

Abstract
Background
Genome-wide association studies for coronary artery disease (CAD)/myocardial infarction (MI) revealed a 58kb risk locus on 9p21.3. Refined genetic analyses revealed unique haplotype blocks conferring susceptibility to atherosclerosis per se vs risk for acute complications in the presence of underlying CAD. The cell proliferation inhibitor locus, CDKN2A, maps just upstream of the MI risk block, is at least partly regulated by the non-coding RNA, ANRIL, overlapping the risk block, and has been associated with platelet (PLT) counts in humans. Thus, we tested the hypothesis that CDKN2A deficiency predisposes to increased PLT production leading to increased PLT activation in the setting of hypercholesterolemia.
Methods and Results
PLT production and activation were measured in B6-Ldlr−/−Cdkn2a+/− mice and a congenic strain carrying the region of homology with the human 9p21.3/CDKN2A locus. The strains exhibit decreased expression of CDKN2A (both p16INK4a and p19ARF) but not CDKN2B (p15INK4b). Compared to B6-Ldlr−/− controls, both Cdkn2a-deficient strains exhibited increased PLT counts and bone marrow megakaryopoiesis. The PLT over-production phenotype was reversed by treatment with cyclin-dependent kinase 4/6 inhibitor, PD0332991/palbociclib, that mimics the endogenous effect of p16INK4a. Western-diet feeding resulted in increased PLT activation, increased thrombin/anti-thrombin complex, and decreased bleeding times in Cdkn2a-deficient mice compared to controls.
Conclusions
Together, the data suggest that one or more Cdkn2a transcripts modulate PLT production and activity in the setting of hypercholesterolemia, amenable to pharmaceutical intervention. Enhanced PLT production and activation may predispose to arterial thrombosis, suggesting an explanation, at least in part, for the association of 9p21.3 and MI.
Keywords: mouse, genetics, platelet, arterial thrombosis
Introduction
Coronary artery disease (CAD) and acute complications such as myocardial infarction (MI) are a leading global cause of morbidity and mortality. Human genome-wide association studies for CAD/MI revealed robust associations with a 58kb risk locus on chromosome 9p21.3.1-4 These early studies defined CAD cases based on adverse events (including MI, coronary artery revascularization, coronary death with family history). Subsequent studies, utilizing refined disease classification and expanded genetic analysis of the region, revealed unique haplotype blocks conferring extent of atherosclerosis per se vs risk for acute complications in the presence of underlying CAD. Angiographically-defined CAD cases showed association of the 58kb region with severity of atherosclerotic disease/number of occluded vessels.5, 6 Meta-analysis, including angiographic data as well as MI status, also showed association of the 58 kb region with severity of disease but not MI.7 Patel et al8 reported differential association of the 58kb region with first versus subsequent CHD events, consistent with a primary effect of the locus on atherogenesis rather than acute complications. More recently, interrogations of an expanded 500kb region at 9p21.3 confirmed the 58kb CAD core block and revealed a unique rs518394-tagged block associated with adverse outcomes (including all-cause mortality and non-fatal MI or stroke)9 and prevalent MI.10 Notably, the rs518394-tagged adverse outcomes/MI block has also been significantly associated with platelet (PLT) reactivity11 and pro-inflammatory mixed PLT:leukocyte aggregates.12
The cell proliferation inhibitor locus, CDKN2A, maps just upstream of the rs518394-tagged block, is at least partly regulated by the non-coding RNA, ANRIL,13, 14 overlapping the risk block, and has been highly associated with PLT counts in humans.15 Both linear and circular isoforms of ANRIL correlate with CDKN2A gene expression in humans16 and inheritance of 9p21.3 risk alleles is associated with decreased ANRIL and CDKN2A transcript levels.17 We have previously-reported moderately increased atherosclerosis in a C57BL/6J-Ldlr−/− (B6-Ldlr−/−) subcongenic strain carrying the region of homology with the human CDKN2A/9p21.3 locus and exhibiting decreased Cdkn2a (p16INK4a and p19ARF) but not Cdkn2b (p15INK4b) transcript levels compared to the B6-Ldlr−/− control strain.18 The phenotype was recapitulated in irradiated mice receiving bone marrow (BM) from gene-targeted Cdkn2a+/− mice and the increase in atherosclerosis was associated with altered proliferation of monocyte/macrophage subpopulations.18 A similar transplantation experiment with BM derived from transcript-specific, homozygous p16INK4a−/− mice, which exhibit compensatory upregulation of the nearby cell proliferation inhibitor locus, Cdkn2b, did not show an effect on atherosclerosis in the B6-Ldlr−/− background.19 Further, homozygous Cdkn2a−/− or p19ARF−/− alleles in the APO*E3 Leiden transgenic background, also leading to compensatory upregulation of Cdk inhibitor genes, resulted in no effect or decreased atherosclerosis.20 These data, along with earlier reports of mRNA compensation in homozygous knockouts in wild-type genetic backgrounds,21, 22 make it difficult to interpret effects of Cdkn2a transcripts on atherogenesis.
Cyclin-dependent kinase (CDK) inhibitors have been shown to play an important role in hematopoietic stem cell (HSC) and progenitor cell self-renewal.23-25 The proteins encoded by CDKN2A are functionally unrelated but potent tumor suppressors: p16INK4a, an inhibitor of CDK4/6, and p19/ARF, a regulator of p53 stability. Both proteins are mediators of cellular senescence and competitive BM transplantation experiments showed improved repopulating ability of HSCs derived from p16INK4a−/− mice compared to controls, in an age-dependent manner.26 Moreover, hematologic toxicity of ionizing radiation in mice was ameliorated through pharmacologic inhibition of CDK4/6, mimicking the role of endogenous p16INK4a.27 Somatic loss of CDKN2A - by deletion, mutation or epigenetic silencing - is the most frequent genetic alteration detected in human tumors. However, association of PLT counts with a common nucleotide variant of CDKN2A in the general population suggests that CDKN2A may play a role in PLT production in healthy individuals. Using two unique mouse models of Cdkn2a deficiency,18 we tested the hypothesis that deficiency of one or more transcripts encoded by the Cdkn2a locus predisposes to increased PLT production leading to increased PLT activation. To more closely mimic the condition in which athero-thrombosis occurs in humans, the experiments were carried out under the condition of hypercholesterolemia.
