

Abstract
This review focuses on recent developments in the use of natural products as therapeutics for Alzheimer’s disease. Compounds span a diverse array of structural classes and are organized according to their mechanism of action, with the focus primarily on the major hypotheses. Overall, the review discusses more than 180 compounds and summarizes 393 references.
1. Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that affects approximately 36 million people worldwide as of 2010.1 The disease was originally described in 1906 by Alois Alzheimer based on the observation of amyloid plaques, neurofibrillary tangles and vascular anomalies during the autopsy of Auguste Deter, a patient who died with severe cognitive defects.2 The pathogenesis of the disease is complex, with both genetic and environmental factors3 likely contributing to varying degrees (reviewed in Jakob-Roetne4) with death occurring approximately 9 years after diagnosis. Genetic factors that correlate with early onset AD include mutations in amyloid precursor protein (APP)5,6 and presenilin 1 & 2 (PS1 & PS2)7 along with APP gene duplication,8 but the causative factors of late onset or sporadic AD are less well understood. A strong correlation does exists though with mutations in the ɛ4 allele of apolipoprotein (APOE),9 which can induce endocytosis of APP.10
Ultimately, the biochemical rationale for targeting a particular pathway stems from pathological, genetic, or epidemiological observations. The observation of amyloid plaques gave rise to the amyloid cascade hypothesis11,12 and compounds designed to reduce the rate of APP processing or abundance of plaques. Ubiquitous hyperphosphorylated neurofibrillary tangles in AD patients resulted in the tau hypothesis13,14 and the development of kinase inhibitors to modulate this aberrant phosphorylation. Microglial activation gave rise to a hypotheses based on prolonged inflammation,15,16 while abnormal levels of calcium,17 glucose,18 metals,19,20 and neurotransmitters, particularly acetylcholine (ACh), in AD patients have sparked treatments designed to restore normal concentrations. Figure 1 summarizes several pathways implicated in AD pathology for which natural product leads have been reported. A full discussion is unfortunately beyond the scope of this review, and interested readers are encouraged to consult the relevant references in this paragraph for detailed discourses.
Figure 1.

Summary of Relevant Alzheimer Disease Pathways.
Currently, there are five prescribed treatments in the United States and Europe for AD. These are symptomatic treatments that do not actually slow or reverse the progression of the disease. Three of these drugs are acetylcholinesterase (AChE) inhibitors, while one modulates N-methyl-D-aspartic acid (NMDA) receptors. Given the prevalence of AD and the lack of effective long-term therapies, there is a pressing need to discover viable leads that can be developed into clinically approved drugs with disease-modifying effects. There has been a heavy focus on developing drugs against amyloid plaques, although ultimately a clinical validation of either plaques or tangles as a target capable of exerting a disease-modifying effect is still lacking. The challenges here are substantial, in part, because of the pharmacokinetic issues associated with central nervous system (CNS) drug therapy (BBB permeability and P-glycoprotein efflux), in part, because of the lengthy clinical trials that are required to observe statistically significant cognitive differences in patients vs controls for neurological diseases, and, in part, because of the uncertainty regarding the role and timing of the two key pathological events, the formation of amyloid plaques and tau tangles. This latter fundamental issue is complicated by the fact that decades usually separate when disease initiation occurs and when clinical symptoms are first manifest; early and accurate validated methods of clinical diagnosis for patients prior to the onset of mild cognitive impairment is still a major unmet need facing the field.
1.1. Scope of the review
This review summarizes the natural products that have been reported as leads in the area of AD. The focus of this review is primarily molecules that were either recently described or in which significant advances have been reported in the last five years. Both terrestrial and marine sources have been considered. In the case of the latter, this review represents one of the first attempts to summarize the relevant literature in a number of years, while for terrestrial sources this manuscript builds on the summaries by Viegas21 and Houghton et al.22 Readers are directed towards the article by Cichewicz and workers in this issue for information on therapeutic leads for other neurodegenerative diseases.
In many instances, there is a close parallel between cancer targets, which have been investigated heavily by the natural product community,23 and neurodegenerative targets, particularly in the area of kinases.24 In those cases, we have chosen to focus on molecules in which a direct application to AD has been described. The underlying etiology of AD is complex, and while significant advances have been made, numerous competing hypotheses still exist.11,13,25–30 Every effort has been made to include compounds relevant to the major targets, but those in which only a few natural product leads have been reported or without significant new developments in the last few years have been omitted.
2. Acetylcholinesterase inhibitors (cholinergic hypothesis)
Acetylcholinesterase (AChE), mainly present in the central nervous system (CNS), catalyzes the hydrolysis of the neurotransmitter acetylcholine (ACh) to choline. This process is necessary to return an activated cholinergic neuron to a resting state. It was deficits in this neurotransmitter, which were noted in AD patients, that led to the cholinergic hypothesis.31 Two of the drugs currently licensed to alleviate cognitive symptoms in dementia are AChE inhibitors derived from natural products (galantamine and rivastigmine). Consequently, extensive research has been directed towards the identification of other AChE inhibitors, with the majority of these arising from the plant kingdom. While structurally diverse, these compounds are primarily alkaloids. There have also been numerous attempts to develop semi-synthetic or synthetic derivatives of these naturally derived AChE inhibitors, with the aim of improving inhibitory potency and selectivity or for some structures, engineering dual modes of action relevant to AD therapy.
2.1. Alkaloids as AChE inhibitors
Physostigmine (eserine) (1) is an alkaloid with a pyrroloindole skeleton from Physostigma venenosum Balf. (Leguminosae) seeds that is a potent, short-acting and reversible inhibitor of AChE.32,33 Reviewed in Houghton et al.,22 physostigmine has been shown to improve cognitive functions in vivo and in both normal and AD patients.34 To improve the pharmacokinetic profile and efficacy of 1, numerous analogs have been investigated, with the most therapeutically successful being the carbamate-type reversible AChE inhibitor rivastigmine (2), now licensed for the symptomatic treatment of mild to moderate dementia in AD or Parkinson’s disease (PD). Not surprisingly, there have been numerous attempts to synthesise AChE inhibitors using 1 as a template, with the aim of developing drugs with therapeutic advantages over 2. Some of these potent and selective AChE inhibitors have been pharmacomodulated for dual modes of action, to target both cognitive and depressive symptoms in AD.35,36 Some analogs of the carbamate derivative xanthostigmine (3) inhibit AChE-induced β-amyloid aggregation37 and a phenylcarbamate derivative of 1, phenserine (4), inhibits AChE and APP,38 suggesting their potential application in modulating AD symptoms and pathology. (−)-Phenserine enhances cognition in vivo39 and in AD patients (20 patients; 30 mg/day),40 but Phase III trials with AD patients did not show different effects from the placebo; the (+)-enantiomer (Posiphen™) has been investigated in Phase I trials but additional clinical studies are not planned.38 Methyl substitution in the C-2′ position of 4 produces tolserine (5), which has improved selectivity for AChE compared to butyrylcholinesterase (BChE).39 More recently, other analogs of 1 with a cyclic alkyl carbamate of eseroline (6 and 7) showed more potent AChE inhibition and selectivity than 4.41 In general, although numerous derivatives of 1 have been developed, few have reached advanced stages of clinical development for AD.
Rutaecarpine (8) and dehydroevodiamine (9), indole alkaloids from Evodia rutaecarpa (Juss.) Benth. (Rutaceae), have been used as templates to synthesise new AChE inhibitors, since the plant extract and 9 inhibit AChE in vitro and reverse scopolamine-induced memory impairment in vivo.42 Some of these synthetic analogs have included structural features of the AChE inhibitor tacrine but disappointingly, showed greater selectivity for BChE,43 whilst other 3-aminoalkanamido-substituted 7,8-dehydrorutaecarpine derivatives (10, 11 and 12) were more potent and showed selectivity for AChE.44 Of four bisindole alkaloids isolated from the root of Tabernaemontana divaricata (L.) R. Br. ex Roem. & Schult. (Apocynaceae), only 19,20-dihydrotabernamine (13) and 19,20-dihydroervahanine A (14) inhibited AChE more potently than galantamine (15) in vitro;45 effects in vivo have not been studied, although a root ethanol extract inhibits cortical AChE activity in vivo.46
Another AChE inhibitory indole alkaloid, geissospermine (16), was considered to largely explain the cognitive effects of an alkaloid fraction from Geissospermum vellosii Allemão (Apocynaceae) stembark, which reduced scopolamine-induced amnesia in vivo.47 Although Catharanthus roseus (L.) G. Don (Apocynaceae) is a source of the anti-cancer drugs vincristine and vinblastine, an AChE inhibitory alkaloid, serpentine (17), from the roots was 10-fold more potent than 1,48 but has not been studied further perhaps because cytotoxicity may limit its clinical development. Other relevant indole alkaloid derivatives (18 and 19) are from the fungus Cortinarius infractus Berk. (Cortinariaceae), which could be promising candidates for development since they inhibit AChE with greater selectivity than galantamine (15) (due to a lack of π-π interactions in BChE), they comply with Lipinski rules and are predicted to cross the blood-brain barrier (BBB).49
Galantamine (15) is produced by Galanthus woronowii Losinsk. and some species of Narcissus and Leucojum aestivum L. (Amaryllidaceae). It is a drug licensed to treat symptoms of mild to moderate dementia in AD, thus has been extensively studied for its AChE inhibitory activity and is reviewed elsewhere.22,34 Numerous synthetic derivatives of 15 have been investigated, with some (heterodimeric alkenyl linked bis-galantamine derivatives) inhibiting AChE more potently than 15,39 although their potential for clinical use is undetermined. Of particular therapeutic relevance is Memogain® (Gln-1062), a prodrug of 15, which has improved cognitive effects in an animal model of amnesia and bioavailability (15-fold) in the brain compared to 15, with fewer adverse gastrointestinal effects.50 Numerous other Amaryllidaceae alkaloids inhibit AChE and are reviewed by Houghton et al.22 and Jin.51 Notably, ungeremine (20), isolated from Nerine bowdenii W.Watson and from species of Galanthus and Narcissus showed stronger AChE inhibition than 15.52–54
Isoquinoline alkaloids from Colchicum speciosum Steven (Colchicaceae) corms are reversible inhibitors of both AChE and BChE in vitro55 and several benzylisoquinoline alkaloids from Coptis (Ranunculaceae) and Corydalis (Papaveraceae) species inhibit AChE.22,56,57 Some of the most potent inhibitors from the latter include epiberberine (21), 13-pseudodehydrocorydaline (22), pseudocoptisine (23) and pseudoberberine (24),58 with 23 and 24 alleviating scopolamine-induced memory impairment in vivo.59,58 The berberine structure has been used as a template to synthesise more potent AChE inhibitors; one derivative (berberine linked with phenol by 4-carbon spacers) was more active than berberine (IC50 0.1, 0.4 μM, respectively) and was suggested to bind to the peripheral anionic site of AChE.60 Some anti-AChE alkaloids from Coptis chinensis Franch. rhizomes display non-competitive β-secretase (BACE1) inhibitory activities (21 and groenlandicine (25)) and are antioxidant (25 and jateorrhizine (26));61 thus, 25 in particular shows multiple activities relevant to AD therapy.
Structure-activity studies with protoberberine alkaloids from Stephania venosa Spreng. (Menispermaceae) show important inhibitory features to be a positive charge at the nitrogen of the tetrahydroisoquinoline portion, steric substitution at the nitrogen and planarity of the molecule or substitutions at C-2, -3, -9, and -10, thus stepharanine (27), cyclanoline (28) and N-methyl stepholidine (29) were more potent inhibitors than stepholidine (30) and corydalmine (31).45 Several quinoline and β-carboline alkaloids, including two new alkaloids (nigellastrines I (32) and II (33)) from the seeds of Peganum nigellastrum Bunge (Zygophyllaceae) showed AChE inhibitory activity in a TLC bioautographic assay, with results suggesting harmine (34), harmaline (35), harmol (36) and harman (37) show similar activity to galantamine (15),62 although more quantitative data is needed. Extracts from aerial parts of Salsola oppositofolia Pall., S. soda L. and S. tragus L. (Chenopodiaceae), inhibited cholinesterase (ChE) in vitro, the latter showing highest AChE inhibitory activity, which could be attributed to the tetrahydroisoquinoline alkaloid content, particularly salsoline (38) and salsolidine (39).63 Although the potency and selectivity of several alkaloids from Stephania venosa, P. nigellastrum and Salsola species have shown promise in these preliminary studies, their pharmacological potential requires further evaluation.
Huperzine A (40) from Huperzia serrata (Thunb.) Trevis. (Lycopodiaceae) is a widely-studied reversible AChE inhibitor which improves cognitive functions in animal studies and in clinical trials with elderly, AD and vascular dementia patients, with limited adverse effects.34 Huperzine A (40), which is also neurotrophic64 and neuroprotective,65 has been used to treat AD symptoms in China and is marketed as powdered H. serrata in the US for memory impairment.66 A recent meta-analysis of the efficacy and safety of 40 showed it to be well-tolerated and to significantly improve cognitive performance and activities of daily living in patients with AD.67 Huperzine B (41), also from H. serrata, is a less potent AChE inhibitor than 40,68 which may explain why it has not been investigated as extensively for potential clinical use. Numerous structural analogs and hybrids based on 40 and 41 have been investigated for their AChE inhibitory effects and are reviewed by Howes & Houghton.34
AChE inhibitors structurally related to the huperzines are carinatumins A (42) and B (43) (IC50 4.6 and 7.0 μM, respectively), isolated from Lycopodium carinatum Desv. ex. Poir. (Lycopodiaceae), but these were not as potent as 40 (IC50 0.8 μM).69 Lycoparin C (44) (which lacks the carboxylic acid at C-15 and the N-methyl groups in the inactive lycoparins A and B) from L. casuarinoides Spring70 and annotine (45) from L. annotinum L.71 also inhibit AChE but, unlike the huperzines, none of these alkaloids appear to have been pursued for therapeutic relevance. Of ten alkaloids isolated from L. annotinum ssp. alpestre (Hartm.) Á. Löve & D. Löve, the most potent AChE inhibitors were anhydrolycodoline (46) and annotine N-oxide (47) but these were still considerably less potent (IC50 191, 404 μM, respectively) than physostigmine (1) (IC50 0.8 μM).72 The low activity was explained by structure-activity studies. Although these alkaloids appeared to fit into the same AChE binding site as 40 and hydrogen-bond acceptors or donors are present, they formed weak interactions with the amino acid residues in this pocket. As 46 was suggested to be more tightly enclosed in the enzyme’s binding site compared to the other alkaloids tested, it is being considered as a template structure to synthesise analogs of increased potency.72
A more relevant therapeutic candidate has been recently discovered from L. japonicum Thunb.; lycojapodine A (48) is a novel C16N-type alkaloid with a 6/6/6/7 tetracyclic ring system that shows comparable AChE inhibitory activity to 40.73 Although a number of the AChE inhibitory alkaloids reported from Lycopodium species are structurally related to the quinolizidines,69–71 cryptadines A (49) and B (50) from L. cryptomerianum Maxim. consist of a piperidine ring and two octahydroquinoline rings. These AChE inhibitors74 closely resemble lycoperine A (51) from L. hamiltonii Spreng.75
Piperidine alkaloids and their synthetic derivatives are less well-documented as AChE inhibitors, compared to other alkaloids such as the indole and isoquinoline structural classes, and thus are generally not as advanced in their development for clinical use. Piperidine alkaloids showing some therapeutic relevance for cognitive disorders are derived from Cassia spectabilis DC. (Leguminosae) and include some semi-synthetic derivatives patented as AChE inhibitors with potential to treat pathologies affecting the cholinergic system.36 The rationale for investigating (−)-spectaline and the (−)-3-O-acetyl derivative from C. spectabilis flowers is that they contain structural features similar to that of ACh; the synthetic derivative (2R,3R,6S)-2-methyl-6-(13-oxotetradecyl)-piperidin-3-yl acetate hydrochloride (LASSBio-767) (52) inhibits rat brain AChE more selectively than BChE and reverses scopolamine-induced amnesia in vivo.76,77 Piperine (53), from Piper species (Piperaceae) improves memory impairment and neurodegeneration in vivo, which are effects associated with increased neuron density and AChE inhibition in the hippocampus.78 It also inhibits monoamine oxidases,79 suggesting it may also alleviate depressive symptoms in dementia.
