

Summary
Pdx1 and Oc1 are co-expressed in multipotent pancreatic progenitors and regulate the pro-endocrine gene Neurog3. Their expression diverges in later organogenesis, with Oc1 absent from hormone+ cells and Pdx1 maintained in mature β cells. In a classical genetic test for cooperative functional interactions, we derived mice with combined Pdx1 and Oc1 heterozygosity. Endocrine development in double-heterozygous pancreata was normal at embryonic day (e)13.5, but defects in specification and differentiation were apparent at e15.5, the height of the second wave of differentiation. Pancreata from double heterozygotes showed alterations in the expression of genes crucial for β-cell development and function, decreased numbers and altered allocation of Neurog3-expressing endocrine progenitors, and defective endocrine differentiation. Defects in islet gene expression and β-cell function persisted in double heterozygous neonates. These results suggest that Oc1 and Pdx1 cooperate prior to their divergence, in pancreatic progenitors, to allow for proper differentiation and functional maturation of β cells.
Introduction
Knowledge gained from developmental biology has been instrumental in deriving glucose-responsive, insulin-secreting pancreatic β cells from embryonic stem (ES) cells or induced pluripotent stem (iPS) cells to generate a cell-based therapy for the treatment of diabetes (Bruin et al., 2015; Pagliuca et al., 2014; Russ et al., 2015). Of particular interest are signaling molecules and transcriptional regulators that direct the β-cell fate or generate fully functional β cells. Many elegant single gene inactivation studies have revealed critical roles for specific transcription factors in different stages of pancreas development and endocrine differentiation. However, few studies have analyzed the functional consequences of combinatorial genetic manipulations of structurally un-related pancreas transcription factors during development (Burlison et al., 2008; Courtney et al., 2013; Shih et al., 2015), Here we report on the genetic and functional cooperativity of the Pdx1 and Oc1 transcription factors and the requirement for a combined threshold of activity in setting up a genetic program for endocrine differentiation and β-cell function.
Pancreatic and duodenal homeobox 1 (Pdx1) is required for pancreas development, endocrine differentiation, and mature β-cell function in mouse and human (Gao et al., 2014; Jonsson et al., 1994; Lammert et al., 2001; Offield et al., 1996; Stoffers et al., 1997b; Stoffers et al., 1997c). Pdx1 is initially expressed in the mouse posterior foregut endoderm at embryonic day (e)8.5, expanding into the antral stomach, rostral duodenum, and common bile duct by e11.5, and maintained at high levels in mature β cells (Guz et al., 1995; Jonsson et al., 1994; Offield et al., 1996; Wu et al., 1997). In addition, the burst of β-cell proliferation that occurs just prior to birth requires Pdx1 (Gannon et al., 2008). Beginning at late gestation and continuing into the early postnatal period, β cells undergo gene expression changes associated with functional maturation, including the acquisition of tightly controlled glucose-stimulated insulin secretion (Artner et al., 2010; Nishimura et al., 2006; Stolovich-Rain et al., 2015). In adult mice, Pdx1 regulates β-cell function and survival (Brissova et al., 2002; Dutta et al., 1998b; Gauthier et al., 2009; Kulkarni et al., 2004; Sachdeva et al., 2009; Waeber et al., 1996). The crucial role for Pdx1 in endocrine-lineage development and postnatal β-cell function is underscored by the identification of diabetes-causing PDX1 mutations in humans (Hani et al., 1999)(Macfarlane et al., 2000b)(Stoffers et al., 1997a).
One-cut 1 (Oc1; also known as hepatic nuclear factor 6; Hnf6) is expressed more broadly in the developing endoderm and plays roles in the developing liver and pancreas (Jacquemin et al., 2000; Jacquemin et al., 2003; Samadani et al., 1996; Zhang et al., 2009). Expression of the endocrine-progenitor transcription factor, neurogenin 3 (Neurog3) is nearly undetectable in Oc1−/− embryos, which are diabetic at birth, with a near complete loss of all pancreatic endocrine cell lineages (Jacquemin et al., 2000). Oc1 binds to an upstream enhancer from the Neurog3 gene (Jacquemin et al., 2000), suggesting that Neurog3 is a direct transcriptional target of Oc1. Unlike Pdx1, Oc1 is not expressed in differentiated, hormone-positive endocrine cells but its expression persists in ducts and acinar cells into adulthood (Pekala et al., 2014; Prevot et al., 2012; Rausa et al., 1997; Zhang et al., 2009). Over-expression of Oc1 in the developing pancreas results in an increase in Neurog3-positive cells (Wilding Crawford et al., 2008). However, its down-regulation in the endocrine lineage is essential: maintained Oc1 expression prevents β-cell maturation, most likely by directly inhibiting expression of the β-cell transcription factor, MafA (Yamamoto et al., 2013), and results in diabetes (Gannon et al., 2000; Tweedie et al., 2006).
Pdx1 and Oc1 are co-expressed in multipotent pancreatic progenitors (MPCs) in the early pancreatic bud and later in the undifferentiated, bipotential duct/endocrine cell pool located within the “trunk” domain of the pancreatic epithelium. Pdx1 and Oc1 each activate Neurog3 expression and our in vitro evidence suggests that a physical interaction between these two factors involving the Pdx1 C-terminus promotes endocrine specification. Pdx1 occupies an evolutionarily conserved Neurog3 enhancer at e13.5 and, in reporter assays, Pdx1 transactivation via this enhancer was significantly enhanced by Oc1. Mice homozygous for a Pdx1 allele with a premature C-terminal truncation (Pdx1ΔC/ΔC) display a global reduction in endocrine lineages and decreased numbers of Neurog3+ progenitors at e13.5 (Oliver-Krasinski et al., 2009).
We hypothesized that the Pdx1-Oc1 interaction is critical at multi- or bipotent stages to promote the specification of pancreatic endocrine progenitors by regulating Neurog3 and other developmentally important genes. To assess the significance of the Pdx1-Oc1 interaction in vivo, we generated animals globally heterozygous for either or both genes. To date, no developmental phenotype for either single heterozygous animal has been reported. At e13.5, double heterozygotes showed normal numbers of glucagon+ and Neurog3+ cells, suggesting that the first wave of endocrine differentiation is unaffected. By e15.5 at the height of the secondary wave of differentiation, the numbers of Neurog3+ endocrine progenitors and insulin+ and glucagon+ cells were reduced. Whole transcriptome analysis at e15.5 revealed a dramatic and unique impact of Pdx1-Oc1 heterozygosity on the endocrine compartment. Later stages of endocrine differentiation and function, well after the normal down-regulation of Oc1 in the endocrine lineage, were also defective in double heterozygotes. Thus, Pdx1 and Oc1 cooperate to promote endocrine specification and subsequent functional maturation of β-cells, most likely by establishing a state of competency in progenitors earlier in development that allows for later steps in endocrine differentiation to be realized.
Results
Combined Pdx1 and Oc1 heterozygosity has a broad effect on the transcriptional network regulating endocrine development
To determine the effect of combined global heterozygosity for Pdx1 and Oc1 on pancreas development, we analyzed the transcriptome of pancreata from control (WT), Pdx1lacZ/+ (which carry one null allele of Pdx1 containing a lacZ cassette (Offield et al., 1996)), Oc1+/−, and Pdx1lacZ/+;Oc1+/− (hereafter: double heterozygous, DH) animals using RNA-Sequencing (RNASeq; see Supplemental Methods). We performed our analysis at e15.5 since all pancreatic lineages are present at this stage and the greatest number of Neurog3+ cells can be detected (Gradwohl et al., 2000). A total of 2331 genes were differentially expressed in at least one of the three experimental genotypes (Fig1A, Table S1). Expression of 102 genes was altered in Pdx1lacZ/+ pancreata (Fig. 1B). Oc1+/− pancreata showed the greatest number of gene expression changes, with more than 2000 genes affected. In contrast to Pdx1 single-heterozygotes (SH), β-cell genes such as Ins1, Ins2, and Iapp were increased in Oc1+/− SH, consistent with a role for Oc1 in suppressing β-cell differentiation (Tweedie et al., 2006).
Figure 1. Combined heterozygous reduction in Pdx1 and Oc1 gene dosage has a broad impact on the transcriptional network of endocrine pancreas progenitors.
