

Abstract
Objective:
To generate a clinical and pathologic phenotype of patients carrying rare loss-of-function mutations in ABCA7, identified in a Belgian Alzheimer patient cohort and in an autosomal dominant family.
Methods:
We performed a retrospective review of available data records, medical records, results of CSF analyses and neuroimaging studies, and neuropathology data.
Results:
The mean onset age of the mutation carriers (n = 22) was 73.4 ± 8.4 years with a wide age range of 36 (54–90) years, which was independent of APOE genotype and cerebrovascular disease. The mean disease duration was 5.7 ± 3.0 years (range 2–12 years). A positive family history was recorded for 10 carriers (45.5%). All patient carriers except one presented with memory complaints. The 4 autopsied brains showed typical immunohistochemical changes of late-onset Alzheimer disease.
Conclusions:
All patients carrying a loss-of-function mutation in ABCA7 exhibited a classical Alzheimer disease phenotype, though with a striking wide onset age range, suggesting the influence of unknown modifying factors.
Alzheimer disease (AD) is the most frequent cause of neurodegenerative dementia.1 Genome-wide association studies (GWAS) have identified over 20 risk genes that are potentially implicated in AD pathogenesis, though their functional role and significance still has to be fully elucidated.2–4 In one risk gene, the ATP-binding cassette subfamily A member 7 (ABCA7), rare loss-of-function (LOF) mutations were reported that were associated with AD in an Icelandic population, and subsequently replicated in AD cohorts from Finland, Germany, Norway, and the United States.5 Independently, rare deleterious coding mutations in ABCA7 were identified in a Caucasian late-onset AD cohort.6
In our Belgian AD cohort, ABCA7 was significantly associated with disease risk and subsequent targeted resequencing identified 7 different LOF mutations in ABCA7 in patients with AD in ABCA7 that were absent in control individuals.7 One frameshift mutation of 7 bp (p.E709fs), leading to a premature stop codon, cosegregated with disease in an extended autosomal dominant AD family, DR170, and was also present in 10 seemingly unrelated patients with AD who all shared the same haplotype, supportive of a genetic relationship to a distant ancestor. Together, the data indicated that haploinsufficiency is the underlying cause of AD in ABCA7 mutation carriers. How this leads to AD remains unclear, but ABCA7 is highly expressed in hippocampal microglial cells and is involved in lipid metabolism, microglial phagocytosis, and β-amyloid precursor protein (APP) processing.8 Here we report on the clinical and pathologic characteristics of the Belgian ABCA7 mutation carriers.
METHODS
Study population.
The Belgian AD cohort consisted of 955 patients (65.9% [n = 629] women, mean onset age 74.6 ± 8.9 years, 50.8% [n = 485] APOE ε4+) selected from a larger prospective memory clinic–based cohort of Belgian patients with AD.7 All patients received a consensus diagnosis of possible, probable, or definite AD according to the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association9 or the National Institute on Aging–Alzheimer's Association diagnostic criteria.10–12 A positive familial history (i.e., at least one first-degree relative with an AD diagnosis) was noted in 15.6% (n = 149) of patients. Based on the onset age (≤65 years), 16.0% (n = 124) were classified as early-onset AD.
A second cohort consisted of 347 Belgian patients with AD (52.2% [n = 181] women, mean age at referral 62.6 ± 10.0 years, 43.5% [n = 151] reported familial history) referred to our molecular diagnostic unit for PSEN1, PSEN2, or APP screening. Of these, 57.9% (n = 201) were classified as early-onset AD. All patients in this cohort were analyzed for mutations in causal AD genes (i.e., PSEN1, PSEN2, APP) and frontotemporal lobar degeneration genes (C9Orf72, MAPT, GRN, VCP) as well as APOE through a MASTR assay or by Sanger sequencing on genomic DNA.
Standard protocol approvals, registrations, and patient consents.
All participants or their legal guardians signed informed consent forms for participation in the clinical and genetic studies. Participants for whom autopsied brain material was available gave written informed consent for inclusion in pathologic studies. All investigations were approved by the Ethics Committee of the University of Antwerp, the Antwerp University Hospital, and the Hospital Network Antwerp (ZNA), Belgium.
Molecular genetic analyses.
