

Abstract
Kahalalide F (1) shows remarkable antitumor activity against different carcinomas and has recently completed phase I clinical trials and is being evaluated in phase II clinical studies. The antifungal activity of this molecule has not been thoroughly investigated. In this report, we focused on acetylation and oxidation of the secondary alcohol of threonine, as well as reductive alkylation of the primary amine of ornithine, and each product was evaluated for improvements in antifungal activity. 1 and analogues do not exhibit antimalarial, antileishmania, or antibacterial activity; however, the antifungal activity against different strains of fungi was particularly significant. This series of compounds was highly active against Fusarium spp., which represents an opportunistic infection in humans and plants. The in vitro cytotoxicity for the new analogues of 1 was evaluated in the NCI 60 cell panel. Analogue 5 exhibited enhanced potency in several human cancer cell lines relative to 1.
Introduction
The kahalalides are a family of natural depsipeptides that were first isolated from the Hawaiian herbivorous marine sacoglossan mollusk Elysia rufescens (Plakobranchidae) and later from its green algal diet of Bryopsis pennata (Bryopsidaceae).1–8 Kahalalide F (KF,a 1) is the largest and most biologically active cyclic peptide of the 13 natural peptides isolated from E. rufescens. Kahalalides are likely secondary metabolites synthesized by an associated microorganism or from the diet of the green algae B. pennata. 1 is a tridecapeptide with the molecular formula C75H124N14O16 and mass 1477.9408 [M + H]+ composed of a cyclized macrolide region and a linear region with a 5-methylhexanoic acid, conjugated with the N-terminus. Investigations of the structure of 1 revealed a NRPS peptide (cyclo[l-Val(1)-Z-Dhb-l-Phe-d-Val(2)-d-allo-Ileu(1)-d-allo-Thr(1)]-d-allo-Ileu(2)-l-Orn-d-Pro-d-Val-(3)-l-Val(4)-l-Thr(2)-d-Val(5)-5-MeHex) (Figure 1).1,9,10
Figure 1.

Kahalalide F and target functional groups for synthetic modification and lead optimization.
1 exhibits significant in vitro and in vivo antitumor activity in various solid tumor models, including colon (HT-29), breast (T-47D), non-small-cell lung (A-549), and prostate cancer (DU-145). The activity of 1 has been investigated in phase I clinical trials for androgen-refractory prostate cancer11 and in phase II clinical studies with patients having liver cancer, non-small-lung cell cancer (NSCLC), and melanoma.12 Recently, the results of the phase II clinical study of kahalalide F in patients with advanced NSCLC, hepatocarcinoma (HC), and advanced malignant melanoma (AMM) have been reported at the 31st European Society for Medical Oncology Congress (ESMO), revealing an excellent tolerability profile with no serious adverse events.13 A positive response and stable disease were observed in a number of patients.
Lysosomes are intracellular targets of 1. Treatment of tumor cells with 1 resulted in larger vacuoles, and target cells became dramatically swollen because of changes in lysosomal membrane.14 Further investigations consistently showed the loss of lysosomal integrity and the induction of cytoplasmic swelling and vacuolization in breast and prostate cancer cells.15 More recent studies have suggested that inhibition of the HER/neu (ErbB) family signaling is a part of the mechanism of kahalalide F’s action.16 The cytotoxic activity of 1 against several tumor types significantly correlated with protein expression levels of the ErbB3 receptor.17 This specific interaction has been described in a translational program that has confirmed a selective down-regulation of ErbB3 expression by 1 treatment. These findings suggest that ErbB3 may be a potential determinant for 1 sensitivity in patients.18 Trastuzumab (145K amu protein), is a humanized monoclonal antibody that acts on the HER2/neu (ErbB2) receptors. Recently, trastuzumab was used in the treatment of HER2-positive metastatic breast cancer as an anticancer therapy. It is believed that in patients whose tumors overexpress HER2 receptors, trastuzumab can turn off HER2–HER2 homodimers. Unlike available marketed drugs for HER1 (cetuximab [146K amu protein], gefitinib [N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine], and erlotinib [N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)-quinazolin-4-amine]), and HER2 (trastuzumab) related cancers, there is no report regarding HER3 (ErbB3) inhibiting drugs.19
As a result, 1 and its anologues would be promising candidates for inhibition of HER3 receptors in tumor cells. The mechanism of action of 1 on fungi may involve targets similar to the human tumor cells.
Because of the limited availability of the natural product, data regarding structure-activity relationship (SAR) studies of 1 have not been reported in the literature and remain limited to the data generated from isolated natural products, as well as solid-phase total synthesis reported for 1.10,20–22 In a continued investigation of biologically active marine natural products and their optimization using semisynthesis, we were particularly interested in structure–activity relationships of 1 analogues against opportunistic fungal infections of humans and plants.23–25
In this study we focused our attention on the secondary alcohol of threonine and the primary amine of ornithine as the key functional groups for modification beginning with the natural product because they play a crucial role in the bioactivity of this class of compounds.26 Additional semisynthetic modifications and biocatalytic studies will be evaluated in due course.21
We evaluated the activity of the parent compound and its derivatives for antiprotozoal, antibacterial, and antifungal activity focusing on Fusarium spp. because of the challenge of treating these opportunistic infectious diseases.27 Immunocompromised patients with invasive fungal infections have increased in recent decades because of a greater number of transplant patients, more aggressive anticancer chemotherapeutic regimens, and the emergence of AIDS. The therapeutic interventions for fungal infections are limited. The most commonly encountered etiologic agents of systemic fungal infections in immunocompromised patients are Candida spp., Aspergillus sp., Cryptococcus neoformans, Fusarium spp., and Trichosporon spp.28 We focused our attention on the SAR of 1 against fungal infections caused by Fusarium spp. because there are a limited number of drugs active against these fungi. It is also noteworthy that peptides are a well established source of antibiotics including gramicidins, polymyxins, bacitracins, vancomycin, teicoplanin, and echinocandins.29 However, among these only the echinocandin family is active against fungi and is a noncompetitive inhibitor of the synthesis of 1,3-β-d-glucan, a major component of the fungal cell wall.30
Results and Discussion
1. Semisynthesis of Kahalalide F Analogues
For spectroscopic and biological comparison, we first converted 1 to kahalalide G (KG, 2) under mild basic conditions. This reaction also provided valuable information regarding stability of the lactone under basic conditions. Kahalalide F was hydrolyzed at the ester linkage using H2O–MeOH (2:1) in the presence of potassium carbonate at room temperature affording cleavage of the macrolactone to the target ring-opened product 2 (Scheme 1).
Scheme 1.

Hydrolysis of Kahalalide F to Kahalalide Ga
a Reagents and conditions: K2CO3, H2O/MeOH (1:2), room temp, 8 h, 95%.
Our studies continued with the acetylation of kahalalide F. Ramirez et al. reported that the complete acetylation of a vast array of secondary and tertiary alcohols could be readily achieved by treatment with acetic anhydride and BF3•OEt2 at room temperature for a just few seconds.31 However, without control of the reaction time, the formation of an array of acetate species occurred.
Treatment of 1 free base with an excess 2:1 volumetric ratio of acetic anhydride to BF3.OEt2 at room temperature after 5 min afforded KF monoacetate (3) with a 68% yield (Scheme 2). Compound 3 showed a molecular [M + H]+ ion peak at m/z 1519.9478 in high-resolution electrospray ionization mass spectrometry (HRESIMS) in positive mode. 1H NMR of the monoacetate 3 shows that the β-proton of l-threonine shifted downfield to 5.00 ppm. This signal along with the β-proton of d-allo-threonine at 5.04 ppm suggested two ester linkages, which were validated by heteronuclear multiple-bond correlation (HMBC) between β-protons of l-threonine and d-allo-threonine with the corresponding carbonyl groups of acetyl at 169.9 ppm and l-Val-1 at 169.8 ppm, respectively. Further confirmation of O-acetylation was secured by the observation of HMBC correlation between the new carbonyl signal at 169.9 ppm and the methyl singlet at 1.92 ppm.
Scheme 2.

Conversion of 1 to the Acetate and Oxo Analoguesa
a Reagents and conditions: (a) Ac2O, BF3•OEt2, CH2Cl2, room temp, 5 min, 68%; (b) Dess–Martin periodinane (DMP), MeCN, room temp, 1 h, 75%.