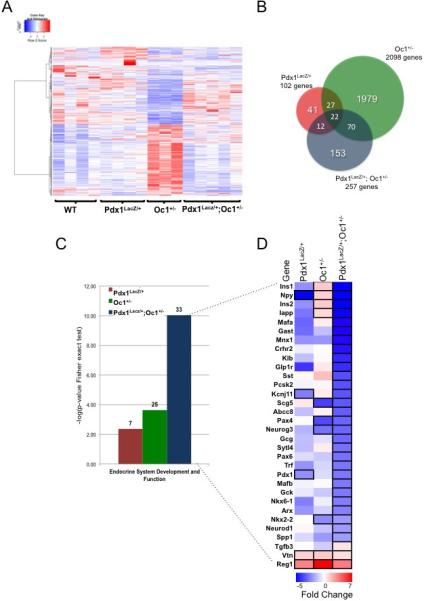
(A) Hierarchical clustering of 2331 differentially expressed genes in individual pancreata at e15.5 from Pdx1LacZ/+ (n=4), Oc1+/− (n=3), and Pdx1LacZ/+;OC1+/− mice (n=5) compared to WT (n=4); (B) Venn diagram depicting the number of altered genes in the Pdx1LacZ/+, Oc1+/− and Pdx1LacZ/+;Oc1+/−; (C) Endocrine system development and function gene ontology category in each genotype, according to negative log of p-value from Fisher exact test. The numbers above each column represent the number of genes enriched in each category; (D) Heat map of endocrine development and function genes. Up- or down-regulated genes with false discovery rate less than 0.1 and fold change higher than 0.5 versus WT are highlighted with bold black borders. (See also Tables S1 and S2)
The transcriptome of Pdx1-Oc1 double heterozygotes showed a pattern of gene dysregulation distinct from either single heterozygote transcriptome. Of the 257 genes affected in DH, 153 genes were specifically altered in DH compared to WT (Fig. 1B), including key transcriptional regulators MafA, MafB, NeuroD1, and Nkx6.1, providing strong support for functional cooperation between these two transcription factors to regulate a distinctive genetic program. Furthermore, the shift of gene expression patterns of Oc1+/− SH versus DH or Pdx1lacZ/+ SH versus DH (Fig. 1A) suggests the Pdx1-Oc1 interaction acts cooperatively or antagonistically at the level of broad categories of genes.
To assess systematic changes in expression of genes involved in canonical signaling pathways, disease and biological function categories, and molecular networks of genes altered in the three experimental genotypes, we performed comparison-enrichment analysis using Ingenuity Pathway Analysis (IPA). The top gene ontology categories ascribed to genes altered in Oc1+/− or in Pdx1lacZ/+ SH pancreata were “cancer”, “embryonic development” and “cellular development” or “gene expression”, respectively (Table S2). The genes altered in DH pancreata clustered primarily in the “endocrine system development and function”, “carbohydrate metabolism”, “endocrine system disorders” categories (Fig. 1C, Table S2). The “endocrine system development and function” category was enriched for genes associated with “quantity of endocrine cells” and “quantity of beta and alpha islet cells” (Table 1), indicating a possible impact on endocrine progenitors and endocrine differentiation. Genes in these categories clustered less strongly in Pdx1lacZ/+ or Oc1+/− SH because the vast majority of genes were specifically altered in the DH dataset and not in SH pancreata (Fig 1C,D and Table 1).
Table 1.
Clustering of genes in each Endocrine System Development and Function GO category in each experimental genotype.
| Endocrine System Development and Function Annotation | p-value | Molecules | |
|---|---|---|---|
| Pdx1Lacz/+ | migration of beta islet cells | 4.38E-03 | Vtn |
| transmembrane potential of beta islet cells | 4.38E-03 | Kcnj11 | |
| development of enteroendocrine cells | 8.74E-03 | Atoh1 | |
| replication of beta islet cells | 1.31E-02 | Reg1a | |
| formation of parathyroid gland | 1.74E-02 | Aldh1a7 | |
| glucuronidation of beta-estradiol | 1.74E-02 | Nr1i2 | |
| differentiation of beta islet cells | 2.17E-02 | Slc2a2 | |
| Oc1+/− | glucose tolerance | 2.34E-04 | ABCB4, ATF4, C19orf10, CBL, CCKBR, CYP2J2, FFAR3, Fxyd2, GRB10, HNF4A, HTR2C, HTR3A, HYOU1, IAPP, INS, IRS2, KLF15, Mir-802, NR0B2, NR1H4, PCBD1, RBP1, SERP1, SREBF1, WNT10B |
| Pdx1Lacz/+; Oc1+/− | quantity of endocrine cells | 9.28E-11 | Arx,Gast,Ins,Ins1,Kcnj11,Mafb,Mnx1,Neurog3,Nkx6-1,Pax4,Pax6,Pcsk2 |
| quantity of beta islet cells | 2.40E-10 | Arx,Ins,Ins1,Kcnj11,Mnx1,Nkx6-1,Pax4,Pcsk2 | |
| quantity of alpha islet cells | 2.79E-06 | Arx,Ins,Ins1,Pcsk2 | |
| entry into cell cycle progression of endocrine cell lines | 2.53E-05 | Ins1,SST,TF | |
| concentration of corticosterone | 2.77E-05 | Crhr2,Gck,Glp1r,Ins,Klb,Npy,Scg5 | |
| glucose tolerance | 1.43E-04 | Abcc8,Glp1r,Iapp,Ins,Mafa,Pdx1,Sytl4,Tgfb3 | |
| replication of beta islet cells | 3.55E-04 | Gast,Reg1a | |
| differentiation of endocrine cells | 1.10E-03 | Nkx6-1,Pax4,Pdx1 | |
| area of islets of Langerhans | 1.17E-03 | Ins,Ins1 | |
| differentiation of beta islet cells | 1.17E-03 | Nkx6-1,Pdx1 | |
| regeneration of islet cells | 1.17E-03 | Nkx6-1,Pcsk2 | |
| formation of islet cells | 2.42E-03 | Pax6,Pdx1 | |
| quantity of delta islet cells | 2.42E-03 | Arx,Pcsk2 | |
| synthesis of hormone | 2.88E-03 | Abcc4,Crhr2,Cyp11a1,Ins,Pax6,Vip | |
| proliferation of beta islet cells | 3.98E-03 | Iapp,Ins,Nkx6-1,Pax4 | |
| quantity of enteroendocrine cells | 6.15E-03 | Arx,Pax4 | |
| steroidogenesis of hormone | 7.54E-03 | Abcc4,Crhr2,Cyp11a1,Ins,Vip | |
| size of beta islet cells | 8.60E-03 | Pcsk2,Pdx1 | |
| synthesis of corticosterone | 8.60E-03 | Apoa1,Cyp11a1 | |
| activation of parathyroid gland | 1.09E-02 | Casr | |
| arrest in organogenesis of pancreas | 1.09E-02 | Pdx1 | |
| binding of hypothalamus | 1.09E-02 | Pyy |
The largest effects of DH on transcription were observed in pancreatic endocrine-hormone expression. Insulin was most affected in the DH pancreata, being significantly reduced compared to both WT and SH animals (Fig. 1D). mRNA levels of other islet hormones (Gcg, Iapp and Sst) were also decreased in the DH animals by 30-50% compared to WT or SH pancreata, suggesting an overall synergistic action of Pdx1 and Oc1 on the entire endocrine compartment. Within the gene set significantly altered in DH pancreata, we also observed an impressive array of transcription factors crucial for development of the endocrine pancreas. The decreased expression of MafA, MafB, Pax4, Pax6, Mnx1, Nkx2.2, and Nkx6.1 suggests that simultaneous reduction in expression of both Pdx1 and Oc1 acts at multiple levels of endocrine differentiation and not just at the level of activation of Neurog3. Expression of Neurog3 and Pax4 was decreased in Oc1 SH and DH, while neuropeptide Y (NPY) was decreased similarly in the Pdx1 SH and DH compared with WT, indicating that some genes show sensitivity to reductions in either Pdx1 or Oc1 gene dosage alone.
In DH pancreata, we also observed large decreases in expression of genes encoding proteins involved in multiple aspects of glucose-stimulated insulin secretion– glucose metabolism (Gck, G6pc2), KATP channel components (Abcc8 and Kcnj11), vesicle trafficking (Scg5 and Sytl4), G protein coupled receptors (Glp1r and Ffar1), and transmembrane proteins (Klb) (Fig. 1D). These findings are likely related to the decreased number of differentiated endocrine cells in DH at this developmental age (see below). Although the majority of changes were associated with down-regulation of gene expression, some genes were up-regulated in all three mutant genotypes, including vitronectin (Vtn), which is increased in delaminating endocrine-committed cells (Cirulli et al., 2000) and inhibits insulin production and secretion (Kaido et al., 2006), and regenerating islet-derived 1 (Reg1), which is associated with islet regeneration (Kobayashi et al., 2000; Terazono et al., 1988).
