

Abstract
Glioblastoma Multiforme (GBM), a uniformly lethal stage IV astrocytoma, is currently treated with a combination of surgical and radiation therapy as well as Temozolomide (TMZ) chemotherapy. Resistance to TMZ is rapidly acquired by GBM cells and overcoming this resistance has been an area of signi?cant research. GBM 'cancer stem cells' (CSC) also known as 'cancer initiating cells' are often positively selected by CD133 expression and TMZ resistance. In this project, we selected GBM CSC from two cell lines based on CD133 expression. CD133+ and CD133− GBM cells showed comparable cell cycle status. The expression of genes within the Sonic Hedgehog Signaling pathway, PTCH1 (SHH receptor/basal signaling repressor) and Gli1 (effector transcription factor) were increased. The recent literature indicated a decreased in PTCH expression by miRNA and this was independent of SHH expression. We analyzed 5 potential PTCH-targeting miRNA and identi?ed an increase in miRNA-9-2. The CD133+ cells showed an increase in the Multiple Drug Resistance 1 gene (MDR1). Knockdown of Gli1 and MDR1 with siRNA enhanced TMZ induced cell death. Taken together, these studies show CD133+ GBM CSCs expressed greater levels of miR-9 and activation of the SHH/PTCH1/MDR1 axis. This axis has been shown to impart TMZ resistance. In the case of the CD133+ cells, the resistance is not acquires but seems to be inherent. Identi?cation of this pathway as well as the identi?cation of miR-9 may allow for the development of miRNA-targeted approach to Cancer Stem Cell therapy in GBM.
Keywords: Cancer stem cell, Glioblastoma, miRNA-9, CD133+ cells, Temozolomide
INTRODUCTION
Glioblastoma Multiforme (GBM) is the most common adult intracranial malignancy, and the most aggressive cancer with rv11-month median survival [1]. The current treatments include surgical resection, radiation and chemotherapy with the frontline temozolomide (TMZ). Bevacizumab was given accelerated approval by the food and drug administration (FDA) only when the patient resisted TMZ treatment. Irinotecan, which does not have FDA approval for GBM, has been shown in a trial to enhance the effectiveness of Bevacizumab.
The cancer stem cell (CSC) hypothesis suggests that the hypoxic region of the tumor contain stem cells, which can undergo self-renewal and tumor initiation. The more differentiated cancer cells cannot sustain the long-term growth of the tumor [2]. Based on these properties of CSCs, it has been proposed that their elimination will halt neoplastic expansion.
Although the field of CSCs is not new, the early report of a well-documented example was reported for leukemia. A small fraction of the leukemia cells express stem-cell markers and showed tumor propagating potential [3]. Functionally similar cells have been isolated from brain tumors and were shown to express CD133 (prominin-1) [4].
GBM generally exhibit chemoresistance [5]. Conventional methods of neoplastic treatment have been shown to spare a small subset of CD133+ GBM cells. These cells were designated CSCs of GBM [6]. The CD133+ GBM cells show constitutive activation of Sonic Hedge-hog (SHH) signaling [7]. The stromal cells of GBM secrete SHH [8]. SHH interacts with the the surface receptor, Patched-1 (PTCH1), to induce cellular activation of GBM [9].
Dysregulation of SHH signaling has been linked to medulloblastoma [10]. Its role in glioblastomas has been less defined. Gli1, the SHH effector protein, was originally discovered in primary glioblastoma samples, as an amplified oncogene [11]. Gli1 has been shown to be highly active in GBM-derived CSCs [12]. The addition, the alkyloid SHH signaling inhibitor, cyclopamine, reduced the activity of Gli in a dose-dependent manner [7]. The surviving GBM cells following SHH blockade lost their ability to form tumors in athymic mice. This suggested that cyclopamine may induce the differentiation of the CSC subset.
