

Abstract
Methylmercury (MeHg) is an ubiquitous environmental pollutant which is transported into the mammalian cells when present as the methylmercury-cysteine conjugate (MeHg–Cys). With special emphasis on hepatic cells, due to their particular propensity to accumulate an appreciable amount of Hg after exposure to MeHg, this study was performed to evaluate the effects of methionine (Met) on Hg uptake, reactive species (RS) formation, oxygen consumption and mitochondrial function/cellular viability in both liver slices and mitochondria isolated from these slices, after exposure to MeHg or the MeHg–Cys complex. The liver slices were pre-treated with Met (250 μM) 15 min before being exposed to MeHg (25 μM) or MeHg–Cys (25 μM each) for 30 min at 37 °C. The treatment with MeHg caused a significant increase in the Hg concentration in both liver slices and mitochondria isolated from liver slices. Moreover, the Hg uptake was higher in the group exposed to the MeHg–Cys complex. In the DCF (dichlorofluorescein) assay, the exposure to MeHg and MeHg–Cys produced a significant increase in DFC reactive species (DFC-RS) formation only in the mitochondria isolated from liver slices. As observed with Hg uptake, DFC-RS levels were significantly higher in the mitochondria treated with the MeHg–Cys complex compared to MeHg alone. MeHg exposure also caused a marked decrease in the oxygen consumption of liver slices when compared to the control group, and this effect was more pronounced in the liver slices treated with the MeHg–Cys complex. Similarly, the loss of mitochondrial activity/cell viability was greater in liver slices exposed to the MeHg–Cys complex when compared to slices treated only with MeHg. In all studied parameters, Met pre-treatment was effective in preventing the MeHg-and/or MeHg–Cys-induced toxicity in both liver slices and mitochondria. Part of the protection afforded by Met against MeHg may be related to a direct interaction with MeHg or to the competition of Met with the complex formed between MeHg and endogenous cysteine. In summary, our results show that Met pre-treatment produces pronounced protection against the toxic effects induced by MeHg and/or the MeHg–Cys complex on mitochondrial function and cell viability. Consequently, this amino acid offers considerable promise as a potential agent for treating acute MeHg exposure.
Keywords: Methylmercury, MeHg–Cys complex, Methionine, Mitochondria, Liver
Introduction
Exposure to methylmercury (MeHg), the most toxic form of mercury (Hg) in the environment, is well recognized as the cause of a series of cellular disorders in several systems, especially in the central nervous system (CNS) (Choi 1991; Sakamoto et al., 1998; Clarkson et al., 2003; Sakaue et al., 2006). However, the exact molecular mechanisms underlying MeHg-induced toxicity in the developing and adult CNS, as well as in other tissues, remain unclear. The methyl mercuric ion (CH3Hg+) does not exist in biological systems as a free, unbound cation (Hughes, 1957), but rather, is found conjugated to thiol-containing biomolecules, such as glutathione (GSH), cysteine (Cys) and homocysteine (Hcy) (Clarkson, 1993). Thus, many of the mechanisms proposed to explain the rapid diffusion of MeHg across membranes and, consequently, the cellular damage induced by MeHg is largely based upon its high affinity for −SH groups. Corroborating these notions, several studies have demonstrated that the absorption and cellular uptake of MeHg are significantly increased when it is present as Cys– or Homocysteine–MeHg conjugates (Ballatori, 2002; Roos et al., 2010). Additionally, experimental evidence supports the idea that the neutral amino acid transport system L is a significant route for MeHg–Cys transmembrane movement (Yin et al., 2008; Roos et al., 2010), since MeHg–Cys complexes are thought to mimic structurally methionine (Met), a substrate for amino acid carriers such as the L-type large neutral amino acid transporters (LATs). The major LATs subtypes (LAT1, LAT2 and LAT3) are widely expressed in organs and tissues of the kidney, placenta, brain and intestinal wall (Palacín et al., 1998; Kanai and Endou, 2001). In the liver, amino acid transporters with system L transport activity have been identified mainly in human hepatoblastoma cell line HepG2 (Sarkar et al., 1999). However, the physiological function as well as the precise subcellular localization of these transporters in normal hepatic cells has yet to be determined (Bode, 2001; Babu et al., 2003; Fukuhara et al., 2007; Wagner et al., 2010).
A number of particular cellular mechanisms and molecules are the primary targets of MeHg cytotoxicity. Disruption of calcium homeostasis and free radicals generation are among the detrimental effects associated with MeHg-induced toxicity (Limke et al., 2003; Ikeda et al., 1999). In this scenario, mitochondria play a crucial role, as these organelles can act as a buffer against cytosolic calcium and can mediate (RS) formation in cells (Norenberg and Rao, 2007; Chacko et al., 2009). It has been shown that mitochondrial dysfunctions induced by MeHg include the failure of energy metabolism, the disruption of calcium homeostasis and the dissipation of the mitochondrial membrane potential, effects which lead to a mitochondrial burst of reactive oxygen species (ROS) production (Kim and Sharma 2003; Kang et al., 2006; Dreiem and Seegal, 2007). ROS are important mediators of damage to cell structures, including lipids and membranes, as well as proteins and nucleic acids (Poli et al., 2004). The detrimental effects of ROS are balanced by the antioxidant action of non-enzymatic antioxidants in addition to antioxidant enzymes (Poli et al., 2004). However, in vivo and in vitro experimental observations have shown that the toxic effects of MeHg are accompanied by a significant deficit of antioxidant defenses, such as the depletion of GSH and the inhibition of GSH peroxidase activity (Farina et al., 2004; Chang and Tsai, 2008; Stringari et al., 2008; Farina et al., 2009). Thus, oxidative stress has been implicated in a number of events involved in MeHg-induced cytotoxicity (Roos et al., 2009).