A number of steroidal alkaloids from Sarcococca and Buxus species (Buxaceae) have shown anti-ChE activities and these, including their structure-activity relationships, have been recently reviewed.80,22,34 Several steroidal alkaloids from Fritillaria species (Liliaceae) inhibit ChE81,82 but pharmacological and clinical efficacy in relation to cognition has not been determined. Arecoline (54), a reduced pyridine alkaloid derivative from Areca catechu L. (Arecaceae) (commonly known as betel nut), has been shown to inhibit AChE in vitro and in the nervous tissue of the mollusc Lymnaea acuminata, but in a separate study, arecoline did not inhibit AChE, even though an extract and fractions from A. catechu did produce inhibition.83 Arecoline (54) improves scopolamine-induced cognitive impairment and passive avoidance performance in vivo84,85 and improves cognitive function and recognition skills in AD patients;86,87 these effects suggest a cholinergic action that may be due to AChE inhibition, although evidence for this is inconclusive, or by binding to muscarinic receptors.88,89
Other alkaloids that are of therapeutic interest include sinapine (55) (an ester of sinapic acid and choline, that occurs in several plants including Raphanus sativus L. (Brassicaceae)), which potently inhibits AChE in vitro and in brain tissue36 and tapsine (56) (a protoalkaloid from Magnolia x soulangiana Soul.-Bod. (Magnoliaceae) leaves) which produces long-acting and concentration-dependent inhibition of AChE (IC50 0.3 μM) and was more potent than galantamine (15) (IC50 3.2 μM).90 Tapsine (56) is suggested to bind closely to the catalytic triad in AChE. This is facilitated by π-stacked interactions between the planar aromatic ligand and Trp84 and Phe330 of AChE, anchoring of the cationic side chain with His444 reaching into the catalytic site, and H-bonding with active site water molecules and Ser122.90 Semi-synthetic derivatives (including 57) of some oxoisoaporphine alkaloids, which occur in Menispermum dauricum DC. (Menispermaceae), are being investigated for their potential to treat AD, since the 1-azabenzanthrone moiety in their chemical structure binds to the peripheral anionic site of AChE, so inhibiting activity.36
Several non-alkaloidal but potent AChE inhibitors have been isolated from fungal sources.22 Recently, a novel alkaloid 16α-hydroxy-5N-acetylardeemin (58) from the fungus Aspergillus terreus has shown AChE inhibitory activity almost as potent as tacrine.91 Zeatin (59) is a cytokinin phytohormone.92 This isopentenyl purine derivative is of therapeutic interest as it inhibits AChE,93 protects against β-amyloid-induced neurotoxicity in vitro and scopolamine-induced cognitive impairments in vivo.94 An alkaloid fraction from Trigonella foenum-graecum L. (Leguminosae) and the component alkaloid trigonelline (60) also inhibit AChE;95 interestingly, intake of coffee (Coffea arabica L., Rubiaceae), which also contains 60, has been associated with a reduced risk of dementia.96
2.2. Terpenoids as AChE inhibitors
Numerous essential oils (or oil absolutes) have shown inhibitory activity against ChE including those from Narcissus poeticus L. (Amaryllidaceae),97 Melaleuca species (Myrtaceae),98 Acorus calamus L. (Acoraceae),99 Eucalyptus camaldulensis Dehnh.,100 Marlierea racemosa Kiaersk. (Myrtaceae),101 Cymbopogon schoenanthus Spreng. (Poaceae)102 and several oils from the Lamiaceae.39,103–106 Many constituents of these oils have been identified as AChE inhibitors including monoterpenoids (e.g. geranial, neral and linalool)103,107,108 and sesquiterpenoids (e.g. caryophyllene oxide, (+)-(S)-ar-tumerone (61))109,110 and some phenylpropanoids (e.g. eugenol, α- and β-asarone).103,99,111,112 AChE structure-activity relationships for monoterpenoids have been discussed previously.22,113 More recently, structure-activity studies with bisabolane-type sesquiterpenoids, derived from Peltophorum dasyrachis Kurz ex Baker (Leguminosae), inhibited AChE in the following order of potency: ketones > alcohols > hydrocarbons; oxidation at C-9 and a single-bond between C10–C11 were also concluded to be important in the bisabolane-type inhibitors.110 Although this study revealed (+)-(S)-ar-tumerone (61) and (+)-(S)-dihydro-ar-tumerone (62) in P. dasyrachis oil were potent inhibitors (IC50 191, 82 μM, respectively), they were not as active as galantamine (15) (IC50 3 μM).110
Although documented as AChE inhibitors, relatively few of these oils and their constituents have been investigated for their potential effects in cognitive disorders. Those studied more extensively include the steam-distilled oils from S. officinalis L. and S. lavandulifolia Vahl. (Lamiaceae), which inhibited AChE in vitro and positively influenced cholinergic function and cognition in vivo.114,115 1,8-Cineole (63) and α-pinene (64) are considered to be the most active AChE inhibitory components of S. lavandulifolia oil (the latter is also an anti-AChE component of S. potentillifolia Boiss. & Heldr. ex Benth. oil),116 although other oil constituents may inhibit AChE, perhaps synergistically.108,109 An extract from the aerial parts of another member of the Lamiaceae, Teucrium polium L. is anti-amnesic in vivo and inhibits AChE in vitro,117 although the compounds responsible have not been determined. Limonene (65) and perillyl alcohol (66), components of Citrus (Rutaceae) essential oils, improve scopolamine-induced memory impairment, which is suggested to be due to AChE inhibition (observed in vitro).118 Sesquiterpenoids from the root of Leontopodium alpinum Cass. (Asteraceae) increased extracellular ACh in rat brains, but only silphiperfolene acetate (67) inhibited AChE activity in vitro, although weakly.119
Numerous diterpenoids inhibit AChE,22,120 however, few have demonstrated potencies that have stimulated further investigation. 5α,8α-(2-Oxokolavenic acid) (68) was the most potent of several clerodane diterpenoids from the fruit of Detarium microcarpum Harms (Leguminosae) to inhibit AChE but it was 10-fold less potent than galantamine (15).121 The ent-kaurane diterpenoid melissoidesin (69), from leaves of Isodon wightii (Benth.) H.Hara (Lamiaceae), inhibits AChE in vitro, but further studies to confirm inhibitory potency are required.122 The isoprimarane diterpenoids 7β-hydroxyisopimara-8,15-dien-14-one and 14α-hydroxyisopimara-7,15-dien-1-one, from the leaves of Hypoestes serpens R.Br. (Acanthaceae), required 50- and 20-fold higher concentrations than 15, respectively, to inhibit AChE in a TLC bioautographic assay.123 Thus, structural modification may be necessary to optimise the AChE inhibitory potency of these less active diterpenoids. A more potent AChE inhibitor (only 2-fold less potent than 15) is the cassane diterpene niloticane (70), from Acacia nilotica subsp. kraussiana (Benth.) Brenan (Leguminosae) bark, which also showed selective inhibition of cyclooxygenase-1 (COX-1),124 an effect that has also been considered relevant in dementia treatment. Of more therapeutic relevance are the first diterpenoids shown to inhibit AChE from Salvia miltiorhiza Bunge (Lamiaceae), particularly cryptotanshinone (71),125 which ameliorates scopolamine-induced learning impairments in rodents.126,127 Although a promising drug candidate, pharmacokinetic studies show penetration of 71 across the BBB may be limited in vivo.128
Compared to alkaloids, relatively fewer triterpenoid or steroidal derivatives have been discovered as AChE inhibitors and in many cases their potency is weak. Nevertheless, weak to moderate AChE inhibitors are frequently used as template structures for the synthesis of more potent and therapeutically relevant inhibitors. Jujubogenin glycosides occur in Bacopa monnieri Wettst. (Scrophulariaceae),129 a plant widely-studied for effects on cognition;130 semi-synthetic derivatives are described in a patent as AChE inhibitors for potential use in AD.36 Other triterpenoid or steroidal derivatives have been described, which are not AChE inhibitors, but may have some therapeutic potential as they possess additional activities relevant to dementia. For example, taraxerol (72) is a triterpenoid from Clitoria ternatea L. (Leguminosae) that inhibits AChE in vitro and in the brain of rodents in vivo.131,132 Extracts from aerial parts and roots of this plant attenuate memory deficits in rats, but this was not directly correlated with AChE inhibition,133 suggesting other modes of action; particularly since 72 was not as potent as physostigmine (1) when tested for AChE inhibition.131,132 Timosaponin AIII (73), a steroidal saponin from Anemarrhena asphodeloides Bunge (Asparagaceae) significantly reversed the scopolamine-induced learning deficits and expression of tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) in the brain, and increased hippocampal ACh levels in vivo.134 Although anti-inflammatory mechanisms could contribute to these observed cognitive effects, AChE inhibition (reported in vitro) is considered the principal mode of action.134
2.3. Shikimate-derived compounds as AChE inhibitors
Compared to alkaloids, a relatively low number of shikimate-derived compounds are documented as AChE inhibitors. One structure-activity relationship study focused on 17 flavonoids, including those from Buddleja davidii Franch. (Buddlejaceae) leaves, but only linarin (74) (shown in a separate study to inhibit AChE)135 and tilianin (75) inhibited AChE, thus a 4′-OMe group, a 7-O-sugar, and the length of the interglycosidic links of the sugar chain were considered important structural features for AChE inhibition.136 Other flavonoid inhibitors of AChE include quercitrin, 3-methoxy quercetin, tiliroside (76) and quercetin (77) from Agrimonia pilosa Ledeb. (Rosaceae); although these flavonols were not as active as tacrine and berberine, 76 and 77 were almost two-fold more active than the alkaloid dehydroevodiamine (9).137,138
Generally, there is limited evidence for in vivo cognitive effects of flavonoids in relation to ChE activity. Luteolin (78), a common flavone, inhibits AChE (IC50 > 0.1 mM) and BChE in vitro139,140 and is anti-amnesic in vivo, protecting against β-amyloid-induced toxicity and inhibiting AChE activity (increasing ACh levels) in the cerebral cortex.141 Although naringenin (79) did not inhibit AChE in the study by Fan et al.,136 in a different study this flavanone dose-dependently inhibited AChE in vitro, perhaps explaining why it also ameliorated scopolamine-induced amnesia in rodents;142 the 7-neohesperidoside, naringin (80), also alleviates cognitive impairment and oxidative stress, and attenuates brain AChE activity in vivo.143 The prenylflavone icariin (81) from Epimedium species (Berberidaceae) improves cognitive impairments in mice, which was attributed to increasing monoamine levels, inhibiting oxidative damage and also decreasing AChE activity.144 The isoflavan glabridin (82) from the roots of Glycyrrhiza species (Leguminosae) antagonised scopolamine-induced amnesia in mice, an effect associated with a reduction of brain ChE activity.145
A diet of soy isoflavones for 16 weeks in aged male rats produced AChE inhibition in the cortex, basal forebrain and hippocampus,146 although other modes of action may explain the cognitive effects observed with these isoflavones.147,148 The potential AChE inhibitory effects of compounds with isoflavone structures have been confirmed by the evaluation of synthetic flavonoid derivatives. Of the derivatives based on chalcone, flavone, flavanone and isoflavone structures, the most potent and selective AChE inhibitors were the isoflavone derivatives; 6,7-dimethoxy-3-[4-(pyrrolidin-1-ylmethyl)-phenyl]-4H-chromen-4-one was even more potent than donepezil.149 Other isoflavones that inhibit AChE are osajin and pomiferin and their iso- derivatives from fruits of Maclura pomifera (Raf.) C.K.Schneid. (Moraceae), but IC50 values were in mM (0.1 – 2.7) concentrations.150
A screen of 45 non-alkaloid natural compounds found six of the seven AChE inhibitors to be xanthones. The most potent (83) had an additional cyclic component and a hydrophobic side-chain (not features of the other active xanthones), considered important structural features for AChE inhibition.151 Other xanthones from Gentiana campestris L. (Gentianaceae) leaf inhibit AChE; optimum inhibitory activity was associated with absence of a glucopyranosyl, and a methoxy group present in position C-3, since the most potent inhibitor, bellidifolin (84), was similar in potency to galantamine (15).152 Macluraxanthone (85) (occurs in some Guttiferae) is a potent non-competitive AChE inhibitor,137 but it also inhibits platelet aggregation,153 therefore therapeutic development could be limited due to the potential for drug interactions with antiplatelet / anticoagulant drugs.
Several coumarins and derivatives inhibit AChE including bergapten (86), scopoletin (87), 4-methylumbelliferone (88), feronielloside (89), marmesin (90) and columbianetin (91).22,34,36 The inhibitory potency of simple coumarins (e.g. 87) and furanocoumarins (e.g. 86) is considered to be moderate to low and large substituents (e.g. benzyloxy) in position C-7 of the coumarin nucleus, or at positions 5 and 8 of the furanocoumarin nucleus, improve inhibitory potency compared to smaller substituents (e.g. hydroxyl and methoxy).154 Few AChE-inhibitory coumarins have been investigated for their cognitive effects in vivo, but psoralen (92), isopsoralen (93) (furanocoumarins from Psoralea species (Leguminosae)), decursin (94) and nodakenin (95) reverse scopolamine-induced cognitive impairments in rodents, correlating with AChE inhibition.34 The coumarin analog ensaculin (96) inhibits AChE, modulates dopaminergic, serotonergic and adrenergic function and is an NMDA receptor antagonist,155 thus is further advanced in clinical development for AD as the HCl salt (KA-672).
2.4. Miscellaneous compounds as AChE inhibitors
Magnolia officinalis Rehder & E.H.Wilson (Magnoliaceae) extracts have shown numerous biological activities relevant to dementia treatment and the component lignans honokiol (97) and magnolol (98) inhibit AChE.156 More recently, an extract and 4-O-methylhonokiol (99) from the bark of this species prevented scopolamine-induced memory impairment and the increase in brain AChE in mice; the latter inhibited AChE in vitro (IC50 12 nM; tacrine: 135.4 nM),157 thus lignans from M. officinalis warrant further study for their therapeutic potential in AD.
Although ferulic acid (100) is a competitive inhibitor of AChE158 it has been subjected to pharmacomodulation to produce tacrine-ferulic acid hybrids for dual anti-AChE (tacrine moiety) and antioxidant (ferulic acid moiety) activities, with some being both antioxidant and more potent AChE inhibitors than tacrine.159 Another phenolic compound that inhibits AChE is hopeahainol A (101) (from Hopea hainanensis Merr. & Chun (Dipterocarpaceae) stem bark), which shows comparable potency to huperzine A (40)160 and is also neuroprotective in vitro,161 providing two mechanisms of interest for AD. Also of interest for further study are procyanidin-containing extracts from lotus (Nelumbo nucifera Gaertn. (Nelumbonaceae)) seeds, and Cornus officinalis Siebold & Zucc. (Cornaceae) fruit extract and its constituent iridoid glycoside, loganin (102), since they could improve cognitive impairments and decrease brain AChE activity in vivo.162–164
Other recently discovered AChE inhibitors include synthetic derivatives of cardanol (a non-isoprenoid phenolic lipid from Anacardium occidentale L. (Anacardiaceae)), which correlate with the AChE binding effects of rivastigmine (2)165 and an unusual polyketide, sporothrin A (103), from an endophytic marine fungus Sporothrix sp. (Ophiostomataceae), which is described as a strong AChE inhibitor (although not compared with a positive control).165
In general, AChE inhibitors of marine, fungal or bacterial origin are less well-documented compared to those from plant origin, and have been reviewed previously.22,166 More recently discovered AChE inhibitors from marine origin include a steroidal alkaloid 4-acetoxy-plakinamine B from the sponge Corticum sp.,34 a cembranoid crassumolide E from the coral Lobophytum sp.,167 the plastoquinones sargaquinoic acid and sargachromenol from the algae Sargassum sagamianum168 and phlorotannins (eckstolonol, eckol, phlorofucofuroeckol A, dieckol, 2- and 7-phloroeckol) from another algae Ecklonia stolonifera,169 but all were either less potent than alkaloid positive controls, or did not report a positive control. However, the pentacyclic pyridoacridine alkaloid petrosamine (104) from the sponge Petrosia n. sp., was a more potent AChE inhibitor than galantamine (15) in vitro,170 so could be of interest for further study. Extracts from the sponge Topsentia ophiraphidites171 and the seaweeds Gracilaria gracilis,172 G. edulis, Ulva reticulata, Hypnea valentiae and Padina gymnospora173 also inhibit AChE but identification of the compounds responsible and any therapeutic potential requires further study.
Although AChE inhibition is one of the major pharmacological targets for AD at present, new therapeutic strategies have emerged (vide infra). Thus, the continued clinical relevance of AChE inhibitors for symptomatic treatment of AD may eventually be superseded by new and more effective disease-modifying drugs targeting other pathophysiological processes.
3. Protease inhibitors (amyloid cascade hypothesis)
The buildup of amyloid plaques is one of the hallmarks of AD. This observation has led to the proposal by Hardy11 that these sessile plaques are critical to the observed neurodegeneration (amyloid cascade hypothesis), which is supported by familial mutations. Recent modification174 of this original hypothesis implicates the precursors (oligomers of Aβ42) as the causative agent. Two distinct therapeutic strategies consistent with this proposal have been investigated, including inhibiting the proteolytic enzymes involved in forming the Aβ42 building blocks (secretase modulation), and reducing the concentration of Aβ42 oligomers either by inhibiting aggregation or increasing the rate that the monomer or oligomers are cleared. Recent in vivo kinetic data suggests this latter issue may be at the heart of the observed Aβ42 build-up, rather than APP overproduction or secretase overactivity, as rates of clearance are essentially half in transgenic AD mice models vs controls.175,176
The two major enzyme targets in the amyloidogenic pathway of Aβ formation are the aspartic proteases, β- and γ-secretases. While aspartic proteases have been successfully targeted as HIV therapeutics, developing β- and γ-secretase inhibitors has been more problematic. The latter have potential target toxicity as NOTCH, which is crucial in cell-cell signaling, is a substrate for γ-secretase, while the former possesses an active site large enough to require molecules that suffer from poor BBB (>500 MW). To date though, several synthetic γ-secretase inhibitors and one inhibitor of β-secretase (Comentis’ CTS-21166) have been evaluated in clinical trials. While the exact structure of this latter clinical candidate has not been revealed yet, it is a transition state analog of peptide hydrolysis and would likely be considered a natural product mimic under the classification system of Newman and Cragg.177 Interestingly, no direct inhibitor of γ-secretase has yet been reported from natural sources, although the fungal metabolite beauverolide modifies this process indirectly.178 As of July 2010, 49 β-secretase inhibitors have been reported (IC50 < 100 μM), of which three have IC50 values of less than 1 μM (bastadin 9 (134), luteolin (78), neocorylin (112)). No natural product has yet displayed the < 100 nM potency of the most promising synthetic pre-clinical candidates though. The vast majority of the reported inhibitors are plant-derived flavonoids, including flavones, and related phenolic compounds. Interestingly, while β-secretase (BACE1) inhibitors derived from synthetic lead discovery programs are competitive inhibitors,179 many of these natural products are non-competitive inhibitors that were active in FRET assays, with few demonstrating potency in cell or animal models. It has been suggested that these compounds might bind to either a β-secretase subsite or to a regulatory domain,180 but more detailed structural work is needed to clarify these issues.
3.1. Shikimate-derived compounds as secretase inhibitors
By far the largest class of natural product BACE1 inhibitors reported is shikimic–acid derived. Shimmyo et al. reported SAR data for a series of related flavonols (myricetin (105), quercetin (77), kaempferol (106), morin (107)) and a flavone (apigenin (108)).181 While in FRET-based enzymatic assays all four flavonols displayed moderate BACE1 inhibition (1.4 μM to 40 μM), only 105 and 77 reduced BACE1 activity (by approximately 20–30%) and Aβ42 production in a cellular system. This led to the hypothesis that the C-3 hydroxyl group was critical for the observed effect, as the corresponding flavone (108) was the weakest inhibitor. Docking studies suggest that this hydroxyl group stabilizes the enzyme-inhibitor complex by hydrogen bonding to Asp32 of BACE1 (one of two catalytic Asp residues involved in the hydrolysis), while hydroxy groups at 5′ and 3′ also participate in hydrogen bonding. In 2009, the flavone luteolin (78), from Perilla frutescens var. acuta (Thunb.) Kudô (Lamiaceae), was discovered to be a BACE1 inhibitor in an enzyme assay. With an IC50 value of 0.5 μM, it is one of the most potent natural product inhibitors of BACE1 to date.180 A comparison of the planar structures of this compound and the inactive 108 indicates the C-5 hydroxyl group is responsible for the two order of magnitude increase observed in potency. Assuming no change in the binding conformation compared to 105–108 above, this increase in potency could be partially attributed to an additional H-bond to the Trp198 residue in the binding pocket. Since the identical structural variation between myricetin (105) and quercetin (77) results in only a marginal increase in activity, the explanation must involve other factors. Counter-screens indicate luteolin (78) does not inhibit serine proteases (trypsin, chymotrypsin), AChE or TNF-α converting enzyme (TACE; a putative γ-secretase) at 100 μM, but additional work is needed to assess selectivity against more structurally relevant aspartic proteases.
Hwang et al. reported a series of lavandulyl flavanones (109–110) isolated from Sophora flavescens Aiton (Fabaceae) that display similar potency (1–10 μM) in an in vitro assay system.182 Biochemical analyses indicated these non-competitive inhibitors reduced soluble APPβ levels (the extracellular BACE1 cleavage product), but did not affect intracellular full-length APP suggesting selective in vivo BACE1 inhibition. Minor structural variations resulted in negligible potency differences, with the exception of hydroxylation at C-4″ of the lavandulyl moiety in 111 which abolished activity.
Neocoylin (112), an isoflavone from the seeds of Psoralea corylifola L. (Fabaceae),139 potently inhibited BACE1 cleavage of APP (0.7 μM) in a FRET assay system. That this compound retains significant activity despite substitution at C-3 suggests an alternative binding conformation or site than the flavonols in which hydroxylation was shown to be critical for binding (105–108; vide supra). Several other phenolic compounds were also isolated although they lack the C-3 2H–chromene moiety and are less active. Soy isoflavones have also attracted attention as an Alzheimer’s treatment. Currently, a pilot study on 60 patients is being conducted to evaluate the potential effects of soy isoflavone supplements (Novasoy®) on cognitive function for men and women with AD (NCT00205179).
Another potent naturally occurring BACE1 inhibitor is (−)-gallocatechin gallate (113), isolated from green tea, Camellia sinensis L. (Theaceae), after an examination of 260 species of herbal drugs.183 Compounds 113–115 were the first examples of non-peptidic natural product β-secretase inhibitors and non-competitively inhibited BACE1 in a dose-dependent manner with Ki values of 0.17, 0.27 and 5.3 mM, respectively. Preliminary SAR data indicates that the pyrogalloyl moiety is essential for activity as removal of one of the hydroxyl groups, as in 116, resulted in an inactive compound. Continued development of these compounds has resulted in on-going Phase II/III clinical trials using 200–800 mg/day of epigallocatechin gallate (EGCG) (115) (NCT00951834), based primarily on the observed neuroprotective effects in cell and animal studies. Multiple pathways are being modulated in addition to BACE1 though, including increasing non-amyloidogenic α-secretase processing, preventing the aggregation of Aβ, as well as antioxidant effects (see section 6) and modulation of mitochondrial function.
Active stilbenoids and phenylpropanoid esters were isolated from the dried rhizomes of Smilax china L. (Smilacaceae).184 These compounds include trans-/cis-resveratrol, oxyresveratrol, veraphenol and cis-scripusin. Again, IC50 values for these non-competitive inhibitors were in the single digit micromolar range. Specifically, trans-resveratrol (117) and cis-scripusin (118) had IC50 values of 7.5 and 10 μM, respectively. All compounds were tested for selectivity against TACE, elastase, chymotrypsin and elastase, and found to be inactive at up to 100 μM.
The need for counterscreens to assess selectivity is illustrated by our report of two related phenylpropanoid esters from Cordia sebestena L. (Boraginaceae), which is commonly known as the Geiger tree.185 Initial biological evaluation indicated a dose-dependent inhibition of BACE1 in a complementation-based enzyme assay for sebestenoids C (119) and D (120). In contrast to the related compound cis-scripusin (118), these compounds also inhibited chymotrypsin in a standard chemiluminescent assay although at slightly lower concentrations. Further testing revealed this inhibition was strongly affected by the addition of detergent as assays performed in the presence of 0.01% Triton X-100 resulted in 4-times lower IC50 values. These results are consistent with the non-competitive inhibition expected from a non-specific aggregation inhibitor as has been outlined elegantly by the Shoichet’s lab.186
Several other complex aromatics have been reported as inhibitors. An investigation of pomegranate husk Punica granatum L. (Lythraceae) identified the complex phenolic ellagic acid and punicalagin as weak inhibitors of BACE1,187 while more potent glucopyranoside galloyl derivatives were isolated from Sanguisorba officinalis L. var. officinalis or var. longifolia (Bertol.) T.T.Yu & C.L.Li..188 Finally, weak (>100 μM) furanocoumarin inhibitors of BACE1 were reported from the roots of Angelica dahurica L. (Apiaceae) and phlorotannin inhibitors were reported from the marine kelp Eisenia bicyclis Setchell (Lessoniaceae) with low micromolar potency.189
3.2. Polyketides as secretase inhibitors
A screen of 256 plant and fungal extracts led to the identification of the polyketide derived hispidin (121) from a fungal mycelium of Phellinus linteus (Hymenochaetales),190 although this compound had been previously isolated from P. pomaceus.191 In the course of a total synthesis, analogs 122 and 123 were generated and tested. These analogs were an order of magnitude less potent than hispidin with IC50 values of 40 and 72 μM respectively, which led the authors to suggest “a catechol moiety might not be necessary for stronger activity.”190 Selectivity was evaluated against chymotrypsin, trypsin, elastase, PEP (prolyl peptidase), TACE, with hispidin displaying equipotency towards BACE1 and PEP.
Taine et al.192 reported four alkylphenolic acids as inhibitors of BACE1. These compounds were isolated from perennial Araceae herbs, widely distributed in southern China, and used as a traditional Chinese medicine. Represented by 124, the other analogs differ in the chain length and positions of oxidation, but the potency is essentially unaffected by these minor variations. The position of these double-bonds is odd perhaps indicating that their biogenesis may begin with a 6-hydroxy-2-methylbenzoic rather than 6-hydroxylbenzoic acid. These 7 μM non-competitive inhibitors are also reminiscent of NP-12 (125), a phenylprenyl derivative isolated from the marine sponge Sarcotragus sp. by Noscira.193 These compounds were put forth as clinical candidates that inhibited both BACE1 and GSK3, the latter phosphorylates tau, at micromolar concentrations, thus being able to simultaneously modulate both major histopathological hallmarks of AD.194 Treatment with NP-12, also known as Nypta or Tideglusib, was tolerated over a 20 week period and produced a positive impact on patients’ cognitive performance in a Phase IIa trial, although given the small sample size this trial did not reach statistical significance, which is not unexpected.195 A larger Phase IIb trial is scheduled for later in 2010 to evaluate the compound more fully.
Investigation of the marine sponge Xestospongia sp. Schmidt (Petrosiidae) yielded xestosaprols D-M that weakly inhibited β-secretase.196,197 The configuration of the hydroxyl group on the D-ring of xestosaprol H (126) was found to be important for the observed inhibition, as the corresponding epimer, xestosaprol F (127) was significantly less active. While the activity of these compounds was weak, given their small size and hydrophobic nature, their BBB permeability is predicted to be high which suggests further evaluation may be warranted.
3.3. Terpenoids as secretase inhibitors
Terpenoid inhibitors of BACE1 are relatively rare. From the roots of the edible herb Aralia cordata Thunb. (Araliaceae), one ent-pimarane (128) and two ent-kaurane-type diterpenes (129–130) were isolated which inhibited BACE1.56 Modest activity was observed for these compounds in an enzyme assay (128: 24.1; 129: 18.6, 130: 23.4 μM). The identification of an inactive analog, possessing an epoxide at C-14/15, hints at the importance of the bicyclic system for observed inhibition.