In this study, we genotyped the p.E709fs mutation in genomic DNA by means of PCR-based amplification followed by Sanger sequencing as previously described.7 A panel of polymorphic simple tandem repeat markers covering the ABCA7 locus, as well as single nucleotide polymorphism genotype data in the locus, were used as previously described to examine allele and haplotype sharing among p.E709fs mutation carriers.7
Clinical analyses.
Medical records of ABCA7 mutation carriers were reviewed. Onset age was defined as onset of the first symptomatology (e.g., memory problems) as reported through history from the patient or the informant. Disease duration was measured from symptom onset until death. Structural or functional neuroimaging was performed and interpreted at the referring neurologic center. CSF AD biomarkers (Aβ1-42, total tau, and p-tau181P) were determined as previously described.13
Neuropathologic analyses.
Available neuropathology data of ABCA7 mutation carriers was reviewed and autopsied brain reexamined. After fixation in 10% buffered formalin, brains were embedded in paraffin. Sections were prepared from the cingulate gyrus, frontal superior gyrus, temporal superior gyrus, hippocampus, area striata, mesencephalon, pons, and cerebellum. Histology was performed using Cresyl violet, hematoxylin & eosin, Bodian, and Klüver-Barrera. Immunohistochemical analysis was performed with Ubiquitin (Dako, Glostrup, Denmark), AT8 (Innogenetics, Zwijnaarde, Belgium), 4G8 (Signet, Dedham, MA), TDP-43 (Proteintech Group Inc., Chicago, IL), FUS (Proteintech Group Inc.), and P62 (BD Diagnostics, Erembodegem, Belgium).
RESULTS
We identified 22 patients carrying a LOF mutation in ABCA7, 18 by sequencing of ABCA7 in the Belgian AD cohort,7 and 4 in this study by targeted mutation screening for the founder mutation, p.E709fs, in a group of patients with AD who were referred for medical genetic testing (tables 1 and 2 and figure e-1 on the Neurology® Web site at Neurology.org). Fifteen patients carried the p.E709fs mutation; the others carried a nonsense (n = 2), frameshift (n = 3), or splicing mutation (n = 2).
Table 1.
ABCA7 loss-of-function mutations in Belgian patients with Alzheimer disease
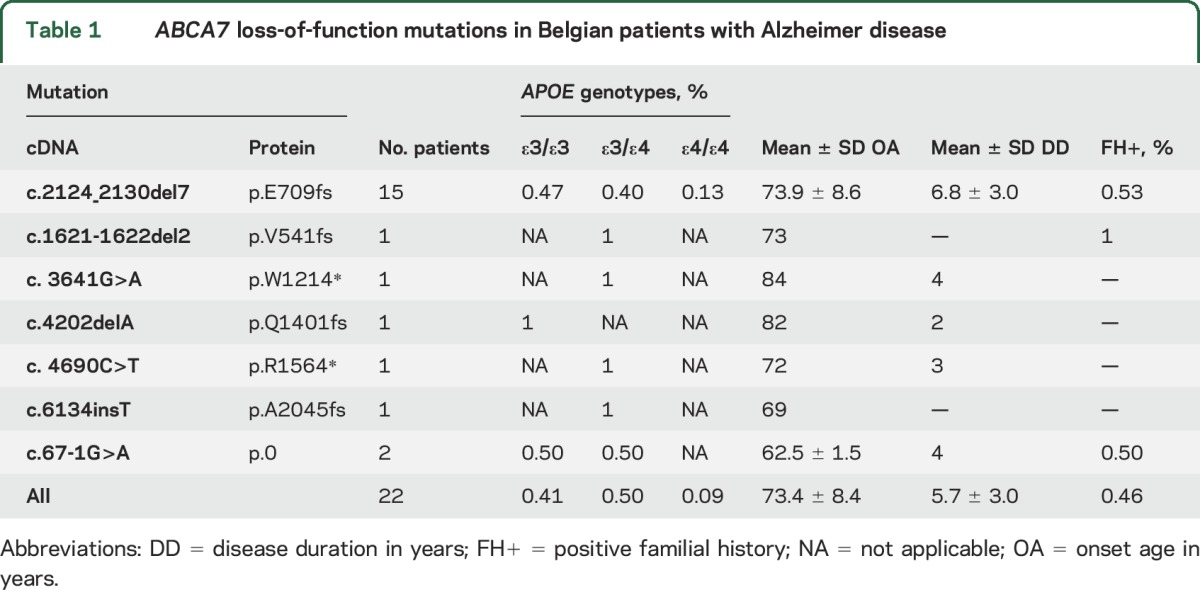
Table 2.