MicroRNAs (miR) are small RNA molecules, which negatively regulate mRNA translation by binding to the 3′ untranslated region (UTR) to suppress translation. Several miRNAs have been shown to mediate GBM chemoresistance [13]. MiR-9 has been shown to target PTCH1, resulting in SHH signaling to impart chemoresistance in GBM [14].
Prominin-1 (CD133) is commonly associated with cells expressing primitive phenotypes [15]. In several cancers, CD133+ cells have been shown to express genes associated with pluripotency [16]. Among these genes are the expression of drug transporters [17]. Thus, CD133+ cells often show resistance to chemotherapeutic agents [18]. In the case of GBMs, there are contradictory reports regarding the chemoresistance of CD133+ cell [15]. This study analyzed a miR-9/PTCH1/MDR1 axis in CD133+ GBM cells.
This study reported on an increase in miR-9 in CD133+ subset of GBM cells. We showed that miR-9 activated SHH signaling by regulating PTCH1 (a repressor) at the level of post-transcription. This observation contrasted with the effects observed on the CD133− subset, which comprised the bulk tumor cells. We identified a role for miR-9 in the upregulation of the MDR1 (ABCB1) gene in the CD133+ cells.
MATERIALS AND METHODS
Cell lines
U87 and T98G cells were purchased from American Type Culture Collection (ATCC; Manassas, VA) and then expanded as per manufacturer’s instructions.
Isolation of CD133+ GBM cells
CD133 expressing GBM cells were positively selected from U87 and T98G cells by Fluorescent Activated Cell Sorting. The cells were labeled with PE-conjugated mouse anti-human CD133 (Miltenyi Biotec, Bergisch Gladbach, Germany). Positive and Negative cells were analyzed post-sort by Flow Cytometry to confirm purity.
Real-time RT-PCR
RNA was extracted with Trizol reagent (Life Technologies Invitrogen). Reverse transcription with 200 ng of cDNA was performed with the High Capacity cDNA Reverse Transcription Kit (Life Technologies Applied Biosystems) in accordance with the manufacturer’s recommendation. Real-time PCR was performed on 7300 Real-Time PCR System (Life Technologies Applied Biosystems) as follows: an initial incubation of 50°C for 2 min followed by 95°C for 10 min. After this, the cycling conditions were as follows: 95°C for 15 s and 60°C for 60 s, for 40 cycles. Primers were purchased from Sigma (St. Louis, MO). The primers for MGMT are as follows: Forward, ggc acc gct gta tta aag ga; Reverse, ata gag caa ggg cag cag cgt ta. The relative expression was calculated using 2(-Delta Delta C(T)), as previously described [19].
Short interference RNA (siRNA) targeted knockdown
PTCH1, Gli1 and MDR1 targeting-siRNA pooled duplexes (Dharmacon) were used to knock down the respective genes in GBM cells. Briefly, GBM cells (104) were seeded in 96-well plates. After 24 h, 30 nM siRNA was delivered with DharmaFECT Transfection reagent. Control non-targeting siRNA duplexes were purchased from Dharmacon.
Cell viability assay
Lactate dehydrogenase release assay (Promega, Madison, WI) was our primary method of analyzing cell death. LDH assays have been reported to confirm cell death 72 h after TMZ treatment [7]. The LDH release cell viability assay uses a coupled reaction in which NAD+ and Lactate are converted to NADH, and pyruvate by LDH. NADH+INT (a tetrazolium salt) uses Diaphorase to produce NAD+ and formazan. The formazan is red and thus can be detected in the solution. Thus, we measured conversion of Formazan (Red) using a Victor3V Multilabel Plate Reader (Perkin Elmer; Waltham, MA, USA) with an emission filter of 490nm. Results were normalized to complete cell lysis by provided lysis buffer.