Withania somnifera (L.) Dunal (Solanaceae) and Centella asiatica (L.) Urb. (Apiaceae) are recommended as memory and intellect enhancers in traditional ayurvedic medicinal systems. Chan et al. investigated the major components. Treatment of neuronal cells with withanolide A (131) and asiatic acid (132) (from W. somnifera and C. asiatica, respectively) significantly affected APP processing.198 In both cases, BACE1 processing was down-regulated while non-amyloidogenic α-secretase processing was increased. While the exact mechanism is unknown, direct inhibition of BACE1 is known to increase non-amyloidogenic processing as these pathways are mutually exclusive. In this case, evidence suggests a direct activation of α-secretase, in addition to activation of Aβ clearance mechanisms, occurs upon treatment in cells. The authors reiterate the proposal that “multifunctional” and “multilevel” activity may be required in an Alzheimer’s drug for true efficacy.199,200
3.4. Alkaloids as secretase inhibitors
Despite the large number of synthetic alkaloids known to inhibit BACE1, few naturally occurring alkaloids have been reported with this activity. Marine sponges belonging to the family Thorectidae, and genus Smenospongia in particular, are well-known sources of indole alkaloids, and an examination of a Panamanian species of S. cerebriformis Duchassaing & Michelotti (Thorectidae) identified an unusual bis-2-amino-imidazolone, dictazole A (133), which weakly inhibited BACE1.201 This compound may be of interest for further development as the 2-imino-imidazolidinone moiety is considered a privileged subunit responsible for the observed activity against BACE1 in many structurally unrelated compounds.202
The marine natural product bastadin 9 (134), isolated from Ianthella basta Pallas (Lanthellidae), is also reported to reduce APP processing via inhibition of BACE1.203 This compound is part of a larger family of metabolites that consists of more than 26 members,204 which have been reported to display a range of biological activities.205–208 Bastadin 9 inhibited BACE1 cleavage of APP with IC50 values of 0.3 and 2.8 μM in enzyme- and cell-based assays, respectively. Although other analogs inhibited BACE1 as well, they were less potent.203 At the time of their isolation, the bastadins were a new structural class of submicromolar BACE1 inhibitors, as no reports of other oxime (C=NOH) based inhibitors had been disclosed against this target. In 2008, Bristol-Myers Squibb patented a series of oxime-based cyclic nanomolar BACE1 inhibitors that resembled the upper half of the bastadins, and were able to permeate the BBB with nanomolar potency.209 These data suggest a critical evaluation of the BACE1 inhibitory effect of the bastadin structural class is warranted.
4. Compounds promoting anti-aggregation & clearance
While Aβ peptides are common in the brains of individuals with or without AD, the physiological role of Aβ is still unclear. It has been implicated in myelin sheath formation in developing cells210 and early lethality has been recently noted in transgenic mice,211 thus a drug that prevents Aβ formation could have unknown consequences. Reducing the concentration of Aβ42 oligomers by either inhibiting aggregation or increasing the rate of clearance of the soluble oligomers or insoluble fibril plaques is another possible therapeutic strategy that has been investigated.212 An ideal anti-aggregation drug would prevent the formation of Aβ aggregates, destabilize plaques, and reduce existing deposits. Although the oligomers are more toxic than their insoluble fibril plaque counterparts, both cause neurodegeneration though through two different apoptotic pathways,213 so an ideal drug would inhibit both processes.
Most of the compounds described below are only able to inhibit fibrillogenisis, but inhibitors of oligomerization would be more valuable. In general, these compounds are proposed to disrupt the weak bonds between residues in the β–sheets fibrils, through interfering with hydrogen bonding or electrostatic interactions. These compounds are often small (low molecular weight) and are either lipophilic or possess a number of polar substituents capable of competing for hydrogen bonds. Few of these small molecules show activity in the μM or nM range, and those that have the greatest activity have other problems (such lack of bioavailability, inability to cross the BBB, or lacking specificity) that prevent them from becoming likely drug candidates. However, many of these compounds are non-toxic and have multiple bioactivities (i.e., antioxidant, anti-inflammatory, enzyme inhibitors) that could provide multi-prong therapeutics for fighting the diverse pathologies of AD.
4.1. Compounds in clinical trials with these mechanisms
One of the first natural products to be investigated with an anti-aggregation mechanism was tramiprosate (135).214 The active ingredient is homotaurine, or 3-aminopropanesulfonic acid, which occurs naturally in seaweed.215 The synthetic compound was evaluated as tramiprosate, Alzhemed™, and Cerebril™. It is a glycosaminoglycan (GAG) mimetic that competes for GAG-binding sites in soluble Aβ and prevents the formation of fibrils. In vivo studies using TgCRND8 transgenic mice showed that tramiprosate specifically binds to soluble Aβ, preventing the β-sheet conformation, as well as reduces the amount of plaque, and the soluble and insoluble forms of Aβ40 and Aβ42 in the brain.214 While tramiprosate (Alzhemed, homotaurine, 3-aminopropanesulfonic acid) potentially inhibits Aβ, it promotes undesired tau aggregation.216 This drug failed Phase III clinical trials in the US, and Phase III trials were halted in the EU due to statistically inconclusive results.217 Ultimately, this compound may not have had the necessary potency to demonstrate a clear effect. In a controversial move, Bellus Health, formerly Neurochem, opted to market homotaurine as the “memory protective” nutraceutical Vivimind™ in Canada and on the internet.218
Scyllo-cyclohexanehexol (136), which occurs naturally in dogwood219 Cornus florida L. Spach (Cornaceae) and coconut palm220 Cocos nucifera L. (Arecaceae), is being evaluated in clinical trials by Transition Therapeutics and Élan, as AZD-103/ELND-005. In 2006, JoAnne McLaurin, Peter St. George-Hyslop, and colleagues at the University of Toronto demonstrated that AZD-103 lowered the amount of insoluble Aβ40 and Aβ42, lowered the amount of soluble Aβ oligomers, and reduced Aβ plaque load in the brains of transgenic mice.221 The resulting reversal of memory deficits was attributed to the inhibition and disaggregation of Aβ oligomers (20–60% decrease).221 In fact, the scyllo-treated mice navigated the water maze just as well as controls after several days of testing. Measurements of synaptic loss and glial inflammation also showed marked improvement. Results of Phase I clinical trials suggested the compound was well-tolerated and resulted in Phase IIa trials at multiple doses for mild to moderate AD (NCT00568776).221 The two-highest dose trials (1000 mg and 2000 mg dosed twice daily) were discontinued earlier this year (2010) due to nine deaths though, while the lower dose trial (250 mg dosed twice daily) continues.222
An active area of development is the use of the vaccines or antibodies to facilitate disruption of aggregates and clearance, which is based on the early work by the Schenk et al. with transgenic mice.223 Bapineuzumab224 is a humanized monoclonal antibody developed by Wyeth and Élan in Phase III clinical trials.225 It is widely considered one of the most promising candidates. Previous clinical trials using β-amyloid vaccinations (AN-1792) were halted when several patients developed aseptic meningoencephalitis.226 Almost no plaques were observed in brain autopsies of these two subjects though despite them reaching severe end-dementia.227 The significance of these findings, that Aβ levels were reduced with AN-1792 treatment, but neurodegeneration continued is still debated. Some argue it signifies the end of the amyloid cascade hypothesis, while others suggest that earlier treatment is necessary to stop the neurodegeneration initially triggered by Aβ. A more recent Phase II study evaluating the efficacy of bapineuzumab showed no significant results, although reversible vasogenic edema occurred more frequently in patients who were on higher doses and had APOEº4 risk factors.228 Based on an a prosteriori analysis, a Phase III clinical trial is currently being conducted in individuals without the APOEº4 mutation, who appeared to show a five point improvement, as compared to a two or three point improvement with AChE inhibitors such as Aricept,229 on the standard Alzheimer’s Disease Assessment Scale-cognitve subscale (ADAS-COG) in the Phase II trial.230 Ponezumab (PF-04360365; Pfizer) is another antibody vaccination that successfully completed two Phase I studies in patients with mild to moderate AD.231 This vaccination is now undergoing Phase II clinical trials.
Colostrinin™ (CLN; ReGen Therapeutics) is a mixture of proline-rich polypeptides (PRP) originally isolated by Polish scientists232 from ovine (sheep) colostrums. This is a form of milk produced by mammalian mammary glands late in a pregnancy. Some peptide components of CLN were homologous to annexin and β-casein, while other peptide components had unique sequences.233 Mechanistically, CLN inhibits Aβ aggregation and facilitates disassembly of existing aggregates by disrupting β-sheets bonding. In aged mice, CLN facilitated spatial learning and improved incidental learning, while having no negative effect on locomotive skills.234 In limited human trials, eight out of 15 Alzheimer’s patients who were given oral tablets showed an improved mental state and developed new memories better than controls.235 More extensive double-blind studies with 105 Alzheimer’s patients over a 15 week period on low dosages demonstrated that CLN helped to maintain cognitive and daily functions in patients with mild to moderate AD, and that it was well-tolerated.236 CLN is currently licensed as a non-medical nutraceutical CogniSure in the US and Australia.237
4.2. Polyketides promoting anti-aggregation & clearance
Rifampicin (137) is a semisynthetic polyketide originally derived from Amycolatopsis rifamycinica Bala (Pseudonocardiaceae). Popular for its use as a treatment for tuberculosis and leprosy, 137 also appears to inhibit Aβ aggregation in vitro.238 Rifampicin is proposed to inhibit the toxicity of Aβ fibrils by binding to the aggregates and preventing the adhesion of the fibrils to the cell surface.238 However, it was recently shown that instead of inhibiting the formation of amyloid fibrils, 137 was inhibiting the interaction of fibrils and thioflavin-T, which is used as an indicator in the assay.239 This result highlights the major difficult in screening for anti-aggregation inhibitors, specificity. Rifampicin also has poor BBB permeability,240 and thus lacks a crucial prerequisite for a viable neurological drug lead. A randomized trial though assessing the effectiveness of combining rifampicin and doxycycline (138) over a three-month period demonstrated a reduction in cognitive decline after six months in patients with mild to moderate AD.240 Doxycycline, a semisynthetic tetracycline antibiotic, was incorporated into the trial due to observations that tetracyclines inhibit the formation and promote disassembly of fibrils (vide infra). Presumably, multiple mechanisms are involved in this effect, including anti-inflammatory and anti-aggregation. Currently, a Phase III clinical trial is underway testing the effect of this combination on AD biomarkers in the cerebral spinal fluid.241
Tetracycline (139) is capable of inhibiting the formation of Aβ fibrils and promoting disassembly of existing Aβ fibrils in vitro.242 Isolated from Streptomyces spp., the antibiotic is structurally analogous to Congo red and iododoxorubicin, which are dyes known to bind to amyloid aggregates. Tetracycline interacts with Aβ fibrils via a combination of hydrogen bonding through its polar substituents and hydrophobic interactions through its aromatic groups.242 While 139 is able to penetrate the BBB, it produces minimal beneficial effects.242 A semi-synthetic derivative called minocycline was better able to inhibit Aβ fibril formation in vitro at a molar ratio of 2.5:1 to Aβ in comparison to tetracycline, which only inhibited when present in a ratio of 8:1. Other results of this same study showed that both tetracycline and minocycline could also inhibit microglial activation.243 Minocycline (140) exhibits neuroprotective effects in both in vitro and in vivo studies,244 as well as reduces capase-3 activation and the development of hyperphosphorylated tau species.245
4.3. Shikimate- and sugar-derived compounds promoting anti-aggregation & clearance
Curcumin (141), or diferuloylmethane, is a well-known ingredient in traditional Indian cuisine from the turmeric plant Curcuma longa L. (Zingiberaceae). This polyphenol effectively inhibits the formation of Aβ oligomers, binds already existing plaques, as well as reduces amyloid in vivo,246 while possessing greater BBB permeability than similar synthetic compounds such as Congo red. Several other beneficial effects have been attributed to 141 including anti-inflammatory and antioxidant properties.247 These mechanisms may be involved in the observed neuroprotective effect by blocking microglial activation.248 Structure-activity relationship studies suggest optimal activity requires two phenyl groups, one of which has polar substituents for hydrogen bonding, linked by a 8–16Ǻ spacer containing less than two sp3-hybridized carbons.249 Owing to numerous attractive qualities, including BBB permeability and minimal toxicity,246 Longvida, a curcumin formulation, is being evaluated in a Phase II Alzheimer’s clinical trial (NCT01001637).
Not surprisingly, a number of flavonoids have been reported as anti-aggregation agents. These compounds include: fisetin (142; 3,3′,4′,7-tetrahydroxyflavone), 77–78, chrysin, 105–106, 3′,4′,7-trihydroxyflavone, 3,3′,4′-trihydroxyflavone, 3,3′,7-trihydroxyflavone, 5-deoxykaempferol and synthetic derivatives.250 Myricetin (105) was the most potent compound while 3,3′,7-trihydroxyflavone, 5-deoxykaempferol, chrysin, and 106 enhanced fibril formation. The key inhibitory pharmacophore in these compounds was the 3′,4′-dihydroxyl group of the B ring,250 which facilitates the preferential reversible binding of myricetin to the neurotoxic oligomers rather than monomers.251 Compound 105 also interferes with Aβ conformational changes, inhibits BACE-1, and displays antioxidant activity.252
Other polyphenols inhibit fibrogenic Aβ (fAβ) formation and increase its degradation. Nordihydroguaiaretic acid (NDGA; 143) found in Larrea divaricata Cav. (Zygophyllaceae), was more potent than rifamycin253 and as an antioxidant can suppress the accumulation of reactive oxygen species (ROS) and Ca2+.254 Rosmarinic acid (144), found in various culinary herbs, has comparable activity to 105, 141, and 143.255 While 144 reduces Aβ accumulation in vitro, Aβ accumulation was noted in an in vivo rodent model.256 Rosmarinic acid also effects many Alzheimer’s related pathways, such as ROS formation, lipid peroxidation, DNA fragmentation, caspase-3 activation, and tau protein hyperphosphorylation.257,246 Tannic acid (145), widely distributed in wood, is more potent in inhibiting fAβ in vitro than 143,258 but with its large molecular weight, it is an unrealistic neurological drug candidate. A comparison of these and other active compounds established inhibition and degradation of the aggregates occurred with the following relative potency: tannic acid (145) = NDGA (143) = curcumin (141) = rosmarinic acid (144) = myricetin (105) > kaempferol (106) = ferulic acid (100) > (+)-catechin = (−)-epicatechin (146) > tetracycline (139).255
Another polyphenol that promotes the decomposition of Aβ aggregates is resveratrol (117; trans-3,4′,5-trihydroxystilbene). Commonly found in grapes, this compound promotes the clearance of intracellular Aβ by activating proteasomal degradation.259 A recent study suggests 117 disrupts Aβ42 hydrogen bonding thus preventing fibril formation, and it can destabilize preformed fAβ42 in vitro,260 but does not prevent oligmerization.260 The ability of 117 to inhibit Aβ42 aggregation ranked amongst the highest of the previously studied antioxidants: 117 > catechin > curcumin (141) > piceid > ginkgolides.260 Resveratrol is currently in Phase III clinical trials as a nutritional supplement in combination with glucose and malatein (Clinical trial # NCT00678431). The underlying rationale is that the glucose and malate prime oxidative metabolism and the Krebs-cycle in the brain, which aids in regenerating the reduced form of resveratrol under normal brain cell metabolism.261
1,2,3,4,6-Penta-O-galloyl-β-D-glucopyranose (147; PGG) is another tannin-type polyphenol that affects Aβ aggregation. Studies have shown that 147 inhibits and destabilizes fAβ40 and fAβ42 both in vitro and in vivo.262 In vivo studies also suggested that it inhibits oligomerization.262 While 147 has activity comparable to curcumin (141) and is non-toxic, its high molecular weight and hydrophilicity indicate poor BBB permeability is a serious issue.
Another sugar derivative with anti-aggregation properties is enoxaparin (148), a heparin found in the intestinal mucosa of pigs. Enoxaparin sodium, also known as Lovenox®, is manufactured by Sanofi-Aventis to prevent deep vein thrombosis. This poly-sulfonated compound was found to reduce Aβ plaque load, reduce the amount of astrocytes surrounding Aβ deposits, and reduce inflammatory effects associated with AD in vitro. However, the authors of this study recognize that this compound is not an effective long-term therapy due to its anticoagulant effect and poor bioavailability.263
The sugar analog α-D-mannosylglycerate (149) is a natural extremolyte found in bacteria and archaea that exist in extremely high temperatures. This analog was shown to inhibit aggregation of Aβ42 in vitro which increased cell survival rates. The authors suggested that 149 had an electrostatic interaction with a lysine residue of the Aβ42 that inhibited its aggregation, but acknowledged further confirmation was needed.264 An advantage to this compound is that it is non-toxic even at high concentrations.
4.4. Terpenoids promoting anti-aggregation & clearance
Terpenoids reported to have anti-aggregation activity are highly lipophilic and include retinol (150), retinal (151), and retinoic acid (152) (components of vitamin A), as well as β-carotene (153). Although vitamin A and 153 could inhibit the aggregation of fAβ40 and fAβ42, and destabilize them in vitro, these compounds could not depolymerize amyloid fibrils into monomers or oligomers.265 While 150 and 151 have activity comparable to NDGA (143), 152 and 153 are less active than 143. Anti-amyloidogenic and fibril-destabilizing activity are in the order of NDGA= retinol = retinal > β-carotene > retinoic acid.265 A similar result was obtained with coenzyme Q10 (154), although it was slight less potent than 143.266
4.5. Alkaloids promoting anti-aggregation & clearance
Only two naturally occurring alkaloids have been reported to directly affect Aβ aggregation. Nicotine (155), found in the Solanaceae family, affects multiple stages of amyloidogenesis in vitro. It was reported that 155 inhibited the formation and extension of fAβ40 and fAβ42, and was able to destabilize preformed fAβ40 and fAβ42. However, it could not break down the aggregates to their respective oligomers and monomers.267 This anti-amyloidogenic activity was attributed to the N-methylpyrrolidine moieties.268 While 155 is less potent than the other inhibitors described above,267 Aβ plaque levels were significantly reduced in transgenic mice after 5.5 months of treatment with 155 versus controls.268 However, in vivo studies also suggest 155 increases the aggregation and phosphorylation of tau, which would be an unwanted side-effect.269 Melatonin (156) is another anti-aggregation alkaloid found in many organisms. By disturbing salt bridges between the histidine and asparagine residues in the β-sheet conformation, 156 promotes random coil conformations of Aβ peptides, which facilitates clearance.270 In contrast to many of the compounds mentioned above, 156 can effectively cross the BBB.
5. Kinase modulators (tau and amyloid hypotheses)
Both activators and inhibitors of kinases have been examined as potential treatments for neurological disorders as kinases regulate a diverse array of cellular functions (reviewed recently by Watterson24). Numerous natural products that interact with kinases have been reported primarily focusing on their anti-cancer potential.271,272 There has been comparatively little direct application of natural product kinase inhibitors to the field of Alzheimer’s drug discovery though.
5.1. Compounds that activate protein kinase C
Protein kinase C (PKC) isoforms are a family of serine/threonine kinases,273 which are central and potentially critical junctions for memory acquisition and loss in both invertebrates and mammals.274,275 For example, the retrieval and formation of long- and short-term memories can be blocked by infusion of PKC inhibitors in an isozyme-dependent manner.276 The first indication of a potential casual role for these proteins in this neurodegenerative disorder was the observation of decreased levels of PKC-α in the brains of Alzheimer’s patients277 and that soluble β-amyloid is involved in the reduction278–280 by increasing non-amyloidogenic APP processing. Both direct and indirect PKC activation of the non-amyloidogenic pathways has been demonstrated. PKC can directly phosphorylate TACE (TNF-α converting enzyme, ADAM17),281,282 which is responsible for regulated α-secretase activity,283,282 or indirectly activate α-secretase through the MAP kinase ERK1/2.284,285
Most known PKC activators also promote tumor formation though. The PKC activating phorbol esters and indole alkaloids lyngbyatoxin A and teleocidin are widely used in biochemical studies, but are potent co-carcinogens preventing clinical applications. Synthetic compounds have been designed to reduce this effect,286,287 and in one case, the tumor promoting aplysiatoxin (157) from marine cyanobacteria, simplified derivatives (158) have been developed that decouple these two effects.288 The most notable exception is the natural product bryostatin, discovered in an anti-cancer screen of marine samples by George Pettit289 from the bryozoan Bugula neritina L. (Bugulidae). Bryostatin (159) enhances α-secretase activation in human fibroblast cells, reduces Aβ42 levels, and reduces mortality of transgenic AD mice.290 It also reverses Aβ42 produced deficits of PKC and ERK1/2 phosphorylation in cellular models of AD. Phase I and II clinical trials, focused on evaluating its anti-cancer potential,291 have confirmed bryostatin is not tumorigenic despite activating PKC. Blood-brain permeability is clearly a serious obstacle for this compound and a number of simplified synthetic analogs, “bryologs”, have been developed which may be able to address this issue.292 A Phase II clinical trial using bryostatin is on-going though.
Naturally-occurring polyunsaturated fatty acids (PUFA) from marine oils have been examined as neuroprotective agents against AD (reviewed by Boudrault293). Alpha-linolenic acid (160), eicosapentaenoic acid (161), and docosahexaenoic acid (162) are the three major dietary fatty acids, with the last two derived primarily from fish oil. Compound 162 is abundant in the human brain, accounting for 8% of the dry weight. PUFA intake has been evaluated in clinical trials (NCT01058941), the results of which suggest little therapeutic benefit as a treatment for AD, although subgroups of patients with mild cognitive impairment were responsive. Long-term preventative intake over a life-time may be a more viable strategy. Numerous mechanisms have been investigated for these compounds spanning neuroprotective to cholesterol lowering (relating to the APOɛ4 mutation). More recently Nishizaki and coworkers294 have demonstrated 160 is capable of selectively activating PKC-ɛ, a novel PKC, in the absence of the other required cofactors (phosphatidylserine and diacylglycerol) facilitating hippocampal synaptic transmission. Cyclopropyl derivatives, such as 163, which showed enhanced potency were prepared separately by Nshizaki294 and Nelson.295 Unlike PKC activators, such as bryostatin and the phorbol esters, that bind to the 1,2-diacylglycerol-binding site and produce prolonged down-regulation, the new activators produced sustained activation of PKC resulting in a 60–70% reduction in APP-overexpression in human neuronal cell cultures. Endothelin-converting enzyme was also significantly activated suggesting that the Aβ-lowering ability of these PKCε activators is caused by increasing the rate of Aβ degradation by endothelin-converting enzyme and not by activating non-amyloidogenic amyloid precursor protein metabolism.295 These intriguing findings further illustrate the complexity of APP processing and Aβ buildup.