Clinical characteristics of Belgian patients carrying an ABCA7 loss-of-function mutation
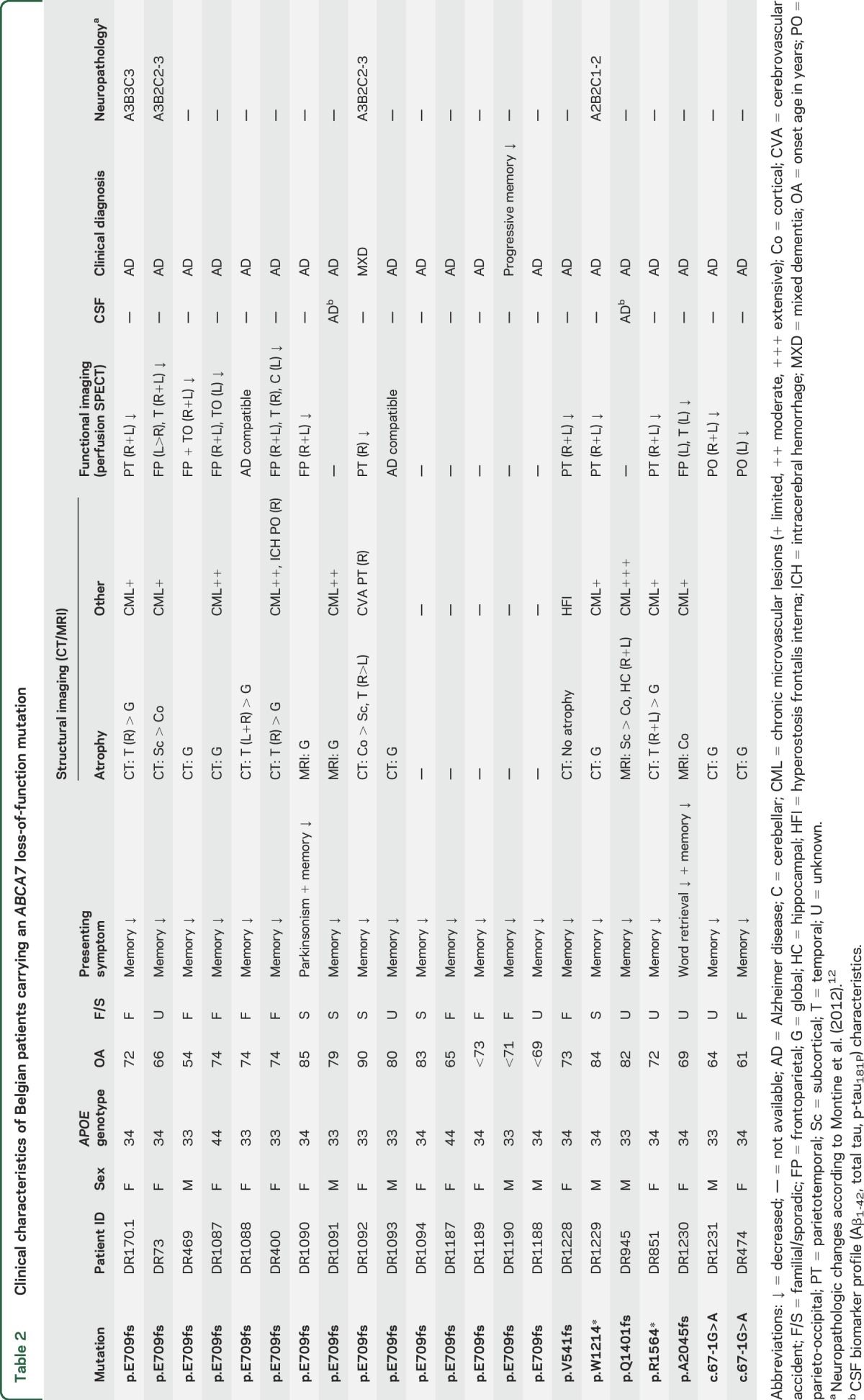
Clinical characteristics.