The addition of the Dess–Martin periodinane (DMP)32 to a solution of 1 in acetonitrile furnished the oxidation of the secondary alcohol to the corresponding oxo-KF (4) in good yield (75%). The presence of a trace amount of an unknown byproduct was observed during repeated HPLC purification, likely due to the reversible formation of a Schiff’s base between the new carbonyl and the NH2 of ornithine. High-resolution TOF-ESI-MS of analogue 4 provided molecular mass of [M + H]+ at m/z 1475.9237, corresponding to the loss of 2 mass units, in comparison with parent molecule. The presence of a C═O resonance at 200.9 ppm in the 13C NMR spectrum and elimination of the CH–OH signal at 66.9 ppm from the 1 spectrum in 13C NMR and DEPT 135° (distortionless enhancement by polarization transfer, 135°) spectra unambiguously revealed the oxidation of the secondary alcohol of threonine to a ketone group. Furthermore, the loss of the OH singlet and associated methine CH–OH in the 1H NMR provides additional evidence for this transformation.
To gain a better understanding of the structure–activity relationships of 1, we extended the range of 1 derivatives by alkylating the primary amine group of the l-ornithine residue. Synthetic approaches were based on the stepwise reductive N-alkylation of the amino group to the corresponding monoalkylor dialkylamino-KF in the presence of a carboxaldehyde and hydride reducing agent.33,34 This stepwise one-pot procedure involves the initial formation of the intermediate carbinol amine, which dehydrates to form an imine. Subsequent in situ reduction of this imine produces the alkylated amine. The reductive alkylation of 1 with aldehydes was best performed under optimized conditions in which the parent molecule was exposed to 5 equiv of aldehyde in methanol for 30 min at room temperature prior to portionwise addition of 2 equiv of sodium triacetoxyborohydride [NaBH(OAc)3] at the same conditions. The reaction time was varied from a few hours to a couple of days and gave the desired products in good yields; however, the formation of dialkylated amines in low yield was found to be a common side reaction (Scheme 3).
Scheme 3.

Synthesis of Monoalkyl and Dialkyl N-Substituted KF Based on Stepwise Reductive Alkylation in the Presence of a Carboxaldehyde and Triacetoxyborohydridea
a Reagents and conditions: aldehydes, Na(OAc)3BH, MeOH, 3 Å molecular sieves, room temp.
Attempts to accomplish a similar reductive alkylation reaction in the presence of 4-pyridinecarboxaldehyde resulted in a lower yield of 4-pyridinylmethylamino-KF (7) and bis(4-pyridinylmethyl)amino-KF (12) relative to other alkylamino derivatives. We were not able to isolate trace amounts of the dialkylamine moiety. Because of the basic nature of 4-pyridinecarboxaldehyde, the cyclic macrolactone appears to be attacked by this base to give an opened-ring species (13–15), which are detectable only with LC-MS (Figure 2).
Figure 2.

Byproducts resulting from reductive alkylation of 1 and 4-pyridinecarboxaldehyde detected by LC-MS (single charge/double charges unit mass).
In order to protect the parent molecule from further basic ring-opening reactions and formation of undesired byproducts, preliminary stirring of the reaction mixture was eliminated for the reductive alkylation of 1 using 4-pyridinecarboxaldehyde and morpholin-4-ylbenzaldehyde.
The structures of N-alkylated KF analogues were confirmed with spectroscopic (1H NMR and DEPT 135°) and high-resolution ESIMS techniques (Table 1 and Supporting Information).
Table 1.
Selected 1H NMR and DEPT 135° Data for 1 and Its N-Alkylated KF Analogues in DMSO-d6a
 | |||||||
|---|---|---|---|---|---|---|---|
| compd | HRESIMS [M + H]+ |
1H NMR | 13C NMR | ||||
| l-ornithine | derivative (R) residue |
l-ornithine | derivative (R) Residue |
||||
| NHR | CH2(δ) | CH2(δ) | NHCH2R | ||||
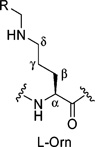
| 1 | 1477.9408 | 7.62 (m, 2H, NH2) | 2.74 (m) | 7.19–7.28 (m, 5H, Ph) | 38.7 | 126.8 (Ph), | |
| 128.5 (2C, Ph), | |||||||
| 129.5 (Ph), | |||||||
| 130.1 (Ph) | |||||||
| 5 | 1585.9769 | 7.60 (m) | 2.91 (m) | 7.18–7.28 (m, 5H, Ph), | 38.5 | 46.6 | 115.9 (Ph), |
| 7.49–7.52 (m, 2H, Ph), | 116.1 (Ph), | ||||||
| 8.77–8.80 (m, 2H, Ph) | 127.0 (Ph), | ||||||
| 128.6 (2C, Ph), | |||||||
| 129.7 (Ph), | |||||||
| 130.3 (Ph), | |||||||
| 132.7 (Ph), | |||||||
| 132.8 (Ph) | |||||||
| 7 | 1568.9814 | 7.63 (m) | 2.93 (m) | 7.21–7.28 (m, 5H, Ph), | 38.5 | 47.6 | 124.7 (2C, Py), |
| 7.49 (m, 2H, Py), | 126.9 (Ph), | ||||||
| 8.66 (s, 2H, Py) | 128.7 (2C, Ph), | ||||||
| 129.7 (Ph), | |||||||
| 130.3 (Ph), | |||||||
| 150.3 (2C, Py) | |||||||
| 8 | 1573.9429 | 7.63 (m) | 2.92 (m) | 7.22–7.29 (m, 5H, Ph), | 38.5 | 47.6 | 126.9 (Ph), |
| 7.10 (m, Th), | 127.8 (Th), | ||||||
| 7.21 (m, Th), | 128.6 (2C, Ph), | ||||||
| 7.57 (m, Th) | 128.7 (Th), | ||||||
| 129.7 (Ph), | |||||||
| 130.2 (Ph), | |||||||
| 130.9 (Th) | |||||||
| 10 | 1562.0334 | 7.59 (m) | 2.85 (bs) | 7.21–7.27 (m, 5H, Ph), | 38.5 | 47.6 | 14.3 (CH3) |
| 0.61–1.55 (m, 13H, aliphatic) | 22.3 (CH2) | ||||||
| 25.8 (CH2) | |||||||
| 26.0 (CH2) | |||||||
| 31.1 (CH2) | |||||||
| 126.9 (Ph), | |||||||
| 128.6 (2C, Ph), | |||||||
| 129.7 (Ph), | |||||||
| 130.2 (Ph) | |||||||
| 11 | 1646.1271 | 2.95 (bs) | 7.18–7.25 (m, 5H, Ph), | 38.4 | 47.6 | 14.3 (CH3) | |
| 0.61–1.55 (m, 26H, aliphatic) | 22.4 (CH2) | ||||||
| 23.4 (CH2) | |||||||
| 23.5 (CH2) | |||||||
| 31.2 (CH2) | |||||||
| 127.0 (Ph), | |||||||
| 128.6 (2C, Ph), | |||||||
| 129.7 (Ph), | |||||||
| 130.2 (Ph) | |||||||
Abbreviations: Ph = phenyl, Py = pyridine, Th = thienyl.
To understand the kinetics and distribution of 1 by fluorescence microscopy, a coumarin-based fluorophore of 1 was prepared through an amidation reaction.35 As shown in Scheme 4, the target fluorescent molecule was synthesized by treatment of DEAC-carboxylic acid and 1 in the presence of EDC and HBTU in DMF at room temperature for 1 h. In this reaction, the primary amine reacted with high chemo- and regioselectivity in the presence of the secondary alcohol and amides giving exclusively the desired DEAC-KF-amide (16) as an intense yellow solid in good yield.
Scheme 4.

Amidation Reaction of Kahalalide F and DEAC Acid in the Presence of a Coupling Agenta
a Reagents and conditions: O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU), N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide (EDC), DMF, room temp, 1 h, 84.9%.
Compound 16 showed a molecular [M + H]+ ion peak at m/z 1721.0282 in HRESIMS. Three additional aromatic protons of this analogue at 6.61 (s, 1H), 6.77 (m, 1H), and 7.67 (m, 1 H) ppm in 1H NMR spectrum along with a chemical shift at 3.48 (m, 4H) ppm for two methylene groups and a characteristic signal at 8.66 (s, 1H) ppm assignable to the 2-pyrone moiety revealed the formation of a new amide linkage between DEAC acid and 1. Further validation of the structure was obtained from the DEPT 135° spectrum at 12.8 (2 × CH3), 44.8 (2 × CH2), 96.4 (Ph), 110.6 (Ph), 132.0 (Ph), 148.2 (pyrone) ppm.