Based on the evidence presented above, it is reasonable to assume that Met, acting as competitive inhibitor of MeHg–Cys transport through system L could prevent or reduce MeHg-induced cytotoxicity. To date, there have been no studies on the efficacy of Met to attenuate mitochondrial MeHg uptake and mitochondrial function. The experimental model employed, namely hepatic cells, possess a particular propensity to accumulate appreciable quantities of Hg after exposure to MeHg (de Freitas et al., 2009). Specifically, we have examined, for the first time, the effects of Met pre-treatment on Hg uptake, RS formation, oxygen consumption and cellular viability in both liver slices and mitochondria isolated from these slices, after exposure to MeHg or the MeHg–Cys complex.
Materials and experimental procedures
Chemicals
MeHgCl and L-Cysteine chloride were obtained from Aldrich (St. Louis, MO). All other chemicals were of analytical reagent grade and were purchased from Merck (Rio de Janeiro, Brazil).
Animals
Adult male Wistar rats from our own breeding colony (200–250 g) were maintained in Plexiglas cages with food and water ad libitum, in a temperature-controlled room (22–25 °C) and on a 12 h-light/dark cycle with lights on at 7:00 a.m. Animals were handled and treated according to the guidelines set forth by the Committee on Care and Use of Experimental Animal Resources of the Federal University of Santa Maria, Brazil.
Preparation of liver slices
Animals were killed by decapitation, and the whole liver was quickly removed and placed on ice. Afterward, the liver was cut into transverse slices 300 μm thick using a McIlwain tissue chopper (Campbell Instruments; The Mickle Laboratory Engineering Co). The slices were placed in Krebs–Ringer buffer (10 mM D-glucose, 129 mM NaCl, 1.25 mM NaHPO4, 22 mM NaHCO3, KCl 3 mM, CaCl2 1.8 mM, MgSO4 1.8 mM, Hepes 5 mM, pH 7.4), which was previously bubbled with O2 95% and CO2 5% for 30 min. Sixty slices (per group) were carefully selected, weighted (30±2 μg each) and randomly placed in buffer (2 mL) for the respective treatments. In the final step of each experiment the total protein content was determined (Peterson, 1977).
Treatment of liver slices
The slices were subdivided to the following groups: (1) control; (2) MeHg (25 μM); (3) Cysteine (25 μM); (4) MeHg–Cys complex (25 μM each); (5) Methionine (250 μM); (6) Met (250 μM)+MeHg (25 μM); and (7) Met (250 μM)+MeHg–Cys complex (25 μM each). The slices were exposed to the different treatments for 30 min at 37 °C, in the presence of O2 (95%) and CO2 (5%). The molar ratio of cysteine to MeHg was 1, and the stoicheometric reaction between cysteine and MeHg was confirmed by Ellman’s reagent (Ellman, 1959). The Methionine groups (250 μM) were pre-treated for 15 min with methionine before being exposed to MeHg or the MeHg–Cys complex. All reagents were dissolved in Krebs–Ringer buffer.
Mitochondrial preparation
Liver mitochondria were isolated as previously described by Brustovetsky and Dubinsky (2000a, 2000b), with some modifications. After treatment, the liver slices were washed three times and manually homogenized in cold buffer I (manitol 225 mM, sucrose 75 mM, K+ EGTA 1 mM, bovine serum albumin (BSA) 0.1% and K+-HEPES 10 mM pH 7.2), using a potter glass (length: 10 cm; diameter: 1 cm). Next, the homogenized slices were centrifuged at 2000×g for 7 min at 4 °C. The pellet was discarded and the supernatant was centrifuged again at 12,000×g for 10 min at 4 °C. Then, the resultant supernatant was discarded, and the pellet was re-suspended in buffer II (manitol 225 mM, sucrose 75 mM, K+ EGTA 1 mM and K+-HEPES 10 mM pH 7.2) and re-centrifuged at 12,000×g for 10 min at 4 °C. Finally, the last supernatant was discarded, and the pellet was re-suspended and maintained in buffer III (sucrose 100 mM, KCl 65 mM, K+-HEPES 10 mM and EGTA 50 μM pH 7.2) for subsequent analyses.
Mercury quantification
Both the aliquot of the homogenate of liver slices and the mitochondrial suspension isolated from liver slices were subjected to Hg analysis, which was carried out by Cold Vapor-Atomic Fluorescence Spectrometry according to the method described by Bergdahl et al., 1998. The total Hg content was determined after acid digestion with HNO3, H2O2, H2SO4 and perchloric acid (Bergdahl et al., 1998).
Evaluation of Reactive Species (RS) formation with DCH (dichlorofluorescein-reactive species, DCH-RS)
RS levels were measured using the oxidant sensing fluorescent probe, 2′,7′-dichlorofluorescein diacetate (DCHF–DA) (Hempel et al., 1999). The oxidation (DCHF–DA) to fluorescent dichlorofluorescein (DCF) was determined at 488 nm for excitation and 525 nm for emission. After being exposed to the reagents, the liver slices were homogenized in buffer I (1 mL), and an aliquot of 10 μL (50 μg/protein, Peterson, 1977) of both the homogenate of the liver slices and the homogenate of the isolated mitochondria was added to 3 mL of buffer III (containing 5 mM glutamate and 5 mM succinate). After 10 s, 10 μM (DCHF–DA) (prepared in ethanol) was added to the mixture; and the fluorescence intensity from DCF was measured for 300 s and expressed as a percentage of the untreated control group.