Mean onset age (n = 19), or when not available age at referral (n = 3), was 73.4 ± 8.4 years with a wide range of 36 (54–90) years. The mean disease duration was 5.7 ± 3.0 years with a range of 10 (2–12) years. All patients presented with progressive memory impairment except for DR1090, who had onset of nonprogressive asymmetric rigidity in the limbs and a festinated gait, which responded to low-dose levodopa, 1 year before onset of memory complaints (table e-1). Clinical criteria for Lewy body dementia were not met.14 In 3 other patients (DR170.1, DR400, and DR1094), mild extrapyramidal signs developed during the disease course. In DR170.1, a mild symmetric rigidity was noted in the advanced dementia stage, whereas DR400 and DR1094 showed mild symmetric extrapyramidal signs after initiation of antipsychotics. In DR1230, the onset of memory problems was accompanied by word-finding difficulties that were the initial predominant clinical feature. Further progression of the disease was typical of AD, with memory impairment dominating the clinical picture.
An extensive neuropsychological examination was available for 17 patient carriers showing marked amnesia as well as at least one other affected cognitive domain. One patient, DR1092, showed a slight asymmetry in her neurocognitive profile, which, in combination with the vascular lesions visualized on brain CT and perfusion SPECT, led to a mixed dementia diagnosis. In DR945, the neurocognitive profile showed evidence of both cortical and subcortical dysfunction. Due to the presence of mild visual hallucinations and delusions, as well as the observation of slightly fluctuating attention, an initial differential diagnosis of Lewy body dementia was made.
Frontal signs in the neurocognitive profile were noted in 7 (7/17, 41.2%) patients, although these were not the presenting symptom. In addition, clinical observation revealed 13 patients (13/17, 76.5%) showing signs of disinhibition (n = 9) or marked behavioral disturbances (n = 9) during their disease course. Frontal release signs at clinical examination were noted in 4 patients (4/17, 23.5%). Language difficulties were present in 10 patients (10/17, 58.8%), most notably in the form of word-finding difficulties (n = 8) or diminished verbal fluency (n = 4). Six patients (6/17, 35.3%) showed marked signs of depression, with one patient exhibiting a pseudobulbar affect with uncontrollable crying. Delusions were present in 4 patients (4/17, 23.5%), with 3 patients (3/17, 17.6%) exhibiting paranoid delusions. Visual hallucinations were present in 3 patients (3/17, 17.6%), although in DR1090 visual impairment due to advanced bilateral cataract was an important differential diagnosis. In 2 patients (2/17, 11.8%), epileptic seizures occurred during the disease course. DR474 had a generalized seizure, presumed to be of a neurodegenerative etiology, in the late-stage dementia phase, whereas in DR400 generalized seizures occurred secondary to an intracerebral hemorrhage.
Neuroimaging characteristics.
Brain CT protocols were available for 17 carriers (table 2). In accordance with disease progression, diffuse (sub)cortical atrophy was noted, in 3 patients (3/17, 17.6%) more striking in the (medial) temporal region. Brain MRI was performed in 4 patients, showing diffuse cerebral atrophy with variable signs of chronic cerebrovascular disease. In DR945, bilateral hippocampal atrophy was evident as well (figure e-2). Brain perfusion SPECT was performed in 15 patients and mainly reflected a relative hypoperfusion in the parietotemporal regions, with relatively preserved perfusion in the sensorimotor cortex. In 10 patients (10/15, 66.7%), the relative hypoperfusion extended to the frontal regions. Of note, SPECT imaging in DR400 revealed a bilateral frontoparietal hypoperfusion, suggestive for a frontal lobe dementia. In addition, asymmetric hypoperfusion in the right-sided temporal lobe and the contralateral cerebellar hemisphere (crossed cerebellar diaschizis), compatible with an old infarction in the right temporal lobe as previously visualized on brain CT, was noted. However, based on the clinical phenotype, the diagnosis of AD was retained.
CSF analysis.