Cell cycle analysis
Cell cycle status was analyzed by flow cytometry. U87 or T98G cells (106) were resuspended in PBS and then incubated with anti-CD133-PE (Clone AC133, Miltenyi Biotec). After this, the cells were labeled with 1 mg/mL of Hoechst dye (Life Technologies Invitrogen) for 2 h. The CD133+ cells were gated and then analyzed for DNA content using the FACSCalibur (BD Biosciences, San Jose, California). The data were analyzed with Cellquest Pro (BD Biosciences).
Statistical analysis
Data were analyzed using the students t-test for two groups (control/experimental). A p value <0.05 was considered significant.
RESULTS
Frequency of CD133+ cells in GBM cell lines
GBM cells were labeled with PE-conjugated anti-CD133, 293C3 clone and the CD133+ cells were sorted using the FACS sorter. The CD133+ cells were <0.2% within the U87 and T98G GBM cells (Figures 1A and 1B, top panels). In order to verify that the sorted cells were indeed CD133+ or CD133− cells by immune-labeling with anti-CD133. The results showed efficient sorting of CD133+ and CD133− cells (Figures 1A and 1B, lower panels).
Figure 1. Subset of CD133+ in GBM cells.

T98G (top panel) and U87 cells were sorted using FACS with a PE-conjugated murine anti-CD133 antibody. The sorted cells added to tissue culture plates. After 24 h, the cells were fixed and then labeled for CD133 (Green), Texas Red-Phalloidin (Red Actin Fibers) and DAPI (Blue nuclei). The images are shown using 20× magnification.
TMZ resistance of CD133+ cells
The reports indicated that CD133+ GBM cells were chemoresistant [4]. Yet, previous research has shown that TMZ inhibited the proliferation of CD133+ GBM cells without inducing cell death [20]. We previously showed that 200 μM of TMZ resulted in chemoresistant cells after 72 h [21]. We therefore asked if there are differences between CD133+ and CD133− GBM cells with respect to TMZ resistance. The subsets of GBM cells were treated with 200 μM of TMZ. After 72 h, cell viability was performed with the LDH release assay, CytoTox 96®. Cell death was significantly (p < 0.05) reduced in the CD133+ cells as compared to CD133− GBM cells (Figure 2, open vs. right diagonal bar). The results indicated that CD133+ GBM cells were more resistant to TMZ than the CD133− subset.
Figure 2. Resistance of CD133+ cells to TMZ.

Sorted U87 and T98G cells were transfected with anti-miR-9 or untransfected. The cells were treated with 200 μM TMZ. After 72 h, cell viability was assayed by CytoTox 96® LDH release. The results are presented as % cell death±SD, n ¼ 4. *p < 0.05 vs. CD133− cells. ** p > 0.05 vs. untransfected CD133+ cells.
Role of miR-9 in the resistance of CD133+ to TMZ
We previously reported on miRNA-9 as a mediator of TMZ resistance [14]. We asked if miR-9 was responsible for the resistance of CD133+ cells to TMZ. WE studied cell viability with CD133+ cells in which we blocked the effect of miR-9 with anti-miR-9 and then treated the cells with 200 μM of TMZ. The results indicated a significant (p < 0.05) reversal of TMZ resistance as compared to cell transfected with control anti-miR (Figure 2, hatched bar). In summary, these results indicated that miR-9 was involved in CD133+ resistance to TMZ.
CD133+ cells do not alter cell cycle activity
Since CD133+ cells have been reported to be the CSCs of GBM, it is expected that these cells would be in cycling quiescence [22]. We therefore asked if the resistance of TMZ could be explained by the slow cycling of CD133+ GBM cells. To address this question we asked if there are differences in the cell cycle status between CD133+ and CD133− cells. We labeled U87 and T98G cells with PE-conjugated anti-CD133-PE and Hoechst dye and then analyzed the cells on the FACS analyzer. The results showed similarities in the cycling status of both CD133− and CD133+ subsets (Figure 3). This suggested that the chemoresistant properties of CD133+ cells could not be explained by changes in cell cycling.
Figure 3. Cell cycle phase of CD133+ U87 and T98G cells.