Embryonic endocrine progenitor specification and endocrine cell maturation during the secondary transition are impaired by double Pdx1-Oc1 heterozygosity
The e15.5 RNASeq data suggested that endocrine lineage commitment and differentiation were impaired in DH pancreata. Specifically, expression of Neurog3, the critical endocrine specification transcription factor, and Pax6, a pan-endocrine-lineage transcription factor downstream of Neurog3, were down-regulated. Pancreas development is asynchronous and multiple developmental stages can be observed at a single time point (Guney and Gannon, 2009). To obtain cellular resolution of gene expression changes, we quantified the number of cells expressing either Neurog3 or Pax6 at e15.5. DH pancreata had approximately 50% fewer Neurog3+ endocrine progenitors than WT (Fig. 2A, C). These data support our hypothesis that combined Pdx1 and Oc1 deficiency leads to reduced endocrine lineage specification. Despite a reduction in Neurog3 mRNA expression in Oc1 SH (Fig. 1D), the number of Neurog3+ cells in Oc1 SH was not statistically significantly affected compared to WT (Fig. 2C), suggesting a decrease in Neurog3 expression per cell in Oc1 SH. Although the reduction in Pax6+ endocrine precursor cells did not achieve statistical significance (Fig. 2B, D), the ratio of Neurog3+ cells to Pax6+ cells at e15.5, was similar among genotypes (Fig. S1), supporting an overall decrease in the number of specified and committed endocrine cells. In contrast, at e13.5 Neurog3+ progenitor numbers were not affected in SH or DH pancreata (Fig. S2A). Pax6+ and glucagon+ cells were also normal at this stage (Fig. S2B, C; too few insulin+ cells were detected to quantify). Thus, the impact of decreased Pdx1-Oc1 dosage appears to be restricted to the second wave of endocrine differentiation that gives rise to cells within the mature islets of Langerhans (Guney and Gannon, 2009).
Figure 2. Reduced number and altered location of Neurog3-expressing endocrine progenitors in DH mice at e15.5.
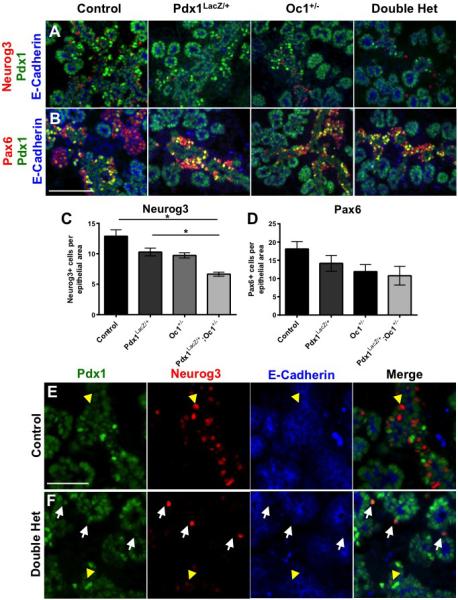
WT, Pdx1LacZ/+, Oc1+/− and DH pancreata were immunolabeled for (A,E,F) Neurog3 or (B) Pax6 (in red), Pdx1 (green), and E-cadherin (blue). (C,D) Quantification for Neurog3 and Pax6. White arrows: delaminated Neurog3+ progenitors; yellow arrowheads: Neurog3+ progenitors within developing trunk. A and B are at 20X magnification. Scale bar represents 100 μm. E and F are at 40X magnification. Scale bar represents 100 μm. p-value for all marked comparisons was <0.05 by One-Way Anova with Tukey correction. *p=0.0019. (See also Figure S1)
During normal pancreas development, Neurog3 expression initiates within a subset of bipotential trunk epithelial cells (Beucher et al., 2012) and becomes elevated in cells destined to undergo commitment to the endocrine lineage. It is thought that the Neurog3hi cells give rise to hormone-expressing cells after delaminating from the ductal epithelium (Villasenor et al., 2008). Closer examination of DH pancreata at e15.5 revealed that, in contrast to WT pancreata, fewer Neurog3+ cells could be found within the pancreatic trunk epithelium. A greater proportion of the Neurog3+ cells present were instead located adjacent to the epithelium in DH compared with the other three genotypes; these extra-truncal Neurog3+ cells seemed to express high levels of Neurog3 (Fig. 2E, F white arrows compared to yellow arrows).
To characterize the impact of Pdx1-Oc1 cooperativity on the endocrine compartment during the definitive second wave of endocrine differentiation, we examined markers of differentiated endocrine lineages at e15.5 including the hormones insulin and glucagon. DH pancreata showed significant decreases in the numbers of both insulin+ and glucagon+ cells (Fig. 3A, B). Only DH pancreata showed a reduction in glucagon+ cell number (Fig. 3B), whereas similar decreases in the number of insulin+ cells were observed in Pdx1 SH and DH (Fig. 3A). While previous studies have shown decreased β-cell function and impaired glucose homeostasis in Pdx1 SH post-weaning, here we show that defects in the β-cell lineage exist in Pdx1 SH during embryonic development. Defective β-cell development could contribute to the susceptibility to mature onset diabetes of the young (MODY) and adult-onset diabetes observed in mice and humans with heterozygous Pdx1 mutations (Dutta et al., 1998b; Hani et al., 1999; Macfarlane et al., 2000a; Sachdeva et al., 2009; Stoffers et al., 1997c; Weng et al., 2001). Taken together, these data reveal a reduction in the numbers of emerging hormone-expressing cells at e15.5 in mice with combined reduction in Pdx1 and Oc1 gene dosage.
Figure 3. Defective differentiation of DH hormone-expressing cells at e15.5.
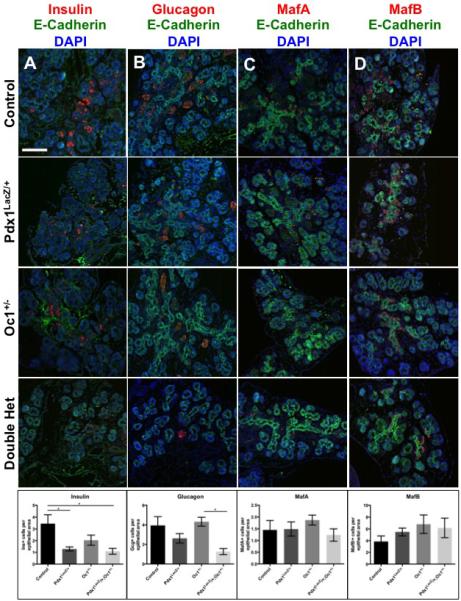
WT, Pdx1LacZ/+, Oc1+/− and DH pancreata were immunolabeled for insulin (A), glucagon (B), MafA (C), or MafB (D) in red, E-Cadherin (green), and DAPI (blue). Images are at 20X magnification. Scale bar represents 100 μm. p-value for all marked comparisons was <0.05 by One-Way Anova with Tukey correction. *p<0.05. (See also Figure S3)
In light of the transcriptional impact of Pdx1 and Oc1 reduction on markers of α and β cell maturation (Fig. 1), we assessed the number of cells expressing the “large Maf” transcription factors, MafA and MafB. MafB is activated soon after endocrine progenitor delamination in both glucagon+ and insulin+ cells, and is down-regulated postnatally in insulin+ cells in mice. MafA expression initiates later, specifically in insulin+ cells and is maintained in these cells. Despite measurable decreases in MafA and MafB transcripts (Fig. 1D), we observed no differences in the total number of MafA+ or MafB+ cells at e15.5 (Fig. 3C, D). Normal cell numbers in spite of reduced transcripts suggests that MafA and MafB expression per cell is decreased. When we compared the numbers of MafA/B+ cells to the number of insulin/glucagon-expressing cells, we found a significant increase in the number of Maf+/hormone- cells in DH pancreata (Fig. S3). These Maf+ cells may derive from Neurog3+ cells generated prior to e13.5 that fail to gain mature hormone expression in a timely fashion. Taken together these data further establish that the simultaneous decrease in Pdx1-Oc1 dosage preferentially affects the endocrine progenitor program during the second wave of endocrine differentiation.