5.2. Compounds that inhibit glycogen-synthase kinase 3 (GSK3)
GSK3 is involved in the phosphorylation of tau, a neuronal microtubule-associated phosphoprotein. Tau phosphorylation by GSK3 regulates the binding of tau to microtubules, its degradation and its aggregation. Hyperphosphorylation of tau is the other hallmark of AD, with many arguing this is the critical event. It has been proposed that β-amyloid promotes GSK activation, resulting in this hyperphosphorylation (reviewed by Henandez296). In addition, a “GSK hypothesis of AD” has been proposed by Lovestone and coworkers in which the over-activity of GKS3 results in memory impairment, tau hyperphosphorylation, increased β-amyloid production, and microglial-mediated inflammation.297 Regardless, of which hypotheses hold true, inhibiting GSK3 may be a viable therapeutic strategy (reviewed in Martinez194).
The first direct reversible inhibitor of GSK3 was lithium, discovered in 1996.298 It reduces GSK3 activity by competing with magnesium ions for binding299 and by increasing the inhibitory N-terminal serine phosphorylation of GSK3.300 Treatment with lithium results in decreased tau phosphorylation which enhances the binding of tau to microtubules and prevents neurotoxicity by reducing the production of β-amyloid in transgenic mouse models.301 Clinical trials in Japan and the US are on-going to evaluate the potential of these compounds.
Originally isolated by Higa from an Okinawan sponge of the genus Haliclona,302 the alkaloid manzamine A (164) was reported as a cellular GSK inhibitor (10 μM) by Hamann and co-workers in collaboration with NeuroPharm.303 Manzamine A effectively decreased tau hyperphosphorylation in human neuroblastoma cell lines thus successfully interfering with tau pathology. Analog semi-synthesis was reported along with structure-activity relationship studies against GSK3, which indicated both the indole and ircinal fragments were required. Selectivity was evaluated against a panel of six kinases, specifically CDK1, PKA, CDK5, MAPK, and GSK3α. Compound 164 non-competitively inhibited GSK3β and CDK5, the two kinases involved in tau hyperphosphorylation. These results suggest that 164 constitutes a promising scaffold from which more potent and selective GSK3 inhibitors could be designed as potential therapeutic agents for AD.
Palinurin (165) is a linear sesterterpene originally isolated from a marine sponge Iricina spp. Polejaeff (Irciniidae) by Alfano and coworkers in 1979.304 These non-ATP competitive inhibitors of GSK3 (4.5 μM) were patented by the Spanish biotechnology company NeuroPharm305 in collaboration with PharmaMar. No details have been reported after the initial patent though. Interestingly, a related difuran analog306 was inactive against both GSK3 and CDK5, suggesting the furanone moiety is crucial for activity.
Hymenaldisine (166) is a marine alkaloid that belongs to a group of natural products containing both bromopyrrole and guanidine moieties from sponges produced by the Agelasidae, Axinellidae, and Halichondriidae families. As part of a larger screen for kinase inhibitors using a purified library of natural products, Meijer and coworkers307 discovered this compound was a competitive ATP inhibitor of GSK3β (35 nM) and CDK5/p35 (28 nM) in vivo. Both kinases contribute to the phosphorylation of 40 of the 85 available sites on tau. Some degree of selectivity was noted against a broad panel of 40 kinases as most IC50 values were greater than 1 μM, although CDK1/cyclin B, CDK2/cyclin A, CDK2/cyclin E, and CDK3/cyclin E were also inhibited at low nanomolar concentrations. A crystal structure was obtained with CDK2, but extending these conclusions to identify common structural features for all inhibited kinases was not possible, so additional crystallographic work is required using GSK3β or CDK5 to better understand hymenaldisine’s selectivity against the AD relevant kinases.
Mejier and coworkers also reported that the indirubins were selective GSK3 inhibitors.308 These compounds 167–168, occurring naturally in gastropod mollusks of the Muricidae and Thaididae families, have been widely used as components of a dye, known as tyrian blue, due to their striking purplish-blue color and are the key ingredient in the traditional Chinese medicine ‘danggui longhui wan’ used to treat chronic myelocytic leukemia. Compound 167 is derived from a spontaneous non-enzymatic dimerization of two common precursors, istatin and indoxyl. Evaluating both naturally occurring and synthetic analogs revealed that 6′-bromosubstitution on the indirubins conferred strong selectivity for both isoforms of GSK3 (IC50 22 nM). The selectivity of these competitive ATP kinase inhibitors can be enhanced by converting the ketone 167 to an oxime 168 such that the ERK, MAPKK, PKC families of kinases were not inhibited up to 10 μM, while CDK-1, -2, -4 were inhibited at approximately 0.3 μM. Crystal structures of 167 and other derivatives bound to GSK3β, CDK5, and CDK2 indicate that the selectivity is due to an unfavorable van der Waal interaction in CDK5 and CDK2 between the 6′ bromine atom and the Phe132 residue. In GSK3, a smaller leucine residue is substituted in this position thus reducing this unfavorable steric interaction. Given the hydrophobic nature of these compounds though, the issue of solubility must be addressed before any clinical application.
A number of other GSK inhibitors have been identified from natural sources over the years (reviewed in Fusetani309 and Alonso305), although as outlined above few have been evaluated in Alzheimer’s models. Given the importance of GSK3 in this degenerative disease, a re-evaluation of many of these structural classes as neurological leads may be warranted.
6. Antioxidant natural products
Although oxidative mechanisms are associated with AD pathology and hypotheses have been proposed,310,311,16 clinical trials investigating antioxidants to alleviate AD symptoms have, to date, not been convincing. Evidence for their ability to delay disease progression is limited, with most studies focusing on antioxidant vitamins rather than phytochemicals.312–316 Since the pathology of AD is complex, it is too simplistic to assume that antioxidant treatment alone might alleviate or delay cognitive decline in dementia. It should be considered however, that there is some epidemiological evidence that diets rich in particular antioxidant phytochemicals may reduce the risk of developing dementia.317 Thus, a multi-targeted therapeutic approach including antioxidants in combination with drugs or phytochemicals that target other pharmacological mechanisms might be a more rational approach to dementia treatment. It is therefore not surprising that those plants that produce some positive effects on cognition in AD patients have frequently shown antioxidant and other activities relevant to dementia pathology when their modes of action have been investigated in vitro and in vivo. Those species with a range of relevant neurobiological activities, including antioxidant, that have shown some promising effects on cognition in dementia patients include Salvia officinalis L. or S. lavandulifolia Vahl., Melissa officinalis L. (Lamiaceae), Crocus sativus L. (Iridaceae) and the extensively studied Ginkgo biloba L. (Ginkgoaceae).96 G. biloba has been the subject of much attention for its antioxidant activity and other modes of action, and for its clinical effects in dementia (recently reviewed by Shi et al.318 and Perry and Howes96). It should be noted however, that a recent Cochrane review concluded that evidence for any predictable or clinically significant benefit of G. biloba in dementia or cognitive impairment is inconsistent and unreliable, although it appears to do no harm.319 The contradictory data for these compounds clearly suggest further trials are needed.
Consumption of curry is associated with improved cognitive function320 and a lower prevalence of AD in some populations is suggested to be due to a curcumin-rich diet.317 Curcumin (141) from turmeric (Curcuma longa L. (Zingiberaceae)) has numerous activities, including antioxidant, which might explain the beneficial cognitive effects. Curcumin (141) is neuroprotective in vitro321 and in addition to other compounds from C. longa (demethoxycurcumin (169), bisdemethoxycurcumin (170), calebin A (171)) and some synthetic analogs, protects PC12 cells from β-amyloid322,323 due to antioxidant effects.322 Other mechanisms324 may also contribute to these protective effects though as curcumin (141) protects against ethanol-induced brain injury, reduces brain lipid peroxide levels and enhances glutathione, thus reducing oxidative damage, in an in vivo dementia model.325–327 It also protects against aluminum-induced cognitive dysfunction and ameliorates memory impairment (prevents and treats) in vivo.111,328 It is reported that the enol structure with the intramolecular hydrogen bond is principally responsible for the free radical scavenging activity of 141, and the phenolic hydrogens are essential for antioxidant activity and free radical kinetics.329 An indirect antioxidant mechanism of 141 is its ability to complex with metal ions (observed in vitro and in vivo),330 suggested to be via the diketone and pairs of phenol and methoxy groups of the structure of 141.331 It is therefore suggested to reduce metal-induced amyloid aggregation or oxidative neurotoxicity in AD.331 A recent finding is that 141 can maintain the catalytic function of protein disulfide isomerase (PDI), which catalyzes maturation of disulfide-bond-containing proteins; 141 prevents PDI-resistant misfolded protein forms that accumulate with oxidative stress and are involved in the pathogenesis of AD.332 Other mechanisms of 141 relevant to modulating AD pathology have been recently reviewed.317 Evidence for efficacy of 141 in dementia patients is lacking, although some clinical studies in AD patients are in progress but outcomes are still unknown.329
Green tea (Camellia sinensis Kuntze (Theaceae)) leaves contain polyphenolic compounds, with catechins as the major constituents, although processing methods influence the chemical composition of tea leaves.333 Epidemiological data suggests green tea polyphenols improve age-related cognitive decline and associates tea drinking with a reduced risk of dementia.334,335 Catechins are well-documented as antioxidants and scavengers of reactive oxygen species (ROS), and they chelate metal ions.317,336,334,337 Tea catechins improve memory acquisition and retention in senescence-accelerated mice (SAM) and suppress oxidative damage to DNA in SAM brain.334 Epigallocatechin-3-gallate (115) is a more potent scavenger of ROS than some other tea catechins, which is attributed to the presence of the trihydroxyl group on the B ring and the gallate moiety at the 3′ position in the C ring;314,336 this is perhaps why subsequent studies investigating the relevance of tea catechins for AD have focused on 115. It is the scavenging of ROS that explained a protective effect of 115 on hippocampal neuronal cells exposed to β-amyloid338 and in vivo, 115 mitigated β-amyloid-induced oxidative stress and reduced hippocampal lipid peroxide.317 One mechanism by which 115 protects against β-amyloid-induced oxidative cell death is by enhancing cellular glutathione by elevating mRNA expression of γ-glutamylcysteine ligase, the rate limiting enzyme in glutathione biosynthesis.339 It is also suggested that 115 is neuroprotective against oxidative stress by stimulating protein kinase C and modulating cell cycle genes.340 Epicatechin (146) increased glutathione levels in astrocytes,341 and it improved memory skills and decreased lipid peroxidation and ROS in rats with β-amyloid-induced hippocampal toxicity.342 Other bioactivities of tea catechins relevant to AD have been recently reviewed.317,336,337
Light to moderate alcohol consumption is associated with a reduced risk of AD and some other types of dementia343 and epidemiological evidence suggests moderate red wine consumption may attenuate clinical dementia in AD.344 Various activities have been associated with wine polyphenols, resveratrol (117) in particular, and thus are suggested to explain the possible preventive effects, and have been investigated for possible therapeutic effects for AD. Resveratrol (117) (trans-3,4′,5-trihydroxystilbene) occurs in various plants including grapes (Vitis vinifera L. (Vitaceae)) and it produces a number of mechanistic effects, including antioxidant, relevant for AD treatment. Specifically, it scavenges ROS (the 4′-OH group is an important structural feature), upregulates cellular antioxidants including glutathione and is neuroprotective against oxidative stress in vitro and in vivo.345,317,346 Piceatannol (172) (3′,4′,3,5-tetrahydroxystilbene) occurs in many resveratrol-containing species but at lower concentrations. It is a metabolite of 117 and has also been considered for application in neurodegenerative diseases. Interestingly, 172 is a more efficient scavenger of ROS than 117, due to the additional 3′-OH group adjacent to the 4′-OH group present in 117; the 3′-OH hydrogen is shared through a strong intramolecular hydrogen bond between O3′ and O4′ making the abstraction and transfer of the 4′-H atom to a free radical easier, and a more stable semiquinone radical is formed.347 Resveratrol (117) protects astrocytes in rat hippocampal slices from H2O2-induced oxidative stress by increasing glutathione levels in addition to other mechanisms.348 It also prevents cognitive impairments and associated oxidative stress in vivo317,349 and reduces plaque formation in a transgenic model of AD.350 It is apparent there is emerging evidence that 117 may modulate AD pathology due to antioxidant effects, or by various other mechanisms (including activation of sirtuin 1, a histone deacetylase which is involved in responding to molecular damage and metabolic imbalances).317,351,352 However, clinical evidence for any potential efficacy on AD progression is still lacking.
There are numerous other reports of structurally diverse phytochemicals that display antioxidant effects and are suggested as relevant to modulate dementia pathology, although the majority of these studies have focused on structures with phenolic components. The effects of a range of dietary polyphenols on events related to AD pathology have recently been reviewed.353,354 There are many flavonoids reported to protect against oxidative stress in neuronal cell-lines and in some brain conditions in vivo.355,356 Antioxidant mechanisms are considered to explain why some flavonoids protect against oxidative stress [quercetin (77), myricetin (105), luteolin (78),357,358 hyperoside,359 fisetin (142)360] and β-amyloid-induced toxicity [puerarin,361 genistein,362 77,363 naringenin (79),364 baicalein, baicalin93] in neuronal cells in vitro, and against β-amyloid-induced cognitive impairments in vivo [silibinin365]. Several synthetic lipophilic alkenylated 2,3-dehydrosilybin analogs of the flavolignan silibinin are neuroprotective against H2O2-induced toxicity in vitro and are being investigated for their pharmacological potential in CNS disorders.366
Also off notable interest are studies in rodents showing dietary supplementation with spinach (Spinacia oleracea L. (Chenopodiaceae)), strawberry (Fragaria ananassa Duchesne (Rosaceae)), or blueberry (Vaccinium myrtillus L. (Ericaceae)) extracts to reverse brain age-related deficits and behavioral function, and blueberry supplementation effectively reversed cognitive declines in object recognition; these observations were not attributed to antioxidant activity alone.367 However, strawberry and blueberry anthocyanins protect against oxidative stress in neuronal cells in vitro368 and in rodent brain in vivo, respectively.369 In a recent preliminary trial, wild blueberry (V. angustifolim Benth.) juice (containing anthocyanins and phenolic compounds, including chlorogenic acid) consumed for 12 weeks produced neurocognitive benefits in older adults with early memory changes, compared to a placebo beverage absent from natural polyphenolic compounds.370 It is apparent that there is some therapeutic potential for antioxidant phytochemicals in cognitive disorders. However, there is a lack of evidence that the activities of these phytochemicals can be translated to clinically relevant effects in the complex pathology of AD.
7. Neuro-regenerative compounds
Allopregnanolone (173) is a progesterone metabolite produced in embryonic and adult CNS. The abundance of neurosteroids, such as allopregnanolone, decreases during aging, but in particular, in AD patients in a manner that parallels the decline in the proliferative ability of neuronal progenitor cells. Treatment with allopregnanolone has been shown to significantly increase proliferation and survival of neural progenitor cells in rodents and human cells in vitro.371 These beneficial effects occur via GABAA receptor and L-type Ca2+ channel-dependent mechanisms leading to an efflux of chloride and an activation of CREB transcription factors.372,371 This activates a key pathway in adult hippocampal neurogenesis that promotes proliferation, survival, and differentiation of neural progenitor cells. Brinton and coworkers recently demonstrated the efficacy of allopregnanolone treatment in a male triple transgenic mouse model of Alzheimer’s.373 In a dose dependent manner, treatment with 173 prior to the onset of AD pathology restored neural progenitor cell proliferation to normal levels and reversed the cognitive deficits, thus restoring normal learning and memory performance. These findings suggest that treatment with allopregnanolone may serve as a regenerative therapeutic for patients with mild cognitive impairment and AD, at a stage before immunodetectable Aβ.373
Other steroids have been evaluated as neuroprotective agents. HF0220 is a 7-hydroxy epiandrosterone (174). These compounds are produced when CYP450 hydroxylates steroids, such as estradiol, at the 7-position which occurs at high levels in the brain. Pringle and coworkers374 have demonstrated that these hydroxylated metabolites significantly reduced neurotoxicity at concentrations of 10 and 100 nM in a rat model of cerebral ischaemia even when administered after onset. HF0220 (174) has been developed further by Hunter-Fleming, which was acquired in 2008 by Newron Pharmaceuticals. This compound has successfully completed safety and tolerability studies in patients with mild to moderate AD. The very high rate of completion of the study by patients, the absence of clinically relevant or statistically significant changes in safety measures, and the very low number of patients experiencing any adverse events, indicate that HF0220 can be safely administered to patients with AD who often experience multiple concomitant illnesses and who are more susceptible to the side-effects of their usual medications.375
Successful axon regeneration in the adult CNS is normally prevented by a number of factors, including the presence of inhibitory molecules that originate from the myelin sheath and glial scars.376 Upon injury to axons, the myelin sheath breaks down releasing inhibitory components, myelin associated inhibitors (MAIs), that contain proteins Nogo-A, myelin-associated glycoproteins (MAG) and oligodendrocyte-myelin glycoproteins.377 A glial scar forms shortly after the injury producing extracellular matrix molecules, such as chondroitin sulfate proteoglycan, that suppress axonal growth.378,379 Only a few compounds have the ability to interrupt these inhibitors in a manner that enhances neurite outgrowth in vivo though. The Discovery Neurosciences division of Wyeth (now Pfizer Research) initiated a screen of 180 diverse natural products and a 1300-compound drug set to identify antagonizers of these inhibitory molecules. Amphotericin B (175) was shown to promote neurite outgrowth and prevent the activities of the major myelin and glial associated inhibitors.380 This intriguing effect was not related to amphotericin’s antifungal activity as the structurally and mechanistically related pore-forming antibiotic nystatin had no effect on neurite outgrowth. Comparative screening of a library of kinase inhibitors revealed, this antagonism was shown to occur via activation of Akt, which suppressed the activity of GSK3β.
Almost 20% of the body’s consumption of glucose occurs in the brain, suggesting metabolic deficits that are noted in the AD patients may significantly contribute to pathogenesis. Attempts to compensate for reduced cerebral metabolic rates by supplementing with glucose and insulin have met with some success, although have notable side effects. An alternative strategy is to administer ketone bodies (acetone, acetoacetic acid, and beta-hydroxybutyrate), either directly or as their metabolic precursors (medium chain triglycerides). Accera developed two products along these lines. In a preliminary study, Ketasyn™ (AC-1202) demonstrated pharmacological activity and statistically significant efficacy in improving short-term memory and attention performance after a single dose in clinical trials. More recently, this formulation, has been renamed Axona (the active ingredient is 176) and it has been approved as a “medical food” intended for the clinical dietary management of the metabolic processes associated with mild-to-moderate AD. Medical foods are specially formulated prescribed foods defined under the US Orphan Drug Act of 1988. These are “intended for the specific dietary management of a disease or condition for which distinctive nutritional requirements, based on recognized scientific principles, are established by medical evaluation.”
8. Modulators of ion channels
Ion channels have a critical role in maintaining proper CNS function, and have long been suspected as a potential factor in AD.381 For example, accumulation of Aβ peptides in neurons has been shown to activate ion channels causing an influx of Ca2+ that disrupts homeostasis, leading to mitochondria malfunction and oxidative stress and apoptosis of neurons.382 To date, the only successful modulator of these targets for the treatment of AD is the synthetic N-methyl-D-aspartate receptor (NMDAR) antagonist, memantine, approved in the EU in 2002 and in the US in 2003383 for patients with moderate-to-severe AD. Evidence suggests it protects against Ca2+ influx without disrupting physiological synaptic activity.384 This beneficial effect is in sharp contrast to other high-affinity NMDAR antagonists that are often neurotoxic.384 A clear rationale to explain these discrepancies is still lacking though.384 Several synthetic compounds, which target L-type Ca2+ (reviewed by Yu385) and nicotinic receptor ion channels (reviewed by Arneric386) are currently undergoing clinical evaluations to determine their efficacy as well.
The first marine natural product to be evaluated as a potential treatment for AD, based on its ability to modulate ion channels, was described by William Kem (reviewed by Kem387). In 1971, Kem reported the discovery of the alkaloid anabaseine (177) from several species of marine worms which prompted an extensive evaluation of this compound and synthetic derivatives. This research has culminated in DMXBA [also known as GTS-21; 178] which recently completed a Phase II clinical trial with Comentis Inc, although for schizophrenia, in which patients demonstrated improvements in cognitive function.23 These beneficial effects are due to the ability of GTS-21 to stimulate selectively α7 nicotinic acetylcholine receptors expressed on CNS neurons and astrocytes.388 GTS-21 counteracts the neurotoxic effects of β-amyloid in neuronal cells from the cerebral cortex, and improves cognitive functions in a variety of animal models. For full details regarding GTS-21, the reader is directed towards the recent review by Toyohara and Hashimoto.389
Natural products that modulate ion channels have been discussed elsewhere,390,391 and as such, will only be briefly mentioned. Talatisamine (179), an alkaloid in Aconitum heterophyllum Wall. (Ranunculaceae), was evaluated for its channel specificity (K+, Na+ and Ca2+) in rat hippocampal neurons.392 Compound 179 specifically inhibited potassium currents (IK) by binding to the external pore entry of the channel with an IC50 value of 146.0 ± 5.8 μM.392 The prenylflavone icariin (81), isolated from Epimedium brevicornu Maxim. (Berberidaceae), was shown to reduce behavioral dysfunction and neurodegeneration in rat models,393 while blocking Ca2+ currents disrupted by Aβ25-35.394 A synthetic derivative of the sponge (Aplysina cavernicola Vacelet (Aplysinidae)) metabolite 11,19-dideoxyfistularin, NP04634 (180), protected neurons from calcium overload and mitochondrial disruption. Specifically, 180 protects neurons from toxicity induced by 30K+/5Ca2+/FPL at a concentration of 10 μM, reduces voltage dependent Ca2+ levels and protects mitochondrial disruption by preventing depolarization.395
Voltage-gated K+ channels (KV) are responsible for the electrical activity in neurons and a reduction of K+ in cells can cause neuronal apoptosis. A study by Yu et al. demonstrated that exposure to Aβ fragments induces cell death by enhancing the delayed rectifier K+ current (IK), which causes an efflux of K+ that consequently leads to apoptosis.396 Since the addition of IK blockers (TEA, 5mM) slowed neuronal death in this study, as opposed to the addition of Ca2+ blockers, it was suggested that the IK plays a pivotal role in neuronal apoptosis. Further evidence that the KV 3.4 ion channel, in particular, is involved in amyloid pathology was provided in the result of overexpression of the KV 3.4 in transgenic mice with the Swedish mutation of APP.397
BDS-I and –II toxins, isolated from Anemonia viridis Forskål (Actiniidae), are peptides that were found to block KV more specifically than talatisamine. Out of the four major potassium channel subfamilies (KV1, KV2, KV3, and KV4), BDS-I and –II were originally reported to inhibit the KV3.4 ion channel with IC50 values of 47nM and 56nM respectively.398 While Diochot et al. reported that BDS-I was able to block KV 3.4 specifically, Yeung et al. was able to show that BDS-I also inhibits other KV 3 subfamilies, KV 3.1 and KV 3.2.399 Since AD distinctively affects the KV 3.4 ion channel, there is still a need for blockers that are specific to the KV 3.4 ion channel.