Methods
Mice
All experiments were carried out in the B6-Ldlr−/− background (www.jaxmice.org stock #002207). B6-Ldlr−/−Cdkn2a+/− and B6-Ldlr−/−.MOLFchr4subD mice were described previously.18 Heterozygous Cdkn2a knockout mice were employed to more closely mimic natural gene expression variation in humans and to avoid the complication of spontaneous tumorigenesis that occurs in homozygous knockouts. B6-Ldlr−/−.MOLFchr4subD is a congenic strain originally developed in an atherosclerosis susceptibility mapping study.18 These mice carry a region of mouse chromosome 4 derived from MOLF/Ei on the B6-Ldlr−/− background. The region confers increased susceptibility to atherosclerosis and contains the region of homology with the human 9p21.3 CAD/MI GWAS locus and the CDKN2A/CDKN2B genes. B6-Ldlr−/−Cdkn2a+/− and B6-Ldlr−/−.MOLFchr4subD mice are deficient in both p16INK4a and p19ARF, but not related Cdk inhibitor transcripts p15INK4b or p18INK4c. The congenic strain exhibits a more pronounced deficiency of Cdkn2a transcripts without the complication of tumorigenesis.18 Mice were fed 1) regular chow, 2) chow + 50 mg/kg PD0332991/palbociclib (CDK4/6 inhibitor, Pfizer) or 3) Western-type diet (WTD, Harlan Teklad #TD88137) as indicated. Since there was no effect of sex on the PLT production phenotypes (Supplemental Figure 1), similar numbers of males and females were used for all experiments, except for the tail vein bleeding assay under the condition of WTD diet feeding, in which there were not enough males available. All procedures were in accordance with Institutional guidelines.
Complete blood count
Whole blood collected from the retro-orbital plexus, with 10% volume of acid-citrate-dextrose (ACD) or EDTA as anticoagulant, was analyzed using a FORCYTE Veterinary Hematology Analyzer (Oxford Science, Inc.).
MkP-CFU assay
Primary BM cells were plated in collagen-based medium (5,000 cells per assay) containing thrombopoietin (TPO, 50 ng/ml) and interleukin-3 (IL-3, 10 ng/ml), and incubated for 12 days according to the manufacturer's protocol (Megacult-C, Stemcell Technologies). The collagen gels were then dehydrated, fixed and stained for acetylcholinesterase activity to identify MkP cells. Nuclei were counterstained with Harris’ hematoxylin. MkP-CFUs were defined by groupings of at least three MkP cells. Large MkP-CFUs were defined by groupings of at least 10 MkP cells.
Immunohistochemistry staining
Bone samples were fixed in 10% formalin for 24 hrs and decalcified with EDTA for 48 hrs prior to sectioning. Paraffin sections were incubated with von Willebrand factor (vWF) antibody (Dako A0082, 1:400 dilution) and visualized using the avidin-biotin complex (ABC) staining method.
Flow cytometry-based hematopoietic stem cell profiles
BM cells from mouse femurs and tibias were stained with a cocktail of antibodies as previously described.28 Briefly, BM cells were incubated with antibodies to lineage-committed cells (CD45R, CD19, CD11b, CD3e, TER-119, CD2, CD8, CD4 and Ly-6C/G, all FITC conjugated; eBioscience), Sca1-Pacific blue (Biolegend) and c-Kit-APC cy7 (eBioscience) to identify LSK (Lin−Sca1+c-Kit+) cells and hematopoietic stem and progenitor cells (HSPC, Lin−Sca1−c-Kit+) cells. Further, another cocktail of antibodies to lineage-committed cells (same as mentioned above) plus Sca-1-FITC, CD16/CD32 (FcγRII/III)-Pacific blue, CD34-PerCP Cy5.5 (BD Biosciences), CD71-phycoerythrin (PE) and CD41-PE cy7 to identify progenitor cell populations including common myeloid progenitor (CMP, Lin−Sca1−c-Kit+CD34intFcγRII/IIIint), granulocyte-macrophage progenitor (GMP, Lin−Sca1−c-Kit+CD34intFcγRII/IIIhi), megakaryocyte-erythroid progenitor (MEP, Lin−Sca1−c-Kit+CD34intFcγRII/IIIlow), erythroid progenitor (ErP, Lin−Sca1−c-Kit+CD34intFcγRII/IIIintCD71+CD41−) and megakaryocyte progenitor (MkP, Lin−Sca1−c-Kit+CD34intFcγRII/IIIintCD71−CD41+). For spleen cells, similar staining and gating strategies were used in separating spleen LSK and HSPC. Spleen MkP was defined as (Lin−Sca1−c-Kit+CD41+). Samples were run on an LSR-II instrument using FACSDiVa software (BD Biosciences). Cell sorting was performed on a BD Influx using BD Sortware 1.2.0.142 (BD Biosciences) and analyzed with FlowJo software. All antibodies were used at 1:50 dilution.
Flow cytometry of platelet-leukocyte aggregates (PLAs)
ACD-whole blood collected from the retro-orbital plexus was incubated with an excess of ice-cold lysis buffer plus 1 μM prostaglandin E1 (PGE1) to prevent PLT activation and in vitro formation of PLAs as described.29 The blood cells were lysed briefly (1 min), spun at 4°C and stained with a cocktail of antibodies at 1:100 dilution: CD45-Pacific blue (eBioscience), CD115-APC (eBioscience), Gr-1-PerCP cy5.5 (Ly6-C/G; BD Biosciences), CD11b-PE Cy7 (eBioscience) and CD41-FITC (eBioscience). Platelet Ly-6Chi monocyte aggregates, platelet Ly-6Clo monocyte aggregates and platelet neutrophil aggregates were defined as CD45+Gr-1hiCD115+CD41+, CD45+Gr-1loCD115+CD41+ and CD45+Gr-1+CD115−CD41+.
Reticulated PLT detection
While both young and mature PLTs are anuclear, young reticulated PLTs have residual RNA content that can be used for detection, providing an indication of relative rates of megakaryopoiesis in the BM. ACD- or EDTA-whole blood collected from the retro-orbital plexus was incubated with thiazole orange fluorescent dye (nucleic acid-binding, final concentration 1 μg/ml) and counter-stained with a CD41-PE antibody (to identify PLTs). Samples were incubated at room temperature for 20 min, fixed with 1% formaldehyde in PBS and analyzed within 45 min of blood collection. Data was acquired using logarithmic amplification of light scatter and fluorescence signals. PE-positive cells were gated in a thiazole orange fluorescence vs side scatter dot plot, reducing interferences due to PLT size and PLT aggregation.30
PLT activity assays
PLT activation was assessed by PLT surface expression of P-selectin, a marker of PLT degranulation, and surface JON/A, which selectively binds to the high affinity conformation of mouse integrin αIIIbβ3. Briefly, ACD-whole blood was diluted in HEPES-buffered, modified Tyrode's buffer (HBMT) plus 2 mM CaCl2. The samples were treated with or without 100 μM PAR4 agonist AYPGKF (Sigma A3227)) stimulation and incubated at room temperature for 15 min with anti JON/A-PE (Emfret M023-2), anti P-selectin-FITC (Emfret M130-1) and anti CD41-APC (Biolegend 133914) as a pan PLT marker. The reactions were stopped by the addition of 400 μl HBMT and analyzed within 30 min.