2. Biological Evaluation of Products
2.A. Activity in Opportunistic Infections
Kahalalide D (KD), 2, 1, and its analogues were evaluated in vitro for their activity against several microorganisms that cause opportunistic infections, including Escherichia coli, Pseudomonas aeruginosa, methicillin-resistant Staphylococcus aureus, Candida albicans, Cryptococcus neoformans, Mycobacterium intracellulare, and Aspergillus fumigatus. The antimicrobial assay was conducted in three steps (primary, secondary, and tertiary assays) in order to determine the IC50 and MIC values. If the compound showed no activity in the primary assay, no continuation in secondary and tertiary assays was completed. These depsipeptides do not demonstrate activity against E. coli, P. aeruginosa, or methicillin-resistant S. aureus. No analogues showed activity against M. intracellulare with the exception of 3, which showed inhibition at an IC50 of 14.32 µM, while 8 inhibited at an IC50 of 11.98 µM (Table 2). These data revealed that introduction of a thienylmethyl substitute to the primary amine and acetylation of the secondary alcohol of kahalalide F alter the spectrum of antibacterial activity. Results revealed that modification of the primary amine with thienylmethyl led to improved activity against M. intracellulare compared to acetylation products.
Table 2.
In Vitro Data of Antimicrobial Activitiesa
|
C. albicans ATCC 90028 (µM) |
C. neoformans ATCC 90113 (µM) |
A. fumigatus ATCC 90906 (µM) |
M. intracellulare ATCC 23068 (µM) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| compd | IC50 | MIC | MFC | IC50 | MIC | MFC | IC50 | MIC | MFC | IC50 | MIC | MBC |
| 1 | 3.02 ± 0.04 | 10 | 20 | 1.53 ± 0.38 | 5 | 5 | 3.21 ± 0.05 | 10 | 20 | >20 | >20 | >20 |
| 2 | >13.38 | NT | NT | >13.38 | NT | NT | >13.38 | NT | NT | >13.38 | NT | NT |
| KD | >33.57 | NT | NT | >33.57 | NT | NT | >33.57 | NT | NT | >33.57 | NT | NT |
| 3 | 3.28 ± 0.02 | 10 | 20 | 1.89 ± 0.01 | 5 | 5 | 3.38 ± 0.09 | 10 | 10 | 14.32 ± 0.33 | >20 | >20 |
| 4 | 12.44 ± 0.07 | >20 | >20 | 4.34 ± 0.44 | 10 | 20 | >13.6 | >20 | >20 | >20 | >20 | >20 |
| 5 | >12.6 | >20 | >20 | 3.77 ± 0.09 | 10 | 10 | >12.6 | >20 | >20 | >20 | >20 | >20 |
| 6 | >11.81 | NT | NT | >11.81 | NT | NT | >11.81 | NT | NT | >11.81 | NT | NT |
| 7 | 4.19 ± 0.11 | >20 | >20 | 2.12 ± 0.37 | 5 | 5 | 4.26 ± 0.1 | 10 | 20 | >20 | >20 | >20 |
| 8 | 3.81 ± 0.14 | 10 | 20 | 1.91 ± 0.23 | 5 | 10 | 3.22 ± 0.06 | 10 | 20 | 11.98 ± 0.57 | >20 | >20 |
| 10 | 5.69 ± 0.15 | 20 | 20 | 1.93 ± 0.07 | 5 | 10 | 3.12 ± 0.02 | 10 | 20 | >20 | >20 | >20 |
| 11 | >12.2 | >20 | >20 | 6.71 ± 0.02 | >20 | >20 | >12.2 | >20 | >20 | >20 | >20 | >20 |
| 16 | >11.62 | NT | NT | >11.62 | NT | NT | >11.62 | NT | NT | >11.62 | NT | NT |
| amphotericin B | 0.25 ± 0.04 | 0.625 | 1.25 | 0.79 ± 0.05 | 0.016 | 0.06 | 1.32 ± 0.06 | 2.5 | 2.5 | NT | NT | NT |
| ciprofloxacin | NT | NT | NT | NT | NT | NT | NT | NT | NT | 0.42 ± 0.07 | 1.0 | >1 |
NT = not tested. Amphotericin B (a polyene antifungal drug) and ciprofloxacin (1-cyclopropyl-6-fluoro-4-oxo-7-piperazin-1-ylquinoline-3-carboxylic acid) are used as positive antifungal and antibacterial controls, respectively. The IC50 is the concentration that affords 50% inhibition of growth. MIC (minimum inhibitory concentration) is the lowest test concentration that yields detectable growth. MFC/MBC (minimum fungicidal/bactericidal concentration) is the lowest test concentration that kills 100% of the organism.
Interestingly, 1 and its analogues exhibited significant activity against a fungus type yeast (C. albicans), dimorphic fungus (C. neoformans), and filamentous fungi (A. fumigatus and Fusarium spp.). The in vitro activity against fungi is shown in Table 2.
Peptide 1 as well as several analogues such as, 3, 8, and 10 exhibited activity against C. albicans, C. neoformans, and A. fumigatus. Analogues 4, 5, 7, and 11 show selectivity against certain types of fungi. Analogue 4 was active against C. albicans and C. neoformans, while 7 inhibited C. albicans, C. neoformans and A. fumigatus. Products 5 and 11 were selective against C. neoformans. Kahalalide D, 2, and analogues 6 and 16 showed no activity in the antimicrobial assays and were not subjected to further evaluation. Vero cells at 4760 ng/mL showed no significant toxicity for 1 and its analogues.
2.B. Antiparasitic Activity
1 and its analogues were subjected to evaluation against Plasmodium falciparum and Leishmania donovani. 1 and analogues 8 and 10 were active against Leishmania donovani with IC50 and IC90 values of 17–19 and 34–40 µg/mL. No compounds from this class showed significant antimalarial activity against P. falciparum D6 clone and W2 clone.
2.C. Bioactivity against Mycobacterium tuberculosis
1 and its analogues were evaluated against M. tuberculosis strain H37Rv. All compounds were evaluated at 16 µg/mL. The MIC was determined for compounds that showed greater than 90% inhibition (Table 3). Analogues 3, 8, and 11 were more active than 1 and produced 91–94% inhibition, respectively, while the other analogues were determined to be inactive at 16 µg/mL. Analogue 11 showed better potency than analogues 3 and 8 against M. tuberculosis. In addition, analogue 11 was less active against M. intracellulare, a relatively nonpathogenic model organism for genus Mycobacterium. This activity difference between analogue 11 and analogues 3 and 8 demonstrates a selectivity of analogue 11 against M. tuberculosis. Overall, the findings indicated that O-acetylation of secondary alcohol or N-alkylation of primary amine can significantly improve the activity and selectivity of the parent molecule to the target.
Table 3.
Minimum Inhibitory Concentration (MIC) of 1 Analogues versus Mycobacterium tuberculosis H37Rv Strainsa
| compd | % inhibition at 16 µg/mL | MIC (µg/mL) |
|---|---|---|
| RMP | 93 | 0.08 |
| 1 | 67 | >16 |
| KD | 49 | NT |
| 2 | 29 | NT |
| 3 | 94 | 15.4 |
| 5 | 77 | NT |
| 7 | 24 | NT |
| 8 | 92 | 15.5 |
| 10 | 88 | >16 |
| 11 | 91 | 9.4 |
| 16 | 46 | NT |
NT = not tested. RMP = rifampcin.
2.D. Bioactivity against Fusarium spp
Our interest in the activity of 1 and its analogues against Fusarium spp. is inspired by the fact that a limited number of antifungal agents are effective against Fusarium, and 1 possesses well established antifungal activity. In addition, there is a growing need to identify drugs to alleviate opportunistic fungal infections in immunocompromised patients who become susceptible to a variety of fungal infections caused by Fusarium spp. Two bioassay methods have been employed to examine the activity of 1 and its analogues against Fusarium spp.
2.D.1. Bioautography Assay
Two bioassay methods have been employed to examine the activity of 1 and its analogues against Fusarium spp. Bioautography was utilized as a preliminary screen to detect the antifungal activity and was followed by a microtiter assay. 1 and its analogues were found to be active against F. proliferatum. Modification of structure does not appear to have an affect on the interaction to specific species of Fusarium (Supporting Information). This data shows that oxidation of the secondary alcohol to 4 broadens the spectrum of activity against Fusarium spp. and that all other 1 analogues are ineffective against F. solani and F. oxysporium (Supporting Information).