Many natural products have been identified as different potassium channel blockers, for instance, compounds isolated from scorpion venom (charybdotoxin, maurotoxin, margatoxin, agitoxin-2, kaliotoxin), sea anemone toxin (ShK toxin), and a Costa Rican tree (correolide) all of which are active at nM and pM concentrations.400 All of these compounds serve as examples of very active ion channel blockers from natural sources; however they also point to the importance of finding a blocker with the affinity to the right type of ion channel, as well as to the desired ion channel subfamily.
9. Conclusion
Several conclusions about natural product Alzheimer’s drug leads are supported by the literature. First, the majority of the compounds examined to date with a direct relevance to AD are primarily from plants, with comparatively fewer molecules derived from marine and microbial sources. Also, to date, the greatest successes have come from plant-based AChE discovery programs, which have resulted in two of the four currently approved drugs for the treatment of AD. This latter fact likely significantly contributes to the former. It is widely accepted that these AChE inhibitors are only effective at treating the symptoms of AD in the short term though and a broader range of therapeutics is needed. Given the diverse pathology of AD these therapeutics might target multiple mechanisms simultaneously. Several compounds noted above, EGCG (115), galantamine (1), resveratrol (117), and withanolide A (131), fulfill these requirements, although further study is needed in all cases. As illustrated by the review, and summarized in Table 1, a number of natural products are currently being evaluated for their clinical effects. Whether the requisite properties can be engineered into these leads, either as individual agents or in combination, remains to be seen.
Table 1.
Clinical Status of Selected Alzheimer’s Natural Products or Derivatives
| Mechanism | Compound | Source | Phase I |
Phase II |
Phase III |
Phase IV |
|---|---|---|---|---|---|---|
| AChE | ||||||
| Galantamine (1) | Plant | |||||
| Rivastigmine (2) | Syn. Analog | |||||
| Huperzine A (40) | Plant | |||||
| Secretase | ||||||
| NP-12 (125) | Marine | |||||
| Aggr./Clear. | ||||||
| Homotaurine (135) | Marine | Dis. | Nutraceutical | |||
| AZD-103 (136) | Plant | |||||
| Bapineuzmab | Antibody | |||||
| Colostrinin | Mammal | Nutraceutical | ||||
| Rifampicin (137) | Bacteria | |||||
| Curcumin (141) | Plant | |||||
| Resveratrol (117) | Plant | |||||
| Kinases | ||||||
| Bryostatin A (159) | Marine | Recruit. | ||||
| Lithium | ||||||
| Neuroprot. | ||||||
| HF-0220 (174) | ||||||
| Axona (176) | Medical Food | |||||
| Ion Chan. | ||||||
| GTS-21 (178) | Syn. Analog. | |||||



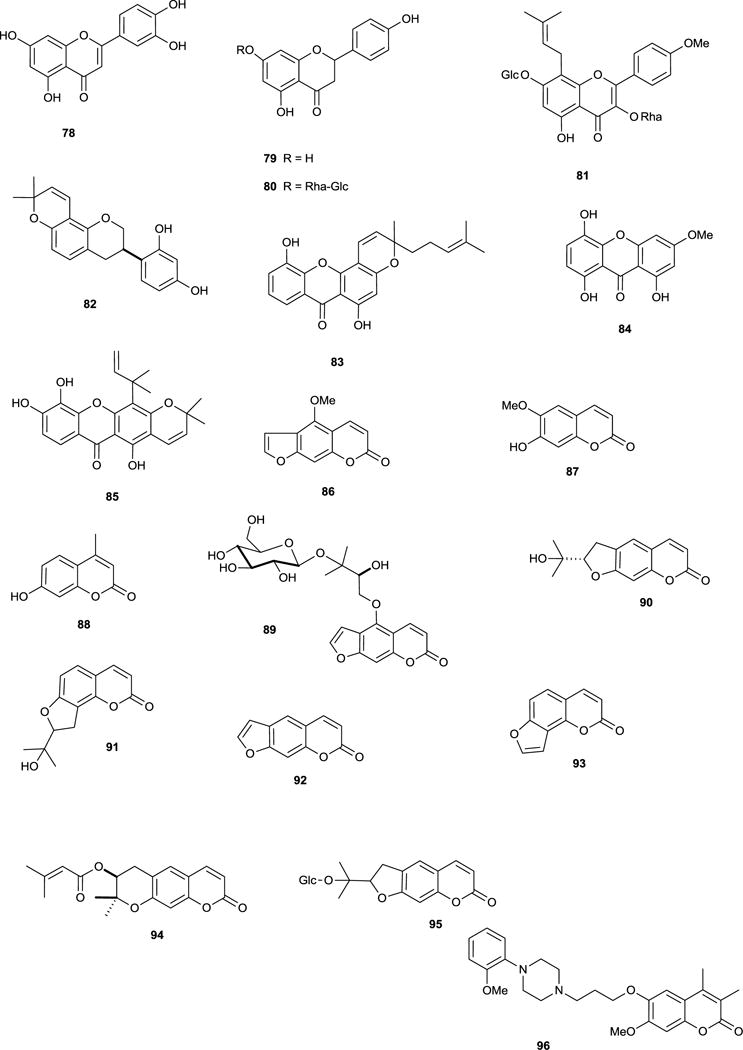

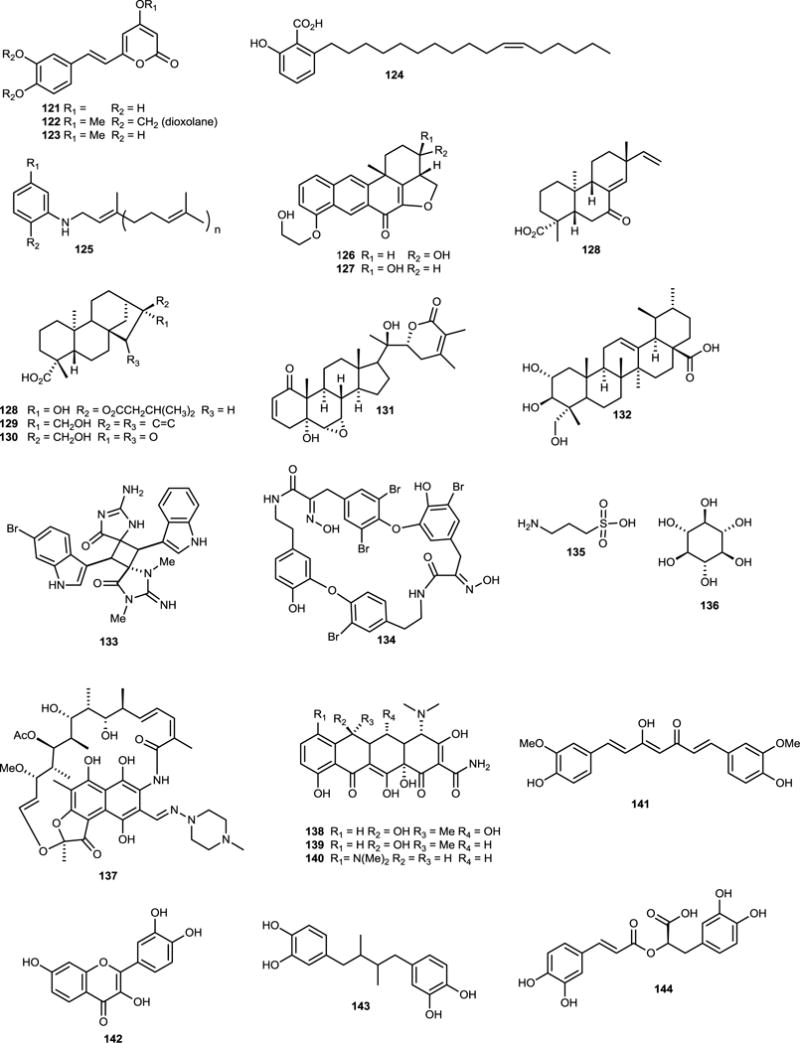



Acknowledgments
Funding for the Williams’ laboratory was provided by the Alzheimer’s Association, Alzheimer’s Drug Discovery Foundation, the Victoria S. and Bradley L. Geist Foundation and the National Institute of Aging (R21AGO32405).
References
- 1.Alzheimer’s Disease International. World Alzheimer Report. 2009:1–24. [Google Scholar]
- 2.Alzheimer A. Centrablat fur Nevenheilkunde Psychiatrie. 1907;30:177–179. [Google Scholar]
- 3.Lahiri DK, Maloney B, Basha MR, Ge YW, Zawia NH. Curr Alzheimer Res. 2007;4:219–228. doi: 10.2174/156720507780362164. [DOI] [PubMed] [Google Scholar]
- 4.Jakob-Roetne R, Jacobsen H. Angew Chem Int Ed. 2009;48:3030–3059. doi: 10.1002/anie.200802808. [DOI] [PubMed] [Google Scholar]
- 5.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 6.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 7.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 8.Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerrière A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 9.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 10.He X, Cooley K, Chung CHY, Dashti N, Tang J. J Neurosci. 2007;27:4052–4060. doi: 10.1523/JNEUROSCI.3993-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardy JA, Higgins GA. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 12.Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 13.Lee VM, Trojanowski JQ. Curr Opin Neurobiol. 1992;2:653–656. doi: 10.1016/0959-4388(92)90034-i. [DOI] [PubMed] [Google Scholar]
- 14.Wolfe MS. Journal of Biological Chemistry. 2009;284:6021–6025. doi: 10.1074/jbc.R800013200. [DOI] [PubMed] [Google Scholar]
- 15.Mandrekar-Colucci S, Landreth GE. CNS Neurol Disord Drug Targets. 2010;9:156–167. doi: 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moreira PI, Smith MA, Zhu X, Santos MS, Oliveira CR, Perry G. Expert Rev Neurother. 2004;4:995–1004. doi: 10.1586/14737175.4.6.995. [DOI] [PubMed] [Google Scholar]
- 17.Deary IJ, Hendrickson AE. Lancet. 1986;1:1219. doi: 10.1016/s0140-6736(86)91205-5. [DOI] [PubMed] [Google Scholar]
- 18.Perry G, Nunomura A, Raina AK, Aliev G, Siedlak SL, Harris PL, Casadesus G, Petersen RB, Bligh-Glover W, Balraj E, et al. Neurochem Res. 2003;28:1549–1552. doi: 10.1023/a:1025678510480. [DOI] [PubMed] [Google Scholar]
- 19.Hung YH, Bush AI, Cherny RA. J Biol Inorg Chem. 2010;15:61–76. doi: 10.1007/s00775-009-0600-y. [DOI] [PubMed] [Google Scholar]
- 20.Crapper DR, Krishnan SS, De Boni U, Tomko GJ. Trans Am Neurol Assoc. 1975;100:154–156. [PubMed] [Google Scholar]
- 21.Viegas C, Jr, Bolzani VS, Barreiro EJ, Fraga M, Carlos A. Mini Rev Med Chem. 2005;5:915–926. doi: 10.2174/138955705774329546. [DOI] [PubMed] [Google Scholar]
- 22.Houghton PJ, Ren Y, Howes M. Nat Prod Rep. 2006;23:181–199. doi: 10.1039/b508966m. [DOI] [PubMed] [Google Scholar]
- 23.Mayer AM, Glaser KB, Cuevas C, Jacobs RS, Kem W, Little RD, McIntosh JM, Newman DJ, Potts BC, Shuster DE. Trends Pharmacol Sci. 2010;31:255–265. doi: 10.1016/j.tips.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 24.Chico LK, Van Eldik LJ, Watterson DM. Nat Rev Drug Discov. 2009;8:892–909. doi: 10.1038/nrd2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H, Zhu X, Nunomura A, Perry G, Smith MA. Curr Alzheimer Res. 2006;3:75–80. doi: 10.2174/156720506775697124. [DOI] [PubMed] [Google Scholar]
- 26.Lee H, Zhu X, Castellani RJ, Nunomura A, Perry G, Smith MA. J Pharmacol Exp Ther. 2007;321:823–829. doi: 10.1124/jpet.106.114009. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen JT, Yamani A, Kiso Y. Curr Pharm Des. 2006;12:4295–4312. doi: 10.2174/138161206778792976. [DOI] [PubMed] [Google Scholar]
- 28.Salloway S, Mintzer J, Weiner MF, Cummings JL. Alzheimers Dement. 2008;4:65–79. doi: 10.1016/j.jalz.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 29.Bencherif M, Lippiello PM. Med Hypothesis. 2010;74:281–285. doi: 10.1016/j.mehy.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Gong G, O’Bryant SE. Alzheimer Dis Assoc Disord. 2010 doi: 10.1097/WAD.0b013e3181d71bc7. [DOI] [Google Scholar]
- 31.Bartus RT, Dean RL, Pontecorvo MJ, Flicker C. Ann N Y Acad Sci. 1985;444:332–358. doi: 10.1111/j.1749-6632.1985.tb37600.x. [DOI] [PubMed] [Google Scholar]
- 32.Julian PL, Pikl J. J Am Chem Soc. 1935;57:755–757. [Google Scholar]
- 33.Kamal MA, Greig NH, Alhomida AS, Al-Jafari AA. Biochem Pharmacol. 2000;60:561–570. doi: 10.1016/s0006-2952(00)00330-0. [DOI] [PubMed] [Google Scholar]
- 34.Howes MR, Houghton P. Int J Biomed Pharm Sci. 2009;3:67–86. [Google Scholar]
- 35.Musiał A, Bajda M, Malawska B. Curr Med Chem. 2007;14:2654–2679. doi: 10.2174/092986707782023217. [DOI] [PubMed] [Google Scholar]
- 36.Tumiatti V, Bolognesi ML, Minarini A, Rosini M, Milelli A, Matera R, Melchiorre C. Expert Opin Ther Patents. 2008;18:387–401. [Google Scholar]
- 37.Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M, Melchiorre C. J Med Chem. 2008;51:347–372. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- 38.Butler MS. Nat Prod Rep. 2008;25:475–516. doi: 10.1039/b514294f. [DOI] [PubMed] [Google Scholar]
- 39.Loizzo MR, Tundis R, Menichini F, Menichini F. Curr Med Chem. 2008;15:1209–1228. doi: 10.2174/092986708784310422. [DOI] [PubMed] [Google Scholar]
- 40.Kadir A, Andreasen N, Almkvist O, Wall A, Forsberg A, Engler H, Hagman G, Lärksäter M, Winblad B, Zetterberg H, Blennow K, Långström B, Nordberg A. Ann Neurol. 2008;63:621–631. doi: 10.1002/ana.21345. [DOI] [PubMed] [Google Scholar]
- 41.Zhan Z, Bian H, Wang J, Shan W. Bioorg Med Chem Lett. 2010;20:1532–1534. doi: 10.1016/j.bmcl.2010.01.097. [DOI] [PubMed] [Google Scholar]
- 42.Park CH, Kim SH, Choi W, Lee YJ, Kim JS, Kang SS, Suh YH. Planta Med. 1996;62:405–409. doi: 10.1055/s-2006-957926. [DOI] [PubMed] [Google Scholar]
- 43.Decker M. Eur J Med Chem. 2005;40:305–313. doi: 10.1016/j.ejmech.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 44.Wang B, Mai Y, Li Y, Hou J, Huang S, Ou T, Tan J, An L, Li D, Gu L, Huang Z. Eur J Med Chem. 2010;45:1415–1423. doi: 10.1016/j.ejmech.2009.12.044. [DOI] [PubMed] [Google Scholar]
- 45.Ingkaninan K, Changwijit K, Suwanborirux K. J Pharm Pharmacol. 2006;58:847–852. doi: 10.1211/jpp.58.6.0015. [DOI] [PubMed] [Google Scholar]
- 46.Chattipakorn S, Pongpanparadorn A, Pratchayasakul W, Pongchaidacha A, Ingkaninan K, Chattipakorn N. J Ethnopharmacol. 2007;110:61–68. doi: 10.1016/j.jep.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 47.Lima JA, Costa RS, Epifânio RA, Castro NG, Rocha MS, Pinto AC. Pharmacol Biochem Behav. 2009;92:508–513. doi: 10.1016/j.pbb.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 48.Pereira DM, Ferreres F, Oliveira JMA, Gaspar L, Faria J, Valentão P, Sottomayor M, Andrade PB. Phytomedicine. 2010;17:646–652. doi: 10.1016/j.phymed.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 49.Geissler T, Brandt W, Porzel A, Schlenzig D, Kehlen A, Wessjohann L, Arnold N. Bioorg Med Chem. 2010;18:2173–2177. doi: 10.1016/j.bmc.2010.01.074. [DOI] [PubMed] [Google Scholar]
- 50.Maelicke A, Hoeffle-Maas A, Ludwig J, Maus A, Samochocki M, Jordis U, Koepke AKE. J Mol Neurosci. 2010;40:135–137. doi: 10.1007/s12031-009-9269-5. [DOI] [PubMed] [Google Scholar]
- 51.Jin Z. Nat Prod Rep. 2009;26:382–445. doi: 10.1039/b718045b. [DOI] [PubMed] [Google Scholar]
- 52.Berkov S, Codina C, Viladomat F, Bastida J. Phytochemistry. 2007;68:1791–1798. doi: 10.1016/j.phytochem.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 53.Labraña J, Machocho AK, Kricsfalusy V, Brun R, Codina C, Viladomat F, Bastida J. Phytochemistry. 2002;60:847–852. doi: 10.1016/s0031-9422(02)00154-1. [DOI] [PubMed] [Google Scholar]
- 54.Rhee IK, Appels N, Hofte B, Karabatak B, Erkelens C, Stark LM, Flippin LA, Verpoorte R. Biol Pharm Bull. 2004;27:1804–1809. doi: 10.1248/bpb.27.1804. [DOI] [PubMed] [Google Scholar]
- 55.Rozengart E, Basova N, Suvorov A. J Evol Biochem Physiol. 2006;42:408–416. [Google Scholar]
- 56.Jung H, Lee E, Kim J, Kang S, Lee J, Min B, Choi J. Arch Pharm Res. 2009;32:1399–1408. doi: 10.1007/s12272-009-2009-0. [DOI] [PubMed] [Google Scholar]
- 57.Kim DK. Arch Pharm Res. 2002;25:817–819. doi: 10.1007/BF02976997. [DOI] [PubMed] [Google Scholar]
- 58.Hung TM, Na M, Dat NT, Ngoc TM, Youn U, Kim HJ, Min B, Lee J, Bae K. J Ethnopharmacol. 2008;119:74–80. doi: 10.1016/j.jep.2008.05.041. [DOI] [PubMed] [Google Scholar]
- 59.Hung TM, Ngoc TM, Youn UJ, Min BS, Na M, Thuong PT, Bae K. Biol Pharm Bull. 2008;31:159–162. doi: 10.1248/bpb.31.159. [DOI] [PubMed] [Google Scholar]
- 60.Huang L, Shi A, He F, Li X. Bioorg Med Chem. 2010;18:1244–1251. doi: 10.1016/j.bmc.2009.12.035. [DOI] [PubMed] [Google Scholar]
- 61.Jung HA, Min B, Yokozawa T, Lee J, Kim YS, Choi JS. Biol Pharm Bull. 2009;32:1433–1438. doi: 10.1248/bpb.32.1433. [DOI] [PubMed] [Google Scholar]
- 62.Zheng X, Zhang Z, Chou G, Wu T, Cheng X, Wang C, Wang Z. Arch Pharm Res. 2009;32:1245–1251. doi: 10.1007/s12272-009-1910-x. [DOI] [PubMed] [Google Scholar]
- 63.Tundis R, Menichini F, Conforti F, Loizzo MR, Bonesi M, Statti G, Menichini F. J Enzym Inhib Med Chem. 2009;24:818–824. doi: 10.1080/14756360802399662. [DOI] [PubMed] [Google Scholar]
- 64.Tang L, Wang R, Tang X. Acta Pharmacol Sin. 2005;26:673–678. doi: 10.1111/j.1745-7254.2005.00130.x. [DOI] [PubMed] [Google Scholar]
- 65.Wang R, Yan H, Tang X. Acta Pharmacol Sin. 2006;27:1–26. doi: 10.1111/j.1745-7254.2006.00255.x. [DOI] [PubMed] [Google Scholar]
- 66.Ma X, Tan C, Zhu D, Gang DR, Xiao P. J Ethnopharmacol. 2007;113:15–34. doi: 10.1016/j.jep.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 67.Wang B, Wang H, Wei Z, Song Y, Zhang L, Chen H. J Neural Transm. 2009;116:457–465. doi: 10.1007/s00702-009-0189-x. [DOI] [PubMed] [Google Scholar]
- 68.He X, Feng S, Wang Z, Shi Y, Zheng S, Xia Y, Jiang H, Tang X, Bai D. Bioorg Med Chem. 2007;15:1394–1408. doi: 10.1016/j.bmc.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 69.Choo CY, Hirasawa Y, Karimata C, Koyama K, Sekiguchi M, Kobayashi J, Morita H. Bioorg Med Chem. 2007;15:1703–1707. doi: 10.1016/j.bmc.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 70.Hirasawa Y, Kato E, Kobayashi J, Kawahara N, Goda Y, Shiro M, Morita H. Bioorg Med Chem. 2008;16:6167–6171. doi: 10.1016/j.bmc.2008.04.044. [DOI] [PubMed] [Google Scholar]
- 71.Halldorsdottir ES, Olafsdottir ES. Planta Med. 2006;72:961. doi: 10.1055/s-0035-1546182. [DOI] [PubMed] [Google Scholar]
- 72.Halldorsdottir ES, Jaroszewski JW, Olafsdottir ES. Phytochemistry. 2010;71:149–157. doi: 10.1016/j.phytochem.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 73.He J, Chen X, Li M, Zhao Y, Xu G, Cheng X, Peng L, Xie M, Zheng Y, Wang Y, Zhao Q. Org Lett. 2009;11:1397–1400. doi: 10.1021/ol900079t. [DOI] [PubMed] [Google Scholar]
- 74.Koyama K, Hirasawa Y, Kobayashi J, Morita H. Bioorg Med Chem. 2007;15:7803–7808. doi: 10.1016/j.bmc.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 75.Hirasawa Y, Kobayashi J, Morita H. Org Lett. 2006;8:123–126. doi: 10.1021/ol052760q. [DOI] [PubMed] [Google Scholar]
- 76.Castro NG, Costa RS, Pimentel LSB, Danuello A, Romeiro NC, Viegas C, Barreiro EJ, Fraga CAM, Bolzani VS, Rocha MS. Eur J Pharmacol. 2008;580:339–349. doi: 10.1016/j.ejphar.2007.11.035. [DOI] [PubMed] [Google Scholar]
- 77.Viegas C, Bolzani VS, Pimentel LSB, Castro NG, Cabral RF, Costa RS, Floyd C, Rocha MS, Young MCM, Barreiro EJ, Fraga CAM. Bioorg Med Chem. 2005;13:4184–4190. doi: 10.1016/j.bmc.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 78.Chonpathompikunlert P, Wattanathorn J, Muchimapura S. Food Chem Toxicol. 2010;48:798–802. doi: 10.1016/j.fct.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 79.Rahman T, Rahmatullah M. Bioorg Med Chem Lett. 2010;20:537–540. doi: 10.1016/j.bmcl.2009.11.106. [DOI] [PubMed] [Google Scholar]
- 80.Devkota KP, Lenta BN, Fokou PA, Sewald N. Nat Prod Rep. 2008;25:612–630. doi: 10.1039/b704958g. [DOI] [PubMed] [Google Scholar]
- 81.Atta-ur-Rahman, Akhtar MN, Choudhary MI, Tsuda Y, Sener B, Khalid A, Parvez M. Chem Pharm Bull. 2002;50:1013–1016. doi: 10.1248/cpb.50.1013. [DOI] [PubMed] [Google Scholar]
- 82.Lin B, Ji H, Li P, Fang W, Jiang Y. Planta Med. 2006;72:814–818. doi: 10.1055/s-2006-947168. [DOI] [PubMed] [Google Scholar]
- 83.Gilani AH, Ghayur MN, Saify ZS, Ahmed SP, Choudhary MI, Khalid A. Life Sci. 2004;75:2377–2389. doi: 10.1016/j.lfs.2004.03.035. [DOI] [PubMed] [Google Scholar]
- 84.Carey GJ, Costall B, Domeney AM, Gerrard PA, Jones DN, Naylor RJ, Tyers MB. Pharmacol Biochem Behav. 1992;42:75–83. doi: 10.1016/0091-3057(92)90449-p. [DOI] [PubMed] [Google Scholar]
- 85.Riekkinen P, Riekkinen M, Sirviö J. J Pharmacol Exp Ther. 1993;267:1484–1492. [PubMed] [Google Scholar]
- 86.Tariot PN, Cohen RM, Welkowitz JA, Sunderland T, Newhouse PA, Murphy DL, Weingartner H. Arch Gen Psychiatry. 1988;45:901–905. doi: 10.1001/archpsyc.1988.01800340023003. [DOI] [PubMed] [Google Scholar]
- 87.Raffaele KC, Asthana S, Berardi A, Haxby JV, Morris PP, Schapiro MB, Soncrant TT. Neuropsychopharmacol. 1996;15:163–170. doi: 10.1016/0893-133X(95)00179-H. [DOI] [PubMed] [Google Scholar]
- 88.Lim D, Kim I. Acta Pharmacol Sin. 2006;72:71–79. doi: 10.1111/j.1745-7254.2006.00233.x. [DOI] [PubMed] [Google Scholar]
