

Abstract
Familial syndromes of hyperparathyroidism, including multiple endocrine neoplasia type 1 (MEN1), multiple endocrine neoplasia type 2A (MEN2A), and the hyperparathyroidism-jaw tumor (HPT-JT), comprise 2 to 5% of primary hyperparathyroidism cases. Familial syndromes of hyperparathyroidism are also associated with a range of endocrine and non-endocrine tumors, including potential malignancies. Complications of the associated neoplasms are the major causes of morbidities and mortalities in these familial syndromes, e.g., parathyroid carcinoma in HPT-JT syndrome, thymic, bronchial and enteropancreatic neuroendocrine tumors in MEN1, and medullary thyroid cancer and pheochromocytoma in MEN2A. Because of the different underlying mechanisms of neoplasia, these familial tumors may have different characteristics compared to their sporadic counterparts. Large-scale clinical trials are frequently lacking due to the rarity of these diseases. With technological advances and the development of new medications, the natural history, diagnosis and management of these syndromes are also evolving. In this article, we summarize the recent knowledge on endocrine neoplasms in three familial hyperparathyroidism syndromes, with an emphasis on disease characteristics, molecular pathogenesis, recent developments in biochemical and radiological evaluation, and expert opinions on surgical and medical therapies. Because these familial hyperparathyroidism syndromes are associated with a wide variety of tumors in different organs, this review is focused on those endocrine neoplasms with malignant potential.
Keywords: multiple endocrine neoplasia type 1 (MEN1), multiple endocrine neoplasia type 2A (MEN2A), hyperparathyroidism-jaw tumor (HPT-JT), malignant tumor, neuroendocrine tumor
INTRODUCTION
Primary hyperparathyroidism has an estimated prevalence of 0.1% in the United States. Familial forms represent some 2 to 5% of primary hyperparathyroidism and are caused by germline genetic mutations (Table 1). Among these, multiple endocrine neoplasia type 1 (MEN1), multiple endocrine neoplasia type 2A (MEN2A), and the hyperparathyroidism-jaw tumor syndrome (HPT-JT) are familial syndromes of hyperparathyroidism that are associated with a spectrum of endocrine tumors and other malignancies. Some of these familial endocrine tumors behave very differently from their sporadic counterparts, showing distinct clinical characteristics and disease courses. Many of these tumors also have a significant malignant potential and require different treatment approaches. Being familiar with their unique features and natural history may facilitate diagnosis and help to establish individualized management to improve outcomes. We herein summarize and update the knowledge on endocrine neoplasms with malignant potential found in familial syndromes of hyperparathyroidism, including 1) HPT-JT associated parathyroid carcinoma; 2) MEN1-associated nonfunctioning pancreatic neuroendocrine tumors (pNET), gastrinoma and insulinoma, thymic and bronchial NET (sometimes referred to as “carcinoid tumors”); and 3) MEN2A-associated medullary thyroid cancer and pheochromocytoma. Because of space limitations we favor the citation of recent review articles, wherein the curious reader can easily find reference to the original studies.
Table 1.