2.D.2. Microtiter Assay
In contrast with bioautography data, the microtiter assay revealed that 1 and its analogues exhibit activity against F. oxysporium, F. proliferatum, and F. solani at the highest concentration tested (30 µM). Table 4 shows the percent inhibition of Fusarium spp. after treatment with compounds at three different concentrations for 48 and 72 h.
Table 4.
Microtiter Analysis of Kahalalide Analogues at Different Concentrations (48 and 72 h)
| % inhibitiona (0.3 µM) | % inhibitiona (3.0 µM) | % inhibitiona (30 µM) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| compd |
Fusarium oxysporum |
Fusarium proliferatum |
Fusarium solani |
Fusarium oxysporum |
Fusarium proliferatum |
Fusarium solani |
Fusarium oxysporum |
Fusarium proliferatum |
Fusarium solani |
| 1 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 9.9 (0) | 100 (99.6) | 99.8 (100) | 98.9 (99.9) |
| 3 | 0 (0) | 0 (0.3) | 0 (0) | 0 (0) | 0 (0.3) | 0 (0) | 78.5 (74.4) | 70.8 (34.7) | 80.9 (96.7) |
| 4 | 1.8 (0) | 1.8 (0) | 0 (0) | 1.8 (0) | 5.0 (0) | 0 (0) | 91.9 (82.2) | 92.7 (73.2) | 90.6 (85.5) |
| 5 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 16.5 (0) | 96.6 (94.3) | 97.5 (96.1) | 97.3 (97.3) |
| 7 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 98.2 (91.2) | 99.1 (95.3) | 99.2 (97.5) |
| 8 | 0 (0) | 0 (0) | 0 (0) | 13.7 (0) | 4.8 (0) | 47.0 (32.9) | 98.8(99.2) | 98.7 (99.0) | 99.0 (99.3) |
| 10 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 93.6 (89.0) | 95.3 (92.8) | 94.3 (94.7) |
| 16 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| azoxystrobin | 22.7(15.3) | 0 (0) | 16.6 (11.7) | 32.3 (7.1) | 21.7 (14.8) | 25.3 (15.0) | 77.2 (32.9) | 53.0 (40.7) | 62.7 (51.7) |
| captan | 0.2 (0) | 6.0 (3.0) | 0 (0) | 7.6 (0) | 12.0 (0) | 5.1 (0) | 99.5 (98.1) | 100 (100) | 90.7 (47.9) |
| amphotericin B | 0 (0) | 0 (0) | 0 (0.1) | 12.5 (0) | 7.9 (0) | 18.1 (0) | 99.7 (87.1) | 99.8 (79.0) | 100 (98.1) |
Sample results only indicate inhibition. Zero (0) does not indicate the degree of stimulation, only that there was no inhibition. Numbers above are 48 h results. Numbers enclosed in parentheses are 72 h results.
At the lowest concentration (0.3 µM) none of the compounds showed activity except for 4 after 48 h of exposure; however, the activity was lost after 72 h with the exception of azoxystrobin [methyl (E)-2-((6-(2-cyanophenoxy)-4-pyrimidinyl)oxy)-α-(methoxymethylene)benzeneacetate], which caused inhibition of F. oxysporum and F. solani.
At 3 µM, compound 5 showed 16% inhibition against F. solani after 48 h but no activity after 72 h. Analogue 8 inhibited all Fusarium spp. tested, with the highest activity against F. solani, which showed 47% inhibition after 48 h and decreased to 32.9% inhibition after 72 h. Amphotericin B and 1 showed 18% and 9.9% inhibition after 48 h, respectively. Analogue 4 showed inhibition against F. oxysporum and F. proliferatum. Analogue 5 revealed selectivity against F. solani after 48 h of exposure, but the activity was diminished at 72 h. All compounds showed no activity after 72 h at 3 µM with the exception of analogue 8 and azoxystrobin, which remained active against F. solani.
At the highest concentration (30 µM), all compounds except for 16 exhibited activity against Fusarium spp. The compounds provided better activity in comparison to azoxystrobin and captan [3a,4,7,7a-tetrahydro-2-[(trichloromethyl)thio]-1H-isoindole-1,3(2H)-dione]. Activity against F. oxysporium declined after 72 h of exposure in treatments with standard antifungals (amphotericin B and azoxystrobin) as well as analogues with 3, 4, and 10. 1, analogue 8, and captan maintained their antifungal activity after 72 h. The active compounds against F. proliferatum showed more than 90% inhibition except for 3 and azoxystrobin. Captan, 1, 5, 8, and 10 remained active against F. proliferatum after 72 h of exposure at 30 µM. Improved antifungal activity was observed for compound 3, which remained active after 72 h of exposure with an inhibition of 96.7%. All active compounds were observed to be more active than azoxystrobin.
Figure 3 presents graphs of the growth response of F. oxysporium, F. proliferatum, and F. solani after 72 h of exposure to the compounds. Growth stimulation of fungi was observed at 0.3 and 3 µM, while inhibition was shown at 30 µM. Growth stimulation due to the presence of drug is a common phenomenon in antifungal therapy. This observation shows how antifungal therapy given in inappropriate doses will be a growth stimulator rather than inhibitor. More than 90% inhibition was observed in F. oxysporum and F. proliferatum in the presence of 1, 5, 7, 8, and 10 comparable to standards (amphotericin B and captan). All analogues except for 16 caused more than 90% inhibition of F. solani comparable to amphotericin B, while captan and azoxystrobin inhibited less than 50%.
Figure 3.

Growth response of F. oxysporium, F. proliferatum, and F. solani at three different concentrations of analogues.
2.D.3. Fungistatic and Fungicidal Activity
The fungistatic and fungicidal effect of each analogue was examined at 30 µM after 72 h using a microtiter plate assay. The solution from each well was grown on an agar plate free of drug to observe the growth of fungus. The results are shown in Table 5.
Table 5.
Fungistatic (FS) and Fungicidal (FC) Activity of 1 and Its Analogues at 30 µM against Fusarium spp.
| antifungal activity at 30 µM | |||
|---|---|---|---|
| compd |
Fusarium oxysporum |
Fusarium proliferatum |
Fusarium solani |
| 1 | FC | FC | FC |
| 3 | FS | FC | FC |
| 4 | FC | FS | FC |
| 5 | FS | FS | FC |
| 7 | FS | FC | FC |
| 8 | FC | FS | FC |
| 10 | FS | FS | FC |
| 16 | NA | NA | NA |
| azoxystrobin | FS | FS | FS |
| captan | FS | FS | FS |
| amphotericin B | FC | FC | FC |
The results showed that the standard amphotericin B acts as a fungicidal agent against Fusarium spp. while captan and azoxystrobin are fungistatic. 1 analogues were fungicidal against F. solani, but 1 was consistently fungicidal against the three species of Fusarium tested. The analogues showed various fungicidal and fungistatic effects against F. oxysporium and F. proliferatum. From these data it suggests that modification of 1 appears to alter its mode of action against Fusarium spp. Unfortunately, modification of 1 with coumarin (16, our fluorescent probe) eliminated the antifungal activity. Our data indicated that analogues 7 and 8 show promising antifungal activity against Fusarium spp. with potency better than that of the gold standard (amphotericin B) for antifungal therapy (Table 4 and Figure 3). Although the full therapeutic window of these 1 analogues has not been addressed, their activity highlights the potential for development of 1 for antifungal therapy.
2.D.4. Fluoresence and Microscopy Analysis
In order to better understand the effect of 1 and its analogues against Fusarium spp., we investigated the mode of transport of the compound through the fungus. The fluorescence probe provided an excellent tool for tracking the compounds in the cell. We started by observing autofluorescence of the conidia; hyphae and mycelia resulted in no autofluorescence observation (Supporting Information). The fungus was grown in the presence of analogue 16 at 20 µg/mL. Although 16 showed no antifungal activity, it resulted in an interesting fluorescence image under fluorescence microscopy (Figure 4). The pictures generated under light and fluorescence microscopy showed the destruction and clear effect of labeled 1 against F. proliferatum after 3 days of exposure. Parts a and b of Figure 4 show a change of mycelia morphology and fungal cell lysis, while parts c and d of Figure 4 show split hyphae and leakage of mycelia. All these processes can be seen very clearly under a fluorescence microscope relative to light microscope.
Figure 4.

Mycelia of F. proliferatum after 3 days of exposure with fluorescent 16: (a, c) under light microscopy (400×); (b, d) under fluorescence microscopy (400×).