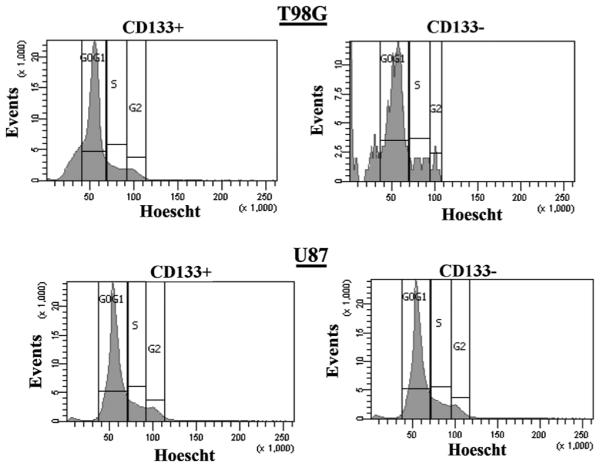
T98G (upper panel) and U87 (lower panel) cells were co-labeled with anti-CD133-PE and Hoechst dye. The CD133+ cells were gated and then studied for cell cycle phase. The results represent four independent experiments.
SHH signaling in CD133+ GBM cells
The SHH signaling has been shown to cause chemoresistance of GBM cells [14]. We therefore asked if the SHH pathway was activated in the CD133+ GBM cells. Real time PCR for Gli1 and PTCH1 in the CD133+ and CD133− sorted cells showed a significant (p < 0.05) decrease in PTCH1 mRNA in the CD133+ cells as compared to the CD133− subset (Figure 4, top/left panel). This pattern of PTCH1 expression contrasted Gli1 mRNA level (Figure 4, top/right panel). Since Gli1 is a downstream target of SHH signaling, this suggested that SHH signaling is active in CD133+ cells, regardless of TMZ exposure.
Figure 4. SHH signaling and ABC transporter in CD133+ cells.

CD133+ sorted U87 and T98G cells were analyzed by real time PCR for Gli1 and PTCH1 (A) and, the ABC transporters, MDR1 and ACBG2 (B).
Increases in MDR1 and ABCG2 in CD133+ cells
Increases in miR9 and Gli1 have been linked to TMZ resistance through increases in the ABC transporter genes [23]. We there studied the expression of xenobiotic drug transporters, MDR1 and ABCG2 by real-time PCR in CD133+ and CD133− U87 and T89G cells. The values obtained for CD133− was normalized to 1 and then used to present the fold change in CD133+. The results indicated significant (p < 0.05) increases for MDR1 in the CD133+ cells but no change for ABCG2 (Figure 4). These results suggested that MDR1 expression may have a role for the innate resistance of CD133+ cells than ABCG2.
MiR-9 expression in TMZ resistant GBM cells
We previously reported that miR-9 can target PTCH1 mRNA to increase SHH signaling [14]. We have identified 5 putative PTCH1-targeting miRNAs. We therefore quantitated them using Taqman® qPCR. The results showed an upregulation of miR-9 in both CD133+ and CD133− cells, although there was significantly (p < 0.05) higher level in the CD133+ cells (Figure 5A).
Figure 5. MiR-9-2 level in CD133+ cells from GBM.

CD133+ cells were sorted. MiRNA was isolated and then analyzed by Taqman® miRNA qPCR for the predicted PTCH1 targeted miRNA (A). Real time was performed for miR-9-2 (B). The results are shown as the fold change from unsorted GBM cells, ±SD, n ¼ 4.
Mature miR-9 may be the result from gene activation on three independent loci on chromosomes 1, 5, and 15 [24]. We therefore studied CD133+ and CD133− GBM cells by real time PCR for all three loci. The results indicated that the miR-9-2 was increased and thus may be responsible for the high level of miR-9 in the CD133+ cells (Figure 5B).