Familial Causes of Hyperparathyroidism
| Disorder | Germline gene mutation | Molecular pathology | Endocrine neoplasms with malignant potential |
|---|---|---|---|
| Familial syndromes leading to primary hyperparathyroidism and including endocrine tumors with malignant potential | |||
| Multiple endocrine neoplasia type 1 syndrome | MEN1 | loss-of-function | malignant duodenopancreatic neuroendocrine tumor, thymic and bronchial neuroendocrine tumors |
| Multiple endocrine neoplasia type 2A syndrome | RET | gain-of-function | medullary thyroid cancer; malignant pheochromocytoma |
| Hyperparathyroidism-jaw tumor syndrome | CDC73 | loss-of-function | parathyroid carcinoma |
|
| |||
| Familial disorders leading to primary hyperparathyroidism only | |||
| Familial hypocalciuric hypercalcemia | CASR | loss-of-function | not applicable |
| Neonatal severe primary hyperparathyroidism | CASR | loss-of-function | not applicable |
| Familial isolated hyperparathyroidism(CDC73, MEN1, CASR mutation testing negative) | unknown | unknown | not applicable |
PARATHYROID CANCER IN THE HYPERPARATHYROIDISM-JAW TUMOR SYNDROME
HPT-JT is an autosomal-dominant familial cancer syndrome caused by inactivating germline mutation of the Cell Division Cycle 73 (CDC73) gene (formerly called HRPT2), with incomplete penetrance and variable expression (Bradley, et al. 2006; Carpten, et al. 2002). Because of its incomplete penetrance, patients with germline CDC73 mutation can present with a spectrum of phenotypes including seemingly sporadic parathyroid cancer, familial isolated hyperparathyroidism (FIHP) with or without parathyroid cancer, or full expression of HPT-JT (Sharretts and Simonds 2010). Patients with HPT-JT may manifest hyperparathyroidism and other medical conditions, including fibro-osseous tumors of the maxilla or mandible (histologically classified as cemento-ossifying fibromas), kidney tumors and uterine lesions. Hypercalcemia and its complications (such as nephrolithiasis) and bone disease are major causes of mortality and morbidity in primary hyperparathyroidism. Patients of HPT-JT also have an increased risk of developing parathyroid carcinoma, ranging from 15% to 37.5% in different case series (Mehta, et al. 2014; Sharretts and Simonds 2010). For comparison, parathyroid carcinoma only accounts for 0.005% of total cancer cases in National Cancer Database Report (Hundahl, et al. 1999). Interestingly, somatic inactivating CDC73 mutations are strongly implicated in sporadic parathyroid carcinoma and have been found in up to 70% of such cancers (Do Cao, et al. 2015; Shattuck, et al. 2003).
HPT-JT is an extremely rare disease. The available literature includes only case reports, case series and retrospective studies of small sample size. The prevalence of this disease in general population is unknown. Cases of HPT-JT have been reported in the Australia, US, Canada, Europe, Japan, India, China, Thailand and France (Bricaire, et al. 2013; Howell, et al. 2003; Khadilkar, et al. 2015; Kong, et al. 2014; Kutcher, et al. 2013; Mathews, et al. 2015; Shibata, et al. 2015; Sriphrapradang, et al. 2014), suggesting that this disease has no ethnic preference. Neither was gender preference found in a large patient cohort (Asare, et al. 2015).
Clinical manifestations
The vast majority of parathyroid carcinomas are functioning tumors that cause clinical symptoms or signs related to severe primary hyperparathyroidism and hypercalcemia, such as bone or joint pain, fatigue, nephrolithiasis, muscle weakness, constipation, reduced bone mass, and psychiatric abnormalities. Distinct from primary hyperparathyroidism due to parathyroid adenoma, patients with parathyroid cancer (either sporadic or in the context of HPT-JT or FIHP) usually present with higher calcium levels and more severe symptoms (Shane 2001; Wei and Harari 2012). Total serum calcium levels are usually more than 13 mg/dl and may be even higher than 16 mg/dl, with parathyroid hormone levels at least three to ten times of the upper normal limit and often in the range of 200–1800 pg/ml (Mehta et al. 2014; Sriphrapradang et al. 2014; Wei and Harari 2012). Non-functioning parathyroid carcinoma has been reported in a normocalcemic CDC73 mutation carrier (Guarnieri, et al. 2006). Patients with HPT-JT may also have or have had silent or clinically evident cemento-ossifying fibromas of the maxilla or mandible or uterine abnormalities.
Parathyroid carcinoma can cause compressive symptoms due to mass effect such as dysphagia, dyspnea, dysphonia or dysarthria (Shane 2001; Wei and Harari 2012). Parathyroid carcinomas are likely to be much bigger than parathyroid adenomas, with a diameter typically 2–3 cm or greater and a weight in the range of 2 –10 grams (Asare et al. 2015; Chiofalo, et al. 2014; Harari, et al. 2011; Siu, et al. 2011; Wei and Harari 2012). Because of their large size and frequently firm texture, parathyroid carcinomas can often be palpated on physical exam (Shane 2001; Wei and Harari 2012). Patients with parathyroid carcinoma may also have complaints related to distant metastasis. The most common sites of metastasis include upper mediastinum, lung, pleura and bone (Do Cao et al. 2015).