Impaired terminal differentiation and function of hormone+ cells with double Pdx1-Oc1 heterozygosity
To determine whether the developmental defects in DH persist after birth, we examined early postnatal stages physiologically and morphologically. Immediately at birth, prior to feeding, there were no significant differences in body weight or blood glucose levels (Fig. S2). However, with the start of feeding at postnatal day 1 (P1) DH animals failed to increase in body weight compared to the other genotypes (Fig. 4A). At P1 DH pups exhibited elevated ad lib blood glucose compared to WT and Oc1+/− animals (Fig. 4B). We therefore analyzed whether DH neonates had reduced α- or β-cell mass, consistent with our observations of decreased insulin+ and glucagon+ cells at e15.5. There was a strong trend toward reduced α- and β-cell mass at P1 in Pdx1 SH and DH, but this was not statistically significant (Fig. 4G, H). Islet morphology (Fig. 4C-F) and α:β cell ratio were unchanged at P1 (Fig. 4I). However, there was a dramatic and significant decrease in total pancreatic insulin protein content in DH when compared with WT or either SH (Fig. 4J). These data suggest reduced insulin production per β cell and indicate a functional β-cell defect in DH mice that persists after birth in a cell population in which Pdx1 and Oc1 no longer co-localize.
Figure 4. Defects in glucose homeostasis and islet gene expression in DH mice at P1.
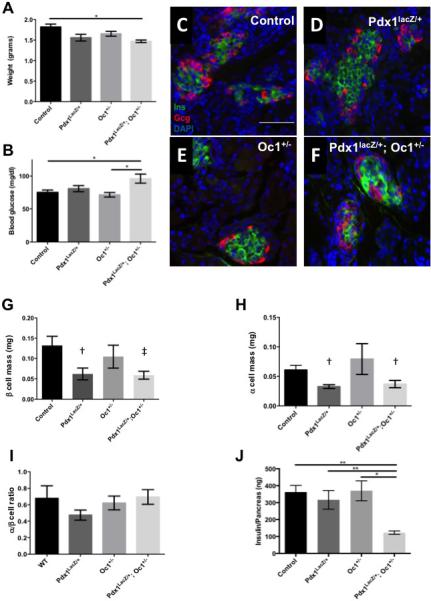
Body weight (A) and ad lib feeding blood glucose measurements (B). (C-F) Insulin (green) and glucagon (red) immunolabeling of pancreatic sections from WT (C), SH (D,E) and double heterozygotes (F). (G) β cell mass, (H) α cell mass, and (I) α/β cell ratio. (J) Total pancreatic insulin content. †p>0.10, ‡p=0.069, *p <0.05, **p<0.01 by One-Way Anova with Tukey correction.
A postnatal functional defect in DH islets is also supported by islet gene expression data at P1 (Fig. 5). TLDA analysis revealed that P1 DH islets had undetectable levels of insulin and glucagon mRNA; expression of these hormones was unaffected in islets from SH (Fig. 5A). Quantitative RT-PCR using primers spanning the insulin and glucagon transcripts confirmed the decrease in insulin and glucagon in DH islets at P1 (Fig. S5, Table S4). Sst mRNA expression was not changed in any genotype at P1. Expression of several key islet transcription factor genes that regulate either insulin or glucagon expression was substantially increased specifically in DH, suggesting attempts at compensation. However, expression of Pax4, a critical β-cell differentiation factor (Sosa-Pineda et al., 1997), was undetectable (Fig. 5B), consistent with the reduction in insulin+ cells and the decrease in Pax4 expression in DH at e15.5 detected by RNASeq. Similarly, expression of genes involved in glucose sensing and hormone-granule exocytosis was also increased (Fig. 5C). The BMP inhibitor Sostdc1 was increased in DH P1 islets (Fig. 5C), possibly contributing to the impaired islet function (Henley et al., 2012), as autocrine BMP activity was shown to be important for glucose-stimulated insulin secretion (Goulley et al., 2007).
Figure 5. Combined Pdx1 and Oc1 heterozygosity leads to dramatic alterations in neonatal islet gene expression.
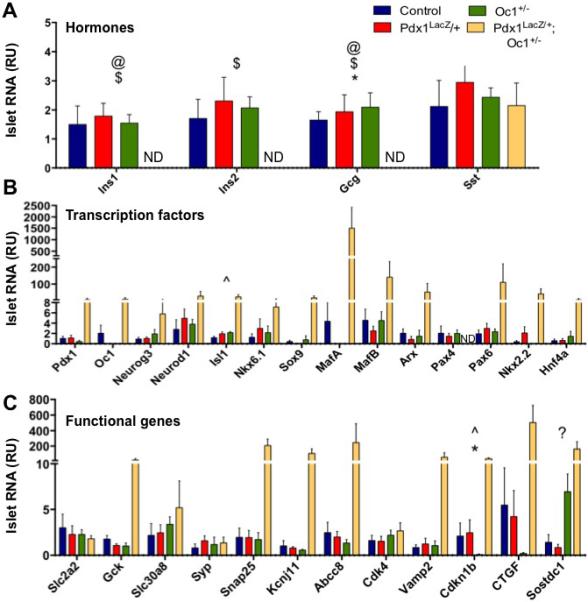
WT, Pdx1LacZ/+, Oc1+/− and Pdx1LacZ/+;Oc1+/− islets were analyzed for gene expression of hormones (A), endocrine-associated transcription factors (B), and secretory functional genes (C). p-value for marked comparisons was <0.05 by Kruskal-Wallace Test followed by two-tailed Student's T-Test. *: WT v. Pdx1LacZ/+;Oc1+/−; #: WT v. Pdx1LacZ/+; ^: WT v. Oc1+/−; @: Pdx1LacZ/+ v. Pdx1LacZ/+;Oc1+/−; $: Oc1+/− v. Pdx1LacZ/+;Oc1+/−; ?: Pdx1LacZ/+ v. Oc1+/−. ND = not detected. (See also Figure S5)
We also observed increased expression of Ctgf, a β-cell-derived growth factor that is critical for embryonic β-cell proliferation and capable of inducing proliferation of embryonic α- and β-cells (Crawford et al., 2009; Guney et al., 2011; Riley et al. 2015) (Fig. 5C). The dramatically increased Ctgf expression in DH suggested increased α- and/or β-cell proliferation as a mechanism for restoring α- and β-cell mass by birth. Indeed, we detected a significant increase in α- and β-cell proliferation specifically in DH pancreata at e18.5 (Fig. 6).
Figure 6. Increased α- and β-cell proliferation at e18.5 in DH pancreata.
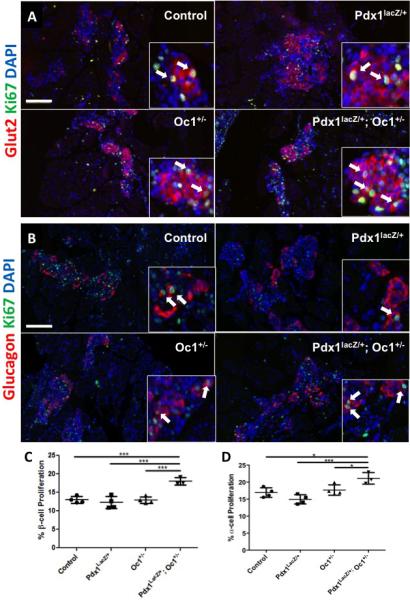
Representative images of proliferating β cells (A; red: Glut2, green: Ki67) and α-cells (B; red: glucagon, green: Ki67) at e18.5. α-cell Proliferation at e18.5. Arrows point to proliferating hormone+ cells. (C) quantification of β-cell proliferation, (D) quantification of α-cell proliferation. Scale bar = 100μm; *: p<0.05; ***: p<0.001.
Restoration of normal glucose homeostasis but persistent gene expression defects in DH animals at weaning
We next examined the DH phenotype at weaning when β cells become functionally fully mature (Nishimura et al., 2006; Stolovich-Rain et al., 2015). At this time point, DH animals had normal body weight (not shown) and normal fasting blood glucose (Fig. S6A). Ad lib blood glucose levels were elevated in Pdx1 SH (Fig. S6B), consistent with the adult phenotype in the literature (Ahlgren et al., 1998; Brissova et al., 2002; Dutta et al., 1998b; Johnson et al., 2003). DH animals showed no statistically significant difference in ad lib feeding blood glucose levels (Fig. S6B). When challenged with glucose during an intraperitoneal glucose tolerance test at three weeks of age, DH animals were not glucose intolerant (Fig. S6C).