2.E. Cytotoxicity
Four new analogues of 1 (5, 7, 8, and 16) were selected and evaluated for in vitro anticancer drug screening in a panel of 60 human cancer cell lines (the NCI-60) as a part of the Developmental Therapeutics Program at the National Cancer Institute (Supporting Information). These 1 analogues exhibited significant activity against selected cell lines, in particular NSCL, colon, ovarian, and breast cancers and especially prostate cancer (Table 6).
Table 6.
In Vitro Activity Data of Compounds 5, 7, 8, and 16 (µM) against Various Cell Linesa
| paclitaxelb | 1b | 5 | 7 | 8 | 16 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cancer | cell line | GI50 | TGI | GI50 | TGI | GI50 | TGI | GI50 | TGI | GI50 | TGI | GI50 | TGI |
| NSCL | A549/ATCC | 0.004 | 25.11 | 0.135 | 0.302 | 0.161 | 0.340 | 0.189 | 0.425 | 0.166 | – | 13.70 | >100 |
| NCI-H322M | 0.013 | 6.310 | 0.191 | 0.372 | 0.203 | 0.429 | 0.131 | 0.238 | 0.167 | 0.304 | 0.165 | 0.490 | |
| leukemia | RPMI-8226 | 0.002 | 5.012 | 1.738 | 6.918 | 0.475 | 1.780 | 0.638 | 1.890 | 1.270 | 2.760 | 40.10 | >100 |
| colon | COLO 205 | 0.003 | 0.316 | – | – | 0.156 | 0.315 | 0.618 | – | 0.243 | – | 4.520 | >100 |
| HCC-2998 | 0.003 | 0.126 | 0.288 | 0.616 | 0.201 | 0.517 | 0.236 | 0.732 | 0.255 | 0.971 | 2.270 | 11.40 | |
| HCT-15 | 0.158 | 15.85 | 0.269 | 0.741 | 0.151 | 0.289 | 0.492 | 1.560 | 0.416 | 1.360 | 0.964 | 17.70 | |
| HT29 | 0.002 | 0.251 | 0.162 | 0.316 | 0.151 | 0.288 | 0.279 | – | 0.187 | – | 2.390 | >100 | |
| KM12 | 0.004 | 15.85 | 0.182 | 0.363 | 0.179 | 0.379 | 0.152 | – | 0.226 | 0.503 | 0.413 | 3.070 | |
| CNS | SNB-75 | 0.004 | 0.126 | 0.224 | 1.905 | 0.265 | 0.655 | 1.290 | 2.610 | 1.010 | 2.190 | 2.110 | 6.870 |
| melanoma | UACC-257 | 0.040 | 25.12 | 1.023 | 2.818 | 1.250 | 0.249 | 1.310 | 2.430 | 1.440 | 2.820 | > 100 | >100 |
| ovarian | SK–OV-3 | 0.008 | 15.85 | 0.191 | 0.355 | 0.183 | 0.387 | 0.374 | 1.360 | 0.223 | 0.485 | 2.550 | 20.20 |
| renal | ACHN | 0.398 | 12.59 | 1.659 | 3.236 | 1.080 | 2.06 | 1.280 | 2.320 | 1.320 | 2.370 | 26.40 | 69.80 |
| prostate | PC-3 | 0.004 | 10.00 | 0.170 | 0.324 | 0.142 | 0.287 | 0.267 | 0.973 | 0.228 | 0.665 | 3.700 | 21.80 |
| DU-145 | 0.005 | 0.794 | – | – | 0.112 | 0.230 | 0.124 | 0.266 | 0.144 | 0.292 | 0.330 | 1.830 | |
| breast | T-47D | – | – | – | – | 0.161 | 0.333 | – | – | 0.191 | 0.394 | 0.518 | 2.830 |
| HS 578T | 0.003 | 0.100 | 0.162 | 0.479 | 0.212 | 0.695 | 0.162 | 0.444 | 0.427 | 2.130 | 3.030 | >100 | |
Dash (–) = no data. GI50 (50% inhibition of cell growth): the concentration needed to reduce the growth of treated cells to half that of untreated (i.e., control) cells. TGI (100% (total) growth inhibition): the concentration required to completely halt the growth of treated cells.
Developmental Thrapeutics Program NCI/NIH (http://dtp.nci.nih.gov).
The total growth inhibition (TGI) of 1 and its analogues exhibited higher cytotoxic potency than paclitaxel against most of the human cell lines.
In particular, 5 showed better activity than 1 against RPMI-8226, HCC-2998, HT29, SK-OV-3, ACHN, and PC3 cell lines. These findings revealed highly promising improvements associated with the modification of 1. Analogues 7, 8, and 16 showed better activity than 1 against NCI-H322M. Analogues 7 and 8 exhibited better potency than 1 against HCC-2998.
Analogue 5 demonstrates significant in vitro cytotoxicity against all human cancer cell lines in the panel, while amidation of 1 with coumarine (analogue 16) resulted in the loss of activity in most cases. Derivatization of primary amine with 4-fluorobenzyl led to better activity than either 4-pyridinylmethyl or 2-thienylmethyl moieties. Compounds 5, 7, and 8 were shown to be nonselective in the inhibition of human cancer cell lines, while 16 had improved selectively for certain types of cell lines (NCI-322M, HCT 15, KM 12, DU-145, T-47D).
Regarding the activity against NSCL, analogues 5, 7, and 8 caused 50% growth inhibition at 0.161, 0.189, and 0.166 µM, respectively, for A549 while analogue 16 provided an inhibition at higher concentration (GI50 = 13.189 µM). In contrast, analogue 16 showed activity against NCI-H322M with a GI50 of 0.165 µM, comparable to analogue 8 (GI50 = 0.167 µM) and better than 5 (GI50 = 0.203 µM). These data indicated the selectivity of 16 against different types of NSCL cell lines. The same phenomenon was observed in the activity against prostate, breast, and colon cancer cell lines. Analogues 5, 7, and 8 showed potent in vitro cytotoxic activity against a panel of human prostate and breast cancer cell lines, with a GI50 ranging from 0.112 µM (DU-145) to 0.427 µM (HS 578T). Analogue 16 exhibited activity against prostate cell line type DU-145 with a GI50 of 0.330 µM and against breast cancer cell line type T-47D with a GI50 of 0.518 µM.
In summary, this work represents the first solution-phase semisynthetic modification and lead-exploration study of 1. This is also the first optimization of 1 as an antifungal agent against filamentous fungi of the genus Fusarium. These limited lead exploration studies led to the discovery of new analogues of 1 with significant in vitro improvements against selected cancer cell lines and fungi compared to the parent molecule.
Experimental Section
General Experimental Procedures
The 1H and 13C NMR spectra were recorded in DMSO-d6 and MeOD on a Bruker DRX NMR spectrometer operating at 400 MHz for 1H and at 100 MHz for 13C NMR. Chemical shift (δ) values are expressed in parts per million (ppm) and are referenced to the residual solvent signals of DMSO-d6 and MeOD at δH/δC 2.50/39.5 and 3.31,4.78/49.1, respectively. UV and IR spectra were respectively obtained using a Perkin-Elmer Lambda 3B UV/vis spectrophotometer and an AATI Mattson Genesis series FTIR instrument. Optical rotations were measured with a JASCO DIP-310 digital polarimeter. The high-resolution ESI-MS spectra were measured using a Bruker Daltonic (GmbH, Germany) micro-TOF series with electrospray ionization. TLC analysis was carried out on precoated silica gel G254 aluminum plates.
Chemicals
Kahalalide F was prepared according to the previously reported methods with some modifications.1 The animals (Elysia rufescens) were collected by snorkeling at low tide near Black Point, O’ahu, in Hawaii. The ethanolic extract of freeze-dried animals was subjected to flash column chromatography on silica gel (EtOAc/MeOH). Preparative HPLC using a Phenomenex 100 mm RP C8 column (250 mm × 100 mm) and a gradient MeCN (0.05% TFA)/H2O followed by further HPLC purification on an amino column (250 mm × 22 mm) using gradient EtOAc/MeOH afforded 1 as a white amorphous powder. All reagents and solvents were obtained from commercial vendors and were utilized without further purification.