In 2 patients (DR1091 and DR945), CSF AD biomarker analyses were performed. Both biomarker profiles were characteristic of AD with decreased CSF Aβ1-42 levels and increased CSF total tau and p-tau181P levels. This profile is known to have a high sensitivity and specificity for AD with diagnostic accuracy exceeding 80% in the setting of established cognitive impairment.13 Of note, the total tau level in DR1091 was very high (1,460 pg/mL), with an increased p-tau181P level of 145.7 pg/mL, but no clinical features suggestive for recent stroke or Creutzfeldt-Jakob disease were present.
Neuropathologic characteristics.
In 4 patient carriers, brain autopsy had been performed (table 2 and figure 1). Three patients, DR170.1, DR1092, and DR73, carried the p.E709fs mutation in ABCA7, whereas DR1229 carried a p.W1214* mutation. Both mutations lead to a premature stop codon and subsequent degradation of the mutant transcript by the nonsense-mediated RNA decay control mechanism.7
Figure 1. Neuropathology findings in ABCA7 loss-of-function mutation carriers.

The 4G8 staining shows extensive amyloid angiopathy in 3 out of 4 patients (A) and extensive amyloid senile plaques (B) in the frontal cortex. Classic neurofibrillary tangles are present in the hippocampal CA1 (C [AT8 stain]). In photographs (D) through (F), the superficial cortical layers of the entorhinal cortex are shown. Multiple neuronal intracytoplasmic inclusions stain with p62 (D), AT8 (E), and TDP-43 (F).
Patients DR170.1, DR1092, and DR1229 had cortical atrophy with neuronal loss in CA1, the subiculum, and entorhinal cortex, which for DR1229 was also evident in CA2 and the transentorhinal cortex. In DR73, severe atrophy with neuronal cell loss was present in the frontal and temporal cortex, as well as the hippocampus and parahippocampal structures.
DR170.1 had high amounts of senile plaques (SP) in the cortex and molecular layer of the cerebellar cortex, with sporadic SP in the reticular formation of the mesencephalon and pons. In DR73, very large amounts of SP in the cortex and to a lesser extent in the cerebellar cortex, mesencephalon, and pons were present. In DR1092, a maximum of diffuse and classic SP was found in the cingulate gyrus, frontal superior gyrus, temporal superior gyrus, hippocampus, and area striata, while SP in the molecular cortical layer of the cerebellum and the reticular formation were less in number. DR1229 had a severe Aβ immunoreactivity with diffuse and neuritic SP in the cortex, cerebellum, and reticular formation of the mesencephalon and pons. A severe amyloid angiopathy was present in DR73 and DR1229, and to a lesser extent in DR1092, but was absent in DR170.1.
Tau pathology was evident in DR170.1 with small neurofibrillary tangles (NFT) and neuronal intracytoplasmic inclusions (NCI) in the upper cortical layers and classic NFT in the deeper cortical layers. In DR1092, typical NFT were also present in the deeper cortical layers, while in the superficial layers, the neuronal inclusions resembled NCI. Sporadic NFT were found in the reticular formation of the mesencephalon. In DR73, a large amount of NFT (and NCI-like) pathology was present in every part of the cortex. The classical NFT as well as the small NCI-like NFT in the upper cortical layers also stain with AT8 and TDP-43. The fused in sarcoma staining was negative. In DR1229, a moderate amount of NFT, neuritic threads (NT), and dystrophic neurites (DN) were found in all examined cortices, more so in the allocortex. The substantia nigra, locus ceruleus, and reticular formation of the mesencephalon and pons were mildly affected. TDP-43 showed occasional NCI, more pronounced in the superficial cortical layers. NCI were seldom found in the dentate gyrus of the hippocampus.
ABCA7 p.E709fs founder mutation.