Plasma T-AT complex ELISA
A dual antibody mouse T-AT complex ELISA kit (Innovative Research, IRAPKT175) was used. Plasma samples were diluted 1:50, plated into T-AT antibody-coated plates and incubated at room temperature for 2 hours. After six plate washes, 50 ul of biotinylated mouse antithrombin antibody was added for an incubation period of 2 hours. After washing, streptavidin-peroxidase conjugate secondary antibody and chromogen substrate were added to the wells. Optical density was measured at 450nm with 570nm correction using a Spectramax M2 plate reader. Final concentration was calculated from a standard curve, generated in parallel, on the same plate.
Tail vein bleeding assay
Mice were anesthetized with isofluorane and a 5mm segment of the distal tail tip was transected with a sharp scalpel. Bleeding was monitored by gently dabbing the tail tip on Whatman paper until the cessation of bleeding, as described.31
Statistics
Three-group analyses were by one-factor ANOVA with Bonferroni correction (single measurements), repeated measures ANOVA (multiple measures) or Kruskal-Wallis with Dunn's multiple comparison (small sample size or skewed data) as indicated. Two-group analyses were by Student's t-test (two-tailed) or Mann-Whitney (small sample size) as indicated. Analyses were performed using Prism 6 (GraphPad Software, Inc). The threshold for significance was p < 0.05. Bar graphs indicate mean ± SD.
Results
Increased PLT production in chow-fed Cdkn2a-deficient strains in the B6-Ldlr−/− background
Complete blood count (CBC) analysis revealed ~30-40% increased circulating PLT counts in B6-Ldlr−/−Cdkn2a+/− and B6-Ldlr−/−.MOLFchr4subD mice compared to B6-Ldlr−/− controls (Figure 1A), and the increases were independent of sex (Supplemental Figure 1A). Total white blood cell (WBC), neutrophil and lymphocyte counts were not altered between the strains (Supplemental Figure 2A). Monocyte counts were modestly elevated in the gene-targeted Cdkn2a-deficient mice compared to controls and both Cdkn2a-deficient strains showed increases in the inflammatory subset characterized by high cell surface expression of Ly6C antigen (Supplemental Figure 2B), consistent with our previous observations in these strains.18 Along with the increases in total PLT counts, percentages of immature reticulated PLTs were increased in blood samples from the two Cdkn2a-deficient strains (Figure 1A, right-hand graph), suggesting increased megakaryopoiesis in the BM. The increases in reticulated PLTs were observed in both males and females of the gene-targeted Cdkn2a-deficient strain but more heavily influenced by males in the subcongenic strain, possibly due to small sample size (Supplemental Figure 1A).
Figure 1.
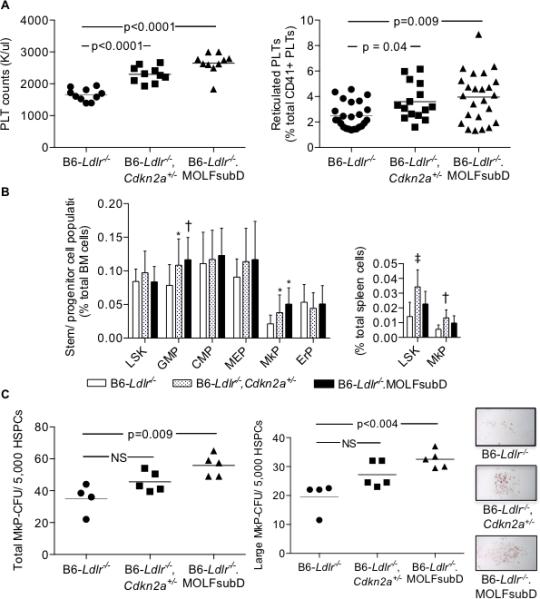
Increased PLT production in chow-fed, Cdkn2a-deficient strains in the B6-Ldlr−/− background. A: PLT counts from EDTA whole blood. Reticulated PLTs from acid-citrate-dextrose blood incubated with CD41-APC and thiazole orange nucleic acid-binding dye and analyzed by flow cytometry: B: LSK and progenitor subpopulations from BM or spleen were measured by flow cytometry. LSK (Lin−Sca1+c-kit+), GMP (Lin−Sca1−c-kit+CD34intFcγRII/IIIhi), CMP (Lin−Sca1−c-kit+CD34intFcγRII/IIIint), MEP (Lin−Sca1−c-kit+CD34intFcγRII/IIIlowCD71−CD41−), MkP (Lin−Sca1−c-kit+CD34intFcγRII/IIIintCD71−CD41+), and ErP (Lin−Sca1−c-kit+CD34intFcγRII/IIIloCD71+CD41−). N = 20 (BM) or 10 (spleen) mice/group. ‡p=0.0003, †p<0.002, and *p<0.05 vs B6-Ldlr−/− control. C: MkP colony-forming units derived from BM cells following 12 days of culture with 50 ng/ml TPO/10 ng/ml IL-3 and stained for acetylcholinesterase activity. One-way ANOVA (A, PLT counts and B); Kruskal-Wallis non-parametric (A, Reticulated PLTs and C). HSPC, hematopoietic stem and progenitor cells; HPF, high-power field.
To test for increased megakaryopoiesis, we looked at HSC and progenitor profiles by flow cytometry and determined megakaryocyte progenitor (MkP) colony-forming potential of the three strains. Analysis of LSK (Lin−Sca1+c-Kit+) and progenitor populations revealed no differences in the percentages of LSK cells in total BM cells between the strains but increased percentages of granulocyte-macrophage progenitors (GMPs), trends toward increased megakaryocyte-erythrocyte progenitors (MEPs) and significantly increased MkPs but not erythrocyte progenitors (ErPs) in the Cdkn2a-deficient strains compared to controls (Figure 1B). The differences in percentages of GMP and MkP cells were independent of sex in both strains (Supplemental Figure 1B). In the spleen, percentages of LSK and MkP cells were significantly increased in the gene-targeted Cdkn2a-deficient strain compared to controls with trends toward increased percentages in the subcongenic strain (Figure 1B, right-hand graph). As a measure of MkP production, we measured MkP-CFUs in BM-derived cells following 12 days of culture in the presence of TPO and IL-3 growth factors. There was a trend toward increased total colony numbers, and colony size, in BM derived from B6-Ldlr−/−Cdkn2a+/− mice, and significantly increased colony numbers and size in BM derived from B6-Ldlr−/−.MOLFchr4subD mice compared to controls (Figure 1C). Finally, immunohistochemical analysis of decalcified bone showed a trend towards increased numbers of von Willebrand factor-positive megakaryocytes (MKs) in B6-Ldlr−/−.MOLFchr4subD mice compared to B6-Ldlr−/− controls (Supplemental Figure 3A). Consistent with these results, we observed decreased Cdkna2, but not related Cdkn2b, transcript levels in MkP cells derived from B6-Ldlr−/−Cdkn2a+/− and B6-Ldlr−/−.MOLFchr4subDmice compared to B6-Ldlr−/− controls (Supplemental Figure 3B), These data indicate a role for one or both Cdkn2a transcripts in regulating megakaryopoiesis and PLT production.