Since the physiological phenotype of DH animals appeared to resolve by weaning, we examined islet gene expression at four weeks of age. Although expression of insulin could be detected in DH islets at this age, it was still significantly reduced compared with all other genotypes (Fig. 7A). At this time point, expression of Sst was also reduced in DH islets. Expression levels of islet transcription factors that were elevated in DH at P1 were normalized at four weeks of age, with the exception of MafA, which remained significantly decreased (Fig. 7B). Expression of genes involved in glucose sensing and insulin secretion was also restored to normal levels in DH islets at four weeks (Fig. 7C). Ctgf expression remained slightly elevated while Sostdc1 levels were normal (Fig. 7C). Expression of the remaining genes that were abnormal at P1 was normalized by weaning.
Figure 7. Early reductions in Pdx1 and Oc1 lead to persistent alterations in islet gene expression at weaning.
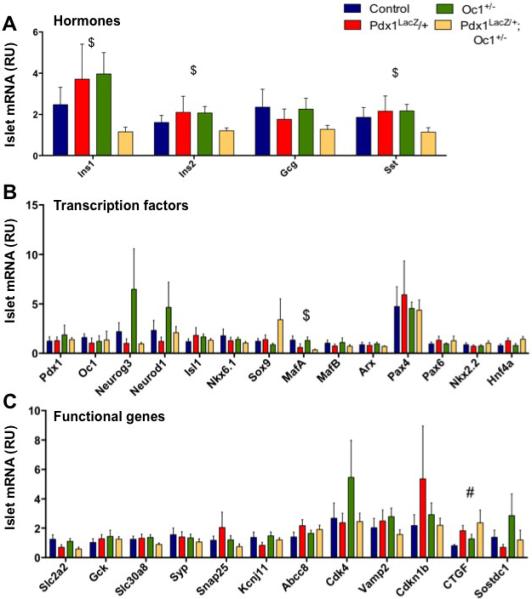
WT, Pdx1LacZ/+, Oc1+/− and DH islets were analyzed for gene expression of hormones (A), endocrine-associated transcription factors (B), and secretory functional genes (C) at P28. p-value for marked comparisons were <0.05 by Kruskal-Wallace Test followed by two-tailed Student's T-Test. #: WT v. Pdx1LacZ/+; $: Oc1+/− v. Pdx1LacZ/+;Oc1+/−. (See also Figure S6)
Oc1 is also expressed in liver and previous studies show that glycogenolysis and gluconeogenesis are impaired in the absence of Oc1 in the liver (Jacquemin et al., 2000; Jacquemin et al., 1999). To determine whether a liver phenotype of Oc1 SH could be contributing to the normalization of glucose tolerance in DH mice by weaning, we examined glycogen deposition. Indeed, Oc1 SH mice have increased glycogen deposition in the liver post-weaning compared to WT. Pdx1 SH have reduced hepatic glycogen compared with WT, likely due to decreased insulin output associated with the known impairment in β-cell function in these animals. DH mice have glycogen deposits that more closely resemble WT animals (Fig. S7). Thus, the effects of the two genotypes on the liver appear to counteract one another, and it is possible that the “normalization” of DH glucose tolerance at weaning reflects compensation by the postnatal liver Oc1 heterozygous phenotype rather than resolution of the endocrine pancreas developmental defect. The persistence of hormone gene expression defects in 4-week old DH islets supports this concept.
Discussion
Pdx1 and Oc1 are co-expressed in MPCs very early in pancreas development and our previous work suggested that these two factors cooperate to regulate transcription of the endocrine progenitor transcription factor Neurog3. We hypothesized that a threshold of cooperative Pdx1 and Oc1 activity is required for realization of the endocrine program from MPCs. Transcriptome analyses of DH pancreas at e15.5 supported our hypothesis, revealing a highly compromised islet differentiation program with reduced expression of several key endocrine lineage transcription factors, including Neurog3 and Pax6, as well as multiple genes involved in mature β-cell function. Complementary morphological and physiological studies point to distinct effects of combined Pdx1 Oc1 reduction in endocrine progenitor specification and maturation with long-term effects on gene expression and function.
Our findings highlight a specific role for combined Pdx1 and Oc1 activity in establishing the endocrine progenitor program during the second wave of endocrine differentiation. Decreased dosage of Pdx1-Oc1 impacted Neurog3+ cell numbers and subsequent endocrine differentiation at e15.5 but not at e13.5. Since the duration of the Neurog3+ state is short (estimated to be ~12 hours) (Bankaitis et al., 2015), the reduced numbers of Neurog3+ cells at e15.5 suggest that endocrine progenitors are more sensitive to Pdx1-Oc1 dosage after e13.5. The normal number of glucagon+ and Pax6+ cells at e13.5, which derive from even earlier Neurog3+ progenitors (Johansson et al., 2007), supports this timeline of events. The relative increase in the proportion of delaminated cells expressing high Neurog3 levels in DH pancreata suggests that Pdx1 and Oc1 synergize to regulate the timing of transition from a Neurog3lo to Neurog3hi cell. The significance of duration of the Neurog3lo state is currently unclear, but may affect subsequent steps in endocrine maturation.
The morphologic and transcriptomic analyses at e15.5 further suggest that Pdx1-Oc1 reduction leads to defective maturation of α- and β-cell lineages. This could occur either directly by decreased dosage of Pdx1 and Oc1, and/or indirectly due to decreased expression of maturational and other endocrine cell markers per cell. The persistence of gene expression defects and elevated blood glucose at P1 indicate that the endocrine maturation program is not being completed successfully by birth in DH embryos.
Our results suggest that the combined activity of two structurally unrelated transcription factors within a progenitor-cell population affects subsequent differentiated cell populations (α and β cells) in which the two factors are not co-expressed. Expression of Pdx1 and Oc1 initially overlaps in MPCs and in bipotential duct/endocrine progenitors in the pancreatic epithelial “trunk”, but they diverge with Oc1 silenced and Pdx1 maintained almost exclusively in the pro-β cell lineage, as endocrine cells become specified. It is possible that the cooperative activity of Pdx1 and Oc1 in progenitors primes the cells for subsequent steps of the differentiation program. This notion that transcription factors can have temporally separated effects on cell behavior is not novel, as deletion of Hnf4α in the embryonic liver affects gene expression in differentiated hepatocytes long after its expression is down-regulated (Kyrmizi et al., 2006). Similarly, the FoxD3 transcription factor, which is expressed in adult, quiescent β cells is required for β-cell proliferation during pregnancy despite being absent from maternal β cells during pregnancy (Plank et al., 2011). Our preferred model is that Pdx1 and Oc1 cooperate in multi- or bi-potent pancreatic progenitors to establish a competency state that is realized in later stages of differentiation. Consistent with the Pdx1-Oc1 interaction occurring during a critical time sensitive period, inactivation of one Oc1 allele in Pdx1 heterozygotes later in development, using a later-acting pancreas-specific Cre driver line, did not impact endocrine function (unpublished observations).
The transcriptome of DH islets at P1 suggests potential mechanisms for the partial recovery of insulin-expressing cells by birth, but also for the persistent defect in β-cell function. The reduced hormone expression could result in a feedback loop that upregulates expression of regulatory transcription factors to promote further endocrine differentiation. Additionally, the Oc1 target Ctgf is essential for embryonic β-cell proliferation; increased Ctgf expression in embryonic β cells induces both α- and β-cell proliferation, resulting in increased α- and β-cell mass at birth (Guney et al., 2011). The dramatic increase in Ctgf expression in DH islets likely increased α- and β-cell proliferation, thereby increasing α- and β-cell mass by P1. It is also possible that decreased insulin expression itself stimulates increased β-cell proliferation during late gestation. Mice with targeted disruption of the both insulin genes in early development exhibit increased β-cell proliferation at e18.5, similar to the DH animals (Duvillie et al., 2002). Reduction in insulin and glucagon expression persists at P1, which likely contributes to impaired glucose homeostasis. In addition, the increase in Sostdc1 could impair β-cell function. Sostdc1 is a BMP inhibitor, and autocrine BMP signaling was suggested to enhance insulin secretion and glucose homeostasis (Goulley et al., 2007). Indeed, inactivation of Sostdc1 enhances glucose-stimulated insulin secretion and glucose homeostasis (Henley et al., 2012). We were surprised to observe that glycemic control is restored in the majority of DH animals by weaning. Sostdc1 expression was no longer elevated and most islet transcription factors, including Pax4, had returned to normal expression levels. However, decreases in insulin and glucagon expression persisted, now along with reduced MafA expression. Loss of MafA is associated with impaired β cell function in adult mice (Artner et al., 2010; Zhang et al., 2005). Thus, it is likely that islets from DH animals still have reduced functionality.