In total, we identified 15 apparently unrelated patients with AD carrying p.E709fs. This mutation segregated in AD family DR170 with autosomal dominant inheritance (table 3 and figure 2).7 All 14 carriers of p.E709fs, unrelated to DR170, shared the same disease haplotype that segregated in family DR170, indicative of a founder effect of p.E709fs in the Belgian population. All p.E709fs carriers received a probable (n = 12) or definite AD (n = 3) diagnosis, except for the 3 at-risk individuals (III-1, III-6, and III-2) of family DR170, who were diagnosed with subjective or mild cognitive impairment (MCI). All 3 presently report memory problems at age 73 (III-1), 78 (III-6), and 74 (III-2) years. For carriers III-1 and III-6, history and cognitive screening was compatible with MCI, with an onset age of 71 (III-1) and 75 (III-6) years. The Montreal Cognitive Assessment (MoCA) showed abnormal results within the MCI range 19–25 for III-1 (MoCA score of 21/30) and III-6 (MoCA score of 23/30), who both made mistakes on the Clock Drawing Test and the delayed recall task. Carrier III-2 scored within the normal range (MoCA score of 28/30), although she lost 2 points on the delayed recall task and made a mistake on the Clock Drawing Test, which was self-corrected. Of note, II-2 and II-4, who are obligate p.E709fs carriers based on the segregation data in family DR170, were also diagnosed with AD with an onset age of 80 (II-2) and 70 (II-4) years. Three other family members (I-1, II-6, and III-4) were diagnosed with AD, at age 80 (I-1), 78 (II-6), and 59 (III-4) years. No biosamples were available and segregation analysis was uninformative to investigate their p.E709fs carrier status. Family member III-5, in whom molecular genetic analysis confirmed the absence of p.E709fs, was diagnosed with Parkinson disease at age 75 years.
Table 3.
Belgian Alzheimer disease (AD) family DR170 segregating the ABCA7 p.E709fs founder mutation
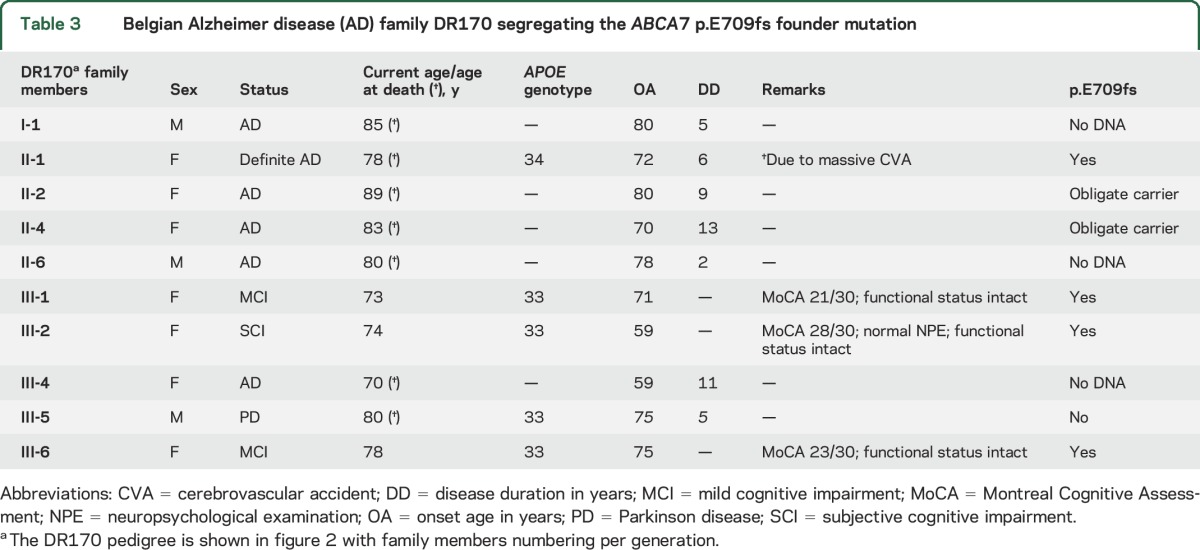
Figure 2. Alzheimer disease (AD) family DR170 segregating the ABCA7 p.E709fs founder mutation.

Dark filled symbols indicate patients with AD, the half-filled symbol indicates mild cognitive impairment (MCI), and the quarter-filled symbol indicates subjective cognitive impairment (SCI). The arrow indicates the proband of family DR170. The 5 individuals for whom we have DNA available are marked by an asterisk. The open circle indicates a patient with Parkinson disease (PD). AAO = age at onset in years.
DISCUSSION
We provide a detailed clinicopathologic description of patients carrying an ABCA7 LOF mutation. The clinical AD diagnosis in the patient carriers appears strong, since most exhibited a classical AD phenotype with conforming technical investigations and strict application of diagnostic criteria.9–12 Almost all patients were seen in a specialized memory clinic by neurologists with extensive experience in cognitive impairment. Moreover, in all 4 carriers in whom brain autopsy was performed, the clinical AD diagnosis was confirmed neuropathologically.