MDR1 knockdown increases TMZ sensitivity in CD133+ GBM cells
In this set of studies, we investigated the relevance of SHH pathway and MDR1 in the chemoresistance in CD133+ cells. We knocked down PTCH1, Gli1 and MDR1 with targeted siRNA or scrambled siRNA. Knockdown was confirmed by real-time PCR for each gene (Figure 6, top panel). Next, we treated the knockdown CD133+ cells with 200 μM TMZ. After 72 h, the viability of the cells was studied with Cell Titer Blue. Here we show that knockdown of PTCH enhanced resistance to TMZ, while Gli1 and MDR1 knockdown sensitized the cells to TMZ.
Figure 6. SHH pathway in CD133+ GBM cells in TMZ resistance.

PTCH1, Gli1 and MDR1 were knocked down with targeted siRNA in U87 and T98G CD133+ cells. The efficiency of the knockdowns was confirmed by real time PCR (left). Knock-down cells were then treated with 200 μM TMZ. After 72 h, cell viability was assayed by CytoTox 96® LDH release (center/right). The results are shown as the mean ± SD, n ¼ 4.
DISCUSSION
In this study, we sorted CD133 expressing cells based on immune methods and categorized them as CD133+ and CD133− cell populations. Approximately 0.2% of the GBM cells expressed CD133. We first studied the chemoresistant nature of the two subsets and found that while the CD133+ cells were resistant to TMZ, this could not be explained by cell cycle alterations (Figures 2 and 3). Real-Time PCR showed the activation of SHH pathway, based on reduced PTCH1 reduction and the increase in Gli1 (SHH transcription factor). The MDR1 gene was also shown upregulated in CD133-expressing cells (Figure 4). Studies involving miRNA indicated that the decrease in PTCH1 in CD133+ GBM cells was caused by miR-9. The results indicated that the increase in miR-9 was caused by the mir-9-2 loci. We have previously shown that miR-9 upregulation activated SHH signaling by the decrease in PTCH1. The importance of the SHH pathway and the implication for increased MDR1 was assessed by targeted siRNA. The results indicated that knockdown of Gli1 and MDR1 enhanced TMZ-induced cell death (Figure 6). In contrast, PTCH1 knockdown (activating SHH signaling) reduced cell death (Figure 6).
GBM is currently one of the most aggressive malignancies of the human central nervous system. Unfortunately, GBM is also the most common primary intracranial tumor. Current treatments have fallen short since the median survival rate is among the lowest among all cancers, with 5% 5-year survival rate. Radiotherapy selectively reduced the bulk tumor but allows for the survival and proliferation of resistant CD133+ GBM CSCs [25]. The CSC hypothesis states that a subset of slow proliferating cells survive within the hypoxic region in the center of the bulk tumor [26]. It is also believed that this small subset of hypoxic resistant cells could ‘fuel’ the tumor. In this regard, a hierarchal organization of tumor cells would allow for the heterogeneity seen in these tumors.
The sensitivity of GBM cells to TMZ has been shown to be significantly associated with the methylation of the O6-Methylguanine-Methyltransferase (MGMT) [27]. We tested if miR-9 caused an increase in MGMT by real time PCR with RNA from GBM cells, transfected with pre-miR-9 or control pre-miR. The results showed no difference in the mRNA for MGMT (Figure S1).
Chemo- and radio-resistance has failed to eradicate cancer. The CSC hypothesis offers insight not only into the origin and maintenance of cancers but the resurgence of tumors following standard treatment. CSCs much like adult stem cells highly express the multi-drug resistance gene MDR1 or ABCB1, which ATP-dependently effluxes a number of drugs such as xenobiotics and chemotherapeutic agents. CSCs also express stem cell pathways, which ensure proper cellular division and pluripotency. These same pathways appear to confer resistance to chemo- and radiotherapy. A better understanding into the activation and persistence of these molecular pathways is warranted. RNA therapeutics is on the cusp of modern therapy and an active area of research. Here we propose a novel method in which GBM stem cells could be maintained; namely, the SHH pathway. The knowledge of the underlying mechanisms may lead new drug targets and interventions for a population of patients, which currently have no other options.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by F.M Kirby to PR.
