 150 Ω). Colonies were plated on LB agar plus chloramphenicol (15 μg/ml).
BAC constructs
DBAC vectors.
PP1α, PL, and EGFP cDNAs in pENTR1A were recombined with DBAC-GW DNA12 maintained in and purified from HH8 bacteria. Recombinants were screened by PCR across the GW cassette and confirmed by field inversion gel electrophoresis (FIGE) analysis (FIGE mapper, BioRad, Hercules, CA) of restriction enzyme digests.
TTA BAC.
KOS-37 BAC, containing the complete strain KOS HSV-1 genome on a BAC,37 was kindly provided by David Leib (Dartmouth Medical School, NH). The HSV unique short (US) region in this BAC is in the reverse orientation relative to the published sequence (positions 132,503–145,393) of HSV-1 KOS38 (GenBank Accession number JQ673480). All modifications of this vector were introduced by Red/ET-mediated Recombination using the Gene Bridges Counter-Selection BAC modification kit with primers designed to generate homology arms for recombination into the BAC (Table 1).
First, KOS-37 BAC was modified by replacement of the internal repeat region of the HSV genome (117,078–131,902 of JQ673480) with a modified GW cassette. An EcoRV fragment containing the GW cassette from Gateway conversion plasmid A (ThermoFisher) was cloned into the EcoRV site of pcDNA3.1 (ThermoFisher Scientific) between the CMV promoter and bGH polyadenylation region. The CMV-GW-bGHpA fragment was PCR amplified with primers containing homology arms targeting the KOS-37 joint region (3′Joint/CMV and 5′Joint/bGHpA). The resultant targeting fragment was recombined into the BAC genome by Red/ET recombination (Gene Bridges, Heidelberg, Germany) using zeocin selection (25 μg/ml). Next, the coding sequence for rat TRPV12 (NM_031982.1) was substituted for the UL23 (thymidine kinase, tk) coding sequence (positions 46,613–47,743 of JQ673480), leaving the tk promoter intact. A counter-selection cassette (kanamycin resistance and streptomycin sensitivity) was PCR amplified with 5′ homology arms targeting UL23 (CS_UL23F and VR1_UL23R) and inserted using the protocol described by the manufacturer (15 μg/ml kanamycin selection). The counter-selection cassette was replaced with a TRPV1 cDNA that had been PCR amplified with primers containing the same homology arms (VR1_UL23F and VR1_UL23R) using streptomycin sensitivity (50 μg/ml streptomycin selection). The final recombinant BACs were referred to as T-GW. A second tk promoter (47,976–47,744 of JQ673480) and TRPV1 coding sequence were inserted in place of the UL41 (host shut-off, vhs) promoter and coding sequence, replacing positions 91,088–92,699 of JQ673480, using the technique described above. The counter-selection cassette was PCR amplified using primers CS_UL41F and CS_UL41R; the tk promoter-driven TRPV1 cDNA was amplified from the previously described T-GW BAC using primers VR1_UL41F and VR1_UL41R. Finally, the ampicillin resistance gene from pcDNA3.1 was recombined into the BAC genome between UL55 and UL56 (after position 116,070 of JQ673480). The targeting fragment was amplified with primers UL55/56_AmpF and UL55/56_AmpR, and selection was done with ampicillin (30 μg/ml). All PCR reactions for BAC recombination were carried out with Accuprime PFX DNA polymerase: 10-ng template DNA and 0.3 μmol/l of each primer at the following cycling conditions: 94°C for 2 minutes; 25 cycles of 94°C for 15 seconds, 58°C for 30 seconds, 68°C for 1 minute per kb; and 68°C for 10 minutes. PCR products were resolved on a 1% agarose gel, visualized by ethidium bromide staining, and gel isolated using the Qiaquick gel extraction kit (Qiagen, Germantown, MD).
BAC library production
The pENTR-based cDNA library derived from a mixture of NGF-differentiated and undifferentiated PC12 cells was previously described.12 TTA BAC DNA was purified using the Qiagen Large Construct Kit (Qiagen). The library was recombined with the TTA BAC Gateway cassette by incubation of 2 μg of BAC DNA and 200 ng of library plasmid DNA with clonase enzyme for 4 hours. Following electroporation into DH10B-T1 bacteria, recombinants were titered by serial dilution on LB plates containing 15 μg/ml of chloramphenicol and 30 μg/ml of ampicillin, demonstrating a total yield of ~200,000 colony forming units. En masse bacterial amplification was performed by overnight incubation at 30°C under shaking in 500 ml LB supplemented with 15 μg/ml of chloramphenicol and 30 μg/ml of ampicillin, with BAC DNA purified using the Qiagen Large Construct Kit. PCR across the GW cassette to assess the diversity of cDNAs recombined into TTA BAC was performed with Taq DNA polymerase and primers CMV-F1 and bGH-R1 (Table 1) using 100 ng BAC DNA, 0.2 μmol/l of each primer, and 0.2 mmol/l of each dNTP (Promega, Madison, WI) at the following cycling conditions: 94°C for 3 minutes; 30 cycles of 94°C for 30 seconds, 58°C for 30 seconds, 72°C for 3 minutes; and 72°C for 10 minutes. PCR products were resolved on a 1% agarose gel and visualized by ethidium bromide staining.