However, 4 (18.2%) patients had an initial differential diagnosis of vascular dementia (DR1092), frontotemporal dementia (DR400), or dementia with Lewy bodies (DR945 and DR1090). In DR1092, autopsy confirmed the presence of SP and NFT, the diagnostic hallmark pathologic lesions of AD. In DR400, an initial differential diagnosis with frontotemporal dementia was made, but during further progression of disease, a diagnosis of AD, with frontal signs, remained most likely. Some degree of frontal dysfunction has been known to occur frequently in the Belgian AD population, with up to 97% of patients with AD exhibiting at least one frontal lobe symptom.15 In patients DR945 and DR1090, dementia with Lewy bodies was considered, in part due to the presence of visual hallucinations, although they were not markedly present during the clinical evaluation. Of note, a recent study in a Belgian cohort of patients with MCI and AD found that psychosis-related symptoms (such as delusions or hallucinations) were present in up to 40% of patients with AD.16 In patient DR1090, the onset of memory problems was also preceded by mild parkinsonian features, which remained stable during the disease course under an unchanging dose of levodopa. A consensus diagnosis of AD was made, although the possibility of a concomitant Lewy body pathology could not be excluded.
The progression of disease in ABCA7 LOF patient carriers (mean disease duration of 5.7 ± 3.0 years) seems to be slightly shorter compared to the complete Belgian AD patient cohort (6.9 ± 3.9 years). However, clinical significance seems unlikely, and the normal life expectancy and late onset in most carriers could have interfered with this observation. However, the observation of a wide range in onset age (36 years) does seem clinically significant. When considering potential modifiers of onset age such as APOE genotype and signs of vascular damage on neuroimaging, we could not provide a clear explanation for this observation. This could be due to another strong modifier or the additive effect of different modifiers with minor effect sizes. For example, patient DR469, who had an early onset age of 54 years, was previously shown to also carry 2 rare mutations in the AD GWAS risk gene clusterin, CLU (p.T445_D447del and p.A309T), though the functional and biological significance of these mutations is not fully elucidated.17 Both CLU and ABCA7 have been linked to lipid metabolism, neuroinflammation, and the amyloid cascade and possibly share a pathogenic pathway. Further investigations are needed to see if an interaction or additive effect of genetic variation in ABCA7 and CLU, or in other AD risk genes, exist and influence disease phenotype.
Neuropathologic findings were typical of AD, showing severe amyloid pathology in the hippocampus and cortex. In addition, 3 out of 4 patients showed a severe amyloid angiopathy although there were no clear signs thereof on antemortem neuroimaging. To a lesser extent—although still severe—tau pathology was present, mainly characterized by NFT, NT, and DN, but in the superficial cortical layers NFT were small and had many similarities with NCI. Interestingly, these NCI have a tendency to costain with TDP-43. Further investigations are needed to see if this is a genuine colocalization. Of note, TDP-43 immunoreactivity was reported to occur in up to 57% of patients with AD.18
Based on our findings, the clinicopathologic findings in ABCA7 LOF mutation carriers seem to conform with a classical AD phenotype. Notwithstanding the later onset age in most ABCA7 carriers, we observed a higher familial load (45.5% with positive familial history for AD) when compared to the complete Belgian AD patient cohort (23%). Therefore, genetic diagnostic testing for ABCA7 mutations in familial late-onset AD analysis might be considered, particularly in the context of a founder effect, as we observed in Belgium. Nonetheless, ABCA7 LOF mutations in AD are rare events with a prevalence in the Belgian AD cohort of 1.2% for the founder mutation and 0.9% for all other pooled LOF mutations. Consequently, the number of ABCA7 mutation carriers we described in this study is low and generalization of the genotype–phenotype findings should be avoided until additional carriers have been studied.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the participants of the study for their cooperation; the personnel of the Genomic Service Facility and the Diagnostic Service Facility at the VIB Department of Molecular Genetics and the Antwerp Biobank of the Institute Born Bunge; and Dr. J. Goethals and the clinical staff of the different neurological centers that are part of the Belgian Neurology (BELNEU) consortium who contributed to the sampling and clinical diagnoses of the participants in this study.