- 89.Yang YR, Chang KC, Chen CL, Chiu TH. Chin J Physiol. 2000;43:23–28. [PubMed] [Google Scholar]
- 90.Rollinger JM, Schuster D, Baier E, Ellmerer EP, Langer T, Stuppner H. J Nat Prod. 2006;69:1341–1346. doi: 10.1021/np060268p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ge H, Peng H, Guo Z, Cui J, Song Y, Tan R. Planta Med. 2010;76:822–824. doi: 10.1055/s-0029-1240726. [DOI] [PubMed] [Google Scholar]
- 92.Bajguz A, Piotrowska A. Phytochemistry. 2009;70:957–969. doi: 10.1016/j.phytochem.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 93.Heo H, Cho H, Hong B, Kim H, Heo T, Kim E, Kim S, Kim C, Shin D. Mol Cells. 2002;13:5–11. [PubMed] [Google Scholar]
- 94.Choi SJ, Jeong C, Choi S, Chun J, Kim YJ, Lee J, Shin D, Heo HJ. J Med Food. 2009;12:271–277. doi: 10.1089/jmf.2007.0678. [DOI] [PubMed] [Google Scholar]
- 95.Satheeshkumar N, Mukherjee PK, Bhadra S, Saha BP. Phytomedicine. 2010;17:292–295. doi: 10.1016/j.phymed.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 96.Perry EK, Howes MR. CNS Neurosci Ther. 2010 doi: 10.1111/j.1755-5949.2010.00202.x. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Okello EJ, Dimaki C, Howes MR, Houghton PJ, Perry EK. Int J Essent Oil Ther. 2008;2:105–110. [Google Scholar]
- 98.Mills C, Cleary BJ, Gilmer JF, Walsh JJ. J Pharm Pharmacol. 2004;56:375–379. doi: 10.1211/0022357022773. [DOI] [PubMed] [Google Scholar]
- 99.Mukherjee P, Kumar V, Mal M, Houghton P. Planta Med. 2007;73:283–285. doi: 10.1055/s-2007-967114. [DOI] [PubMed] [Google Scholar]
- 100.Siramon P, Ohtani Y, Ichiura H. J Wood Sci. 2009;55:41–46. [Google Scholar]
- 101.Souza A, Silva MC, Cardoso-Lopes EM, Cordeiro I, Sobral MEG, Young MCM, Moreno PRH. Nat Prod Commun. 2009;4:1143–1146. [PubMed] [Google Scholar]
- 102.Khadri A, Serrakheiro MLM, Nogueria JMF, Neffati M, Smiti S, Araujo MEM. Food Chem. 2008;109:630–637. [Google Scholar]
- 103.Dohi S, Terasaki M, Makino M. J Agric Food Chem. 2009;57:4313–4318. doi: 10.1021/jf804013j. [DOI] [PubMed] [Google Scholar]
- 104.Houghton PJ, Howes M, Lee CC, Steventon G. J Ethnopharmacol. 2007;110:391–400. doi: 10.1016/j.jep.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 105.Ferreira A, Proença C, Serralheiro M, Araújo M. J Ethnopharmacol. 2006;108:31–37. doi: 10.1016/j.jep.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 106.Loizzo MR, Menichini F, Conforti F, Tundis R, Bonesi M, Saab AM, Statti GA, Cindio B, Houghton PJ, Menichini F, Frega NG. Food Chem. 2009;117:174–180. [Google Scholar]
- 107.Picollo MI, Toloza AC, Mougabure Cueto G, Zygadlo J, Zerba E. Fitoterapia. 2008;79:271–278. doi: 10.1016/j.fitote.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 108.Perry NS, Houghton PJ, Theobald A, Jenner P, Perry EK. J Pharm Pharmacol. 2000;52:895–902. doi: 10.1211/0022357001774598. [DOI] [PubMed] [Google Scholar]
- 109.Savelev S, Okello E, Perry NSL, Wilkins RM, Perry EK. Pharmacol Biochem Behav. 2003;75:661–668. doi: 10.1016/s0091-3057(03)00125-4. [DOI] [PubMed] [Google Scholar]
- 110.Fujiwara M, Yagi N, Miyazawa M. J Agric Food Chem. 2010;58:2824–2829. doi: 10.1021/jf9042387. [DOI] [PubMed] [Google Scholar]
- 111.Kumar A, Dogra S, Prakash A. Behav Brain Res. 2009;205:384–390. doi: 10.1016/j.bbr.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 112.Menichini F, Tundis R, Loizzo MR, Bonesi M, Marrelli M, Statti GA, Menichini F, Conforti F. Fitoterapia. 2009;80:297–300. doi: 10.1016/j.fitote.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 113.Miyazawa M, Yamafuji C. J Agric Food Chem. 2005;53:1765–1768. doi: 10.1021/jf040019b. [DOI] [PubMed] [Google Scholar]
- 114.Perry N, Court G, Bidet N, Court J, Perry E. Int J Geriatr Psychiatry. 1996;11:1063–1069. [Google Scholar]
- 115.Perry NSL, Houghton PJ, Jenner P, Keith A, Perry EK. Phytomedicine. 2002;9:48–51. doi: 10.1078/0944-7113-00082. [DOI] [PubMed] [Google Scholar]
- 116.Kivrak I, Duru ME, Ozturk M, Mercan N, Harmandar M, Topcu G. Food Chem. 2009;116:470–479. [Google Scholar]
- 117.Orhan I, Aslan M. J Ethnopharmacol. 2009;122:327–332. doi: 10.1016/j.jep.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 118.Zhou W, Fukumoto S, Yokogoshi H. Nutr Neurosci. 2009;12:57–64. doi: 10.1179/147683009X388832. [DOI] [PubMed] [Google Scholar]
- 119.Hornick A, Schwaiger S, Rollinger JM, Vo NP, Prast H, Stuppner H. Biochem Pharmacol. 2008;76:236–248. doi: 10.1016/j.bcp.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Riaz N, Nawaz SA, Mukhtar N, Malik A, Afza N, Ali S, Ullah S, Muhammad P, Choudhary MI. Chem Biodivers. 2007;4:72–83. doi: 10.1002/cbdv.200790008. [DOI] [PubMed] [Google Scholar]
- 121.Cavin A, Hay A, Marston A, Stoeckli-Evans H, Scopelliti R, Diallo D, Hostettmann K. J Nat Prod. 2006;69:768–773. doi: 10.1021/np058123q. [DOI] [PubMed] [Google Scholar]
- 122.Thirugnanasampandan R, Jayakumar R, Narmatha Bai V, Martin E, Rajendra Prasad KJ. Nat Prod Res. 2008;22:681–688. doi: 10.1080/14786410801990625. [DOI] [PubMed] [Google Scholar]
- 123.Rasoamiaranjanahary L, Guilet D, Marston A, Randimbivololona F, Hostettmann K. Phytochemistry. 2003;64:543–548. doi: 10.1016/s0031-9422(03)00154-7. [DOI] [PubMed] [Google Scholar]
- 124.Eldeen I, Van Heerden F, Van Staden J. J Ethnopharmacol. 2010;128:555–560. doi: 10.1016/j.jep.2010.01.057. [DOI] [PubMed] [Google Scholar]
- 125.Ren Y, Houghton PJ, Hider RC, Howes MR. Planta Med. 2004;70:201–204. doi: 10.1055/s-2004-815535. [DOI] [PubMed] [Google Scholar]
- 126.Kim DH, Jeon SJ, Jung JW, Lee S, Yoon BH, Shin BY, Son KH, Cheong JH, Kim YS, Kang SS, Ko KH, Ryu JH. Eur J Pharmacol. 2007;574:140–147. doi: 10.1016/j.ejphar.2007.07.042. [DOI] [PubMed] [Google Scholar]
- 127.Wong KK, Ho MT, Lin HQ, Lau K, Rudd JA, Chung RC, Fung K, Shaw P, Wan DC. Planta Med. 2010;76:228–234. doi: 10.1055/s-0029-1186084. [DOI] [PubMed] [Google Scholar]
- 128.Yu X, Lin S, Chen X, Zhou Z, Liang J, Duan W, Chowbay B, Wen J, Chan E, Cao J, Li C, Zhou S. Curr Drug Metabol. 2007;8:365–377. doi: 10.2174/138920007780655441. [DOI] [PubMed] [Google Scholar]
- 129.Sivaramakrishna C, Rao CV, Trimurtulu G, Vanisree M, Subbaraju GV. Phytochemistry. 2005;66:2719–2728. doi: 10.1016/j.phytochem.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 130.Howes MR, Houghton PJ. Herbal Drugs: Ethnomedicine to Modern Medicine. New York: K. G. Ramawat; 2009. pp. 239–291. [Google Scholar]
- 131.Kumar V, Mukherjee BC, Pal BC, Houghton PJ, Mukherjee PK. Planta Med. 2007;73 doi: 10.1055/s-2007-967114. DOI: 10.1055. [DOI] [PubMed] [Google Scholar]
- 132.Lee JH, Lee KT, Yang JH, Baek NI, Kim DK. Arch Pharm Res. 2004;27:52–56. doi: 10.1007/BF02980046. [DOI] [PubMed] [Google Scholar]
- 133.Taranalli AD, Cheeramkuzhy TC. Pharm Biol. 2000;38:51–56. doi: 10.1076/1388-0209(200001)3811-BFT051. [DOI] [PubMed] [Google Scholar]
- 134.Lee B, Jung K, Kim D. Pharmacol Biochem Behav. 2009;93:121–127. doi: 10.1016/j.pbb.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 135.Oinonen PP, Jokela JK, Hatakka AI, Vuorela PM. Fitoterapia. 2006;77:429–434. doi: 10.1016/j.fitote.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 136.Fan P, Hay A, Marston A, Hostettmann K. Pharm Biol. 2008;46:596–601. [Google Scholar]
- 137.Khan MTH, Orhan I, Senol FS, Kartal M, Sener B, Dvorská M, Smejkal K, Slapetová T. Chem Biol Interact. 2009;181:383–389. doi: 10.1016/j.cbi.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 138.Jung M, Park M. Molecules. 2007;12:2130–2139. doi: 10.3390/12092130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Choi Y, Yon G, Hong K, Yoo D, Choi C, Park W, Kong J, Kim Y, Ryu S. Planta Med. 2008;74:1405–1408. doi: 10.1055/s-2008-1081301. [DOI] [PubMed] [Google Scholar]
- 140.Conforti F, Rigano D, Formisano C, Bruno M, Loizzo MR, Menichini F, Senatore F. J Enzym Inhib Med Chem. 2010;25:97–104. doi: 10.3109/14756360903018260. [DOI] [PubMed] [Google Scholar]
- 141.Liu R, Gao M, Qiang G, Zhang T, Lan X, Ying J, Du G. Neuroscience. 2009;162:1232–1243. doi: 10.1016/j.neuroscience.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 142.Heo HJ, Kim M, Lee J, Choi SJ, Cho H, Hong B, Kim H, Kim E, Shin D. Dement Geriatr Cogn Disord. 2004;17:151–157. doi: 10.1159/000076349. [DOI] [PubMed] [Google Scholar]
- 143.Kumar A, Prakash A, Dogra S. Food Chem Toxicol. 2010;48:626–632. doi: 10.1016/j.fct.2009.11.043. [DOI] [PubMed] [Google Scholar]
- 144.He X, Zhou W, Bi M, Du G. Brain Res. 2010;1334:73–83. doi: 10.1016/j.brainres.2010.03.084. [DOI] [PubMed] [Google Scholar]
- 145.Cui Y, Ao M, Li W, Yu L. Planta Med. 2008;74:377–380. doi: 10.1055/s-2008-1034319. [DOI] [PubMed] [Google Scholar]
- 146.Lee YB, Lee HJ, Won MH, Hwang IK, Kang TC, Lee JY, Nam SY, Kim KS, Kim E, Cheon H, Sohn HS. J Nutr. 2004;134:1827–1831. doi: 10.1093/jn/134.7.1827. [DOI] [PubMed] [Google Scholar]
- 147.File S, Jarrett N, Fluck E, Duffy R, Casey K, Wiseman H. Psychopharmacology. 2001;157:430–436. doi: 10.1007/s002130100845. [DOI] [PubMed] [Google Scholar]
- 148.Kim H, Xu J, Su Y, Xia H, Li L, Peterson G, Murphy-Ullrich J, Barnes S. Biochem Soc Trans. 2001;29:216–222. doi: 10.1042/0300-5127:0290216. [DOI] [PubMed] [Google Scholar]
- 149.Sheng R, Lin X, Zhang J, Chol KS, Huang W, Yang B, He Q, Hu Y. Bioorg Med Chem. 2009;17:6692–6698. doi: 10.1016/j.bmc.2009.07.072. [DOI] [PubMed] [Google Scholar]
- 150.Orhan I, Senol FS, Kartal M, Dvorská M, Zemlicka M, Smejkal K, Mokrý P. Food Chem Toxicol. 2009;47:1747–1751. doi: 10.1016/j.fct.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 151.Brühlmann C, Marston A, Hostettmann K, Carrupt P, Testa B. Chem Biodivers. 2004;1:819–829. doi: 10.1002/cbdv.200490064. [DOI] [PubMed] [Google Scholar]
- 152.Urbain A, Marston A, Queiroz EF, Ndjoko K, Hostettmann K. Planta Med. 2004;70:1011–1014. doi: 10.1055/s-2004-832632. [DOI] [PubMed] [Google Scholar]
- 153.Jantan I, Yasim YHM, Jalil J, Murad S, Idris MS. Pharm Biol. 2009;47:1090–1095. [Google Scholar]
- 154.Fallarero A, Oinonen P, Gupta S, Blom P, Galkin A, Mohan CG, Vuorela PM. Pharmacol Res. 2008;58:215–221. doi: 10.1016/j.phrs.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 155.Teismann P, Ferger B. Eur J Pharmacol. 2000;398:247–250. doi: 10.1016/s0014-2999(00)00290-9. [DOI] [PubMed] [Google Scholar]
- 156.Howes MR, Houghton PJ. Pharmacol Biochem Behav. 2003;75:513–527. doi: 10.1016/s0091-3057(03)00128-x. [DOI] [PubMed] [Google Scholar]
- 157.Lee YK, Yuk DY, Kim TI, Kim YH, Kim KT, Kim KH, Lee BJ, Nam S, Hong JT. J Nat Med. 2009;63:274–282. doi: 10.1007/s11418-009-0330-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kumar P, Singh VK, Singh DK. Phytother Res. 2009;23:172–177. doi: 10.1002/ptr.2578. [DOI] [PubMed] [Google Scholar]
- 159.Fang L, Kraus B, Lehmann J, Heilmann J, Zhang Y, Decker M. Bioorg Med Chem Lett. 2008;18:2905–2909. doi: 10.1016/j.bmcl.2008.03.073. [DOI] [PubMed] [Google Scholar]
- 160.Ge HM, Zhu CH, Shi DH, Zhang LD, Xie DQ, Yang J, Ng SW, Tan RX. Chemistry. 2008;14:376–381. doi: 10.1002/chem.200700960. [DOI] [PubMed] [Google Scholar]
- 161.Shi DH, Wu JH, Ge HM, Tan R. Environ Toxicol Pharmacol. 2009;28:30–36. doi: 10.1016/j.etap.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 162.Xu J, Rong S, Xie B, Sun Z, Zhang L, Wu H, Yao P, Zhang Y, Liu L. Phytother Res. 2009;23:1742–1747. doi: 10.1002/ptr.2837. [DOI] [PubMed] [Google Scholar]
- 163.Oh JH, Choi BJ, Chang MS, Park SK. Neurosci Lett. 2009;461:41–44. doi: 10.1016/j.neulet.2009.05.045. [DOI] [PubMed] [Google Scholar]
- 164.Lee KY, Sung SH, Kim SH, Jang YP, Oh TH, KIm YC. Arch Physiol Biochem. 2009;32:677–683. doi: 10.1007/s12272-009-1505-6. [DOI] [PubMed] [Google Scholar]
- 165.de Paula A, Martins J, dos Santos M, Nascente LDC, Romeiro L, Areas T, Vieira K, Gambôa N, Castro N, Gargano R. Eur J Med Chem. 2009;44:3754–3759. doi: 10.1016/j.ejmech.2009.03.045. [DOI] [PubMed] [Google Scholar]
- 166.Alonso D, Castro A, Martinez A. Expert Opin Ther Patents. 2005;15:1377–1386. [Google Scholar]
- 167.Bonnard I, Jhaumeer-Laulloo SB, Bontemps N, Banaigs B, Aknin M. Mar Drugs. 2010;8:359–372. doi: 10.3390/md8020359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Choi BW, Ryu G, Park SH, Kim ES, Shin J, Roh SS, Shin HC, Lee BH. Phytother Res. 2007;21:423–426. doi: 10.1002/ptr.2090. [DOI] [PubMed] [Google Scholar]
- 169.Yoon NY, Chung HY, Kim HR, Choi JE. Fisheries Sci. 2008;74:200–207. [Google Scholar]
- 170.Nukoolkarn VS, Saen-oon S, Rungrotmongkol T, Hannongbua S, Ingkaninan K, Suwanborirux K. Bioorg Med Chem. 2008;16:6560–6567. doi: 10.1016/j.bmc.2008.05.027. [DOI] [PubMed] [Google Scholar]
- 171.Sepcić K, Kauferstein S, Mebs D, Turk T. Mar Drugs. 2010;8:1550–1566. doi: 10.3390/md8051550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Natarajan S, Shanmugiahthevar KP, Kasi PD. Nat Prod Res. 2009;23:355–369. doi: 10.1080/14786410802156036. [DOI] [PubMed] [Google Scholar]
- 173.Suganthy N, Karutha Pandian S, Pandima Devi K. Neurosci Lett. 2010;468:216–219. doi: 10.1016/j.neulet.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 174.Hardy J. J Alzheimers Dis. 2006;9:151–153. doi: 10.3233/jad-2006-9s317. [DOI] [PubMed] [Google Scholar]
- 175.Cirrito JR, May PC, O’Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bateman RJ, Klunk WE. Neurotherapeutics. 2008;5:381–390. doi: 10.1016/j.nurt.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 178.Witter DP, Chen Y, Rogel JK, Boldt GE, Wentworth P. ChemBioChem. 2009;10:1344–1347. doi: 10.1002/cbic.200900139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tian XY, Zhao Y, Yu SS, Fang WS. Chem Biodivers. 2010;7:984–992. doi: 10.1002/cbdv.200900280. [DOI] [PubMed] [Google Scholar]
- 180.Choi S, Hur J, Yang E, Jun M, Park H, Lee K, Moon E, Song K. Arch Pharm Res. 2008;31:183–187. doi: 10.1007/s12272-001-1139-9. [DOI] [PubMed] [Google Scholar]
- 181.Shimmyo Y, Kihara T, Akaike A, Niidome T, Sugimoto H. BBA-Gen Subjects. 2008;1780:819–825. doi: 10.1016/j.bbagen.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 182.Hwang EM, Ryu YB, Kim HY, Kim DG, Hong SG, Lee JH, Curtis-Long MJ, Jeong SH, Park JY, Park KH. Bioorg Med Chem. 2008;16:6669–6674. doi: 10.1016/j.bmc.2008.05.080. [DOI] [PubMed] [Google Scholar]
- 183.Jeon S, Bae K, Seong Y, Song K. Bioorg Med Chem Lett. 2003;13:3905–3908. doi: 10.1016/j.bmcl.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 184.Jeon SY, Kwon SH, Seong YH, Bae K, Hur JM, Lee YY, Suh DY, Song KS. Phytomedicine. 2007;14:403–408. doi: 10.1016/j.phymed.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 185.Dai J, Sorribas A, Yoshida WY, Williams PG. Phytochemistry. 2010 doi: 10.1016/j.phytochem.2010.09.008. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Feng BY, Shoichet BK. Nat Protoc. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kwak H, Jeon S, Sohng B, Kim J, Lee J, Lee K, Jeong H, Hur J, Kang Y, Song K. Arch Pharm Res. 2005;28:1328–1332. doi: 10.1007/BF02977896. [DOI] [PubMed] [Google Scholar]
- 188.Lee H, Seong Y, Bae K, Kwon S, Kwak H, Nho S, Kim K, Hur J, Lee K, Kang Y, Song K. Arch Pharm Res. 2005;28:799–803. doi: 10.1007/BF02977345. [DOI] [PubMed] [Google Scholar]
- 189.Jung HA, Oh SH, Choi JS. Bioorg Med Chem Lett. 2010;20:3211–3215. doi: 10.1016/j.bmcl.2010.04.093. [DOI] [PubMed] [Google Scholar]
- 190.Park I, Jeon S, Lee H, Kim S, Song K. Planta Med. 2004;70:143–146. doi: 10.1055/s-2004-815491. [DOI] [PubMed] [Google Scholar]
- 191.Klaar M, Wolfgang S. Chem Ber. 1997;110:1058–1062. [Google Scholar]
- 192.Tian X, Zhao Y, Yu S, Fang W. Chem Biodivers. 2010;7:984–992. doi: 10.1002/cbdv.200900280. [DOI] [PubMed] [Google Scholar]
- 193.Lopez Ogalla and Munoz Ruiz, 2007
- 194.Martinez A, Perez DI. J Alzheimers Dis. 2008;15:181–191. doi: 10.3233/jad-2008-15204. [DOI] [PubMed] [Google Scholar]
- 195.Newswire PR. Noscira Presents Results of Phase IIa Clinical Trial with Nypta(R) (Tideglusib) on Alzheimer’s Disease. 2010 http://www.prnewswire.com/news-releases/noscira-presents-results-of-phase-iia-clinical-trial-with-nyptar-tideglusib-on-alzheimers-disease-at-international-conference-on-alzheimers-disease-icad-98521119.html.