Transfection of individual BAC DNAs
All transfections were performed with Lipofectamine LTX (ThermoFisher Scientific). DBAC-based recombinants were transfected into 7B cells. All TTA-derived BAC DNAs were transfected into Vero cells. DNA was diluted in 500 μl of Opti-MEM (ThermoFisher Scientific), incubated with 3 μl of Plus Reagent for 5 minutes at RT, 9 μl of LTX reagent was added and the mixture was incubated for 30 minutes at RT. The transfection mix was added to cells and the combination was incubated at 37°C for 6 hours before the supernatant was removed and fresh media (Dulbecco’s Modified Eagle’s medium (DMEM) + 5% FBS) was added. Following transfection, cells were cultured at 37°C overnight, shifted to 33°C, and monitored for cytopathic effect for virus collection. Virus was collected from the supernatant by the addition of 0.45 M NaCl (final concentration), incubation at RT with shaking for 60 minutes, removal of cells by centrifugation at 3,000g for 10 minutes at 4°C, and final filtration through a 0.8 μm filter.
BAC library transfection
U2OS cells were seeded at 3 × 105 cells per well in a six-well dish and grown overnight. Purified BAC library DNA was introduced by transfection with Lipofectamine LTX, as follows. For each well, 2 μg of DNA was diluted in 500 μl of Opti-MEM and incubated with 2.5 μl of Plus Reagent for 5 minutes at RT. 6.25 μl of LTX reagent was then added and the mixture was incubated for 30 minutes at RT, added to the cells, and the combination was incubated at 37°C for 4 hours before removal of the transfection mix and addition of fresh Dulbecco’s Modified Eagle’s medium (DMEM) + 10% FBS. A total of 24 μg of BAC DNA was used to generate the virus library.
Virus titration
Vero cells were seeded in 24-well dishes (1 × 105 cells/well) and confluent monolayers of cells were infected 24 hours later (MOI = 0.01). Supernatants were collected at 48 hpi, the cells were scraped and sonicated, and the two fractions were combined and titered by 10-fold serial dilution on Vero cells. Titers were calculated from the averages of duplicate experiments.
Statistics
Data were analyzed using Microsoft Excel software, and the data sets were statistically compared by using Student’s t-test (two-tailed), assuming unequal variances for the two sets (heteroscedastic).
Acknowledgments
We thank X Zhang for assistance with calcium imaging studies, M Zhang for assistance with animal studies, M S Gold for discussions about the work and valuable comments on the manuscript, and D Leib for KOS-37 BAC. This work was supported by NIH grants DK044935 and NS064988 to J.C.G. B.R. was supported in part by an NIH T32 postdoctoral training grant in Molecular Microbial Persistence and Pathogenesis. B.R., W.F.G., J.R.G., J.B.C., and J.C.G. designed research; B.R., W.F.G., A.H., S.C., and J.R.G. performed research; W.C.D. and N.Y. contributed new reagents/analytical tools; B.R., W.F.G., W.C.D., J.B.C., and J.C.G analyzed data; and B.R., W.F.G., J.B.C., and J.C.G wrote the paper.
J.C.G. is a founder in Switch Bio, LLC, a company interested in neuromodulation.