Footnotes
SUPPLEMENTAL INFORMATION U87 were stably transfected with pCMV-MIR-9 or control vector (Origene, Rockville, MD). RNA was isolated and then analyzed by real-time PCR for MGMT. The normalized results for transfectants with control miRNA were assigned 1. The miR-9 transfectants were presented as fold change over the control miRNA. The results are the mean ± SD of four independent experiments.
CONFLICT OF INTEREST STATEMENT
None of the authors report a conflict of interest.
REFERENCES
- [1].Poulsen HS, Urup T, Michaelsen SR, Staberg M, Villingshoj M, Lassen U. The impact of bevacizumab treatment on survival and quality of life in newly diagnosed glioblastoma patients. Cancer Manag Res. 2014;6:373–387. doi: 10.2147/CMAR.S39306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lara-Padilla E, Caceres-Cortes JR. On the nature of the tumor-initiating cell. Curr Stem Cell Res Ther. 2012;7:26–35. doi: 10.2174/157488812798483412. [DOI] [PubMed] [Google Scholar]
- [3].Dick JE. Immune-deficient mice as models of normal and leukemic human hematopoiesis. Cancer Cells. 1991;3:39–48. [PubMed] [Google Scholar]
- [4].Choy W, Nagasawa DT, Trang A, Thill K, Spasic M, Yang I. CD133 as a marker for regulation and potential for targeted therapies in glioblastoma multiforme. Neurosurg Clinics of North America. 2012;23:391–405. doi: 10.1016/j.nec.2012.04.011. [DOI] [PubMed] [Google Scholar]
- [5].Stopschinski BE, Beier CP, Beier D. Glioblastoma cancer stem cells - From concept to clinical application. Cancer Lett. 2013;338:32–40. doi: 10.1016/j.canlet.2012.05.033. [DOI] [PubMed] [Google Scholar]
- [6].Jackson M, Hassiotou F, Nowak A. Glioblastoma stem-like cells: At the root of tumor recurrence and a therapeutic target. Carcinogenesis (In press) doi: 10.1093/carcin/bgu243. [DOI] [PubMed] [Google Scholar]
- [7].Ulasov IV, Nandi S, Dey M, Sonabend AM, Lesniak MS. Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133(C) glioma stem cells to temozolomide therapy. Mol Med. 2011;17:103–112. doi: 10.2119/molmed.2010.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yan GN, Lv YF, Yang L, Yao XH, Cui YH, Guo DY. Glioma stem cells enhance endothelial cell migration and proliferation via the Hedgehog pathway. Oncol Lett. 2013;6:1524–1530. doi: 10.3892/ol.2013.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Santoni M, Burattini L, Nabissi M, Morelli MB, Berardi R, Santoni G, Cascinu S. Essential role of Gli proteins in glioblastoma multiforme. Curr Protein Pept Sci. 2013;14:133–140. doi: 10.2174/1389203711314020005. [DOI] [PubMed] [Google Scholar]
- [10].Kool M, Jones D, Jager N, Northcott P, Pugh T, Hovestadt V, Piro R, Esparza LA, Markant S, Remke M, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kinzler KW, Bigner SH, Bigner DD, Trent JM, Law ML, O’Brien SJ, Wong AJ, Vogelstein B. Identification of an amplified, highly expressed gene in a human glioma. Science. 1987;236:70–73. doi: 10.1126/science.3563490. [DOI] [PubMed] [Google Scholar]
- [12].Zbinden M, Duquet A, Lorente-Trigos A, Ngwabyt S-N, Borges I, Altaba A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010;29:2659–2674. doi: 10.1038/emboj.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liu Q, Zou R, Zhou R, Gong C, Wang Z, Cai T, Tan C, Fang J. miR-155 Regulates Glioma Cells Invasion and Chemosensitivity by p38 Isforms in vitro. J Cell Biochem (In press) doi: 10.1002/jcb.25073. [DOI] [PubMed] [Google Scholar]