Oxygen consumption of liver slices
The oxygen consumption of the liver slices was measured using an oxymeter (Hansatech model with a Clark-type electrode) at 30 °C. Two slices, weighting approximately 30 μg (30±2 μg) each, were selected and placed in 2 mL Krebs–Ringer buffer. Fifteen minutes after methionine addition, glutamate/succinate (5 mM each) was placed in the medium to increase the respiratory state. After 30 min, either the MeHg solution or the MeHg–Cys complex solution was added. The respiratory ratio and oxygen consumption were determined and compared among groups.
Cell viability/mitochondrial activity
Cell viability and mitochondrial activity were measured by dehydrogenase activity using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay (Mosmann, 1983). After 30, 60 and 120 min of exposure to the respective treatment, four liver slices were selected and incubated with MTT (5 μg/mL) for 20 min. The MTT reduction reaction was stopped by the addition of 1.5 mL of dimethylsulfoxide (DMSO). The formazan color and the colorimetric intensity were determined by the difference in absorbance readings at 570–630 nm, using an UV 2450 Shimadzu spectrophotometer. The ratio values were standardized to protein content and expressed as a percentage of the untreated control group.
Protein determination
All experiments were standardized to protein concentrations (Peterson, 1977), and, when appropriate, were expressed as a percentage of untreated control values.
Statistical analysis
Data were analyzed statistically by one-way ANOVA, followed by Duncan’s multiple range tests when appropriate. The significance between the respiratory rates (Table 1) was analyzed statistically by t-test. Differences between groups were considered to be significant when P<0.05.
Table 1.
Effects of MeHg or MeHg–Cys complex exposure on respiratory rates of rat liver slices.
| Rate 1
|
Rate 2
|
Rate 3
|
|
|---|---|---|---|
| Respiration of slices (nmol O2/mL/min) | Respiration with succ. (nmol O2/mL/min) | Respiration with MeHg or MeHg–Cys (nmol O2/mL/min) | |
| Control | 2.27±0.22 | 6.59±0.37 | 5.32±0.47 |
| MeHg | 2.26±0.47 | 6.09±0.32 | 3.49±0.52a,b |
| MeHg–Cys | 2.10±0.38 | 5.97±0.18 | 2.34±0.05a,b,c |
| With methionine | |||
| Control | 2.42±0.27 | 6.17±0.98 | 5.57±0.75 |
| MeHg | 2.38±0.10 | 6.55±0.53 | 5.45±0.35d |
| MeHg–Cys | 2.37±0.18 | 6.10±0.34 | 3.43±0.26a,b,d |
Slices were pre-treated for 15 min with Met (250 μM) and after exposed for 30 min to MeHg (25 μM) or MeHg–Cys complex (25 μM each).
Indicates p<0.05, rate 3 compared to 2.
Indicates p<0.05 from control.
Indicates p<0.05 from MeHg.
Indicates p<0.05 from MeHg–Cys complex, n=5 mean±S.E).
Results
Hg levels in liver slices and mitochondria isolated from liver slices
The first set of experiments was designated to analyze the Hg content in liver slices and mitochondria isolated from liver slices. The Fig. 1 shows that treatment with MeHg alone caused a significant increase in the Hg concentration in both liver slices (A) and in mitochondria isolated from liver slices (B) and that the content of Hg was further increased in the group exposed to the MeHg–Cys complex when compared to the group treated with MeHg alone (Figs. 1A and B). The data in Fig. 1 also reveal that pre-treatment with Met was effective in reducing the Hg levels of the slices exposed to MeHg or the MeHg–Cys complex (Fig. 1A). However, in mitochondria isolated from these liver slices, the Met pre-treatment effect was observed only in the MeHg–Cys complex group (Fig. 1B).
Fig. 1.
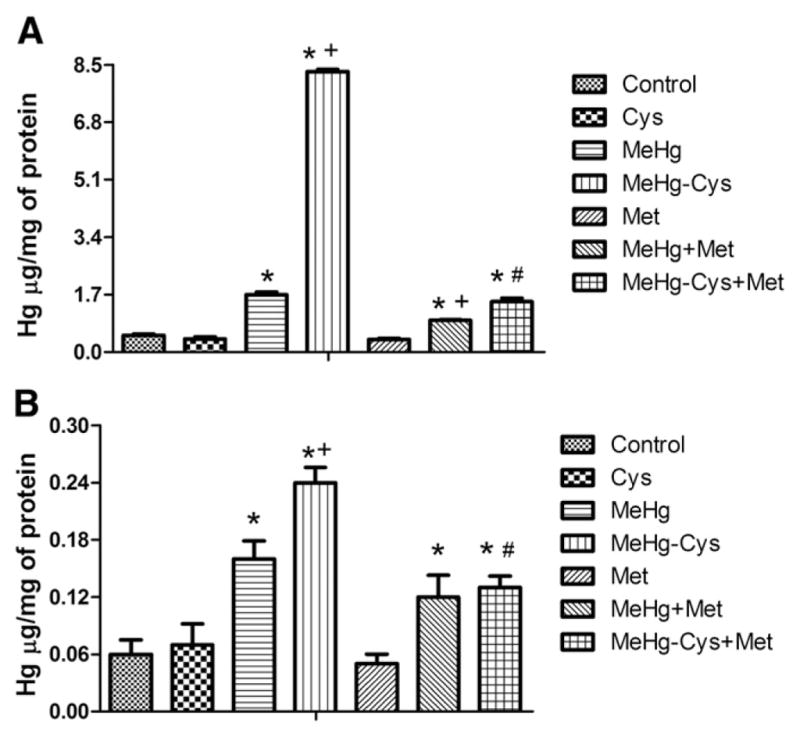
Effects of Met pre-treatment on Hg uptake in rat liver slices (A) and mitochondria (B) exposed to MeHg or the MeHg–Cys complex. Slices were pre-treated for 15 min with Met (250 μM) and then exposed for 30 min to MeHg (25 μM) or the MeHg–Cys complex (25 μM each); (*Indicates p<0.05 from control; +Indicates p<0.05 from MeHg; #Indicates p<0.05 from the MeHg–Cys complex; n=6 mean±S.E.).