Taken together, our results suggest that Pdx1 and Oc1 cooperate within pancreatic MPCs or bi-potential trunk cells to promote endocrine specification and to establish a permissive state that allows for later steps of endocrine differentiation and function. Together, these two transcription factors initiate a network of gene expression beyond simple activation of the endocrine progenitor determinant, Neurog3. The concerted action of Oc1 and Pdx1 is critical for the timely functional maturation of endocrine cells. Their cooperative role in ES or iPS cell differentiation toward functional β cells has not been explored and should be considered.
Experimental Procedures
Mutant and transgenic mice
Pdx1XSLacZ (Pdx1LacZ) animals are described in (Offield et al. 1996). Oc1 floxed mice are described in (Zhang et al., 2009). The Pdx1-Cre and Protamine-Cre (Prm-Cre) transgenes are described in (Hingorani et al., 2003; O'Gorman et al., 1997). The Rosa26-EYFP allele is described in (Srinivas et al., 2001). Mice were on a mixed genetic background, were maintained on a 12-hour light/dark cycle and provided food and water ad libitum (except where indicated). All mouse experiments were approved by the Institutional Animal Care and Use Committee of Vanderbilt University Medical Center. Genotyping was performed using tail or ear punch DNA and the primer sets listed in Table S3.
Tissue dissection, preparation, and histology
The morning of the vaginal plug was defined as e0.5. Digestive organs were fixed for 1-4 hours in 4% paraformaldehyde (PFA) at 4°C, dehydrated, cleared in Citrisolv (Fisher) or xylenes, and embedded in paraffin. Livers were fixed 24 hours in 4% PFA at 4°C, dehydrated, cleared in xylenes and embedded in paraffin. For frozen embedding, tissue was fixed as above and placed in 30% sucrose overnight. Following 30 minutes in 50/50 FSC22 (Frozen Section Compound, Leica) and 30% sucrose, frozen tissues were embedded in 100% FSC22 and frozen on dry ice. Paraffin embedded and FSC22 embedded tissues were cut at 5 μm and 7μm, respectively. Paraffin-embedded tissues were deparaffinzed in Citrisolv or xylenes and rehydrated; frozen tissues were allowed to thaw for 30 minutes and permeabilized in 0.1% Triton 2 X 15 minutes in 1X PBS. Detection of MafA, MafB, glucagon, Pax6, and synaptophysin required antigen retrieval. Neurog3 immunolabeling required amplification with PerkinElmer (Waltham, MA) Tyramide tissue amplification. Antibody information is found in Supplemental Methods. X-gal staining was performed as previously described (Wu et al., 1997). Periodic Acid Schiff staining was performed following manufacturer's protocol (Sigma-Aldrich). Fluorescent and bright field images were captured using an Olympus BX41 microscope, the Aperio ScanScope microscope and slide scanner (Vista, CA), or Nikon 600. Digital images were captured and quantified using MagnaFire software (Optronics Engineering, Goleta, CA), ImageScope software of the Aperio software suite (Vista, CA) for the insulin, glucagon, MafA, and MafB, or MetaMorph software for the Neurog3 and Pax6.
β cell mass, α/β cell area and α:β ratio
β cell mass was analyzed as in (Riley et al., 2015). For α- or β-cell area, whole pancreata were serially sectioned at 5 μm and sections every 250 μm immunolabeled for insulin and glucagon. At least 1-2% of the entire pancreas was imaged. Slides were imaged using MetaMorph or a macro built in Genie (Aperio System, Vista, CA). Proportional insulin/glucagon-positive area was calculated by adding the insulin-positive and glucagon-positive area of each section and dividing it by the total pancreas area of each section. α:β cell ratio was calculated by dividing the number of glucagon-positive cells by the number of insulin-positive cells. Total endocrine area was calculated by measuring total area staining positive for synaptophysin as in (Zhang et al., 2009).
Quantitative Real-Time PCR and TaqMan Low Density Array
Islet isolation required collagenase digestion of whole pancreas at 37°C and hand-picking of islets from exocrine tissue. Islets were placed immediately in 500 μl Trizol reagent or RNALater (Ambion), lysed by vortexing or homogenized using a Tissuemiser (Fisher Scientific), and RNA was isolated using the RNAqueous (Ambion) or RNeasy Micro/Mini kits (Qiagen). RNA concentration and integrity were assessed using a ND-1000 Spectrophotometer (NanoDrop) and the 2100 Electrophoresis Bioanalyzer (Agilent) at Vanderbilt Technologies for Advanced Genomics (VANTAGE) Core. cDNA generated from neonatal islets required amplification with the SMARTer Pico PCR cDNA synthesis kit (Clontech). cDNA was prepared from 50-350 ng islet or pancreas RNA using the Superscript III First-Strand synthesis system (Invitrogen). Real-time reactions were carried out in technical duplicates with iQ SYBR Green supermix (Bio-Rad) on a CFX Real-Time PCR Detection system (Bio-Rad) in the Vanderbilt Molecular and Cellular Biology Resource Core. Primers used for hormone gene expression are listed in Supplementary Table 3. TLDA required 150-300ng cDNA. Genes were analyzed using TaqMan Universal PCR Mastermix (with UNG, Applied Biosystems) on custom-designed TLDA cards using a 7900HT Fast Real-Time PCR PCR system. Data were analyzed using SDS RQ Study software (Applied Biosystems, Life Technologies). All samples were run in triplicate.
Pancreatic insulin content and glucose homeostasis
Pancreatic insulin content was measured as described in (Zhang et al., 2009). Intra-peritoneal glucose tolerance tests (IPGTT) were performed as in (Henley et al., 2012).
Statistics
Results are expressed as mean + SEM. Statistical significance was calculated by Student's T Test, One-way Anova with Tukey correction, or Two-way ANOVA where applicable. P < 0.05 was considered significant.
Supplementary Material
Acknowledgements
We thank Matthew Maulis and Jennifer Dunn for technical support and Michael Ray for Neurog3 antibodies. KDH was supported by the Vanderbilt Molecular Endocrinology Training Program (T32 DK07563) and a pre-doctoral fellowship from the American Heart Association (11PRE7960022). DES was supported by the Pediatric Scientist Development Award (K12-HD000850) and Diabetes Career Development Award (5K12-DK094723). PAK was supported by the Vanderbilt University Training Program in Stem Cell and Regenerative Developmental Biology (T32 HD05702). CVEW was supported by U01 DK 089570. DAS was supported by R01DK105689, R01 DK068157 and U01 DK089540. MG was supported by U01 DK089540, R01DK105689 and a VA Merit award (1BX000990–01A1). RNASeq was performed by the Functional Genomics Core of the University of Pennsylvania Diabetes Research Center (DK019525). This study made use of the Vanderbilt Hormone Assay Core and the Islet Procurement and Analysis Core (supported by DRTC grant DK20593), and the VANTAGE Core (supported by P30 CA58648, P30 EY08126, and G20 RR030956).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contribution:
KDH, DES and PAK contributed equally to the work presented. KDH and PAK planned and conducted experiments, analyzed the results and contributed to manuscript writing under the supervision of MG. DES planned and conducted experiments, analyzed the results and contributed to manuscript writing under the supervision of DAS. KJW performed the bioinformatic analysis of the RNASeq data set and supported the manuscript preparation. CVEW provided conceptual input and data interpretation and helped prepare the manuscript. DAS and MG guided the project and wrote the manuscript.
Accession Number:
RNASeq data was deposited in Gene Expression Omnibus (GEO) database repository (GSE77896 -http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc= GSE7 7896).