- 196.Millán-Aguiñaga N, Soria-Mercado IE, Williams P. Tetrahedron Lett. 2010;51:751–753. [Google Scholar]
- 197.Dai J, Sorribas A, Yoshida WY, Kelly M, Williams PG. J Nat Prod. 2010;76:1188–1197. doi: 10.1021/np100203x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Patil SP, Maki S, Khedkar SA, Rigby AC, Chan C. J Nat Prod. 2010;73:1196–1202. doi: 10.1021/np900633j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Csermely P, Ágoston V, Pongor S. Trends Pharmacol Sci. 2005;26:178–182. doi: 10.1016/j.tips.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 200.Youdim MB, Buccafusco JJ. Trends Pharmacol Sci. 2005;26:27–35. doi: 10.1016/j.tips.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 201.Dai J, Jimeìnez JI, Kelly M, Williams PG. J Org Chem. 2010;75:2399–2402. doi: 10.1021/jo902566n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hills ID, Vacca JP. Curr Opin Drug Discov Devel. 2007;10:383–391. [PubMed] [Google Scholar]
- 203.Williams P, Sorribas A, Liang Z. Curr Alzheimer Res. 2010;7:210–213. doi: 10.2174/156720510791050812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Carroll AR, Kaiser SM, Davis RA, Moni RW, Hooper JNA, Quinn RJ. J Nat Prod. 2010;73:1173–1176. doi: 10.1021/np100010z. [DOI] [PubMed] [Google Scholar]
- 205.Greve H, Kehraus S, Krick A, Kelter G, Maier A, Fiebig H, Wright AD, König GM. J Nat Prod. 2008;71:309–312. doi: 10.1021/np070373e. [DOI] [PubMed] [Google Scholar]
- 206.Kotoku N, Hiramatsu A, Tsujita H, Hirakawa Y, Sanagawa M, Aoki S, Kobayashi M. Arch Pharm (Weinheim) 2008;341:568–577. doi: 10.1002/ardp.200700231. [DOI] [PubMed] [Google Scholar]
- 207.Masuno MN, Pessah IN, Olmstead MM, Molinski TF. J Med Chem. 2006;49:4497–4511. doi: 10.1021/jm050708u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Zieminska E, Lazarewicz JW, Couladouros EA, Moutsos VI, Pitsinos EN. Bioorg Med Chem Lett. 2008;18:5734–5737. doi: 10.1016/j.bmcl.2008.09.080. [DOI] [PubMed] [Google Scholar]
- 209.Y. Wu, S. Gerritz, S. Shi, and S. Zhu, 2010.
- 210.Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- 211.Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels L, Camacho IE, Marjaux E, Craessaerts K, Roebroek AJM, Schwake M, D’Hooge R, Bach P, Kalinke U, Moechars D, Alzheimer C, Reiss K, Saftig P, De Strooper B. J Biol Chem. 2005;280:30797–30806. doi: 10.1074/jbc.M505249200. [DOI] [PubMed] [Google Scholar]
- 212.Citron M. Nat Rev Drug Discov. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 213.Picone P, Carrotta R, Montana G, Nobile MR, San Biagio PL, Di Carlo M. Biophys J. 2009;96:4200–4211. doi: 10.1016/j.bpj.2008.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gervais F, Paquette J, Morissette C, Krzywkowski P, Yu M, Azzi M, Lacombe D, Kong X, Aman A, Laurin J, Szarek WA, Tremblay P. Neurobiol Aging. 2007;28:537–547. doi: 10.1016/j.neurobiolaging.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 215.Ito K, Miyazawa K, Matsumoto F. Hiroshima Daigaku Suichikusangakubu Kiyo. 1977;16:77–90. [Google Scholar]
- 216.Santa-Maria I, Hernández F, Del Rio J, Moreno FJ, Avila J. Mol Neurodegener. 2007;2:17–29. doi: 10.1186/1750-1326-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Alzforum. FDA Deems US Alzhemed Trial Results Inconclusive. 2007 http://www.alzforum.org/new/detail.asp?id=1647.
- 218.Neurochem. Memory Loss? VIVIMIND™ Protects Memory Function. 2010 http://www.vivimind.com/home.aspx.
- 219.Hann R, Sando C. J Biol Chem. 1926;68:399–402. [Google Scholar]
- 220.Muller H. J Chem Soc. 1907;91:1767. [Google Scholar]
- 221.McLaurin J, Kierstead ME, Brown ME, Hawkes CA, Lambermon MHL, Phinney AL, Darabie AA, Cousins JE, French JE, Lan MF, Chen F, Wong SSN, Mount HTJ, Fraser PE, Westaway D, George-Hyslop PS. Nat Med. 2006;12:801–808. doi: 10.1038/nm1423. [DOI] [PubMed] [Google Scholar]
- 222.Elan. Elan and Transition Therapeutics Announce Modifications to ELND005 Phase II Clinical Trials in Alzheimer’s Disease. 2009 http://newsroom.elan.com/phoenix.zhtml?c=88326&p=irol-newsArticle&ID=1365793&highlight=
- 223.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 224.Kerchner GA, Boxer AL. Expert Opin Biol Ther. 2010;10:1121–1130. doi: 10.1517/14712598.2010.493872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.ClinicalTrials. A Long-Term Safety and Tolerability Study of Bapineuzumab in Alzheimer Disease Patients. 2010 http://www.clinicaltrials.gov/ct2/show/NCT00996918?term=Bapineuzumab&rank=2.
- 226.Senior K. Lancet Neurol. 2002;1:3. doi: 10.1016/s1474-4422(02)00023-6. [DOI] [PubMed] [Google Scholar]
- 227.Town T. CNS Neurol Disord Drug Targets. 2009;8:114–127. doi: 10.2174/187152709787847306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, van Dyck CH, Mulnard R, Barakos J, Gregg KM, Liu E, Lieberburg I, Schenk D, Black R, Grundman M. Neurology. 2009;73:2061–2070. doi: 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.ClinicalTrials. Bapineuzumab in Patients With Mild to Moderate Alzheimer’s Disease (ApoE4 Non-Carrier) 2007 http://clinicaltrials.gov/ct2/show/NCT00574132?term=Bapineuzumab&rank=1.
- 230.Kolibas E, Korinkova V, Novotny V, Vajdickova K, Hunakova D. Bratisl Lek Listy. 2000;101:598–602. [PubMed] [Google Scholar]
- 231.ClinicalTrials. Single Dose Escalation Study of PF-04360365 In Subjects With Mild To Moderate Alzheimer’s Disease. 2009 http://clinicaltrials.gov/ct2/show/NCT00733642?term=PF-04360365&rank=1.
- 232.Janusz M, Lisowski J, Franĕk F. FEBS Lett. 1974;49:276–279. doi: 10.1016/0014-5793(74)80529-6. [DOI] [PubMed] [Google Scholar]
- 233.Kruzel M, Janusz M, Lisowski J, Fischleigh R, Georgiades J. J Mol Neurosci. 2001;17:379–389. doi: 10.1385/JMN:17:3:379. [DOI] [PubMed] [Google Scholar]
- 234.Popik P, Bobula B, Janusz M, Lisowski J, Vetulani J. Pharmacol Biochem Behav. 1999;64:183–189. doi: 10.1016/s0091-3057(99)00101-x. [DOI] [PubMed] [Google Scholar]
- 235.Leszek J, Inglot AD, Janusz M, Lisowski J, Krukowska K, Georgiades JA. Arch Immunol Ther Exp (Warsz) 1999;47:377–385. [PubMed] [Google Scholar]
- 236.Bilikiewicz A, Gaus W. J Alzheimers Dis. 2004;6:17–26. doi: 10.3233/jad-2004-6103. [DOI] [PubMed] [Google Scholar]
- 237.ReGen Therapeutics. Colostrinin™. 2010 http://www.regentherapeutics.com/regenplc/products/colostrinin/
- 238.Tomiyama T, Kaneko H, i Kataoka K, Asano S, Endo N. Biochem J. 1997;322:76–83. doi: 10.1042/bj3220859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Meng F, Marek P, Potter KJ, Verchere CB, Raleigh DP. Biochemistry. 2008;47:6016–6024. doi: 10.1021/bi702518m. [DOI] [PubMed] [Google Scholar]
- 240.Loeb MB, Molloy DW, Smieja M, Standish T, Goldsmith CH, Mahony J, Smith S, Borrie M, Decoteau E, Davidson W, Mcdougall A, Gnarpe J, O’donnell M, Chernesky M. J Am Geriatr Soc. 2004;52:381–387. doi: 10.1111/j.1532-5415.2004.52109.x. [DOI] [PubMed] [Google Scholar]
- 241.ClinicalTrials. Effects of Doxycycline and Rifampicin on Biomarkers of Alzheimer’s Disease in the Cerebrospinal Fluid. 2010 http://clinicaltrials.gov/ct2/show/NCT00439166?term=NCT00439166&rank=1.
- 242.Forloni G, Colombo L, Girola L, Tagliavini F, Salmona M. FEBS Lett. 2001;487:404–407. doi: 10.1016/s0014-5793(00)02380-2. [DOI] [PubMed] [Google Scholar]
- 243.Familian A, Boshuizen RS, Eikelenboom P, Veerhuis R. Glia. 2006;53:233–240. doi: 10.1002/glia.20268. [DOI] [PubMed] [Google Scholar]
- 244.Choi Y, Kim H, Shin KY, Kim E, Kim M, Kim H, Park CH, Jeong YH, Yoo J, Lee J, Chang K, Kim S, Suh Y. Neuropsychopharmacology. 2007;32:2393–2404. doi: 10.1038/sj.npp.1301377. [DOI] [PubMed] [Google Scholar]
- 245.Noble W, Garwood C, Stephenson J, Kinsey AM, Hanger DP, Anderton BH. FASEB J. 2009;23:739–750. doi: 10.1096/fj.08-113795. [DOI] [PubMed] [Google Scholar]
- 246.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 247.Aggarwal BB, Kumar A, Bharti AC. Anticancer Res. 2003;23:363–398. [PubMed] [Google Scholar]
- 248.Lee HS, Jung KK, Cho JY, Rhee MH, Hong S, Kwon M, Kim SH, Kang SY. Pharmazie. 2007;62:937–942. [PubMed] [Google Scholar]
- 249.Reinke AA, Gestwicki JE. Chem Biol Drug Design. 2007;70:206–215. doi: 10.1111/j.1747-0285.2007.00557.x. [DOI] [PubMed] [Google Scholar]
- 250.Akaishi T, Morimoto T, Shibao M, Watanabe S, Sakai-Kato K, Utsunomiya-Tate N, Abe K. Neurosci Lett. 2008;444:280–285. doi: 10.1016/j.neulet.2008.08.052. [DOI] [PubMed] [Google Scholar]
- 251.Hirohata M, Hasegawa K, Tsutsumi-Yasuhara S, Ohhashi Y, Ookoshi T, Ono K, Yamada M, Naiki H. Biochemistry. 2007;46:1888–1899. doi: 10.1021/bi061540x. [DOI] [PubMed] [Google Scholar]
- 252.Shimmyo Y, Kihara T, Akaike A, Niidome T, Sugimoto H. J Neurosci Res. 2008;86:368–377. doi: 10.1002/jnr.21476. [DOI] [PubMed] [Google Scholar]
- 253.Ono K, Hasegawa K, Yoshiike Y, Takashima A, Yamada M, Naiki H. J Neurochem. 2002;81:434–440. doi: 10.1046/j.1471-4159.2002.00904.x. [DOI] [PubMed] [Google Scholar]
- 254.Goodman Y, Steiner MR, Steiner SM, Mattson MP. Brain Res. 1994;654:171–176. doi: 10.1016/0006-8993(94)91586-5. [DOI] [PubMed] [Google Scholar]
- 255.Ono K, Yamada M. J Neurochem. 2006;97:105–115. doi: 10.1111/j.1471-4159.2006.03707.x. [DOI] [PubMed] [Google Scholar]
- 256.Hamaguchi T, Ono K, Murase A, Yamada M. Am J Pathol. 2009;175:2557–2565. doi: 10.2353/ajpath.2009.090417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Iuvone T, De Filippis D, Esposito G, D’Amico A, Izzo AA. J Pharmacol Exp Ther. 2006;317:1143–1149. doi: 10.1124/jpet.105.099317. [DOI] [PubMed] [Google Scholar]
- 258.Ono K, Hasegawa K, Naiki H, Yamada M. BBA-Mol Basis Dis. 2004;1690:193–202. doi: 10.1016/j.bbadis.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 259.Marambaud P, Zhao H, Davies P. J Biol Chem. 2005;280:37377–37382. doi: 10.1074/jbc.M508246200. [DOI] [PubMed] [Google Scholar]
- 260.Feng Y, Wang X, Yang S, Wang Y, Zhang X, Du X, Sun X, Zhao M, Huang L, Liu R. Neurotoxicology. 2009;30:986–995. doi: 10.1016/j.neuro.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 261.ClinicalTrials. Randomized Trial of a Nutritional Supplement in Alzheimer’s Disease. 2010 http://clinicaltrials.gov/ct2/show/NCT00678431?term=NCT00678431&rank=1.
- 262.Fujiwara H, Tabuchi M, Yamaguchi T, Iwasaki K, Furukawa K, Sekiguchi K, Ikarashi Y, Kudo Y, Higuchi M, Saido TC, Maeda S, Takashima A, Hara M, Yaegashi N, Kase Y, Arai H. J Neurochem. 2009;109:1648–1657. doi: 10.1111/j.1471-4159.2009.06069.x. [DOI] [PubMed] [Google Scholar]
- 263.Bergamaschini L, Rossi E, Storini C, Pizzimenti S, Distaso M, Perego C, DeLuigi A, Vergani C, Grazia De Simoni M. J Neurosci. 2004;24:4181–4186. doi: 10.1523/JNEUROSCI.0550-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Ryu J, Kanapathipillai M, Lentzen G, Park CB. Peptides. 2008;29:578–584. doi: 10.1016/j.peptides.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 265.Ono K, Yoshiike Y, Takashima A, Hasegawa K, Naiki H, Yamada M. Exp Neurol. 2004;189:380–392. doi: 10.1016/j.expneurol.2004.05.035. [DOI] [PubMed] [Google Scholar]
- 266.Ono K, Hasegawa K, Naiki H, Yamada M. Biochem Bioph Res Co. 2005;330:111–116. doi: 10.1016/j.bbrc.2005.02.132. [DOI] [PubMed] [Google Scholar]
- 267.Ono K, Hasegawa K, Yamada M, Naiki H. Biol Psych. 2002;52:880–886. doi: 10.1016/s0006-3223(02)01417-8. [DOI] [PubMed] [Google Scholar]
- 268.Nordberg A, Hellstrom-Lindahl E, Lee M, Johnson M, Mousavi M, Hall R, Perry E, Bednar I, Court J. J Neurochem. 2002;81:655–658. doi: 10.1046/j.1471-4159.2002.00874.x. [DOI] [PubMed] [Google Scholar]
- 269.Oddo S, Caccamo A, Green KN, Liang K, Tran L, Chen Y, Leslie FM, LaFerla FM. Proc Natl Acad Sci USA. 2005;102:3046–3051. doi: 10.1073/pnas.0408500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Pappolla M, Bozner P, Soto C, Shao H, Robakis NK, Zagorski M, Frangione B, Ghiso J. J Biol Chem. 1998;273:7185–7188. doi: 10.1074/jbc.273.13.7185. [DOI] [PubMed] [Google Scholar]
- 271.Bailly C. Biochem Pharmacol. 2009;77:1447–1457. doi: 10.1016/j.bcp.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 272.Gali-Muhtasib H. Lead Molecules from Natural Products – Discovery and New Trends. Vol. 2. Elsevier; 2006. pp. 155–167. [Google Scholar]
- 273.Webb BL, Hirst SJ, Giembycz MA. Br J Pharmacol. 2000;130:1433–1452. doi: 10.1038/sj.bjp.0703452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Olds JL, Alkon DL. Acta Neurobiol Exp (Wars) 1993;53:197–207. [PubMed] [Google Scholar]
- 275.Hongpaisan J, Alkon DL. Proc Natl Acad Sci USA. 2007;104:19571–19576. doi: 10.1073/pnas.0709311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Vianna MR, Barros DM, Silva T, Choi H, Madche C, Rodrigues C, Medina JH, Izquierdo I. Psychopharmacology (Berl) 2000;150:77–84. doi: 10.1007/s002130000396. [DOI] [PubMed] [Google Scholar]
- 277.Cole G, Dobkins KR, Hansen LA, Terry RD, Saitoh T. Brain Res. 1988;452:165–174. doi: 10.1016/0006-8993(88)90021-2. [DOI] [PubMed] [Google Scholar]
- 278.Favit A, Grimaldi M, Nelson TJ, Alkon DL. Proc Natl Acad Sci USA. 1998;95:5562–5567. doi: 10.1073/pnas.95.10.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Pakaski M, Balaspiri L, Checler F, Kasa P. Neurochem Int. 2002;41:409–414. doi: 10.1016/s0197-0186(02)00026-8. [DOI] [PubMed] [Google Scholar]
- 280.Desdouits F, Buxbaum JD, Desdouits-Magnen J, Nairn AC, Greengard P. J Biol Chem. 1996;271:24670–24674. doi: 10.1074/jbc.271.40.24670. [DOI] [PubMed] [Google Scholar]
- 281.Alfa Cissé M, Sunyach C, Slack BE, Fisher A, Vincent B, Checler F. J Neurosci. 2007;27:4083–4092. doi: 10.1523/JNEUROSCI.5293-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Cissé MA, Gandreuil C, Hernandez J, Martinez J, Checler F, Vincent B. Biochem Biophys Res Commun. 2006;347:254–260. doi: 10.1016/j.bbrc.2006.06.065. [DOI] [PubMed] [Google Scholar]
- 283.Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 284.Díaz-Rodríguez E, Montero JC, Esparís-Ogando A, Yuste L, Pandiella A. Mol Biol Cell. 2002;13:2031–2044. doi: 10.1091/mbc.01-11-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Robinson MJ, Cobb MH. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 286.Kishi Y, Rando RR. Acc Chem Res. 1998;31:163–172. [Google Scholar]
- 287.Kozikowski AP, Chen Y, Subhasish T, Lewin NE, Blumberg PM, Zhong Z, D’Annibale MA, Wang W, Shen Y, Langley B. ChemMedChem. 2009;4:1095–1105. doi: 10.1002/cmdc.200900045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Nakagawa Y, Yanagita RC, Hamada N, Murakami A, Takahashi H, Saito N, Nagai H, Irie K. J Am Chem Soc. 2009;131:7573–7579. doi: 10.1021/ja808447r. [DOI] [PubMed] [Google Scholar]
- 289.Pettit GR, Herald CL, Doubek DL, Herald DL, Arnold E, Clardy J. J Am Chem Soc. 1982;104:6846–6848. [Google Scholar]
- 290.Etcheberrigaray R, Tan M, Dewachter I, Kuipéri C, Van der Auwera I, Wera S, Qiao L, Bank B, Nelson TJ, Kozikowski AP, Van Leuven F, Alkon DL. Proc Natl Acad Sci USA. 2004;101:11141–11146. doi: 10.1073/pnas.0403921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Lam AP, Sparano JA, Vinciguerra V, Ocean AJ, Christos P, Hochster H, Camacho F, Goel S, Mani S, Kaubisch A. Am J Clin Oncol. 2010;33:121–124. doi: 10.1097/COC.0b013e3181a31920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Wender PA, Dechristopher BA, Schrier AJ. J Am Chem Soc. 2008;130:6658–6659. doi: 10.1021/ja8015632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Boudrault C, Bazinet RP, Ma DW. J Nutr Biochem. 2009;20:1–10. doi: 10.1016/j.jnutbio.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 294.Kanno T, Yamamoto H, Yaguchi T, Hi R, Mukasa T, Fujikawa H, Nagata T, Yamamoto S, Tanaka A, Nishizaki T. J Lipid Res. 2006;47:1146–1156. doi: 10.1194/jlr.M500329-JLR200. [DOI] [PubMed] [Google Scholar]
- 295.Nelson TJ, Cui C, Luo Y, Alkon DL. J Biol Chem. 2009;284:34514–34521. doi: 10.1074/jbc.M109.016683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Hernández F, Gómez de Barreda E, Fuster-Matanzo A, Lucas JJ, Avila J. Exp Neurol. 2010;223:322–325. doi: 10.1016/j.expneurol.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 297.Hooper C, Killick R, Lovestone S. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Klein PS, Melton DA. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Ryves WJ, Harwood AJ. Biochem Biophys Res Commun. 2001;280:720–725. doi: 10.1006/bbrc.2000.4169. [DOI] [PubMed] [Google Scholar]
- 300.De Sarno P, Li X, Jope RS. Neuropharmacology. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- 301.Phiel CJ, Wilson CA, Lee VM, Klein PS. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 302.Sakai R, Higa T, Jefford CW, Bernardinelli G. J Am Chem Soc. 1986;108:6404–6405. [Google Scholar]
- 303.Hamann M, Alonso D, Martín-Aparicio E, Fuertes A, Pérez-Puerto MJ, Castro A, Morales S, Navarro ML, Del Monte-Millán M, Medina M, Pennaka H, Balaiah A, Peng J, Cook J, Wahyuono S, Martínez A. J Nat Prod. 2007;70:1397–1405. doi: 10.1021/np060092r. [DOI] [PubMed] [Google Scholar]
- 304.Alfano G, Cimino G, De Stefano S. Experientia. 1979;35:1136–1137. [Google Scholar]
- 305.D. Alonso Gordillo, I. Dorronsoro Diaz, A. Martinez Gil, G. Panizo Del Pliego, A. Fuertes Huerta, J. Perez Puerto, E. Martin Aparicio,, D. Perez Navarro, and M. Medina Padilla, 2004.