Inhibition of CDK4/6 reverses the PLT over-production phenotype of Cdkn2a-deficient strains in the B6-Ldlr−/− background
The Cdkn2a-deficient strains employed in this study are partially deficient in both transcripts encoded by the locus: p16INK4a and p19/ARF. The p16INK4a protein blocks cell proliferation by interfering with the interaction of CDK4 and cyclin D resulting in hypophosphorylation of the retinoblastoma oncogene, Rb, and subsequent G1 arrest. To test the specific role of p16INK4a in megakaryopoiesis and PLT production, we treated mice with the specific CDK4/6 inhibitor, PD0332991/palbociclib, that mimicks the effect of endogenous p16INK4a. The PD0332991 inhibitor was incorporated into regular chow to provide an effective dose of 50 mg/kg.27 Mice were bled at baseline and following 7 and 16 days of treatment. As previously demonstrated with chow feeding, both Cdkn2a-deficient strains had increased circulating PLT counts compared to B6-Ldlr−/− mice at baseline. At 7 days of treatment, PLT counts were decreased in all three strains with only B6-Ldlr−/−.MOLFchr4subD mice exhibiting increased counts compared to controls (Figure 2A). At 16 days of treatment, there were no further decreases in PLT counts and the differences between the strains were eliminated (Figure 2A). Flow cytometry analysis of BM cell profiles at 21 days of treatment revealed no differences between the strains in the percentages of LSK or progenitor subpopulations in total BM cells, including MEP and MkP cells (Figure 2B). The MkP-CFU assay showed that the number and size of MkP colonies derived from both Cdkn2a–deficient strain BM were reduced to at least that of the B6-Ldlr−/− controls following PD0332991 treatment (Figure 2C). Finally, we measured an in vivo marker of thrombin generation, thrombin/anti-thrombin (T-AT) complex, as a potential functional consequence of augmented PLT production. Baseline levels of T-AT complex were elevated in B6-Ldlr−/−Cdkn2a+/−, but not B6-Ldlr−/−.MOLFchr4subD, mice compared to B6-Ldlr−/− controls (Figure 2D). Treatment with PD0332991 reduced T-AT complex levels in B6-Ldlr−/−Cdkn2a+/− mice to that of the controls (Figure 2D). These data are consistent with a transcript-specific role of p16INK4a in megakaryopoiesis, PLT production and thrombin generation in these mouse models.
Figure 2.
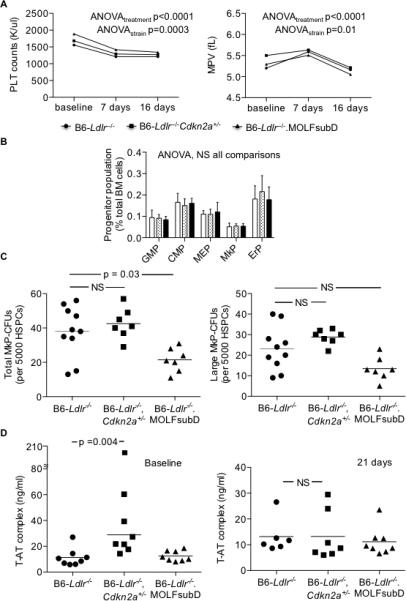
Pharmaceutical inhibition of CDK4/6 reverses the PLT over-production phenotype of Cdkn2a-deficient strains. Chow-fed mice were bled at baseline and following 7, 16 or 21 days of 50 mg/kg PD0332991 treatment in chow diet. BM was collected at 21 days of treatment. A: PLT counts and mean platelet volume (MPV) from EDTA whole blood. N = 8 mice/group. B: Stem cell progenitor subpopulations from BM measured by flow cytometry. GMP (Lin−Sca1−c-kit+CD34intFcγRII/IIIhi), CMP (Lin−Sca1−c-kit+CD34intFcγRII/IIIint), MEP (Lin−Sca1−c-kit+CD34intFcγRII/IIIloCD71−CD41+), MkP (Lin−Sca1−c-kit+CD34intFcγRII/IIIloCD71−CD41+), and ErP (Lin−Sca1−c-kit+CD34intFcγRII/IIIloCD71+CD41−). N = 7-8 mice per group. C: MkP-CFUs derived from BM cells following 12 days of culture with 50 ng/ml TPO/10 ng/ml IL-3 and stained for acetylcholinesterase activity. D: Thrombin/anti-thrombin (T-AT) complex measured in sodium citrate plasma by commercially-available ELISA. Repeated measures (A) or one-way (B) ANOVA; Kruskal-Wallis non-parametric (C-D). NS, not significant.
Cdkn2a deficiency promotes PLT activation in B6-Ldlr−/− strains fed WTD
PLT over-production has been shown to lead to PLT activation in Abcg4- and Abcc6-deficient mouse models.28, 32 Thus, we next studied markers of PLT activation in the Cdkn2a-deficient strains. In vivo ligation of the PLT surface receptor, protease-activated receptor (PAR) 4, by thrombin, results in increased PLT surface expression of cell adhesion molecules that facilitate PLT-leukocyte interactions, PLT-endothelial interactions and intercellular signaling. Ex vivo treatment of whole blood with synthetic peptide AYPGKF, a PAR4 agonist, resulted in increased mean fluorescence intensity of surface P-selectin in chow-fed B6-Ldlr−/−.MOLFchr4subD (1.4-fold), but not B6-Ldlr−/−Cdkn2a+/−, compared to B6-Ldlr−/− controls (Figure 3A). We also observed increased levels of sP-selectin in blood from B6-Ldlr−/−.MOLFchr4subD mice (Figure 3B). In addition, both Cdkn2a-deficient strains exhibited decreased bleeding times compared to controls in a tail vein bleeding assay (Figure 3C). Overall, partial Cdkn2a deficiency in the B6-Ldlr−/− background resulted in modest effects on PLT activation with chow feeding.
Figure 3.