DFC-RS (Diflouroscein-Reactive Species) formation
The second set of experiments was performed to analyze the effect of Met treatment on RS production caused by MeHg in liver slices and mitochondria isolated from liver slices. Fig. 2 illustrates the levels of DFC-RS production in liver slices (A) and mitochondria isolated from liver slices (B) after 45 min of exposure to Met (50–250 μM). The data show that Met pre-treatment, at all concentrations tested, did not cause any effect on DFC-RS production when compared to control values (Figs. 2A and B).
Fig. 2.

Effects of Met pre-treatment on DFC-RS production in rat liver slices (A) and mitochondria (B). Slices were pre-treated for 45 min with Met (50, 100 and 250 μM). The tracings of figure are representative lines of 3 independent experiments.
Fig. 3 shows the effects of exposure to MeHg or the MeHg–Cys complex on DFC-RS generation in liver slices (A) and mitochondria isolated from liver slices (B). In liver slices, the levels of DFC-RS production were slightly enhanced by exposure to MeHg or the MeHg–Cys complex. However, this difference was not statistically significant (Fig. 3A). In contrast, in the mitochondria isolated from these liver slices, MeHg exposure produced a significant increase on DFC-RS production when compared to levels found in the control group (Fig. 3B). Furthermore, the DFC-RS production levels were significantly higher in the mitochondria isolated from liver slices that were treated with the MeHg–Cys complex, when compared to mitochondria isolated from slices exposed to MeHg alone (Fig. 3B). Notably, Met pre-treatment was effective in reducing DFC-RS production only in the mitochondria isolated from slices treated with the MeHg–Cys complex (Fig. 4).
Fig. 3.

Effects of exposure to MeHg or the MeHg–Cys complex on DFC-RS production in rat liver slices (A) and mitochondria (B). Slices were exposed for 30 min to MeHg (25 μM) or the MeHg–Cys complex (25 μM each). (*Indicates p<0.05 from control; +Indicates p<0.05 from MeHg; n=5 mean±S.E.). The tracings of Figs. 3A and B are representative and averaged lines respectively.
Fig. 4.

Effects of Met pre-treatment on DFC-RS production in mitochondria exposed to MeHg or the MeHg–Cys complex. Slices were pre-treated for 15 min with Met (250 μM) and then exposed for 30 min to MeHg (25 μM) or the MeHg–Cys complex (25 μM each). Insets in Fig. 4 represent statistical analysis. (*Indicates p<0.05 from control; +Indicates p<0.05 from MeHg; #Indicates p<0.05 from MeHg–Cys complex; n=6 mean±S.E.). The tracings of Fig. 4 are representative lines.
Oxygen consumption
The third set of experiments was designed to verify mitochondrial viability by determining the oxygen consumption by the liver slices. Fig. 5A shows that MeHg exposure significantly decreased the oxygen consumption of liver slices as compared to the control group, and that this effect was more pronounced in the liver slices treated with the MeHg–Cys complex. Interestingly, Met pre-treatment effectively prevented the reduction of oxygen consumption in both slices treated with MeHg and slices treated with the MeHg–Cys complex (Fig. 5B) when compared to control slices (Fig. 5A). A synopsis of MeHg, MeHg–Cys and Met modulation of mitochondria respiration is depicted in Table 1.
Fig. 5.

Effects of exposure to MeHg or the MeHg–Cys complex on oxygen consumption in rat liver slices (A). Effects of Met pre-treatment on oxygen consumption in rat liver slices (B) exposed to MeHg or the MeHg–Cys complex. Slices were pre-treated for 15 min with Met (250 μM) and then exposed for 30 min to MeHg (25 μM) or the MeHg–Cys complex (25 μM each); (n=5 mean±S.E.).
Cell viability/mitochondrial activity
The final set of experiments was performed to evaluate the cell viability/mitochondria activity in liver slices. Fig. 6 shows that treatment with MeHg alone caused a significant decrease in mitochondrial activity at all tested times (30, 60 and 120 min. Figs. 6A, B and C, respectively) when compared to the control group. At 30 and 60 min, the loss of mitochondrial activity was higher in liver slices exposed to the MeHg–Cys complex when compared to those treated only with MeHg (Figs. 6A and B, respectively). At all times tested, Met pre-treatment prevented mitochondrial dysfunction induced by both MeHg and MeHg–Cys complex exposure (Figs. 6A, B and C).
Fig. 6.

Effects of Met pre-treatment on mitochondrial function of cells exposed to MeHg or the MeHg–Cys complex. Slices were pre-treated for 15 min with Met (250 μM) and then exposed for 30, 60 or 120 min to MeHg (25 μM) or the MeHg–Cys complex (25 μM each) (Figs. 6A, B, and C respectively). (*Indicates p<0.05 from control; +Indicates p<0.05 from MeHg; #Indicates p<0.05 from MeHg–Cys complex; n=6 mean±S.E).