Molecular pathophysiology
The CDC73 gene encodes the 531 amino acid protein parafibromin (also known as protein CDC73 or CDC73p) (Bradley et al. 2006; Carpten et al. 2002). Parafibromin is a presumed tumor suppressor protein because germline-inactivating mutation of the CDC73 gene can result in full or partial expression of the clinical syndrome of HPT-JT. Parafibromin contains both nuclear and nucleolar localization signals (Bradley, et al. 2007; Dehghani, et al. 2005; Hahn and Marsh 2005, 2007), and its C-terminal region bears significant homology to the C-terminal region of yeast protein Cdc73p (Newey, et al. 2010). Like yeast Cdc73p, human parafibromin physically interacts with the components of the Polymerase Associated Factor 1 (PAF1) complex. The PAF1 complex plays a role in transcriptional regulation of RNA polymerase II (Jaehning 2010; Newey, et al. 2009). In eukaryotes, parafibromin and the PAF1 complex play roles in the recruitment and activation of histone modification factors, transcriptional elongation, chromatin remodeling, and the recruitment of 3′ end processing factors necessary for accurate termination of transcription, such as cleavage and polyadenylation specificity factor (CPSF) and cleavage stimulation factor (CStF) (Jaehning 2010; Newey et al. 2010). Downregulation of parafibromin increases levels of c-Myc, a proto-oncogene and established regulator of cell growth (Lin, et al. 2008), and enhances expression of c-Myc target genes (Jaenicke, et al. 2016). Despite this knowledge of the cellular functions of parafibromin, our understanding of the necessary links between the loss of parafibromin function and the development of parathyroid cancer remains incomplete.
Diagnosis
When a patient presents with severe symptomatology of primary hyperparathyroidism and significantly elevated serum calcium and parathyroid hormone levels, it should trigger a high index of suspicion for parathyroid cancer and possible germline CDC73 mutation. Such a presentation should prompt careful elaboration of the family medical history for benign or malignant parathyroid disease and/or fibro-osseous jaw tumors. In patients with suspected or proven parathyroid cancer, genetic investigation for germline CDC73 mutation and jaw imaging should be considered to rule out HPT-JT. Besides CDC73 mutations that can be detected by conventional mutational analysis, intragenic deletion of CDC73 has also been reported in patients with familial hyperparathyroidism and parathyroid carcinoma (Korpi-Hyovalti, et al. 2014). In a French cohort, one third of patients were found to have germline gross deletion of CDC73 gene (Bricaire et al. 2013). Therefore, intragenic and gross gene deletion should be assessed as a part of genetic analysis in patients with a high index of suspicion but negative CDC73 mutational testing. Partial or complete gene deletion can be detected by methods such as exon array comparative genome hybridization. Patients with parathyroid carcinoma have a high risk of lymph node (15–30%) and distal metastasis (33%) at their initial presentation (Wei and Harari 2012). Therefore, multiple imaging modalities are recommended in the initial workup.
No single radiological modality has sufficient sensitivity and specificity for localization of parathyroid carcinoma. In one series, technetium-99m sestamibi scanning made the correct localization in only 46.2% cases (Mehta et al. 2014). In another series, the sensitivity of sestamibi scan and ultrasonography for parathyroid carcinoma were 79% and 69%, respectively (Kebebew, et al. 2001). The combination of sestamibi scan and neck ultrasound is recommended in the initial workup (Wei and Harari 2012) (Figure 1). On ultrasound, parathyroid carcinoma is typically found as a solid mass posterior to the thyroid gland. Compared to benign parathyroid lesions, on ultrasound parathyroid carcinomas usually appear larger, more hypoechoic, hypervascular, heterogeneous, lobulated with irregular borders, and are associated with a higher depth/width ratio and infiltration into surrounding tissues (Do Cao et al. 2015; Hara, et al. 2001; Sidhu, et al. 2011).
Figure 1.

Pre-operative images of a parathyroid carcinoma located in the inferior anterior left neck of a HPT-JT patient. A: Ultrasonographic left longitudinal view showed a large 3.5 × 2 cm, hypoechoic, and heterogeneous lesion with irregular border suspicious for parathyroid carcinoma (arrow). B: Doppler image showed that the lesion is hypervascular. C: Neck CT image showed a heterogeneous mass lesion, marked by the arrow, with internal cystic/necrotic changes located inferiorly to the left lower pole of the thyroid gland. D: Technetium-99m pertechnetate scan of the neck showed normal appearing thyroid glands. E: Sestamibi scan of the neck showed an area of excess increased uptake seen just below the left lobe of the thyroid (arrow). F: Sestamibi-technetium subtraction image showed an increased uptake in a parathyroid lesion that was later proven to be a parathyroid carcinoma by surgical pathology (arrow).