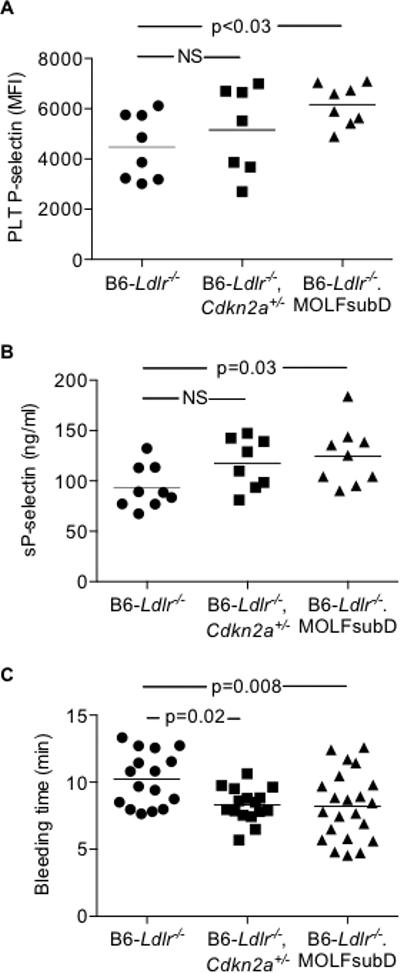
Modest effects of Cdkn2a deficiency on PLT activation with chow feeding. A: PLT surface P-selectin levels in acid-citrate-dextrose whole blood stimulated with PAR4 agonist AYPGKF (100 uM) and analyzed by flow cytometry. B: Circulating soluble P-selectin (sP-selectin) in PLT-poor plasma by commercially-available ELISA. C, Time to cessation of tail vein bleeding following tail clip injury. One-way ANOVA (A-C). MFI, mean fluorescent intensity; NS, not significant.
To more closely mimic the conditions under which athero-thrombosis occurs in humans, we next studied PLT activation in WTD-fed mice. The 8-wk timepoint represents a period of active atherogenesis18, 33 but precedes age-dependent decline in HSC functions.26, 34 As demonstrated with chow feeding, B6-Ldlr−/−Cdkn2a+/− and B6-Ldlr−/−.MOLFchr4subD mice had increased circulating PLT counts, and increased percentages of young, reticulated PLTs, compared to B6-Ldlr−/− mice (Supplemental Figure 4A-B). However, there was no evidence of monocytosis or neutrophilia (Supplemental Figure 5), phenotypes observed in other models with WTD feeding.35 We measured surface expression of both P-selectin and active integrin αIIIbβ3 (JON/A). In the resting state, no differences between the strains were observed for levels of PLT P-selectin or JON/A (Supplemental Figure 6A-B). Following stimulation with PAR-4 agonist AYPGKF, whole blood from both Cdkn2a-deficient strains exhibited markedly increased surface levels of P-selectin (Figure 4A) and JON/A (Figure 4B). For P-selectin, the increases were 8.5- and 39-fold for B6-Ldlr−/−Cdkn2a+/− and B6-Ldlr−/−.MOLFch4subD relative to the controls. For JON/A, the increases were 4- and 9-fold for B6-Ldlr−/−Cdkn2a+/− and B6-Ldlr−/−.MOLFch4subD relative to the controls. To test for a direct role of Cdkn2a/p16 deficiency on PLT activation, we measured PAR-4-stimulated P-selectin and JON/A expression following ex vivo treatment of whole blood with PD0332991. Little to no differences were observed between treated and untreated samples, and no differential strain effects were observed (Supplemental Figure 7). Together, the data are consistent with a primary effect of Cdkn2a deficiency on PLT over-production leading to increased PLT activation.
Figure 4.
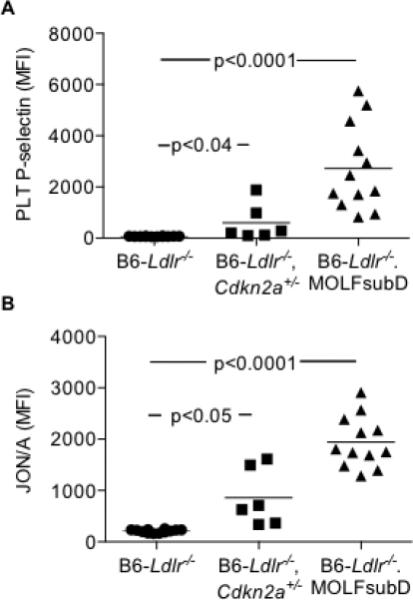
Increased markers of PLT activation following PAR4 agonist stimulation in Western-diet (WTD)-fed Cdkn2a-deficient strains. Acid-citrate-dextrose whole blood stimulated with PAR4 agonist AYPGKF (100 μM) and analyzed by flow cytometry. A: MFI of PLT surface P-selectin. B: MFI of PLT surface activated form of integrin αIIIbβ3 (JON/A). Kruskal-Wallis (A-B).
In addition, we observed increases in PLT:leukocyte aggregates with Cdkn2a deficiency, especially in the B6-Ldlr−/−.MOLFch4subD strain. The percentages of PLT:neutrophil and PLT:monocyte aggregates in total WBCs were increased in B6-Ldlr−/−.MOLFchr4subD mice, with a trend towards increased PLT:neutrophil aggregates in B6-Ldlr−/−Cdkn2a+/− mice, compared to controls (Figure 5A-B). The increase in total PLT:monocyte aggregates in the B6-Ldlr−/−.MOLFchr4subD strain was due to increases in both PLT:Ly6Chi monocyte and PLT:Ly6Clo monocyte aggregates (Figure 5C-D).
Figure 5.
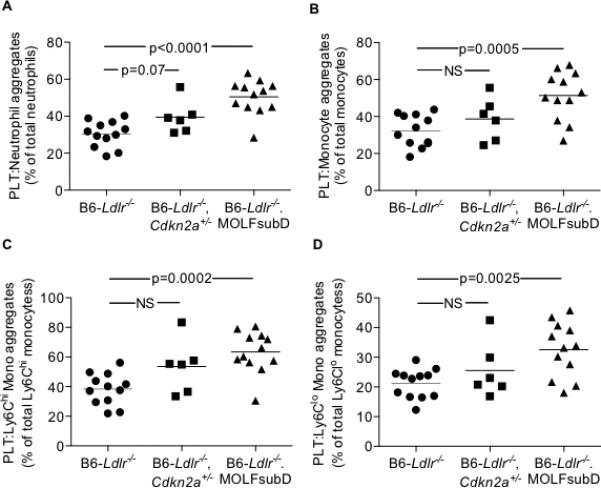
Mixed PLT:leukocyte aggregates in the circulation of WTD-fed mice. A: Platelet:neutrophil aggregates. B: Platelet:monocyte aggregates. C: Platelet:Ly-6Chi monocyte aggregates. D: Platelet:Ly-6Clo monocyte aggregates. One-way ANOVA (A-D).