Discussion
It has been postulated that MeHg is transported as a MeHg–Cys complex by the ubiquitous L-type large neutral amino acid transporters (LATs) and that transport of this conjugate represents the main pathway through which MeHg exerts its toxicity in many tissues (Kerper et al., 1992; Kajiwara et al., 1996; Simmons-Willis et al., 2002; Adachi 2006; Yin et al., 2008). Corroborating this hypothesis, our group recently reported that mice chronically treated with the MeHg–Cys complex show enhanced Hg uptake, especially in the liver, when compared to other organs, such as the brain and kidney (Roos et al., 2010). These results are most likely due to the fact that the liver is a central organ of protein metabolism and receives amino acids absorbed at the intestinal levels as well as those derived from other organs and systems (Duarte, 2003). Although hepatic cells contain some of the same carriers that have been implicated in the transport of Hg in other organs, the precise mechanisms underlying the MeHg uptake across the membrane into normal hepatocytes as well as the influence of the MeHg–Cys complex on Hg uptake and hepatoxi- city have not previously been well defined. Consequently, our study was primarily designed to investigate the Hg content in hepatic cells, at both cytosolic and mitochondrial levels after exposure to MeHg or the MeHg–Cys complex. Several previous studies have investigated and reported on the toxicology of MeHg, but, to date, only chelating agents have been employed to facilitate the removal of Hg from the body (Pingree et al., 2001; Carvalho et al., 2007). However, these drugs are of limited use because of their adverse side effects. In the present study, we have tested the possible use of Met as an efficacious agent capable of protecting against the deleterious effects of MeHg. We observed that the Hg concentration in liver slices and in the mitochondria isolated from liver slices was higher after exposure to the MeHg–Cys complex (Fig. 1).
Notably, we observed that Met decreased MeHg uptake by liver slices (Fig. 1). These results are different from those reported by Adachi (2006) after exposure of mice to MeHg. Adachi reported that Met can increase the hepatic deposition of Hg 2 h after intravenously administration of MeHg and/or methionine. Since we have used only a single time-point of exposure of liver slices to MeHg (30 min) and/or Met (45 min), we cannot disregard the possibility that uptake of MeHg could be increased in the presence of Met. Alternatively, the decrease in Hg uptake in the slices by Met may be, at least in part, related to the relatively high concentration of Met in the medium and, consequently, to direct interaction between MeHg and Met, thus lowering the effective free concentration of MeHg. Accordingly, we can posit that the effect observed in the presence of Met may be related to a direct interaction of the sulfur atom and/or amino end of Met with MeHg (Rabenstein and Fairhurst 1975). Alternatively, Met may be reducing the uptake of MeHg complexed with endogenous cysteine in liver slices. In addition, here we have worked with an in vitro system derived from rats. It is feasible that results obtained after in vivo exposure may be modified by changes in the amino acids and/or MeHg–Cys complex distribution and metabolism in other animal species.
Thus, our results corroborate that (1) the MeHg–Cys complex is a substrate for the neutral amino acid carrier L-type in the liver and (2) Met prevents the hepatoxicity induced by MeHg, reflecting its ability to reduce MeHg uptake as well as cytotoxicity in liver slices and mitochondria isolated from liver slices treated with the MeHg–Cys complex. Regarding the mechanisms which underlie the MeHg-mediated hepatoxicity, we found that exposure to MeHg or the MeHg–Cys complex increased DFC-RS formation, particularly in mitochondria isolated from liver slices. These results are consistent with previous reports from our group, which have shown that MeHg increases ROS production in cortical brain slices only at high concentrations (100 μM) and after long-term exposure (2 h) (Roos et al., 2009; Wagner et al., 2010). These data also suggest that mitochondria are more sensitive to low MeHg concentrations. In agreement with the present data, it has been previously reported that MeHg, at a concentration of 5 μM, increases ROS levels in mitochondria isolated from rat brain slices (Dreiem and Seegal, 2007; Wagner et al., 2010,). It is noteworthy that in our experimental protocol, MeHg and/or the MeHg–Cys complex reduced mitochondrial activity. These effects are likely related, since ROS can react rapidly with cellular macromolecules and induce mitochondrial damage (Puntel et al., 2010; Colquhoun, 2010; Forkink et al., 2010). Furthermore, because MeHg can cause a pronounced disruption of calcium homeostasis (Stavrovskaya and Kristal, 2010), it is plausible that alterations in Ca2+ homeostasis could lead to the collapse of the inner mitochondrial membrane potential, as well as the opening of the mitochondrial permeability pore, events that ultimately result in the loss of mitochondrial function, ROS formation and cell death (Puntel et al., 2010; Colquhoun, 2010; Forkink et al., 2010). Thus, it is reasonable to assume that mitochondria are the primary molecular target for MeHg- and MeHg–Cys-induced cytotoxicity. In addition, we assessed mitochondrial function by analyzing the oxygen consumption of liver slices treated with MeHg or the MeHg–Cys complex. We observed that MeHg exposure attenuated mitochondrial respiration and that this effect was greater in the slices treated with the MeHg–Cys complex. This is in agreement with a recent study, which has demonstrated that dietary MeHg causes a significant decrease in both state 3 of mitochondrial respiration and cytochrome c oxidase activity in mitochondria from contaminated zebrafish muscle fibers (Cambier et al., 2009); and inhibits the activity of the mitochondrial complexes II–III, IV, as well as mitochondrial creatine kinase (Glaser et al., 2010). Furthermore, this work has shown that MeHg exposure induces a decoupling of mitochondrial oxidative phosphorylation in the skeletal muscles of the zebrafish (Cambier et al., 2009).