- 306.Erdogan-Orhan I, Sener B, De Rosa S, Perez-Baz J, Lozach O, Leost M, Rakhilin S, Meijer L. Nat Prod Res. 2004;18:1–9. doi: 10.1080/1478641031000111534. [DOI] [PubMed] [Google Scholar]
- 307.Meijer L, Thunnissen A, White AW, Garnier M, Nikolic M, Tsai LH, Walter J, Cleverley KE, Salinas PC, Wu YZ, et al. Chem Biol. 2000;7:51–63. doi: 10.1016/s1074-5521(00)00063-6. [DOI] [PubMed] [Google Scholar]
- 308.Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, et al. Chem Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 309.Nakao Y, Fusetani N. J Nat Prod. 2007;70:689–710. doi: 10.1021/np060600x. [DOI] [PubMed] [Google Scholar]
- 310.Gibson GE. Free Radic Biol Med. 2002;32:1061–1070. doi: 10.1016/s0891-5849(02)00802-x. [DOI] [PubMed] [Google Scholar]
- 311.Perry VH, Nicoll JAR, Holmes C. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 312.Aliev G, Obrenovich ME, Reddy VP, Shenk JC, Moreira PI, Nunomura A, Zhu X, Smith MA, Perry G. Mini Rev Med Chem. 2008;8:1395–1406. doi: 10.2174/138955708786369582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Bonda DJ, Lee H, Lee H, Friedlich AL, Perry G, Zhu X, Smith MA. Curr Opin Drug Discov Devel. 2010;13:235–246. [PMC free article] [PubMed] [Google Scholar]
- 314.Mecocci P, Mariani E, Polidori MC, Hensley K, Betterfield DA. Cent Nerv Syst Agents Med Chem. 2008;8:28–63. [Google Scholar]
- 315.Kamat CD, Gadal S, Mhatre M, Williamson KS, Pye QN, Hensley K. J Alzheimers Dis. 2008;15:473–493. doi: 10.3233/jad-2008-15314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Saddichha S, Pandey V. Am J Alzheimers Dis Other Demen. 2008;23:150–161. doi: 10.1177/1533317507312957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Kim J, Lee HJ, Lee KW. J Neurochem. 2010;112:1415–1430. doi: 10.1111/j.1471-4159.2009.06562.x. [DOI] [PubMed] [Google Scholar]
- 318.Shi C, Liu J, Wu F, Yew DT. Int J Mol Sci. 2010;11:107–123. doi: 10.3390/ijms11010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Birks J, Grimley Evans J, Iakovidou V, Tsolaki M, Holt FE. Cochrane Database Syst Rev. 2009:CD001191. doi: 10.1002/14651858.CD001191. [DOI] [PubMed] [Google Scholar]
- 320.Ng T, Chiam P, Lee T, Chua H, Lim L, Kua E. Am J Epidemiol. 2006;164:898–906. doi: 10.1093/aje/kwj267. [DOI] [PubMed] [Google Scholar]
- 321.Koo B, Lee W, Chung K, Ko J, Kim C. Life Sci. 2004;75:2363–2375. doi: 10.1016/j.lfs.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 322.Kim DSHL, Kim JY. Bioorg Med Chem Lett. 2001;11:2541–2543. doi: 10.1016/s0960-894x(01)00489-9. [DOI] [PubMed] [Google Scholar]
- 323.Park S, Kim DSHL. J Nat Prod. 2002;65:1227–1231. doi: 10.1021/np010039x. [DOI] [PubMed] [Google Scholar]
- 324.Park S, Kim H, Cho E, Kwon B, Phark S, Hwang K, Sul D. Food Chem Toxicol. 2008;46:2881–2887. doi: 10.1016/j.fct.2008.05.030. [DOI] [PubMed] [Google Scholar]
- 325.Rajakrishnan V, Viswanathan P, Rajasekharan KN, Menon VP. Phytother Res. 1999;13:571–574. doi: 10.1002/(sici)1099-1573(199911)13:7<571::aid-ptr494>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 326.Agrawal R, Mishra B, Tyagi E, Nath C, Shukla R. Pharmacol Res. 2010;61:247–252. doi: 10.1016/j.phrs.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 327.Yaari R, Kumar S, Tariot PN. Expert Opin Drug Discov. 2008;3:745–760. doi: 10.1517/17460441.3.7.745. [DOI] [PubMed] [Google Scholar]
- 328.Awasthi H, Tota S, Hanif K, Nath C, Shukla R. Life Sci. 2010;86:87–94. doi: 10.1016/j.lfs.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 329.Pari L, Tewas D, Eckel J. Arch Physiol Biochem. 2008;114:127–149. doi: 10.1080/13813450802033958. [DOI] [PubMed] [Google Scholar]
- 330.Hatcher H, Planalp R, Cho J, Torti FM, Torti SV. Cell Mol Life Sci. 2008;65:1631–1652. doi: 10.1007/s00018-008-7452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Baum L, Ng A. J Alzheimers Dis. 2004;6:367–377. doi: 10.3233/jad-2004-6403. discussion 443–449. [DOI] [PubMed] [Google Scholar]
- 332.Pal R, Cristan EA, Schnittker K, Narayan M. Biochem Biophys Res Commun. 2010;392:567–571. doi: 10.1016/j.bbrc.2010.01.071. [DOI] [PubMed] [Google Scholar]
- 333.Chen Z, Wang H, Qing X, Xu N. Bioactivity and Therapeutic Potential. Taylor & Francis; London: 2002. Y. S. Zhen. [Google Scholar]
- 334.Unno K, Hoshino M. Cent Nerv Syst Agents Med Chem. 2007;7:109–114. [Google Scholar]
- 335.Schulz V. Phytotherapie. 2009;30:22–23. [Google Scholar]
- 336.Mandel SA, Amit T, Weinreb O, Reznichenko L, Youdim MBH. CNS Neurosci Ther. 2008;14:352–365. doi: 10.1111/j.1755-5949.2008.00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Weinreb O, Amit T, Mandel S, Youdim M. Genes Nutr. 2009;4:283–296. doi: 10.1007/s12263-009-0143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Frank B, Gupta S. Ann Clin Psychiatry. 2005;17:269–286. doi: 10.1080/10401230500296428. [DOI] [PubMed] [Google Scholar]
- 339.Kim C, Lee C, Park GH, Jang J. Arch Pharmacal Res. 2009;32:869–881. doi: 10.1007/s12272-009-1609-z. [DOI] [PubMed] [Google Scholar]
- 340.Zhao B. Neurochem Res. 2009;34:630–638. doi: 10.1007/s11064-008-9900-9. [DOI] [PubMed] [Google Scholar]
- 341.Bahia PK, Rattray M, Williams RJ. J Neurochem. 2008;106:2194–2204. doi: 10.1111/j.1471-4159.2008.05542.x. [DOI] [PubMed] [Google Scholar]
- 342.Cuevas E, Limón D, Pérez-Severiano F, Díaz A, Ortega L, Zenteno E, Guevara J. Eur J Pharmacol. 2009;616:122–127. doi: 10.1016/j.ejphar.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 343.Collins MA, Neafsey EJ, Mukamal KJ, Gray MO, Parks DA, Das DK, Korthuis RJ. Alcohol Clin Exp Res. 2009;33:206–219. doi: 10.1111/j.1530-0277.2008.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Rocha-González HI, Ambriz-Tututi M, Granados-Soto V. CNS Neurosci Ther. 2008;14:234–247. doi: 10.1111/j.1755-5949.2008.00045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Jang J, Surh Y. Free Radic Biol Med. 2003;34:1100–1110. doi: 10.1016/s0891-5849(03)00062-5. [DOI] [PubMed] [Google Scholar]
- 346.Rossignol E, Debiton E, Fabbro D, Moreau P, Prudhomme M, Anizon F. Anti-Cancer Drugs. 2008;19:789–792. doi: 10.1097/CAD.0b013e32830ce4d8. [DOI] [PubMed] [Google Scholar]
- 347.Rossi M, Caruso F, Opazo C, Salciccioli J. J Agric Food Chem. 2008;56:10557–10566. doi: 10.1021/jf801923j. [DOI] [PubMed] [Google Scholar]
- 348.de Almeida LMV, Leite MC, Thomazi AP, Battu C, Nardin P, Tortorelli LS, Zanotto C, Posser T, Wofchuk ST, Leal RB, Gonçalves C, Gottfried C. Arch Biochem Biophys. 2008;480:27–32. doi: 10.1016/j.abb.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 349.Kumar A, Naidu P, Seghal N, Padi S. Pharmacology. 2007;79:17–26. doi: 10.1159/000097511. [DOI] [PubMed] [Google Scholar]
- 350.Karuppagounder SS, Pinto JT, Xu H, Chen H, Beal MF, Gibson GE. Neurochem Int. 2009;54:111–118. doi: 10.1016/j.neuint.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Vingtdeux V, Dreses-Werringloer U, Zhao H, Davies P, Marambaud P. BMC Neurosci. 2008;9(Suppl 2):S6. doi: 10.1186/1471-2202-9-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Pallàs M, Casadesús G, Smith MA, Coto-Montes A, Pelegri C, Vilaplana J, Camins A. Curr Neurovasc Res. 2009;6:70–81. doi: 10.2174/156720209787466019. [DOI] [PubMed] [Google Scholar]
- 353.Darvesh AS, Carroll RT, Bishayee A, Geldenhuys WJ, Van der Schyf CJ. Expert Rev Neurother. 2010;10:729–745. doi: 10.1586/ern.10.42. [DOI] [PubMed] [Google Scholar]
- 354.Sun AY, Wang Q, Simonyi A, Sun GY. Neuromol Med. 2008;10:259–274. doi: 10.1007/s12017-008-8052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Dajas F, Arredondo F, Echeverry C, Ferreira M, Morquio A, Rivera F. Curr Neuropharmacology. 2010;3:193–205. [Google Scholar]
- 356.Ishige K, Schubert D, Sagara Y. Free Radic Biol Med. 2001;30:433–446. doi: 10.1016/s0891-5849(00)00498-6. [DOI] [PubMed] [Google Scholar]
- 357.Jang YJ, Kang NJ, Lee KW, Lee HJ. Ann N Y Acad Sci. 2009;1171:170–175. doi: 10.1111/j.1749-6632.2009.04720.x. [DOI] [PubMed] [Google Scholar]
- 358.Pavlica S, Gebhardt R. Life Sci. 2010;86:79–86. doi: 10.1016/j.lfs.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 359.Liu R, Barkhordarian H, Emadi S, Park CB, Sierks MR. Neurobiol Dis. 2005;20:74–81. doi: 10.1016/j.nbd.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 360.Maher P. Genes Nutr. 2009;4:297–307. doi: 10.1007/s12263-009-0142-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Zhang H, Liu Y, Wang H, Xu J, Hu H. Cell Biol Int. 2008;32:1230–1237. doi: 10.1016/j.cellbi.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 362.Zeng H, Chen Q, Zhao B. Free Radic Biol Med. 2004;36:180–188. doi: 10.1016/j.freeradbiomed.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 363.Ansari MA, Abdul HM, Joshi G, Opii WO, Butterfield DA. J Nutr Biochem. 2009;20:269–275. doi: 10.1016/j.jnutbio.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Heo HJ, Kim D, Shin SC, Kim MJ, Kim BG, Shin D. J Agric Food Chem. 2004;52:1520–1525. doi: 10.1021/jf035079g. [DOI] [PubMed] [Google Scholar]
- 365.Lu P, Mamiya T, Lu LL, Mouri A, Zou L, Nagai T, Hiramatsu M, Ikejima T, Nabeshima T. Br J Pharmacol. 2009;157:1270–1277. doi: 10.1111/j.1476-5381.2009.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Yang LX, Huang KX, Li HB, Gong JX, Wang F, Feng YB, Tao QF, Wu YH, Li XK, Wu XM, Zeng S, Spencer S, Zhao Y, Qu J. J Med Chem. 2009;52:7732–7752. doi: 10.1021/jm900735p. [DOI] [PubMed] [Google Scholar]
- 367.Shukitt-Hale B, Lau FC, Joseph JA. J Agric Food Chem. 2008;56:636–641. doi: 10.1021/jf072505f. [DOI] [PubMed] [Google Scholar]
- 368.Heo HJ, Lee CY. J Agric Food Chem. 2005;53:1984–1989. doi: 10.1021/jf048616l. [DOI] [PubMed] [Google Scholar]
- 369.Rahman MM, Ichiyanagi T, Komiyama T, Sato S, Konishi T. J Agric Food Chem. 2008;56:7545–7550. doi: 10.1021/jf800930s. [DOI] [PubMed] [Google Scholar]
- 370.Krikorian R, Shidler MD, Nash TA, Kalt W, Vinqvist-Tymchuk MR, Shukitt-Hale B, Joseph JA. J Agric Food Chem. 2010;58:3996–4000. doi: 10.1021/jf9029332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Wang JM, Johnston PB, Ball BG, Brinton RD. J Neurosci. 2005;25:4706–4718. doi: 10.1523/JNEUROSCI.4520-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Wang JM, Brinton RD. BMC Neurosci. 2008;9(Suppl 2):S11. doi: 10.1186/1471-2202-9-S2-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Wang JM, Singh C, Liu L, Irwin RW, Chen S, Chung EJ, Thompson RF, Brinton RD. Proc Natl Acad Sci USA. 2010;107:6498–6503. doi: 10.1073/pnas.1001422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Pringle AK, Schmidt W, Deans JK, Wulfert E, Reymann KG, Sundstrom LE. Eur J Neurosci. 2003;18:117–124. doi: 10.1046/j.1460-9568.2003.02734.x. [DOI] [PubMed] [Google Scholar]
- 375.ClinicalTrials. European Study of HF0220 in Mild to Moderate Alzheimer’s Disease Patients. 2010 http://clinicaltrials.gov/show/NCT00357357.
- 376.Berry M. Bibl Anat. 1982:1–11. [PubMed] [Google Scholar]
- 377.Liu BP, Cafferty WBJ, Budel SO, Strittmatter SM. Philos Trans R Soc Lond, B, Biol Sci. 2006;361:1593–1610. doi: 10.1098/rstb.2006.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Asher RA, Morgenstern DA, Moon LD, Fawcett JW. Prog Brain Res. 2001;132:611–619. doi: 10.1016/S0079-6123(01)32106-4. [DOI] [PubMed] [Google Scholar]
- 379.Rudge JS, Silver J. J Neurosci. 1990;10:3594–3603. doi: 10.1523/JNEUROSCI.10-11-03594.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Gao Y, Deng K, Cao Z, Graziani EI, Gilbert AM, Koehn FE, Wood A, Doherty P, Walsh FS. J Neurochem. 2010;113:1331–1342. doi: 10.1111/j.1471-4159.2010.06704.x. [DOI] [PubMed] [Google Scholar]
- 381.Cotman C, Monaghan D. Ann N Y Acad Sci. 1989;568:138–148. doi: 10.1111/j.1749-6632.1989.tb12501.x. [DOI] [PubMed] [Google Scholar]
- 382.Smith MA, Perry G, Pryor WA. Free Radic Biol Med. 2002;32:1049. doi: 10.1016/s0891-5849(02)00793-1. [DOI] [PubMed] [Google Scholar]
- 383.Mount C, Downton C. Nat Med. 2006;12:780–784. doi: 10.1038/nm0706-780. [DOI] [PubMed] [Google Scholar]
- 384.Xia P, Chen HV, Zhang D, Lipton SA. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Yu J, Chang RC, Tan L. Prog Neurobiol. 2009;89:240–255. doi: 10.1016/j.pneurobio.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 386.Arneric SP, Holladay M, Williams M. Biochem Pharmacol. 2007;74:1092–1101. doi: 10.1016/j.bcp.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 387.Kem W. Med Chem Alzheimer Dis. 2008:135–139. [Google Scholar]
- 388.Kem WR, Mahnir VM, Prokai L, Papke RL, Cao X, LeFrancois S, Wildeboer K, Prokai-Tatrai K, Porter-Papke J, Soti F. Mol Pharmacol. 2004;65:56–67. doi: 10.1124/mol.65.1.56. [DOI] [PubMed] [Google Scholar]
- 389.Toyohara J, Hashimoto K. Open Med Chem J. 2010;4:37–56. doi: 10.2174/1874104501004010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Folmer F, Houssen WE, Scott RH, Jaspars M. Curr Opin Drug Discov Devel. 2007;10:145–152. [PubMed] [Google Scholar]
- 391.Appendino G, Minassi A, Pagani A, Ech-Chahad A. Curr Pharm Des. 2008;14:2–17. doi: 10.2174/138161208783330781. [DOI] [PubMed] [Google Scholar]
- 392.Song M, Liu H, Jiang H, Yue J, Hu G, Chen H. Neuroscience. 2008;155:469–475. doi: 10.1016/j.neuroscience.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 393.Li F, Gong Q, Wu Q, Lu Y, Shi J. Pharmacol Biochem Behav. 2010;96:301–305. doi: 10.1016/j.pbb.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 394.Li L, Tsai H, Li L, Wang X. Amer J Chin Med. 2010;38:113–125. doi: 10.1142/S0192415X10007701. [DOI] [PubMed] [Google Scholar]
- 395.Valero T, del Barrio L, Egea J, Cañas N, Martínez A, García AG, Villarroya M, López MG. Eur J Pharmacol. 2009;607:47–53. doi: 10.1016/j.ejphar.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 396.Yu SP, Farhangrazi S, Ying H, Yeh C, Choi DW. Neurobiol Dis. 1998:81–88. doi: 10.1006/nbdi.1998.0186. [DOI] [PubMed] [Google Scholar]
- 397.Angulo E, Noé V, Casadó V, Mallol J, Gomez-Isla T, Lluis C, Ferrer I, Ciudad CJ, Franco R. J Neurochem. 2004;91:547–557. doi: 10.1111/j.1471-4159.2004.02771.x. [DOI] [PubMed] [Google Scholar]
- 398.Diochot S, Schweitz H, Béress L, Lazdunski M. J Biol Chem. 1998;273:6744–6749. doi: 10.1074/jbc.273.12.6744. [DOI] [PubMed] [Google Scholar]
- 399.Yeung SYM, Thompson D, Wang Z, Fedida D, Robertson B. J Neurosci. 2005;25:8735–8745. doi: 10.1523/JNEUROSCI.2119-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Valverde P, Kawai T, Taubman MA. J Dent Res. 2005;84:488–499. doi: 10.1177/154405910508400603. [DOI] [PubMed] [Google Scholar]