Under the condition of WTD feeding, the in vivo marker of thrombin generation, T-AT complex, was significantly elevated in the circulation of both Cdkn2a-deficient strains by ~2-fold compared to B6-Ldlr−/− controls (Figure 6A). In addition, both Cdkn2a-deficient strains exhibited significantly decreased tail vein bleeding times (Figure 6B), indicating an effect on hemostasis. Together, the data indicate that partial Cdkn2a deficiency promotes PLT overproduction leading to PLT activation in the setting of hypercholesterolemia.
Figure 6.
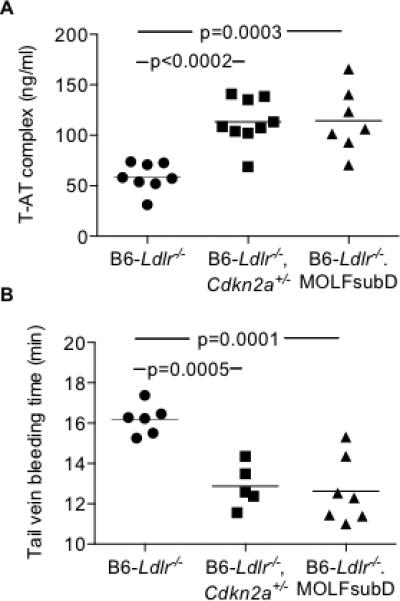
Increased T-AT complex and decreased tail vein bleeding times in WTD-fed Cdkn2a-deficient strains. A: T-AT complex measured in sodium citrate plasma by commercially-available ELISA. B: Time to cessation of tail vein bleeding following tail clip injury.One-way ANOVA (A-B).
Discussion
In the present study, we have identified an important role for Cdkn2a in PLT production leading to PLT activation in hypercholesterolemic B6-Ldlr−/− mice. Gene-targeted heterozygous deficiency of Cdkn2a and naturally-occurring inbred strain variation observed in B6-Ldlr−/−.MOLFch4subD mice, both resulting in decreased expression of both p16INK4a and p19ARF but not p15INK4b transcripts, led to increased PLT counts in the circulation and augmented megakaryopoiesis in the BM. These phenotypes were reversed by pharmacologic treatment with the CDK4/6 inhibitor, PD0332991, consistent with a transcript-specific effect of p16INK4a on PLT production that warrants further testing. While some strain effects on PLT activation were observed with chow diet feeding, Cdkn2a deficiency in the setting of WTD feeding resulted in more robust effects on PLT activation and downstream functional effects such as PLT:leukocyte aggregate formation, T-AT complex generation and decreased bleeding times. Together, the data indicate that Cdkn2a deficiency promotes PLT over-production leading to PLT activation.
In humans and mice, complete deficiency of CDKN2A/Cdkn2a, including both p16INK4a and p19ARF, leads to spontaneous development of a wide spectrum of tumors due to increased mitogenic activity of HSPCs. Pre-clinical studies with the specific CDK4/6 inhibitor, PD0332991, in mice results in tumor regression36 due to decreased proliferative capacity of early myeloid progenitors without effects on apoptosis.37 Analysis of progenitor subpopulations revealed decreased Ki67 staining of multiple progenitor cell types including GMP and MEP leading to modest decreases in erythroid, PLT and myeloid lineages in the circulation.27 Our previous studies in B6-Ldlr−/− mice transplanted with B6-Ldl+/−Cdkn2a+/− or B6-Ldlr−/−.MOLFch417Mb BM, carrying partial deficiencies of Cdkn2a in BM, revealed increased atherogenesis associated with increased ratios of inflammatory Ly6Chi:Ly6Clo monocytes in the circulation. Moreover, the B6-Ldl+/−Cdkn2a+/− recipients exhibited increased cell proliferation of circulating Ly6Chi monocytes and tissue macrophages compared to controls transplanted with B6-Ldlr+/− BM. In the current study utilizing whole-body Cdkn2a deficiency models, we have observed an additional effect of partial Cdkn2a deficiency in the B6-Ldlr−/− background – a pronounced effect on circulating PLT counts. Although an effect on PLT counts is not surprising, the amplitude of the effect in the models described herein suggests that hypercholesterolemia may exaggerate the effect of Cdkn2a deficiency on PLT production. Moreover, the effects of Cdkn2a deficiency on PLT over-production were associated with increased PLT activation, which was enhanced with WTD feeding. PLT production and activation have been implicated in inflammatory pathways contributing to both thrombosis and atherosclerosis development.38, 39 Thus, it is possible that PLT over-production may have contributed to the augmented atherogenesis observed in our previous BM transplantation study, however, we did not measure PLT production in that model. It will be interesting to determine whether Cdkn2a-deficiency has a greater effect on arterial thrombosis.
Hypercholesterolemia has been shown to increase PLT activation by multiple mechanisms, including increased uptake of oxidized lipoproteins via PLT surface scavenger receptors.40, 41 We observed increased markers of PLT activation by multiple measures in WTD-fed Cdkn2a-deficient strains compared to chow-fed mice. Whereas chow-fed B6-Ldlr−/−.MOLFch4subD mice exhibited elevated markers of PLT activation (Figure 3), both strains fed WTD exhibited significantly elevated PLT surface P-selectin and JON/A, following PAR4 stimulation (Figure 4). Further, WTD-feeding resulted in increased levels of circulating PLT:leukocyte aggregates in B6-Ldlr−/−.MOLFch4subD (Figure 5) and elevated T-AT levels in both strains (Figure 6A), indicative of a pro-thrombotic phenotype. Consistent with these findings, we previously reported a PLT over-production phenotype with BM-specific Abcg4 deficiency that was also associated with WTD feeding.28 These data strongly suggest an augmented effect of hypercholesterolemia on Cdkn2a-dependent PLT over-production leading to PLT activation.
While both Cdkn2a-deficient strains studied exhibited enhanced megakaryopoiesis, PLT over-production and increased PLT activation, the phenotype of the congenic B6-Ldlr−/−.MOLFch4subD strain tended to be more robust than that of the gene-targeted B6-Ldlr−/−Cdkn2a+/− strain. The strain-specific differences in the PLT over-production phenotype are likely due to differences in underlying genetic variation at the Cdkn2a locus. We previously reported 3-8-fold vs 2.5-4.5-fold decreases in expression levels of p16INK4a in B6-Ldlr−/−.MOLFch4subD vs B6-Ldlr−/−Cdkn2a+/− mice compared to B6-Ldlr−/− controls, dependent on the tissue/condition queried.18 Thus, the more dramatic phenotype of B6-Ldlr−/−.MOLFch4subD mice is likely due, at least in part, to differences in the level of gene expression.