Kahalalide G (2)
A solution of kahalalide F free base (30 mg, 20 µmol) and potassium carbonate (0.2 g) in H2O/MeOH (1:2) (10 mL) was stirred at room temperature for 8 h. The mixture was partitioned between CHCl3/IPA (2:1) and water. The organic layer was dried over Na2SO4, and the solvent was evaporated under reduced pressure. Reversed-phase HPLC using gradient MeCN (0.05% TFA)/water afforded 28.4 mg (95%) of 2 as a white powder.2 Yield 95.0%; [α]25D −12.3° (c 0.20, MeOH); UV λmax (MeOH) 195 nm; IR neat (NaCl) 3281 (s, br), 2966 (s), 2937 (s), 1732 (s), 1636 (s), 1541 (s), 1456 (s), 1204 (s), 1139 (s) cm−1; HRESIMS m/z calcd for C75H127N14O17 [M + H]+ 1519.9498, found 1519.9466.
Monoacetylated-KF (3)
To a suspension of kahalalide F free base (30 mg, 20 µmol) in anhydrous dichloromethane (5 mL) were added acetic anhydride (0.4 mL) and boron trifluoride–diethyl etherate (0.2 mL) at room temperature under a nitrogen atmosphere. The mixture was stirred for 5 min, poured into a cool saturated NaHCO3 solution (20 mL), and extracted with CHCl3/IPA (2:1) (2 × 10 mL). The combined organic layers were dried over Na2SO4, and the solvent was evaporated under reduced pressure. The purification was carried out with HPLC using a Phenomenex Luna RP C8 column (250 mm × 22 mm) eluted with a gradient water/MeCN (0.05% TFA) to give 20.7 mg (68%) of 3. Yield 68.0%; [α]25D −10.5° (c 0.25, MeOH); UV λmax (MeOH) 194, 222 nm; IR neat (NaCl) 3283 (s, br), 2967 (s), 2936 (s), 1735 (s), 1654 (s),1458 (s), 1389 (s), 1140 (s) cm−1; HRESIMS m/z calcd for C77H127N14O17 [M + H]+ 1519.9498, found 1519.9478.
Oxo-KF (4)
To a solution of Dess–Martin periodinane (22 mg, 48 µmol) in anhydrous acetonitrile (10 mL) was added kahalalide F (60 mg, 40 µmol) in one portion, and the mixture was stirred at room temperature for 1 h under nitrogen atmosphere. The reaction mixture was quenched with 10% Na2S2O3/saturated aqueous NaHCO3 (v/v 1:1, 20 mL) and extracted with CHCl3/IPA (2:1) (2 × 20 mL). The combined organic layers were dried over Na2SO4, and the solvents were evaporated under reduced pressure. The residue was subjected to preparative HPLC using a Phenomenex Luna RP C8 column (250 mm × 22 mm) eluted with gradient water/MeCN (0.05% TFA) to afford 44.2 mg (75%) of pure oxidized compound 4 as a white powder. Yield 75.0%; [α]25D −5.4° (c 0.10, MeOH); UV λmax (MeOH) 195 nm; IR neat (NaCl) 3319 (s, br), 2961 (s), 2925 (s), 1731 (s), 1645 (s), 1531 (s), 1455 (s), 1392 (s) cm−1; HRESIMS m/z calcd for C75H123N14O16 [M + H]+ 1475.9236, found 1475.9237.
General Preparation of Compounds 5–11
To a solution of kahalalide F (29.5 mg, 20 µmol) and aldehyde in anhydrous methanol (5 mL) were added 3 Å molecular sieves (2 g), and the mixture was stirred for 30 min at room temperature under argon followed by portionwise addition of sodium triacetoxyborohydride (8.5 mg, 40 µmol) over a 20 min period. The reaction mixture was stirred for period of time described below, quenched with water (20 mL), and extracted with IPA/CHCl3 (1:2) (2 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, and the solvent was removed under vacuum. The resulting residue was purified with preparative HPLC using a Phenomenex Luna RP C8 column (250 mm × 22 mm) and eluted with gradient MeCN (0.05% TFA)/water to yield the corresponding monoalkyl-KF (major) and dialkyl-KF (minor) products as colorless powders.
4-Fluorobenzylamino-KF (5)
Starting material 4-fluorobenzaldehyde (10.6 µL, 100 µmol) was used, and the reaction mixture was stirred for 18 h. Yield 64.5%; [α]25D −11.9° (c 0.25, MeOH); UV λmax (MeOH) 196 nm; IR neat (NaCl) 3281 (s, br), 2966 (s), 2935 (s), 1774 (s), 1655 (s), 1538 (s), 1450 (s), 1204 (s) cm−1; HRESIMS m/z calcd for C82H130FN14O16 [M + H]+ 1585.9768, found 1585.9769.
Bis(4-fluorobenzyl)amino-KF (6)
Yield 3.1%; HRESIMS m/z calcd for C89H134F2N14O16 [M + H]+ 1694.0148, found 1694.0140.
4-Pyridinylmethylamino-KF (7)
Starting material 4-pyridinecarboxaldehyde (9.5 µL, 100 µmol) was used, and the reaction mixture was stirred for 2 days. The preliminary stirring of the reaction mixture for 30 min was not completed. Yield 53.1%; [α]25D −9.1° (c 0.19, MeOH); UV λmax (MeOH) 193 nm; IR neat (NaCl) 3281 (s, br), 2965 (s), 2923 (s), 1741 (s), 1647 (s), 1541 (s), 1523 (s), 1457 (s), 1203 (s), 1139 (s) cm−1; HRESIMS m/z calcd for C81H130N15O16 [M + H]+ 1568.9815, found 1568.9814.
2-Thienylmethylamino-KF (8)
Starting material 2-thiophenecarboxaldehyde (9.2 µL, 100 µmol) was used, and the reaction mixture was stirred for 3 days. Yield 52.3%; [α]25D −8.0° (c 0.25, MeOH); UV λmax (MeOH) 193 nm; IR neat (NaCl) 3283 (s, br), 2966 (s), 2936 (s), 1734 (s), 1648 (s), 1541 (s), 1524 (s), 1458 (s), 1203 (s) cm−1; HRESIMS m/z calcd for C80H129N14O16S [M + H]+ 1573.9426, found 1573.9429.
Bis(2-thienylmethyl)amino-KF (9)
Yield 12.6%; HRESIMS m/z calcd for C85H133N14O16S2 [M + H]+ 1669.9465, found 1669.9461.
n-Hexylamino-KF (10)
Starting material n-hexanal (12.3 µL, 100 µmol) was used, and the reaction mixture was stirred for 1 h. Yield 45.1%; [α]25D −7.2° (c 0.10, MeOH); UV λmax (MeOH) 195 nm; IR neat (NaCl) 3280 (s, br), 2964 (s), 2934 (s), 1732 (s), 1651 (s), 1539 (s), 1456 (s), 1203 (s) cm−1; HRESIMS m/z calcd for C81H137N14O16 [M + H]+ 1562.0331, found 1562.0334.
Di-n-hexylamino-KF (11)
Yield 25.3%; [α]25D −18.2° (c 0.25, MeOH); UV λmax (MeOH) 195 nm; IR neat (NaCl) 3282 (s, br), 2964 (s), 2935 (s), 1732 (s), 1646 (s), 1540 (s), 1457 (s), 1204 (s), 1137 (s) cm−1; HRESIMS m/z calcd for C87H149N14O16 [M + H]+ 1646.1270, found 1646.1271.
DEAC-KF-amide (16)
7-Diethylaminocoumarin-3-carboxylic acid (5.3 mg, 20.3 µmol), HBTU [O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate] (9.2 mg, 24.4 µmol), and EDC [N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide] (4.3 µL, 24.4 µmol) were dissolved in anhydrous DMF (5 mL). A solution of kahalalide F (30.0 mg, 20.3 µmol) in anhydrous DMF (2 mL) was added portionwise over 10 min at 0 °C, followed by stirring for 1 h at room temperature. The reaction was quenched by adding water (10 mL) followed by extraction with IPA/CHCl3 (1:2) (2 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The resulting residue was purified with HPLC using a Phenomenex Luna RP C8 column (250 mm × 22 mm) and eluted with gradient MeCN (0.05% TFA)/water to afford 16 as a white powder. Yield 84.9%; [α]25D −9.1° (c 0.20, MeOH); UV λmax (MeOH) 201, 422 nm; IR neat (NaCl) 3319 (s, br), 2959 (s), 2924 (s), 1731 (s), 1644 (s), 1514 (s), 1455 (s), 1233 (s), 1135 (s) cm−1; HRESIMS m/z calcd for C89H138N15O19 [M + H]+ 1721.0287, found 1721.0282.