When sestamibi scan is negative, thin-cut CT may be helpful in localizing disease (Harari, et al. 2008). In patients with locally advanced or metastatic parathyroid carcinoma, CT or MRI of the neck/mediastinum/chest/abdomen, technetium bone scan, and 18F-fluorodeoxyglucose PET/CT scanning can all be used as a part of metastasis and recurrence workup (Do Cao et al. 2015; Wei and Harari 2012).
Parathyroid biopsy is not recommended as an initial step to establish the diagnosis if parathyroid cancer is suspected, due to possibility of tumor rupture and seeding. In addition, it is technically difficult to differentiate benign from malignant tumor in histopathology exam using the small sample obtained through fine needle biopsy. Misdiagnosis and confusion between parathyroid carcinoma and Hürthle cell thyroid lesion have been reported (Sriphrapradang et al. 2014). However, in patients with metastatic tumors, when seeding is of less concern, biopsy of metastatic lesions can help establish the diagnosis by histological examination and determination of the parathyroid hormone level in the aspirated tissue (Harari et al. 2011).
Upon surgical pathologic review, certain features, including fibrous bands forming trabecular architecture, capsular invasion, vascular or lymph node invasion, and increased mitotic activity and aneuploidy, are strongly indicative of parathyroid carcinoma (Gao, et al. 2010; Schantz and Castleman 1973). Immunohistochemical staining of parathyroid carcinoma is typically positive for chromogranin A and PTH, and negative for neuropeptide, calcitonin and thyroglobulin (Sriphrapradang et al. 2014). A recent study of 24 parathyroid cancers and 14 benign adenomas provided evidence that employment of an immunohistochemical panel has better sensitivity and specificity than any single marker to diagnose parathyroid cancer, when used in conjunction with classical histopathology (Truran, et al. 2014). For instance, immunohistochemical analysis of parathyroid cancers usually demonstrates positive staining for PGP9.5, galectin-3 and Ki67, but negative staining for nuclear parafibromin (Truran et al. 2014).
Management
Proper screening for HPT-JT associated tumors is recommended to start at age five to ten in asymptomatic patients with germline CDC73 gene mutation found through genetic testing (Jackson MA 2015; Pichardo-Lowden, et al. 2011). Screening includes biochemical testing every 6–12 months, and panoramic dental imaging and renal ultrasound every 5 years (Jackson MA 2015). The follow up schedule should be individualized. First-degree relatives of patients with parathyroid cancer or HPT-JT should also be genetically screened for CDC73 mutation (when mutation is known) or periodically screened for primary hyperparathyroidism.
Surgery is the primary treatment modality for parathyroid carcinoma. In general, patients should be treated in specialized centers with a high volume of endocrine surgical cases. Primary parathyroid carcinoma should be removed by en bloc resection. Due to the high recurrence rate of hyperparathyroidism (about 20%) in HPT-JT patients, some experts recommend en bloc resection of parathyroid tumors with bilateral neck exploration (Mehta et al. 2014). Other authors recommend ipsilateral thyroidectomy and complete central neck dissection for malignant tumors (Pichardo-Lowden et al. 2011), or use of radiological evidence of multiglandular involvement to guide subtotal parathyroidectomy and/or bilateral neck exploration (Haciyanli, et al. 2011; Pichardo-Lowden et al. 2011). For initial surgery, we would recommend en bloc resection for a lesion highly suspicious for parathyroid carcinoma, with strong consideration for bilateral neck exploration guided by radiological findings for other candidate lesions. The risk of nerve injury or other potential surgical complications must be weighed against the risk of recurrence and the likelihood of achieving surgical remission.
After the initial surgery, remission is defined as maintaining normal serum calcium and PTH level for more than 6 months, persistent disease as the return of hypercalcemia within 6 months of operation, and recurrence disease as recurrent hypercalcemia following 6 months of normocalcemia (Mehta et al. 2014).
The calcimimetic cinacalcet can be used in parathyroid carcinoma patients as a palliative or therapeutic treatment for hypercalcemia. Using the dose ranging from 30 mg twice daily to 90 mg four times daily, the greatest reduction of hypercalcemia were observed in patients with higher calcium levels (Silverberg, et al. 2007).