- [14].Munoz JL, Rodriguez-Cruz V, Ramkissoon SH, Ligon KL, Greco SJ, Rameshwar P. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget (In press) doi: 10.18632/oncotarget.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Irollo E, Pirozzi G. CD133: to be or not to be, is this the real question? Am J Transl Res. 2013;5:563–581. [PMC free article] [PubMed] [Google Scholar]
- [16].Li Z. CD133: a stem cell biomarker and beyond. Exp Hematol Oncol. 2013;2:17. doi: 10.1186/2162-3619-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine. 2012;7:597–615. doi: 10.2217/nnm.12.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu J, Mao Z, Huang J, Xie S, Liu T, Mao Z. Blocking the NOTCH pathway can inhibit the growth of CD133-positive A549 cells and sensitize to chemotherapy. Biochem Biophys Res Comm. 2014;444:670–675. doi: 10.1016/j.bbrc.2014.01.164. [DOI] [PubMed] [Google Scholar]
- [19].Lim PK, Bliss SA, Patel SA, Taborga M, Dave MA, Gregory LA, Greco SJ, Bryan M, Patel PS, Rameshwar P. Gap junction-mediated import of MicroRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011;71:1550–1560. doi: 10.1158/0008-5472.CAN-10-2372. [DOI] [PubMed] [Google Scholar]
- [20].Auffinger B, Tobias AL, Han Y, Lee G, Guo D, Dey M, Lesniak MS, Ahmed AU. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014;21:1119–1131. doi: 10.1038/cdd.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Munoz JL, Rodriguez-Cruz V, Greco SJ, Ramkissoon SH, Ligon KL, Rameshwar P. Temozolomide resistance in glioblastoma cells occurs partly through epidermal growth factor receptor-mediated induction of connexin 43. Cell Death Dis. 2014;5:e1145. doi: 10.1038/cddis.2014.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brescia P, Ortensi B, Fornasari L, Levi D, Broggi G, Pelicci G. CD133 is essential for glioblastoma stem cell maintenance. Stem Cells. 2013;31:857–869. doi: 10.1002/stem.1317. [DOI] [PubMed] [Google Scholar]
- [23].Munoz JL, Rodriguez-Cruz V, Greco SJ, Nagula V, Scotto KW, Rameshwar P. Temozolomide induces the production of epidermal growth factor to regulate MDR1 expression in glioblastoma cells. Mol Cancer Ther. 2014;13:2399–2411. doi: 10.1158/1535-7163.MCT-14-0011. [DOI] [PubMed] [Google Scholar]
- [24].Laneve P, Gioia U, Andriotto A, Moretti F, Bozzoni I, Caffarelli E. A minicircuitry involving REST and CREB controls miR-9-2 expression during human neuronal differentiation. Nucleic Acids Res. 2010;38:6895–6905. doi: 10.1093/nar/gkq604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tamura K, Aoyagi M, Ando N, Ogishima T, Wakimoto H, Yamamoto M, Ohno K. Expansion of CD133− positive glioma cells in recurrent de novo glioblastomas after radiotherapy and chemotherapy. J Neurosurg. 2013;119:1145–1155. doi: 10.3171/2013.7.JNS122417. [DOI] [PubMed] [Google Scholar]
- [26].Kim Y, Lin Q, Glazer PM, Yun Z. Hypoxic tumor microenvironment and cancer cell differentiation. Curr Mol Med. 2009;9:425–434. doi: 10.2174/156652409788167113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Thon N, Kreth S, Kreth FW. Personalized treatment strategies in glioblastoma: MGMT promoter methylation status. Onco Targets Ther. 2013;6:1363–1372. doi: 10.2147/OTT.S50208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.