Pathogen Production of Fusarium
F. solani (F-0007), F. oxysporium (F-0001), and F. proliferatum (F0029-1) were obtained from USDA-ARS Laboratory, Natural Products Utilization Research Unit, National Center for Natural Products Research, The University of Mississippi. The isolates were isolated from orchid species by D. E. Wedge and identified by Dr. W. H. Elmer, Connecticut Agricultural Experiment Station. The cultures were stored on silica gel in the USDA-NPURU repository. The isolates were found to be pathogenic to orchid species. Fusarium spp. cultures were initiated on potato dextrose agar (PDA, Difco, Detroit, MI) from spores. Conidia were harvested from 7 day old culture by flooding plates with 10 mL of sterile distilled water and dislodging using an L-shape glass rod. Conidial suspensions were filtered through sterile Miracloth (Calbiochem-Novabiochem Corp., La Jolla, CA) to remove mycelia. Concentration of conidia was determined photometrically at 625 nm from a standard curve. The concentration of conidia was 3 × 105 conidia/mL. The conidia suspension was used for one-dimensional direct bioautography and other experiments.
Bioautography Assay
Each compound was dissolved in methanol at 1 mg/mL. An amount of 100 µL of compounds was spotted on a glass thin layer chromatography plate (TLC plate, silica gel GF, 250 µm, 10 cm × 20 cm, Uniplate, Analtech, Inc., Newark, DE) and allowed to dry. The initial concentration of each compound tested was 100 µg/mL. After drying, the plates were sprayed with spore suspensions of Fusarium spp. Conidial solution was prepared with potato dextrose broth 12 g/500 mL, 0.1% bacto agar, and 0.1% Tween-80 and contained 3 × 105 conidia/mL of each Fusarium spp. Plates were placed in a moisture chamber (Pioneer Plastic, Inc., Dixon, KY) and incubated for 3 days at 26 °C. After incubation, plates were removed from the moisture chamber, dried at room temperature, and exhaustively sprayed with 0.25% MTT (3-(4,5-dimethylthiazl-2-yl)-2,5-diphenyltetrazolium bromide, Sigma) prepared in phosphate buffer. The plates were placed back into a moisture chamber and incubated at 26 °C for 1 day and allowed to develop. The active compounds were visualized by clear zones of fungal growth inhibition on TLC plates with purple background. Amphotericin B, captan, benomyl, and azoxystrobin were used as positive controls to evaluate the activity of 1 and its analogues.
Microtiter Assay
1 and its analogues were evaluated for a dose response effect against fungi using a 96-well microdilution broth assay (Nunc, Micro Well, Roskilde, Denmark) at 0.3, 3, and 30 µM against F. proliferatum, F. solani, and F. oxysporium according to the procedure published by Wedge et al.36 Microtiter plates were covered with plastic lids and incubated in a growth chamber at 24 ± 1 °C and 12 h photoperiod under 60 ± 5 µmol m−2 s−1. Fungal growth was monitored photometrically by measuring absorbance at 620 nm at 24, 48, and 72 h. Mean absorbance values and standard error were used to evaluate fungal growth.
Fungicidal and Fungistatic Testing
The fungicidal or fungistatic assay was conducted to determine the action of each compound at 30 µM. After 72 h of incubation in a microtiter assay, 20 µL of solution of each well was cultured onto a 90 mm Petri plate containing 10 mL of potato dextrose agar free of 1 and its analogues. The plates were incubated at 28 °C for 3 days. The fungicidal effect was defined if no fungal growth was observed on the subcultured plate free of compounds, and the fungistatic activity was defined if fungal growth was observed on the subcultured plate.
Fluoresence Assay
In order to observe the effect of 1 and its analogues on Fusarium spp., we tested our fluorescence probe against the fungi. In a 20 mL test tube containing potato dextrose broth and conidia of Fusarium spp. the fluorescence compound was added at 20 µg/mL. The culture was then incubated at 24 ± 1 °C. The microscopic observation was initiated at 4, 8, 16, and 24 h and continued at 48 and 72 h. Photomicrographs of hyphae or mycelia were taken with an Olympus BX-51 microscope equipped with epifluoresence. Pictures were taken at 400× and 1000× magnification.
Mycobacterium Tuberculosis Assay
In vitro evaluation of antimycobacterial activity was conducted with Mycobacterium tuberculosis H37Rv ATCC 27294 in BACTEC 12B medium using microplate Alamar blue assay (MABA). Primary screening is evaluated at 16 µg/mL. Compounds that showed inhibition less than 90% were not tested further. Compounds that show inhibition of greater than 90% are retested at a lower concentration to determine the MIC values.37
Supplementary Material
Acknowledgments
We are grateful to Melissa Jacob, Shabana Khan, and Babu Tekwani from the National Center for Natural Products Research for the antimicrobial, antimalarial, and antileishmania assays. The authors thank Linda Robertson and Jesse Johnson for their assistance in the handling and preparation of Fusarium culture and Harris Dewayne for his assistance in microtiter assay and data processing. We acknowledge Kully Woodruff and April Wright for their assistance in preparation of 1 analogues and Karumanchi V. Rao and Jiangtao Gao for purification of the peptide. We also thank Jennifer Cook, Jeffrey A. Diers, Eustace Winn, and Erin Mayo for their help with collecting the samples. This work was supported by National Institutes of Health (NIAID) Grant 1RO1AI36596, CDC, and NOAA. This investigation was conducted in a facility constructed with support from Research Facilities Improvements Program (Grant C06 RR-14503-01) from the National Center for Research Resources, NIH. Antimicrobial testing was supported by the NIH, NIAID, Division of AIDS, Grant AI 27094, and the USDA Agricultural Research Service Specific Cooperative Agreement No. 58-6408-2-0009.
Footnotes
aAbbreviations: KF, kahalalide F; KG, kahalalide G; KD, kahalalide D; SAR, structure–activity relationship; NSCLC, non-small-cell lung cancer; HC, hepatocarcinoma; AMM, advanced malignant melanoma; CNS, central nervous system; FS, fungistatic; FC, fungicidal; MABA, microplate Alamar blue assay; RMP, rifampcin; Amp B, amphotericin B; Capt, captan; azoxy, azoxystrobin.
Supporting Information Available: Spectroscopic data for 1 and its analogues; activity data of 60 NCI cell lines for compounds 5, 7, 8, and 16; fluorescence images of DEAC-carboxylic acid as a control; and bioautogram images of analogues 3, 4, and 5. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hamann MT, Scheuer PJ. Kahalalide F: a bioactive depsipeptide from the sacoglossan mollusk Elysia rufescens and the green alga Bryopsis sp. J. Am. Chem. Soc. 1993;115:5825–5826. [Google Scholar]
- 2.Hamann MT, Otto CS, Scheuer PJ, Dunbar DC. Kahalalides: bioactive peptides from a marine mollusk Elysia rufescens and its algal diet Bryopsis sp. J. Org. Chem. 1996;61:6594–6600. doi: 10.1021/jo960877+. [DOI] [PubMed] [Google Scholar]
- 3.Goetz G, Nakao Y, Scheuer PJ. Two acyclic kahalalides from the sacoglossan mollusk Elysia rufescens. J. Nat. Prod. 1997;60:562–567. [Google Scholar]
- 4.Horgen FD, de los Santos DB, Goetz G, Sakamoto B, Kan Y, Nagai H, Scheuer PJ. A new depsipeptide from the sacoglossan mollusk Elysia ornata and the green alga Bryopsis species. J. Nat. Prod. 2000;63:152–154. doi: 10.1021/np990402o. [DOI] [PubMed] [Google Scholar]
- 5.Hill RT, Hamann MT, Enticknap J, Rao KV. Kahalalide-producing bacteria. WO 2005/042720. PCT Int. Appl. 2005