References
- Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes & Development. 1998;12:1763–1768. doi: 10.1101/gad.12.12.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, Magnuson MA, Stein R. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes. 2010;59:2530–2539. doi: 10.2337/db10-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis ED, Bechard ME, Wright CV. Feedback control of growth, differentiation, and morphogenesis of pancreatic endocrine progenitors in an epithelial plexus niche. Genes & Development. 2015;29:2203–2216. doi: 10.1101/gad.267914.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beucher A, Martin M, Spenle C, Poulet M, Collin C, Gradwohl G. Competence of failed endocrine progenitors to give rise to acinar but not ductal cells is restricted to early pancreas development. Dev. Biol. 2012;361:277–285. doi: 10.1016/j.ydbio.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brissova M, Shiota M, Nicholson WE, Gannon M, Knobel SM, Piston DW, Wright CV, Powers AC. Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J. Biol. Chem. 2002;277:11225–11232. doi: 10.1074/jbc.M111272200. [DOI] [PubMed] [Google Scholar]
- Bruin JE, Saber N, Braun N, Fox JK, Mojibian M, Asadi A, Drohan C, O'Dwyer S, Rosman-Balzer DS, Swiss VA, et al. Treating diet-induced diabetes and obesity with human embryonic stem cell-derived pancreatic progenitor cells and antidiabetic drugs. Stem Cell Reports. 2015;4:605–620. doi: 10.1016/j.stemcr.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlison JS, Long Q, Fujitani Y, Wright CV, Magnuson MA. Pdx-1 and Ptf1a concurrently determine fate specification of pancreatic multipotent progenitor cells. Dev. Biol. 2008;316:74–86. doi: 10.1016/j.ydbio.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli V, Beattie GM, Klier G, Ellisman M, Ricordi C, Quaranta V, Frasier F, Ishii JK, Hayek A, Salomon DR. Expression and function of alpha(v)beta(3) and alpha(v)beta(5) integrins in the developing pancreas: roles in the adhesion and migration of putative endocrine progenitor cells. J. Cell Biol. 2000;150:1445–1460. doi: 10.1083/jcb.150.6.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney M, Gjernes E, Druelle N, Ravaud C, Vieira A, Ben-Othman N, Pfeifer A, Avolio F, Leuckx G, Lacas-Gervais S, et al. The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS Genetics. 2013;9:e1003934. doi: 10.1371/journal.pgen.1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford LA, Guney MA, Oh YA, Deyoung RA, Valenzuela DM, Murphy AJ, Yancopoulos GD, Lyons KM, Brigstock DR, Economides A, et al. Connective tissue growth factor (CTGF) inactivation leads to defects in islet cell lineage allocation and beta-cell proliferation during embryogenesis. Mol. Endocrinol. 2009;23:324–336. doi: 10.1210/me.2008-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S, Bonner-Weir S, Montminy M, Wright C. Regulatory factor linked to late-onset diabetes? [letter]. Nature. 1998;392:560. doi: 10.1038/33311. [DOI] [PubMed] [Google Scholar]
- Duvillie B, Currie C, Chrones T, Bucchini D, Jami J, Joshi RL, Hill DJ. Increased islet cell proliferation, decreased apoptosis, and greater vascularization leading to beta-cell hyperplasia in mutant mice lacking insulin. Endocrinology. 2002;143:1530–1537. doi: 10.1210/endo.143.4.8753. [DOI] [PubMed] [Google Scholar]
- Gannon M, Ables ET, Crawford L, Lowe D, Offield MF, Magnuson MA, Wright CV. pdx-1 function is specifically required in embryonic beta cells to generate appropriate numbers of endocrine cell types and maintain glucose homeostasis. Dev. Biol. 2008;314:406–417. doi: 10.1016/j.ydbio.2007.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon M, Ray MK, Van Zee K, Rausa F, Costa RH, Wright CV. Persistent expression of HNF6 in islet endocrine cells causes disrupted islet architecture and loss of beta cell function. Development. 2000;127:2883–2895. doi: 10.1242/dev.127.13.2883. [DOI] [PubMed] [Google Scholar]
- Gao T, McKenna B, Li C, Reichert M, Nguyen J, Singh T, Yang C, Pannikar A, Doliba N, Zhang T, et al. Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metabolism. 2014;19:259–271. doi: 10.1016/j.cmet.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier BR, Wiederkehr A, Baquie M, Dai C, Powers AC, Kerr-Conte J, Pattou F, MacDonald RJ, Ferrer J, Wollheim CB. PDX1 deficiency causes mitochondrial dysfunction and defective insulin secretion through TFAM suppression. Cell Metabolism. 2009;10:110–118. doi: 10.1016/j.cmet.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulley J, Dahl U, Baeza N, Mishina Y, Edlund H. BMP4-BMPR1A signaling in beta cells is required for and augments glucose-stimulated insulin secretion. Cell Metabolism. 2007;5:207–219. doi: 10.1016/j.cmet.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci. USA. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guney MA, Gannon M. Pancreas cell fate. Birth Defects Research Part C, Embryo today : reviews. 2009;87:232–248. doi: 10.1002/bdrc.20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guney MA, Petersen CP, Boustani A, Duncan MR, Gunasekaran U, Menon R, Warfield C, Grotendorst GR, Means AL, Economides AN, et al. Connective tissue growth factor acts within both endothelial cells and beta cells to promote proliferation of developing beta cells. Proc. Natl. Acad. Sci. USA. 2011;108:15242–15247. doi: 10.1073/pnas.1100072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guz Y, Montminy MR, Stein R, Leonard J, Gamer LW, Wright CV, Teitelman G. Expression of murine STF-1, a putative insulin gene transcription factor, in beta cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development. 1995;121:11–18. doi: 10.1242/dev.121.1.11. [DOI] [PubMed] [Google Scholar]
- Hani EH, Stoffers DA, Chevre JC, Durand E, Stanojevic V, Dina C, Habener JF, Froguel P. Defective mutations in the insulin promoter factor-1 (IPF-1) gene in late-onset type 2 diabetes mellitus. J. Clin. Invest. 1999;104:R41–48. doi: 10.1172/JCI7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley KD, Gooding KA, Economides AN, Gannon M. Inactivation of the dual Bmp/Wnt inhibitor Sostdc1 enhances pancreatic islet function. AJP Endocrinol. Metab. 2012;303:E752–761. doi: 10.1152/ajpendo.00531.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- Jacquemin P, Durviaux SM, Jensen J, Godfraind C, Gradwohl G, Guillemot F, Madsen OD, Carmeliet P, Dewerchin M, Collen D, et al. Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol. Cell. Biol. 2000;20:4445–4454. doi: 10.1128/mcb.20.12.4445-4454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemin P, Lannoy VJ, Rousseau GG, Lemaigre FP. OC-2, a novel mammalian member of the ONECUT class of homeodomain transcription factors whose function in liver partially overlaps with that of hepatocyte nuclear factor-6. J. Biol. Chem. 1999;274:2665–2671. doi: 10.1074/jbc.274.5.2665. [DOI] [PubMed] [Google Scholar]
- Jacquemin P, Lemaigre FP, Rousseau GG. The Onecut transcription factor HNF-6 (OC-1) is required for timely specification of the pancreas and acts upstream of Pdx-1 in the specification cascade. Devel. Biol. 2003;258:105–116. doi: 10.1016/s0012-1606(03)00115-5. [DOI] [PubMed] [Google Scholar]
- Johansson KA, Dursun U, Jordan N, Gu G, Beermann F, Gradwohl G, Grapin-Botton A. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Developmental Cell. 2007;12:457–465. doi: 10.1016/j.devcel.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Ahmed NT, Luciani DS, Han Z, Tran H, Fujita J, Misler S, Edlund H, Polonsky KS. Increased islet apoptosis in Pdx1+/− mice. J. Clin. Invest. 2003;111:1147–1160. doi: 10.1172/JCI16537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371:606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- Kaido T, Yebra M, Cirulli V, Rhodes C, Diaferia G, Montgomery AM. Impact of defined matrix interactions on insulin production by cultured human beta-cells: effect on insulin content, secretion, and gene transcription. Diabetes. 2006;55:2723–2729. doi: 10.2337/db06-0120. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Akiyama T, Nata K, Abe M, Tajima M, Shervani NJ, Unno M, Matsuno S, Sasaki H, Takasawa S, et al. Identification of a receptor for reg (regenerating gene) protein, a pancreatic beta-cell regeneration factor. J. Biol. Chem. 2000;275:10723–10726. doi: 10.1074/jbc.275.15.10723. [DOI] [PubMed] [Google Scholar]