Overexpression of p16INK4a resulted in increased venous thrombosis in a murine vascular injury model,42 in contrast to our hypothesis that Cdkn2a deficiency promotes a pro-thrombotic phenotype. However, there are substantial pathobiologic differences between venous and arterial thrombosis.43, 44 Venous thrombosis occurs with stasis and hypercoagulability, and fibrin and red blood cells are the main constituents of venous thrombi. On the other hand, endothelial damage, PLTs, lipids and inflammation play a much larger role in arterial thrombosis. The disease risk genes identified for arterial thrombosis are often distinct from those for venous thrombosis.45 For example, inherited thrombophilia factors that are strongly associated with high risk for venous thrombosis are not associated, or less strongly, with arterial thrombosis.45 In a mouse model of fibronectin haploinsufficiency, delayed thrombus formation was observed in arterioles but not venules.46 Thus, it is highly plausible that Cdkn2a/p16INK4a may have different effects in arterial vs venous thrombosis.
PLT activation is strongly associated with MI risk in humans. Moreover, therapeutic interventions that inhibit PLT activation, such as aspirin and clopidigrel, decrease risk of atherothrombotic events.47 Association of the 9p21.3 adverse outcomes/MI block with PLT reactivity suggests an underlying mechanism for risk of disease.11, 12 While most treatment regimens focus on anti-PLT drugs, there is increasing evidence suggesting that therapies directed at abnormal hematopoiesis, including excessive PLT production, may be beneficial in preventing atherothrombosis/MI.39 The increased PLT production, PLT activation, and increased T-AT complex phenotype of hypercholesterolemic Cdkn2a-deficient mice suggests a pro-thrombotic phenotype. While mouse models of atherosclerosis are highly resistant to thrombosis, vascular injury models have been used to verify gene-specific effects on thrombosis that are often relevant to human thrombosis. Together, these data may provide a causal explanation, at least in part, for the association of 9p21.3 and MI.
Supplementary Material
Clinical Perspective.
Common genetic variants on chromosome 9p21.3 are associated with angiographic coronary artery disease and myocardial infarction. Different haplotype blocks confer susceptibility to atherosclerosis per se vs risk for acute complications in the presence of underlying CAD. Platelet reactivity also maps to the acute complications block. Using mouse models, we tested the cell proliferation inhibitor locus, CDKN2A, which maps near the acute complications block and contains a common variant associated with platelet counts in humans. Cdkn2a-deficiency in a hypercholesterolemic murine background resulted in a robust PLT over-production phenotype including increased bone marrow megakaryopoiesis as well as increased PLT counts, markers of PLT activation, PLT:leukocyte aggregates and thrombin/anti-thrombin (T-AT) complex levels in the circulation. Enhanced PLT production and activation may predispose to arterial thrombosis, suggesting an explanation, at least in part, for the association of 9p21.3 and myocardial infarction.
Acknowledgments
Sources of Funding: This project was funded by NIH grant HL102206 (CLW). The data reported herein made use of the Columbia University CCTI Flow Cytometry Core, supported in part by the Office of the Director, NIH, under awards S10RR027050 and S10OD020056. The content is solely the responsibility of authors and does not necessarily represent the official views of the NIH.
Footnotes
Disclosures: None.
References
- 1.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 2.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Consortium WTCC Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dandona S, Stewart AF, Chen L, Williams K, So D, O'Brien E, et al. Gene dosage of the common variant 9p21 predicts severity of coronary artery disease. J Am Coll Cardiol. 2010;56:479–486. doi: 10.1016/j.jacc.2009.10.092. [DOI] [PubMed] [Google Scholar]
- 6.Patel RS, Su S, Neeland IJ, Ahuja A, Veledar E, Zhao J, et al. The chromosome 9p21 risk locus is associated with angiographic severity and progression of coronary artery disease. Eur Heart J. 2010;31:3017–3023. doi: 10.1093/eurheartj/ehq272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horne BD, Carlquist JF, Muhlestein JB, Bair TL, Anderson JL. Association of variation in the chromosome 9p21 locus with myocardial infarction versus chronic coronary artery disease. Circ Cardiovasc Genet. 2008;1:85–92. doi: 10.1161/CIRCGENETICS.108.793158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel RS, Asselbergs FW, Quyyumi AA, Palmer TM, Finan CI, Tragante V, et al. Genetic variants at chromosome 9p21 and risk of first versus subsequent coronary heart disease events: a systematic review and meta-analysis. J Am Coll Cardiol. 2014;63:2234–2245. doi: 10.1016/j.jacc.2014.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gong Y, Beitelshees AL, Cooper-DeHoff RM, Lobmeyer MT, Langaee TY, Wu J, et al. Chromosome 9p21 haplotypes and prognosis in white and black patients with coronary artery disease. Circ Cardiovasc Genet. 2011;4:169–178. doi: 10.1161/CIRCGENETICS.110.959296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan M, Dandona S, McPherson R, Allayee H, Hazen SL, Wells GA, et al. Two chromosome 9p21 haplotype blocks distinguish between coronary artery disease and myocardial infarction risk. Circ Cardiovasc Genet. 2013;6:372–380. doi: 10.1161/CIRCGENETICS.113.000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Musunuru K, Post WS, Herzog W, Shen H, O'Connell JR, McArdle PF, et al. Association of single nucleotide polymorphisms on chromosome 9p21.3 with platelet reactivity: a potential mechanism for increased vascular disease. Circ Cardiovasc Genet. 2010;3:445–453. doi: 10.1161/CIRCGENETICS.109.923508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gianfagna F, Tamburrelli C, Vohnout B, Crescente M, Izzi B, Pampuch A, et al. Heritability, genetic correlation and linkage to the 9p21.3 region of mixed platelet-leukocyte conjugates in families with and without early myocardial infarction. Nutr Metab Cardiovasc Dis. 2013;23:684–692. doi: 10.1016/j.numecd.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 13.Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38:662–674. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, et al. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene. 2011;30:1956–1962. doi: 10.1038/onc.2010.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gieger C, Radhakrishnan A, Cvejic A, Tang W, Porcu E, Pistis G, et al. New gene functions in megakaryopoiesis and platelet formation. Nature. 