- 6.Dmitrenok A, Iwashita T, Nakajima T, Sakamoto B, Namikoshi M, Nagai H. New cyclic depsipeptides from the green alga Bryopsis species; application of a carboxypeptidase hydrolysis reaction to the structure determination. Tetrahedron. 2006;62:1301–1308. [Google Scholar]
- 7.Ashour M, Edrada R, Ebel R, Wray V, Wätjen W, Padmakumar K, Müller WEG, Lin WH, Proksch P. Kahalalide derivatives from the Indian sacoglossan mollusk Elysia grandifolia. J. Nat. Prod. 2006;69:1547–1553. doi: 10.1021/np060172v. [DOI] [PubMed] [Google Scholar]
- 8.Tilvi S, Naik CG. Tandem mass spectroscopy of kahalalides: identification of two new cyclic depsipeptides, kahalalide R and S from Elysia grandifolia. J. Mass Spectrom. 2006;42:70–80. doi: 10.1002/jms.1140. [DOI] [PubMed] [Google Scholar]
- 9.Bonnard I, Manzanares I, Rinehart KL. Stereochemistry of kahalalide F. J. Nat. Prod. 2003;66:1466–1470. doi: 10.1021/np030334c. [DOI] [PubMed] [Google Scholar]
- 10.Lopez-Macia A, Jimenez JC, Royo M, Giralt E, Albericio F. Synthesis and structure determination of kahalalide F. J. Am. Chem. Soc. 2001;123:11398–11401. doi: 10.1021/ja0116728. [DOI] [PubMed] [Google Scholar]
- 11.Rademaker JM, Horenblas S, Meinhardt W, Stokvis E, de Reijke TM, Jimeno JM, Lopez-Lazaro S, Lopez, Martin JA, Beijnen JH, Schellens JHM. Phase I clinical and pharmacokinetic study of kahalalide F in patients with advanced androgen refractory prostate cancer. Clin. Cancer Res. 2005;11:1854–1862. doi: 10.1158/1078-0432.CCR-04-1534. [DOI] [PubMed] [Google Scholar]
- 12.Hamann MT. Technology evaluation: kahalalide F (PharmaMar) Curr. Opin. Mol. Ther. 2004;6:657–665. [PMC free article] [PubMed] [Google Scholar]
- 13.PharmaMar Biopharmaceutical Co. ( www.pharmamar.com). Presented at the 31st European Society for Medical Oncology Congress (ESMO); Istanbul, Turkey. September 29 through October 3, 2006. [Google Scholar]
- 14.Garcia-Rocha M, Bonay P, Avila J. The antitumoral compound kahalalide facts on cell lysosomes. Cancer Lett. 1996;99:43–50. doi: 10.1016/0304-3835(95)04036-6. [DOI] [PubMed] [Google Scholar]
- 15.Suarez Y, Gonzalez L, Cuadrado A, Berciano M, Lafarga M, Munoz A. Kahalalide F, a new marine-derived compound, induces oncosis in human prostate and breast cancer cells. Mol. Cancer Ther. 2003;2:863–872. [PubMed] [Google Scholar]
- 16.Faircloth GT, Smith B, Grant W, Jimeno JM, Garcia-Gravalos L, Scotto K, Shtil A. Selective antitumor activity of kahalalide F, a marine-derived cyclic depsipeptide. Proc. Am. Assoc. Cancer Res. 2001;42:213. [Google Scholar]
- 17.Janmaat M, Rodriguez J, Jimeno J, Kruyt F, Giaccone G. Kahalalide F induces necrosis-like cell death that involves depletion of ErbB3 and inhibition of Akt signaling. Mol. Pharmacol. 2005;68:502–510. doi: 10.1124/mol.105.011361. [DOI] [PubMed] [Google Scholar]
- 18.Jimeno J, Aracil M, Tercero J. Adding pharmacogenomics to the development of new marine-derived anticancer agents. J. Transl. Med. 2006;4:1–9. doi: 10.1186/1479-5876-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Jarvis LM. Battling breast cancer by targeting multiple HER-family receptors, small-molecule drugs could fill the therapeutic gaps left by Herceptin. Chem. Eng. News. 2006;84(32):21–27. [Google Scholar]; (b) Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 2003;100(15):8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Albericio PF, Giralt LE, Jiménez GJ-C, Lopez MA, Manzanares I, Rodrigues I, Royo EM. Kahalalide compounds. WO 01/58934. PCT Int. Appl. 2001
- 21.Albericio PF, Fernandez DA, Giralt LE, Gracia CC, Lopez RP, Varon CS, Cuevas MC, Lopez MA, Francesca SA, Jiménez GJ-C, Royo EM. New antitumoral compounds. WO 2005/023846. PCT Int. Appl. 2005
- 22.Gracia C, Isidro-Llobet A, Cruz LJ, Acosta GA, Alvarez M, Cuevas C, Giralt E, Albericio F. Convergent approaches for the synthesis of the antitumoral peptide, kahalalide F. Study of orthogonal protecting groups. J. Org. Chem. 2006;71:7196–7204. doi: 10.1021/jo060976f. [DOI] [PubMed] [Google Scholar]
- 23.Faircloth GT, Elices M, Sasak H, Aviles Marin PM, Cuevas Marchante MDC. Preparation of kahalalide antitumoral compounds. WO 2004/035613. PCT Int. Appl. 2004
- 24.Peng J, Shen X, El Sayed KA, Dunbar DC, Perry TL, Wilkins SP, Hamann MT, Bobzin S, Huesing J, Camp R, Prinsen M, Krupa D, Wideman MA. Marine natural products as prototype agrochemical agents. J. Agric. Food Chem. 2003;51:2246–2252. doi: 10.1021/jf0207880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheuer PJ, Hamann MT, Gravalos DG. Cytotoxic and antimicrobial activities of kahalalide F from Elysia rufescens. 6274551. U.S. Patent. 2001
- 26.(a) Patai S. The Chemistry of Amino, Nitroso, Nitro and Related Groups. Wiley; New York: 1996. [Google Scholar]; (b) Salvatore RN, Yoon CH, Jung KW. Synthesis of secondary amines. Tetrahedron. 2001;57:7785–7811. [Google Scholar]; (c) Insaf SS, Witiak DT. Facile non-racemizing route for the N-alkylation of hindered secondary amines. Synthesis. 1999;3:435. [Google Scholar]
- 27.Arikan S, Rex JH. New agents for the treatment of systemic fungal infections—current status. Expert Opin. Emerging Drugs. 2002;7:3–32. doi: 10.1517/14728214.7.1.3. [DOI] [PubMed] [Google Scholar]
- 28.De Lucca AJ. Antifungal peptides: potential candidates for the treatment of fungal infections. Expert Opin. Invest. Drugs. 2000;9:273–299. doi: 10.1517/13543784.9.2.273. [DOI] [PubMed] [Google Scholar]
- 29.Hancock REW, Chapple DS. Peptide antibiotics. Antimicrob. Agents Chemother. 1999;43:1317–1323. doi: 10.1128/aac.43.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanden Bossche H. Echinocandins, an update. Expert Opin. Ther. Pat. 2002;12:151–167. [Google Scholar]
- 31.Martinez-Pascual R, Vinas-Bravo O, Meza-reyes S, Iglesias-Arteaga MA, Sandoval-Ramirez J. A fast and convenient procedure for the acetylation of alcohols. Synth. Commun. 2004;34:4591–4596. [Google Scholar]
- 32.Meyer SD, Schreiber SL. Acceleration of the Dess–Martin oxidation by water. J. Org. Chem. 1994;59:7549–7552. [Google Scholar]
- 33.(a) Borch RF, Bernstein MD, Durst HD. Cyanohydridoborate anion as a selective reducing agent. J. Am. Chem. Soc. 1971;93:2897–2904. [Google Scholar]; (b) Bomann MD, Guch IC, DiMare M. A mild, pyridine-borane-based reductive amination protocol. J. Org. Chem. 1996;60:5995–5996. [Google Scholar]; (c) Szardenings AK, Burkoth TS, Look GC, Campbell DA. A reductive alkylation procedure applicable to both solution- and solid-phase syntheses of secondary amines. J. Org. Chem. 1996;61:6720–6722. doi: 10.1021/jo960124n. [DOI] [PubMed] [Google Scholar]
- 34.(a) Gribble GW, Ferguson DC. Reactions of sodium borohydride in acidic media. Selective reduction of aldehydes with sodium triacetoxyborohydride. J. Chem. Soc., Chem. Commun. 1975:535–536. [Google Scholar]; (b) Nutaitis CF, Gribble GW. Chemoselective reduction of aldehydes with tetra-n-butylammonium triacetoxyborohydride. Tetrahedron Lett. 1983;24:4287. [Google Scholar]; (c) Gribble GW. In: Encyclopedia of Reagents for Organic Synthesis. Paquette LA, editor. Vol. 7. New York: John Wiley and Sons; 1995. p. 4649. [Google Scholar]
- 35.Sandler JS, Fenical W, Gulledge BM, Chamberlin AR, La Clair JJ. Fluorescent profiling of natural product producers. J. Am. Chem. Soc. 2005;127:9320–9321. doi: 10.1021/ja042756u. [DOI] [PubMed] [Google Scholar]
- 36.Wedge DE, Kuhajek JM. A microbioassay for fungicide discovery. SAAS Bull. Biochem. Biotechnol. 1998;11:1–7. [Google Scholar]
- 37.Collins L, Franzblau SG. Microplate Alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.