The U.S. Food and Drug Administration approved Denosumab in 2010 for osteoporosis in postmenopausal women, as well as for prevention/treatment of skeleton-related events in patients with bone metastasis from solid tumors. Denosumab binds to and inhibits the receptor activator of nuclear factor kappa-B ligand (RANKL) and is a potent inhibitor of bone resorption. There is no clinical trial that has studied the efficacy and risk of denosumab in patients with parathyroid carcinoma. Several case reports found denosumab led to successful control of hypercalcemia in patients with advanced parathyroid cancer and intractable hypercalcemia, but long-term follow up is still lacking (Bowyer, et al. 2013; Jumpertz von Schwartzenberg, et al. 2015; Karuppiah, et al. 2014).
Parathyroid carcinoma is not sensitive to radiation therapy or chemotherapy. Successful immunotherapy using intradermal injection of immunogenic peptides to stimulate antibodies against endogenous PTH has been reported (Betea, et al. 2004; Horie, et al. 2010), but clinical trials are lacking and its risks and efficacy still need to be further tested.
Prognosis
Median overall survival of HPT-JT patients ranges from 8.9 years (Mehta et al. 2014) to 14.3 years(Harari et al. 2011) from the date of diagnosis. Based on a retrospective review from 1985 to 2006 in a national cancer database, the 5- and 10-year relative survival rates of parathyroid carcinoma were 82.3% and 66%, respectively (Asare et al. 2015). Several risk factors, including large tumor size, older age at diagnosis and male gender, are associated with shorter survival. In contrast, lymph node status or radical parathyroid surgery was not found to be significant in predicting survival (Asare et al. 2015). Approximately 50% of parathyroid carcinoma patients developed persistent or recurrent disease (Wei and Harari 2012). The 5-year survival rate after recurrence was only about 50% (Silverberg et al. 2007), compared to 82.3% in all parathyroid carcinoma cases.
NONFUNCTIONING PANCREATIC NEUROENDOCRINE TUMORS IN MEN1
MEN1 is a rare autosomal-dominant familial cancer syndrome caused by germline loss-of-function mutation of the MEN1 tumor suppressor gene (Marx, et al. 1999). The syndrome is characterized by a predisposition to endocrine tumors in pituitary, parathyroid and enteropancreatic endocrine cells, although tumors in several other endocrine and non-endocrine tissues are also associated with the syndrome (Schussheim, et al. 2001). The common malignancies in MEN1 include functional and non-functional enteropancreatic neuroendocrine tumors, as well as thymic and bronchial NET.
Pancreatic neuroendocrine tumors (pNET) are clinically evident in 30–75% of MEN1 patients and present in 80 – 100% at postmortem examination, exhibiting an age-dependent penetrance (Goudet, et al. 2010; Norton, et al. 2015). In MEN1, the prevalence of nonfunctioning pNET is much higher than functioning pNET such as gastrinoma, insulinoma and glucagonoma (Table 2) (Yates, et al. 2015). The penetrance of nonfunctioning pNET is 34% at age 50, establishing it as the most frequent enteropancreatic tumor in MEN 1 patients (Triponez, et al. 2006).
Table 2.