References
- Laing, RJ and Dhaka, A (2016). ThermoTRPs and pain. Neuroscientist 22: 171–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina, MJ, Schumacher, MA, Tominaga, M, Rosen, TA, Levine, JD and Julius, D (1997). The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389: 816–824. [DOI] [PubMed] [Google Scholar]
- Rashid, MH, Inoue, M, Bakoshi, S and Ueda, H (2003). Increased expression of vanilloid receptor 1 on myelinated primary afferent neurons contributes to the antihyperalgesic effect of capsaicin cream in diabetic neuropathic pain in mice. J Pharmacol Exp Ther 306: 709–717. [DOI] [PubMed] [Google Scholar]
- Hong, S and Wiley, JW (2005). Early painful diabetic neuropathy is associated with differential changes in the expression and function of vanilloid receptor 1. J Biol Chem 280: 618–627. [DOI] [PubMed] [Google Scholar]
- Christoph, T, Gillen, C, Mika, J, Grünweller, A, Schäfer, MK, Schiene, K et al. (2007). Antinociceptive effect of antisense oligonucleotides against the vanilloid receptor VR1/TRPV1. Neurochem Int 50: 281–290. [DOI] [PubMed] [Google Scholar]
- Chuang, HH, Prescott, ED, Kong, H, Shields, S, Jordt, SE, Basbaum, AI et al. (2001). Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411: 957–962. [DOI] [PubMed] [Google Scholar]
- Price, TJ, Jeske, NA, Flores, CM and Hargreaves, KM (2005). Pharmacological interactions between calcium/calmodulin-dependent kinase II alpha and TRPV1 receptors in rat trigeminal sensory neurons. Neurosci Lett 389: 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra, DP and Nau, C (2005). Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. J Biol Chem 280: 13424–13432. [DOI] [PubMed] [Google Scholar]
- Srinivasan, R, Huang, S, Chaudhry, S, Sculptoreanu, A, Krisky, D, Cascio, M et al. (2007). An HSV vector system for selection of ligand-gated ion channel modulators. Nat Methods 4: 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunieda, K, Someya, A, Horie, S, Ajioka, H and Murayama, T (2005). Lafutidine-induced increase in intracellular ca(2+) concentrations in PC12 and endothelial cells. J Pharmacol Sci 97: 67–74. [DOI] [PubMed] [Google Scholar]
- Someya, A, Kunieda, K, Akiyama, N, Hirabayashi, T, Horie, S and Murayama, T (2004). Expression of vanilloid VR1 receptor in PC12 cells. Neurochem Int 45: 1005–1010. [DOI] [PubMed] [Google Scholar]
- Wolfe, D, Craft, AM, Cohen, JB and Glorioso, JC (2010). A herpes simplex virus vector system for expression of complex cellular cDNA libraries. J Virol 84: 7360–7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sanz, N, Fernández-Carvajal, A, Morenilla-Palao, C, Planells-Cases, R, Fajardo-Sánchez, E, Fernández-Ballester, G et al. (2004). Identification of a tetramerization domain in the C terminus of the vanilloid receptor. J Neurosci 24: 5307–5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krisky, DM, Wolfe, D, Goins, WF, Marconi, PC, Ramakrishnan, R, Mata, M et al. (1998). Deletion of multiple immediate-early genes from herpes simplex virus reduces cytotoxicity and permits long-term gene expression in neurons. Gene Ther 5: 1593–1603. [DOI] [PubMed] [Google Scholar]
- Mitchell, K, Bates, BD, Keller, JM, Lopez, M, Scholl, L, Navarro, J et al. (2010). Ablation of rat TRPV1-expressing Adelta/C-fibers with resiniferatoxin: analysis of withdrawal behaviors, recovery of function and molecular correlates. Mol Pain 6: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss, JR, Gold, MS and Glorioso, JC (2009). HSV vector-mediated modification of primary nociceptor afferents: an approach to inhibit chronic pain. Gene Ther 16: 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan, R, Wolfe, D, Goss, J, Watkins, S, de Groat, WC, Sculptoreanu, A et al. (2008). Protein kinase C epsilon contributes to basal and sensitizing responses of TRPV1 to capsaicin in rat dorsal root ganglion neurons. Eur J Neurosci 28: 1241–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walder, RY, Radhakrishnan, R, Loo, L, Rasmussen, LA, Mohapatra, DP, Wilson, SP et al. (2012). TRPV1 is important for mechanical and heat sensitivity in uninjured animals and development of heat hypersensitivity after muscle inflammation. Pain 153: 1664–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, K, Ye, Y, Viet, CT, Dang, D and Schmidt, BL (2015). TRPV1 expression level in isolectin B₄-positive neurons contributes to mouse strain difference in cutaneous thermal nociceptive sensitivity. J Neurophysiol 113: 3345–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore, P, Wismer, CT, Mikusa, J, Zhu, CZ, Zhong, C, Gauvin, DM et al. (2005). A-425619 [1-isoquinolin-5-yl-3-(4-trifluoromethyl-benzyl)-urea], a novel transient receptor potential type V1 receptor antagonist, relieves pathophysiological pain associated with inflammation and tissue injury in rats. J Pharmacol Exp Ther 314: 410–421. [DOI] [PubMed] [Google Scholar]