GLOSSARY
- ABCA7
ATP-binding cassette subfamily A member 7
- AD
Alzheimer disease
- APP
β-amyloid precursor protein
- DN
dystrophic neurites
- GWAS
genome-wide association studies
- LOF
loss of function
- MCI
mild cognitive impairment
- MoCA
The Montreal Cognitive Assessment
- NCI
neuronal intracytoplasmic inclusions
- NFT
neurofibrillary tangles
- NT
neuritic threads
- SP
senile plaques
Footnotes
Contributor Information
Collaborators: Belgian Neurology Consortium, Dirk Nuytten, Patrick Santens, Jan de Bleeker, Bart Dermaut, Olivier Deryck, Bruno Bergmans, Alex Michotte, Jan Versijpt, Christiana Willems, Jean Delbeck, Adrian Ivanoiu, and Eric Salmon
AUTHOR CONTRIBUTIONS
Conception and design of the study: T.V.d.B., K.S., C.V.B. Acquisition and analysis of data: T.V.d.B., K.S., E.C., S.E., A.S., A.D.R., C.V.C., S.V., M.V.d.B., A.L., K.P., M.M., M.V., R.V., J.-J.M., P.P.D.D., P.C., C.V.B., and the members of the BELNEU consortium. Drafting of the manuscript: T.V.d.B., K.S., C.V.B.
STUDY FUNDING
The sponsors of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The research was funded in part by the European Commission Seventh Framework Programme for research, technological development, and demonstration under grant agreement 305299 (AgedBrainSYSBIO), the Belgian Science Policy Office Interuniversity Attraction Poles program, the Alzheimer Research Foundation (SAO-FRA), the Flemish government-initiated Flanders Impulse Program on Networks for Dementia Research (VIND), the Flemish government-initiated Methusalem Excellence Program, the Research Foundation Flanders (FWO), the Agency for Innovation by Science and Technology Flanders (IWT), and the University of Antwerp Research Fund. The FWO provided a senior clinical investigator mandate to A. Sieben and the IWT a PhD fellowship to E. Cuyvers. The Article Processing Charge was paid by the Flemish Government: Flanders Impulse Program on Networks for Dementia Research (VIND).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement 2013;9:63–75. [DOI] [PubMed] [Google Scholar]
- 2.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 2011;43:429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bettens K, Sleegers K, Van Broeckhoven C. Genetic insights in Alzheimer's disease. Lancet Neurol 2013;12:92–104. [DOI] [PubMed] [Google Scholar]
- 4.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinberg S, Stefansson H, Jonsson T, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet 2015;47:445–447. [DOI] [PubMed] [Google Scholar]
- 6.Vardarajan BN, Ghani M, Kahn A, et al. Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann Neurol 2015;78:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuyvers E, De Roeck A, Van den Bossche T, et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer's disease patients: a targeted resequencing study. Lancet Neurol 2015;14:814–822. [DOI] [PubMed] [Google Scholar]
- 8.Rosenthal SL, Kamboh MI. Late-onset Alzheimer's disease genes and the potentially implicated pathways. Curr Genet Med Rep 2014;2:85–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 10.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engelborghs S, De Vreese K, Van de Casteele T, et al. Diagnostic performance of a CSF-biomarker panel in autopsy-confirmed dementia. Neurobiol Aging 2008;29:1143–1159. [DOI] [PubMed] [Google Scholar]
- 14.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 15.Van der Mussele S, Le Bastard N, Vermeiren Y, et al. Behavioral symptoms in mild cognitive impairment as compared with Alzheimer's disease and healthy older adults. Int J Geriatr Psychiatry 2013;28:265–275. [DOI] [PubMed] [Google Scholar]
- 16.Van der Mussele S, Marien P, Saerens J, et al. Psychosis associated behavioral and psychological signs and symptoms in MCI and Alzheimer's dementia. Aging Ment Health 2015;19:818–828. [DOI] [PubMed] [Google Scholar]
- 17.Bettens K, Brouwers N, Engelborghs S, et al. Both common variations and rare non-synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk. Mol Neurodegener 2012;7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Josephs KA, Murray ME, Whitwell JL, et al. Staging TDP-43 pathology in Alzheimer's disease. Acta Neuropathol 2014;127:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.