2011;480:201–208. doi: 10.1038/nature10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6:e1001233. doi: 10.1371/journal.pgen.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Mohlke KL, et al. INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS One. 2009;4:e5027. doi: 10.1371/journal.pone.0005027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuo CL, Murphy AJ, Sayers S, Li R, Yvan-Charvet L, Davis JZ, et al. Cdkn2a Is an Atherosclerosis Modifier Locus That Regulates Monocyte/Macrophage Proliferation. Arterioscler Thromb Vasc Biol. 2011;31:2483–2492. doi: 10.1161/ATVBAHA.111.234492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wouters K, Cudejko C, Gijbels MJ, Fuentes L, Bantubungi K, Vanhoutte J, et al. Bone marrow p16INK4a-deficiency does not modulate obesity, glucose homeostasis or atherosclerosis development. PLoS One. 2012;7:e32440. doi: 10.1371/journal.pone.0032440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim JB, Deluna A, Mungrue IN, Vu C, Pouldar D, Civelek M, et al. Effect of 9p21.3 coronary artery disease locus neighboring genes on atherosclerosis in mice. Circulation. 2012;126:1896–1906. doi: 10.1161/CIRCULATIONAHA.111.064881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krimpenfort P, Ijpenberg A, Song JY, van der Valk M, Nawijn M, Zevenhoven J, et al. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature. 2007;448:943–946. doi: 10.1038/nature06084. [DOI] [PubMed] [Google Scholar]
- 22.Ramsey MR, Krishnamurthy J, Pei XH, Torrice C, Lin W, Carrasco DR, et al. Expression of p16Ink4a compensates for p18Ink4c loss in cyclin-dependent kinase 4/6-dependent tumors and tissues. Cancer Res. 2007;67:4732–4741. doi: 10.1158/0008-5472.CAN-06-3437. [DOI] [PubMed] [Google Scholar]
- 23.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 24.Walkley CR, Fero ML, Chien WM, Purton LE, McArthur GA. Negative cell-cycle regulators cooperatively control self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol. 2005;7:172–178. doi: 10.1038/ncb1214. [DOI] [PubMed] [Google Scholar]
- 25.Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat Cell Biol. 2004;6:436–442. doi: 10.1038/ncb1126. [DOI] [PubMed] [Google Scholar]
- 26.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 27.Johnson SM, Torrice CD, Bell JF, Monahan KB, Jiang Q, Wang Y, et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest. 2010;120:2528–2536. doi: 10.1172/JCI41402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy AJ, Bijl N, Yvan-Charvet L, Welch CB, Bhagwat N, Reheman A, et al. Cholesterol efflux in megakaryocyte progenitors suppresses platelet production and thrombocytosis. Nat Med. 2013;19:586–594. doi: 10.1038/nm.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang W, Burg N, Vootukuri S, Coller BS. Increased Smad2/3 phosphorylation in circulating leukocytes and platelet-leukocyte aggregates in a mouse model of aortic valve stenosis: Evidence of systemic activation of platelet-derived TGF-beta1 and correlation with cardiac dysfunction. Blood Cells Mol Dis. 2016;58:1–5. doi: 10.1016/j.bcmd.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matic GB, Rothe G, Schmitz G. Flow cytometric analysis of reticulated platelets. Curr Protoc Cytom. 2001 Aug; doi: 10.1002/0471142956.cy0710s17. Chapter 7:Unit 7.10. doi: 10.1002/0471142956.cy0710s17. [DOI] [PubMed] [Google Scholar]
- 31.Sonkar VK, Kulkarni PP, Dash D. Amyloid beta peptide stimulates platelet activation through RhoA-dependent modulation of actomyosin organization. FASEB J. 2014;28:1819–1829. doi: 10.1096/fj.13-243691. [DOI] [PubMed] [Google Scholar]
- 32.Murphy AJ, Sarrazy V, Wang N, Bijl N, Abramowicz S, Westerterp M, et al. Deficiency of ATP-binding cassette transporter B6 in megakaryocyte progenitors accelerates atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:751–758. doi: 10.1161/ATVBAHA.113.302613. [DOI] [PubMed] [Google Scholar]
- 33.Seidelmann SB, Kuo C, Pleskac N, Molina J, Sayers S, Li R, et al. Athsq1 is an atherosclerosis modifier locus with dramatic effects on lesion area and prominent accumulation of versican. Arterioscler Thromb Vasc Biol. 2008;28:2180–2186. doi: 10.1161/ATVBAHA.108.176800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tie G, Messina KE, Yan J, Messina JA, Messina LM. Hypercholesterolemia induces oxidant stress that accelerates the ageing of hematopoietic stem cells. J Am Heart Assoc. 2014;3:e000241. doi: 10.1161/JAHA.113.000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011;121:4138–4149. doi: 10.1172/JCI57559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–1438. [PubMed] [Google Scholar]
- 37.Baughn LB, Di Liberto M, Wu K, Toogood PL, Louie T, Gottschalk R, et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 2006;66:7661–7667. doi: 10.1158/0008-5472.CAN-06-1098. [DOI] [PubMed] [Google Scholar]
- 38.Nording HM, Seizer P, Langer HF. Platelets in inflammation and atherogenesis. Front Immunol. 2015;6:98. doi: 10.3389/fimmu.2015.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy AJ, Tall AR. Disordered haematopoiesis and athero-thrombosis. Eur Heart J. 2016;37:1113–1121. doi: 10.1093/eurheartj/ehv718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magwenzi S, Woodward C, Wraith KS, Aburima A, Raslan Z, et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood. 2015;125:2693–2703. doi: 10.1182/blood-2014-05-574491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cardenas JC, Owens AP, 3rd, Krishnamurthy J, Sharpless NE, Whinna HC, Church FC. Overexpression of the cell cycle inhibitor p16INK4a promotes a prothrombotic phenotype following vascular injury in mice. Arterioscler Thromb Vasc Biol. 2011;31:827–833. doi: 10.1161/ATVBAHA.110.221721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenberg RD, Aird WC. Vascular-bed--specific hemostasis and hypercoagulable states. N Engl J Med. 1999;340:1555–1564. doi: 10.1056/NEJM199905203402007. [DOI] [PubMed] [Google Scholar]
- 44.Previtali E, Bucciarelli P, Passamonti SM, Martinelli I. Risk factors for venous and arterial thrombosis. Blood Transfus. 2011;9:120–138. doi: 10.2450/2010.0066-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Voetsch B, Loscalzo J. Genetic determinants of arterial thrombosis. Arterioscler Thromb Vasc Biol. 2004;24:216–229. doi: 10.1161/01.ATV.0000107402.79771.fc. [DOI] [PubMed] [Google Scholar]
- 46.Matuskova J, Chauhan AK, Cambien B, Astrof S, Dole VS, Piffath CL, et al. Decreased plasma fibronectin leads to delayed thrombus growth in injured arterioles. Arterioscler Thromb Vasc Biol. 2006;26:1391–1396. doi: 10.1161/01.ATV.0000216282.58291.c6. [DOI] [PubMed] [Google Scholar]
- 47.Bhatt DL, Fox KA, Hacke W, Berger PB, Black HR, Boden WE, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med. 2006;354:1706–1717. doi: 10.1056/NEJMoa060989. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.