- Swanson, DM, Dubin, AE, Shah, C, Nasser, N, Chang, L, Dax, SL et al. (2005). Identification and biological evaluation of 4-(3-trifluoromethylpyridin-2-yl)piperazine-1-carboxylic acid (5-trifluoromethylpyridin-2-yl)amide, a high affinity TRPV1 (VR1) vanilloid receptor antagonist. J Med Chem 48: 1857–1872. [DOI] [PubMed] [Google Scholar]
- Tang, L, Chen, Y, Chen, Z, Blumberg, PM, Kozikowski, AP and Wang, ZJ (2007). Antinociceptive pharmacology of N-(4-chlorobenzyl)-N’-(4-hydroxy-3-iodo-5-methoxybenzyl) thiourea, a high-affinity competitive antagonist of the transient receptor potential vanilloid 1 receptor. J Pharmacol Exp Ther 321: 791–798. [DOI] [PubMed] [Google Scholar]
- Gentry, C, Stoakley, N, Andersson, DA and Bevan, S (2010). The roles of iPLA2, TRPM8 and TRPA1 in chemically induced cold hypersensitivity. Mol Pain 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriens, J, Nilius, B and Voets, T (2014). Peripheral thermosensation in mammals. Nat Rev Neurosci 15: 573–589. [DOI] [PubMed] [Google Scholar]
- McNamara, CR, Mandel-Brehm, J, Bautista, DM, Siemens, J, Deranian, KL, Zhao, M et al. (2007). TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci USA 104: 13525–13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, M, Carli, G, Raboisson, P and Reeh, P (2014). The interphase of the formalin test. Pain 155: 511–521. [DOI] [PubMed] [Google Scholar]
- Cohen, PT (2002). Protein phosphatase 1–targeted in many directions. J Cell Sci 115: 241–256. [DOI] [PubMed] [Google Scholar]
- Heroes, E, Lesage, B, Görnemann, J, Beullens, M, Van Meervelt, L and Bollen, M (2013). The PP1 binding code: a molecular-lego strategy that governs specificity. FEBS J 280: 584–595. [DOI] [PubMed] [Google Scholar]
- Docherty, RJ, Yeats, JC, Bevan, S and Boddeke, HW (1996). Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflugers Arch 431: 828–837. [DOI] [PubMed] [Google Scholar]
- Pearce, LV, Toth, A, Ryu, H, Kang, DW, Choi, HK, Jin, MK et al. (2008). Differential modulation of agonist and antagonist structure activity relations for rat TRPV1 by cyclosporin A and other protein phosphatase inhibitors. Naunyn Schmiedebergs Arch Pharmacol 377: 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majima, T, Funahashi, Y, Takai, S, Goins, WF, Gotoh, M, Tyagi, P et al. (2015). Herpes simplex virus vector-mediated gene delivery of poreless TRPV1 channels reduces bladder overactivity and nociception in rats. Hum Gene Ther 26: 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura, H, Ugawa, S, Ueda, T, Morita, A and Shimada, S (2008). TRPM8 activation suppresses cellular viability in human melanoma. Am J Physiol Cell Physiol 295: C296–C301. [DOI] [PubMed] [Google Scholar]
- Miller, BA (2006). The role of TRP channels in oxidative stress-induced cell death. J Membr Biol 209: 31–41. [DOI] [PubMed] [Google Scholar]
- Shapovalov, G, Lehen’kyi, V, Skryma, R and Prevarskaya, N (2011). TRP channels in cell survival and cell death in normal and transformed cells. Cell Calcium 50: 295–302. [DOI] [PubMed] [Google Scholar]
- Goss, JR, Cascio, M, Goins, WF, Huang, S, Krisky, DM, Clarke, RJ et al. (2011). HSV delivery of a ligand-regulated endogenous ion channel gene to sensory neurons results in pain control following channel activation. Mol Ther 19: 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X, Koronowski, KB, Li, L, Freeman, BA, Woodcock, S and de Groat, WC (2014). Nitro-oleic acid desensitizes TRPA1 and TRPV1 agonist responses in adult rat DRG neurons. Exp Neurol 251: 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierasch, WW, Zimmerman, DL, Ward, SL, Vanheyningen, TK, Romine, JD and Leib, DA (2006). Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J Virol Methods 135: 197–206. [DOI] [PubMed] [Google Scholar]
- Macdonald, SJ, Mostafa, HH, Morrison, LA and Davido, DJ (2012). Genome sequence of herpes simplex virus 1 strain KOS. J Virol 86: 6371–6372. [DOI] [PMC free article] [PubMed] [Google Scholar]