- Kulkarni RN, Jhala US, Winnay JN, Krajewski S, Montminy M, Kahn CR. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J. Clin. Invest. 2004;114:828–836. doi: 10.1172/JCI21845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrmizi I, Hatzis P, Katrakili N, Tronche F, Gonzalez FJ, Talianidis I. Plasticity and expanding complexity of the hepatic transcription factor network during liver development. Genes & Development. 2006;20:2293–2305. doi: 10.1101/gad.390906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammert E, Cleaver O, Melton D. Induction of pancreatic differentiation by signals from blood vessels. Science. 2001;294:564–567. doi: 10.1126/science.1064344. [DOI] [PubMed] [Google Scholar]
- Macfarlane WM, Frayling TM, Ellard S, Evans JC, Allen LI, Bulman MP, Ayers S, Shepherd M, Clark P, Millward A, et al. Missense mutations in the insulin promoter factor-1 gene predispose to type 2 diabetes. J. Clin. Invest. 2000a;106:717. [PubMed] [Google Scholar]
- Macfarlane WM, Frayling TM, Ellard S, Evans JC, Allen LI, Bulman MP, Ayers S, Shepherd M, Clark P, Millward A, et al. Missense mutations in the insulin promoter factor-1 gene predispose to type 2 diabetes [In Process Citation]. J. Clin. Invest. 2000b;106:717. [PubMed] [Google Scholar]
- Nishimura W, Kondo T, Salameh T, El Khattabi I, Dodge R, Bonner-Weir S, Sharma A. A switch from MafB to MafA expression accompanies differentiation to pancreatic beta-cells. Devel. Biol. 2006;293:526–539. doi: 10.1016/j.ydbio.2006.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Gorman S, Dagenais NA, Qian M, Marchuk Y. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc. Natl. Acad. Sci. USA. 1997;94:14602–14607. doi: 10.1073/pnas.94.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL, Wright CV. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122:983–995. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- Oliver-Krasinski JM, Kasner MT, Yang J, Crutchlow MF, Rustgi AK, Kaestner KH, Stoffers DA. The diabetes gene Pdx1 regulates the transcriptional network of pancreatic endocrine progenitor cells in mice. J. Clin. Invest. 2009;119:1888–1898. doi: 10.1172/JCI37028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekala KR, Ma X, Kropp PA, Petersen CP, Hudgens CW, Chung CH, Shi C, Merchant NB, Maitra A, Means AL, et al. Loss of HNF6 expression correlates with human pancreatic cancer progression. Lab. Invest. 2014;94:517–527. doi: 10.1038/labinvest.2014.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plank JL, Frist AY, LeGrone AW, Magnuson MA, Labosky PA. Loss of Foxd3 results in decreased beta-cell proliferation and glucose intolerance during pregnancy. Endocrinology. 2011;152:4589–4600. doi: 10.1210/en.2010-1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevot PP, Simion A, Grimont A, Colletti M, Khalaileh A, Van den Steen G, Sempoux C, Xu X, Roelants V, Hald J, et al. Role of the ductal transcription factors HNF6 and Sox9 in pancreatic acinar-to-ductal metaplasia. Gut. 2012;61:1723–1732. doi: 10.1136/gutjnl-2011-300266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausa F, Samadani U, Ye H, Lim L, Fletcher CF, Jenkins NA, Copeland NG, Costa RH. The cut-homeodomain transcriptional activator HNF-6 is coexpressed with its target gene HNF-3 beta in the developing murine liver and pancreas. Devel. Biol. 1997;192:228–246. doi: 10.1006/dbio.1997.8744. [DOI] [PubMed] [Google Scholar]
- Riley KG, Pasek RC, Maulis MF, Peek J, Thorel F, Brigstock DR, Herrera PL, Gannon M. Connective Tissue Growth Factor Modulates Adult beta-Cell Maturity and Proliferation to Promote beta-Cell Regeneration in Mice. Diabetes. 2015;64:1284–1298. doi: 10.2337/db14-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, Guo T, Puri S, Haataja L, Cirulli V, et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015;34:1759–1772. doi: 10.15252/embj.201591058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdeva MM, Claiborn KC, Khoo C, Yang J, Groff DN, Mirmira RG, Stoffers DA. Pdx1 (MODY4) regulates pancreatic beta cell susceptibility to ER stress. Proc. Natl. Acad. Sci. USA. 2009;106:19090–19095. doi: 10.1073/pnas.0904849106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samadani U, Qian X, Costa RH. Identification of a transthyretin enhancer site that selectively binds the hepatocyte nuclear factor-3 beta isoform. Gene Expression. 1996;6:23–33. [PMC free article] [PubMed] [Google Scholar]
- Shih HP, Seymour PA, Patel NA, Xie R, Wang A, Liu PP, Yeo GW, Magnuson MA, Sander M. A Gene Regulatory Network Cooperatively Controlled by Pdx1 and Sox9 Governs Lineage Allocation of Foregut Progenitor Cells. Cell Reports. 2015;13:326–336. doi: 10.1016/j.celrep.2015.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature. 1997;386:399–402. doi: 10.1038/386399a0. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Devel. Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1 [letter]. Nature Genetics. 1997a;17:138–139. doi: 10.1038/ng1097-138. [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Stanojevic V, Habener JF. Insulin promoter factor-1 gene mutation linked to early-onset type 2 diabetes mellitus directs expression of a dominant negative isoprotein. J. Clin. Invest. 1998;102:232–241. doi: 10.1172/JCI2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffers DA, Thomas MK, Habener JF. Homeodomain protein IDX-1: a master regulator of pancreas development and insulin gene expression. Trends in Endocrinol. and Metab. 1997b;8:145–151. doi: 10.1016/s1043-2760(97)00008-8. [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nature Genetics. 1997c;15:106–110. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- Stolovich-Rain M, Enk J, Vikesa J, Nielsen FC, Saada A, Glaser B, Dor Y. Weaning Triggers a Maturation Step of Pancreatic beta Cells. Developmental Cell. 2015 doi: 10.1016/j.devcel.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Terazono K, Yamamoto H, Takasawa S, Shiga K, Yonemura Y, Tochino Y, Okamoto H. A novel gene activated in regenerating islets. J. Biol. Chem. 1988;263:2111–2114. [PubMed] [Google Scholar]
- Tweedie E, Artner I, Crawford L, Poffenberger G, Thorens B, Stein R, Powers AC, Gannon M. Maintenance of hepatic nuclear factor 6 in postnatal islets impairs terminal differentiation and function of beta-cells. Diabetes. 2006;55:3264–3270. doi: 10.2337/db06-0090. [DOI] [PubMed] [Google Scholar]
- Villasenor A, Chong DC, Cleaver O. Biphasic Ngn3 expression in the developing pancreas. Dev. Dyn. 2008;237:3270–3279. doi: 10.1002/dvdy.21740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waeber G, Thompson N, Nicod P, Bonny C. Transcriptional activation of the GLUT2 gene by the IPF-1/STF-1/IDX-1 homeobox factor. Mol. Endocrinol. 1996;10:1327–1334. doi: 10.1210/mend.10.11.8923459. [DOI] [PubMed] [Google Scholar]
- Weng J, Macfarlane WM, Lehto M, Gu HF, Shepherd LM, Ivarsson SA, Wibell L, Smith T, Groop LC. Functional consequences of mutations in the MODY4 gene (IPF1) and coexistence with MODY3 mutations. Diabetologia. 2001;44:249–258. doi: 10.1007/s001250051608. [DOI] [PubMed] [Google Scholar]
- Wilding Crawford L, Tweedie Ables E, Oh YA, Boone B, Levy S, Gannon M. Gene expression profiling of a mouse model of pancreatic islet dysmorphogenesis. PloS One. 2008;3:e1611. doi: 10.1371/journal.pone.0001611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KL, Gannon M, Peshavaria M, Offield MF, Henderson E, Ray M, Marks A, Gamer LW, Wright CV, Stein R. Hepatocyte nuclear factor 3beta is involved in pancreatic beta-cell-specific transcription of the pdx-1 gene. Mol. Cell. Biol. 1997;17:6002–6013. doi: 10.1128/mcb.17.10.6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Matsuoka TA, Kawashima S, Takebe S, Kubo F, Miyatsuka T, Kaneto H, Shimomura I. A novel function of Onecut1 protein as a negative regulator of MafA gene expression. J. Biol. Chem. 2013;288:21648–21658. doi: 10.1074/jbc.M113.481424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K, et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol. 2005;25:4969–4976. doi: 10.1128/MCB.25.12.4969-4976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ables ET, Pope CF, Washington MK, Hipkens S, Means AL, Path G, Seufert J, Costa RH, Leiter AB, et al. Multiple, temporal-specific roles for HNF6 in pancreatic endocrine and ductal differentiation. Mech. Dev. 2009;126:958–973. doi: 10.1016/j.mod.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.