Interestingly, we observed the ability of Met to afford protection against the deleterious effects of MeHg and/or the MeHg–Cys complex. In fact, Met decreased DFC-RS production and prevented the inhibition of mitochondrial respiration and cell viability induced by exposure to MeHg and/or the MeHg–Cys complex. These data show, for the first time, Met’s effectiveness in both reducing the bioavailability of MeHg in hepatocytes, as well as its modulation of mitochondrial function. In terms of molecular mechanisms, it is reasonable to assume that the protective effects of Met are linked to its structural similarities with the MeHg–Cys complex. This idea is in agreement with the existence of a mitochondrial neutral amino acid transport (Raymond et al., 1977), which is likely responsible for the uptake of MeHg (as MeHg–Cys complex) into mitochondria. Based on our results, it is possible to state that LAT is not only important for the transport of MeHg into the cell, but also for the transport of MeHg within cellular organelles, allowing for the occurrence of mitochondrial toxicity probably due to the direct effects of MeHg in mitochondrial proteins.
In summary, the results obtained in this study demonstrate that Met prevents the toxic effects of MeHg and the MeHg–Cys conjugate on mitochondrial function and cell viability. Furthermore, the results suggest the possible use of this amino acid as a therapeutic agent for treating acute MeHg exposure. Additional studies to determine the efficacy of Met in reducing the gastrointestinal absorption of MeHg as well as its ability to accelerate MeHg excretion in animal models of MeHg exposure are well warranted.
Acknowledgments
The financial support by FINEP Research Grant “Rede Instituto Brasileiro de Neurociência (IBN-Net)” # 01.06.0842-00, FAPERGS/ Pronex, CAPES/SAUX, VITAE Foundation, INCT-CNPq-Excitotoxicity and Neuroprotection and CNPq is gratefully acknowledged. J.B.T.R, M. F. and N.B.V.B are the recipients of CNPq fellowships. Michael Aschner was supported in part by NIEHS ES-07331.
References
- Adachi T. Characteristic effects of L-methionine on tissue distribution of methylmercury in mice. J Health Sci. 2006;52:174–179. [Google Scholar]
- Babu E, Kanai Y, Chairoungdua A, Kim DK, Iribe Y, Tangtrongsup S, Jutabha P, Yuewei L, Ahmed N, Sakamoto S, Anzai N, Nagomori S, Endou H. Identification of a novel system L amino acid transporter structurally distinct from heterodimeric amino acid transporters. J Biol Chem. 2003;278:43838–43845. doi: 10.1074/jbc.M305221200. [DOI] [PubMed] [Google Scholar]
- Ballatori N. Transport of toxic metals by molecular mimicry. Environ Health Perspect. 2002;110:689–694. doi: 10.1289/ehp.02110s5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergdahl IA, Schütz A, Ahlqwist M, Bengtsson C, Lapidus L, Lissner L, Hulten B. Methylmercury and inorganic mercury in serum–correlation to fish consumption and dental amalgam in a cohort of women born in 1922. Environ Res. 1998;77:20–24. doi: 10.1006/enrs.1997.3820. [DOI] [PubMed] [Google Scholar]
- Bode BP. Recent molecular advances in mammalian glutamine transport. J Nutr. 2001;131:2475–2485. doi: 10.1093/jn/131.9.2475S. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Dubinsky JM. Limitations of cyclosporin A inhibition of the permeability transition in CNS mitochondria. J Neurosci. 2000a:8229–8237. doi: 10.1523/JNEUROSCI.20-22-08229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N, Dubinsky JM. Dual responses of CNS mitochondria to elevated calcium. J Neurosci. 2000b:103–113. doi: 10.1523/JNEUROSCI.20-01-00103.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier S, Bénard G, Mesmer-Dudons N, Gonzalez P, Rossignol R, Brèthes D, Bourdineaud JP. At environmental doses, dietary methylmercury inhibits mitochondrial energy metabolism in skeletal muscles of the zebra fish (Danio rerio) Int J Biochem Cell Biol. 2009;41:791–799. doi: 10.1016/j.biocel.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Carvalho MC, Franco JL, Ghizoni H, Kobus K, Nazari EM, Rocha JBT, Nogueira CW, Dafre AL, Müller YM, Farina M. Effects of 2, 3-dimercapto-1-propanesulfonic acid (DMPS) on methylmercury-induced locomotor deficits and cerebellar toxicity in mice. Toxicology. 2007;239:195–203. doi: 10.1016/j.tox.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Chacko BK, Srivastava A, Chang MJ, Johnson MS, Ye YZ, Benavides GA, Zelickson BR, Jhala NC, Kalyanaraman B, Murphy MP, Darley-Usmar V. Mitochondria-targeted ubiquinone ameliorates ethanol-induced hepatic steatosis and prevents protein nitration. Free Radical Biol Med. 2009;47:138–138. [Google Scholar]
- Chang JY, Tsai PF. Prevention of methylmercury-induced mitochondrial depolarization, glutathione depletion and cell death by 15-deoxy-delta-12, 14 prostaglandin J2. Neuro Toxicol. 2008;29:1054–1061. doi: 10.1016/j.neuro.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BH. Effects of methylmercury on neuroepithelial germinal cells in the developing telencephalic vesicles of mice. Acta Neuropathol. 1991;81:356–365. doi: 10.1007/BF00293454. [DOI] [PubMed] [Google Scholar]
- Clarkson TW. Molecular and ionic mimicry of toxic metals. Annu Rev Pharmacol Toxicol. 1993;33:545–571. doi: 10.1146/annurev.pa.33.040193.002553. [DOI] [PubMed] [Google Scholar]
- Clarkson TW, Magos L, Myers GJ. Human exposure to mercury: the three modern dilemmas. J Trace Elem Exp Med. 2003;16:321–343. [Google Scholar]
- Colquhoun A. Lipids, mitochondria and cell death: implications in neuro-oncology. Molec Neurobiol. 2010;42:76–88. doi: 10.1007/s12035-010-8134-4. [DOI] [PubMed] [Google Scholar]
- de Freitas AS, Funck VR, Rotta M, dos S, Bohrer D, Mörschbächer V, Puntel RL, Nogueira CW, Farina M, Aschner M, Rocha JB. Diphenyl diselenide, a simple organoselenium compound, decreases methylmercury-induced cerebral, hepatic and renal oxidative stress and mercury deposition in adult mice. Brain Res Bull. 2009;6:77–84. doi: 10.1016/j.brainresbull.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Dreiem A, Seegal RF. Methylmercury-induced changes in mitochondrial function in striatal synaptosomes are calcium-dependent and ROS independent. Neuro Toxicol. 2007;28:720–726. doi: 10.1016/j.neuro.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte CA. Editora Axcel Books do Brasil. 2003. Semiologia Imunológica Nutricional. [Google Scholar]
- Ellman GL. Tissue sulphydryl groups. Arch Biochem Biophys. 1959;82:70. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Komachi H, Sato I, Himi T, Yuasa T, Murota S. Induction of neuronal nitric oxide synthase by methylmercury in the cerebellum. J Neurosci Res. 1999;55:352–356. doi: 10.1002/(SICI)1097-4547(19990201)55:3<352::AID-JNR10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Farina M, Campos F, Vendrell I, Berenguer J, Barzi M, Pons S, Suñol C. Probucol increases glutathione peroxidase-1 activity and displays long-lasting protection against methylmercury toxicity in cerebellar granule cells. Toxicol Sci. 2009;112:416–426. doi: 10.1093/toxsci/kfp219. [DOI] [PubMed] [Google Scholar]
- Farina M, Soares FAA, Zeni G, Souza DO, Rocha JBT. Additive pro-oxidative effects of methylmercury and ebselen in liver from suckling rat pups. Toxicol Lett. 2004;146:227–235. doi: 10.1016/j.toxlet.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Forkink M, Smeitink JAM, Brock R, Willems PHG, Koopman WMJH. Detection and manipulation of mitochondrial reactive oxygen species in mammalian cells. Biochim Biophys Acta Bioenerg. 2010;1797:1034–1044. doi: 10.1016/j.bbabio.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Fukuhara D, Kanai Y, Chairoungdua A, Ellappan J, Babu E, Fumio B, Kawano T, Akimoto Y, Endou H, Yan K. Protein characterization of Na-independent system L amino acid transporter 3 in Mice. Am J Pathol. 2007;170:888–898. doi: 10.2353/ajpath.2007.060428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser V, Nazari EM, Müller YM, Feksa L, Wannmacher CM, Rocha JBT, de Bem AF, Farina M, Latini A. Effects of inorganic selenium administration in methylmercury-induced neurotoxicity in mouse cerebral cortex. Int J Dev Neurosci. 2010;28:631–637. doi: 10.1016/j.ijdevneu.2010.07.225. [DOI] [PubMed] [Google Scholar]
- Hempel SL, Buettner GRO, Malley YQ, Wessels DA, Flaherty DM. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: comparison with 2′, 7′-dichloro dihydrofluorescein diacetate, 5 (and 6)-carboxy-2′, 7′-dichloro dihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radical Biol Med. 1999;27:146–159. doi: 10.1016/s0891-5849(99)00061-1. [DOI] [PubMed] [Google Scholar]
- Hughes WL. A physiochemical rationale for the biological activity of mercury and its compounds. Ann NY Acad Sci. 1957;65:454–460. doi: 10.1111/j.1749-6632.1956.tb36650.x. [DOI] [PubMed] [Google Scholar]
- Kajiwara Y, Yasutake A, Adachi T, Hirayama K. Methylmercury transport across the placenta via neutral amino acid carrier. Arch Toxicol. 1996;70:310–314. doi: 10.1007/s002040050279. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Endou H. Heterodimeric amino acid transporters: molecular biology and pathological and pharmacological relevance. Curr Drug Metab. 2001;2:339–354. doi: 10.2174/1389200013338324. [DOI] [PubMed] [Google Scholar]
- Kang MS, Jeong JY, Seo JH, Jeon HJ, Jung KM, Chin MR, Moon CK, Bonventre JV, Jung SY, Dae KK. Methylmercury-induced toxicity is mediated by enhanced intracellular calcium through activation of phosphatidylcholine specific phospholipase C. Toxicol Appl Pharmacol. 2006;216:206–215. doi: 10.1016/j.taap.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Kerper LE, Ballatori N, Clarkson TW. Methylmercury cross the blood-brain by an amino acid carrier. Am J Physiol. 1992;262:761–765. doi: 10.1152/ajpregu.1992.262.5.R761. [DOI] [PubMed] [Google Scholar]
- Kim SH, Sharma RP. Cytotoxicity of inorganic mercury in murine T and B lymphoma cell lines: involvement of reactive oxygen species, Ca2+ homeostasis, and cytokine gene expression. Toxicol In Vitro. 2003;17:385–395. doi: 10.1016/s0887-2333(03)00040-7. [DOI] [PubMed] [Google Scholar]
- Limke TL, Otero-Montanez JKL, Atchison WD. Evidence for interactions between intracellular calcium stores during methylmercury-induced intracellular calcium dysregulation in rat cerebellar granule neurons. J Pharmacol Exp Ther. 2003;304:949–958. doi: 10.1124/jpet.102.042457. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival—application to proliferation and cytotoxicity assays. J Immunol Meth. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Norenberg MD, Rao KV. The mitochondrial permeability transition in neurologic disease. Neurochem Int. 2007;50:983–997. doi: 10.1016/j.neuint.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacin M, Estevez R, Bertran J, Zorzano A. Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev. 1998;78:969–1954. doi: 10.1152/physrev.1998.78.4.969. [DOI] [PubMed] [Google Scholar]
- Peterson GR., Jr A simplification of the protein assay method of Lowry et al. Which is more generally applicable. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- Pingree SD, Simmonds PL, Woods JS. Effects of 2, 3-dimercapto-1 propanesulfonic acid (DMPS) on tissue and urine mercury levels following prolonged methylmercury exposure in rats. Toxicol Sci. 2001;61:224–233. doi: 10.1093/toxsci/61.2.224. [DOI] [PubMed] [Google Scholar]
- Poli G, Leonarduzzi G, Biasi F, Chiarpotto E. Oxidative stress and cell signalling. Curr Med Chem. 2004;11:1163–1182. doi: 10.2174/0929867043365323. [DOI] [PubMed] [Google Scholar]
- Puntel RL, Roos DH, Folmer V, Nogueira CW, Galina A, Aschner M, Rocha JBT. Mitochondrial dysfunction induced by different organochalchogens is mediated by thiol oxidation and is not dependent of the classical mitochondrial permeability transition pore opening. Toxicol Sci. 2010;117:133–143. doi: 10.1093/toxsci/kfq185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabenstein DL, Fairhurst MT. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. XI The binding of methylmercury by sulfhydryl-containing amino acids and by glutathione. J Am Chem Soc. 1975;16:2086–2092. doi: 10.1021/ja00841a015. [DOI] [PubMed] [Google Scholar]
- Raymond L, Cybulski R, Ronald RF. Mitochondrial neutral amino acid transport: evidence for a carrier mediated mechanism. Biochemistry. 1977;16:5116–5120. doi: 10.1021/bi00642a026. [DOI] [PubMed] [Google Scholar]
- Roos DH, Puntel RL, Santos MM, Souza DOG, Farina M, Nogueira CW, Aschner M, Burger ME, Barbosa NBV, Rocha JBT. Guanosine and synthetic organoselenium compounds modulate methylmercury-induced oxidative stress in rat brain cortical slices: involvement of oxidative stress and glutamatergic system. Toxicol In Vitro. 2009;23:302–307. doi: 10.1016/j.tiv.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Roos DH, Puntel RL, Logukenski TH, Ineu RP, Bohrer D, Burger ME, Franco JL, Farina M, Aschner M, Barbosa NBV, Rocha JBT. Complex methylmercury-cysteine alters mercury accumulation in different tissues of mice. Bas Clin Pharmacol Toxicol. 2010;107:789–792. doi: 10.1111/j.1742-7843.2010.00577.x. [DOI] [PubMed] [Google Scholar]
- Sakamoto M, Wakabayashi K, Kakita A, Hitoshi T, Adachi T, Nakano A. Widespread neuronal degeneration in rats following oral administration of methylmercury during the postnatal developing phase: a model of fetal-type minamata disease. Brain Res. 1998;16:351–354. doi: 10.1016/s0006-8993(97)01400-5. [DOI] [PubMed] [Google Scholar]
- Sakaue M, Adachi T, Okazaki M, Nakamura H, Mori N, Hara S, Sakabe K. Effects of sodium selenite on methylmercury-induced cell death and on mercury accumulation in rat cerebellar neurons in primary culture. Bull Environ Contam Toxicol. 2006;77:779–784. doi: 10.1007/s00128-006-1131-7. [DOI] [PubMed] [Google Scholar]
- Sarkar D, Kambe F, Hirata A, Iseki A, Ohmori S, Seo H. Expression of E16/ CD98LC/hLAT1 is responsive to 2, 3, 7, 8 tetrachlorodibenzo-p-dioxin. FEBS Lett. 1999;462:430–434. doi: 10.1016/s0014-5793(99)01574-4. [DOI] [PubMed] [Google Scholar]
- Simmons-Willis TA, Koh AS, Clarkson TW, Ballatori N. Transport of a neurotoxicant by molecular mimicry: the methylmercury-L-cysteine complex is a substrate for human L-type large neutral amino acid transporter (LAT) 1 and LAT2. Biochem J. 2002;367:239–246. doi: 10.1042/BJ20020841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrovskaya IG, Kristal BS. The powerhouse takes control of the cell: Is the mitochondrial permeability transition a viable therapeutic target against neuronal dysfunction and death? Free Radical Biol Med. 2010;38:687–697. doi: 10.1016/j.freeradbiomed.2004.11.032. [DOI] [PubMed] [Google Scholar]
- Stringari J, Nunes AK, Franco JL, Bohrer D, Garcia SC, Dafre AL, Milatovic D, Souza DO, Rocha JBT, Aschner M, Farina M. Prenatal methylmercury exposure hampers glutathione antioxidant system ontogenesis and causes long-lasting oxidative stress in the mouse brain. Toxicol Appl Pharmacol. 2008;227:147–154. doi: 10.1016/j.taap.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner C, Vargas PV, Roos DH, Morel FA, Farina M, Nogueira CW, Aschner M, Rocha JBT. Comparative study of quercetin and its two glycoside derivatives quercitrin and rutin against methylmercury (MeHg)-induced ROS production in rat brain slices. Arch Toxicol. 2010;84:89–97. doi: 10.1007/s00204-009-0482-3. [DOI] [PubMed] [Google Scholar]
- Yin Z, Jiang H, Syversen T, Rocha JBT, Farina M, Aschner M. The methylmercury-L-cysteine conjugate is a substrate for the L-type large neutral amino acid transporter. J Neurochem. 2008;107:1083–1090. doi: 10.1111/j.1471-4159.2008.05683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]