Clinical features of endocrine neoplasms in MEN1 and MEN2A
| MEN1 | MEN2A | ||||||
|---|---|---|---|---|---|---|---|
| Nonfunctioning pNET | Gastrinoma | Insulinoma | Thymic NET | Bronchial NET | MTC | Pheochromocytoma | |
| Prevalence (%) | 96% | 30% | 18% | 2–8.2% | 13% | 95% | 50% |
| Average onset (year old) | 44.7 | 40–50 | 34.8 | NA (dominant in male smokers) | NA | second decades (variable based on genetic risks) | 32 |
| Malignancy rate | 50% | 23% | ~10% | 25% | NA | ~100% | <1% |
| Clinical symptoms/signs | nonspecific | Zollinger-Ellison syndrome | Whipple Triad; adrenergic and neuroglycopenic symptoms | cough, chest discomfort | cough, chest discomfort | neck mass or pain | elevated BP, headache, sweating, palpitations |
| Major biochemical tests | CgA | Gastrin | Insulin, proinsulin, C-peptide, glucose | NA | gastrin(+/−) | calcium/calcitonin/CEA | urine and plasma catecholamine and metanephrine |
| Major radiological tests (sensitivity if available) | EUS(~100%), CT(81%) & MRI(88%) | EUS, CT & MRI | Ca-stimulated angiography(>90%), intraoperative US(98%) | CT & MRI (~100% for lesion>1cm) | CT & MRI (~100% for lesion>1cm) | US | CT, MRI, MIBG |
| Surgical intervention | observe (<1cm); resection with LND (>2–2.5cm) | observation or surgical resection proceeded by parathyroidectomy | distal pancreatecomy or enucleation, duodenopancreatecomy based on locations | prophylactic thymectomy with parathyrodectomy. En bloc resection for tumor | observe or surgical resection for functioning or large tumors | prophylactic thyroidectomy based on risk; | consider partial or cortical sparing adrenalectomy |
| Medical intervention | SST analog; cytotoxic, sunitinib, everolimus for progressive/metastatic. | potent PPI w/escalated dose | diet, diazoxide, SST analog | may consider SST analog | may consider SST analog | EBRT, SST analog, TKIs (vandetanib, cabozantinib) | α, β-adrenergic receptor blocker |
| 10 year survival rate | 50% | 85% | 25–36% | 71% | variable | likely not to be affected | |
CEA: carcinoembryonic antigen; CgA: chromogranin A; CT: computed tomography; EBRT: external beam radiotherapy; EUS: endoscopic ultrasound; LND: lymph node dissection; MEN1: multiple endocrine neoplasia type 1; MEN2A: multiple endocrine neoplasia type 2A; MIBG: metaiodobenzylguanidine scan; MRI: magnetic resonance imaging; MTC: medullary thyroid cancer; NA: not available or not applicable; NET: neuroendocrine tumor; pNET: pancreatic neuroendocrine tumor; PPI: proton pump inhibitor; SRS: somatostatin receptor scintigraphy; SST analog: somatostatin analogue; TKI: tyrosine kinase inhibitor; US: ultrasound
The natural history of MEN1 has been significantly impacted by the changes in the epidemiology of pNET over the past decade. Studies in France and the US have shown that the causes of death in MEN1 have shifted from functioning pNET to nonfunctioning malignant pNET and thymic tumors (Goudet et al. 2010; Ito, et al. 2013a). The five- and ten-year survival rates for nonfunctioning pNET are 75% and 50%, compared to 90% and 85% for functioning pNET (Jensen, et al. 2008). The median overall survival is significantly longer for MEN1 patients with functioning pNET versus those with nonfunctioning tumors (Kouvaraki, et al. 2006).
Clinical features
Nonfunctioning pNET may be clinically silent in MEN1 patients and detected only by abdominal imaging. Symptoms of malignant nonfunctioning pNET are related to local invasion, lymph node involvement and distant metastases. Common locations for metastasis include liver (67%), lymph node (14%), and lung (10%) (Triponez et al. 2006).
Molecular pathophysiology
The human MEN1 gene encodes the ubiquitously expressed 610 amino acid protein menin (Chandrasekharappa, et al. 1997). Because germline frameshift, nonsense, missense, and deletion mutations in MEN1 expected to cause loss-of-function result in the clinical syndrome of MEN1, it is operationally a tumor suppressor. The menin protein contains multiple nuclear localization signals and can function as an epigenetic activator or repressor of gene transcription in different cellular contexts through interaction with distinct chromatin-modifying protein complexes (Yates et al. 2015). The crystal structure of human menin has been solved and supports its role as a scaffolding protein that can have opposite effects on transcription (Huang, et al. 2012).
Menin interacts with a number of proteins that are involved in transcriptional regulation, genome stability, cell division and cellular proliferation. These proteins include the transcriptional regulators c-Jun and JunD, members of the NF-κB family of transcription factors, Smad family transcription factors including Smad3 and the bone morphogenetic protein-2 regulated Smad1 and Smad5, the osteoblast regulator Runx2, and the forkhead transcription factor FOXN3 (CHES1) involved in the DNA damage response (Yates et al. 2015). Despite these molecular insights, an understanding of the critical links between the loss of such protein-protein interactions in MEN1 and tumorigenesis in susceptible neuroendocrine cells is lacking.
Diagnosis
The mean time from the diagnosis of MEN1 to recognition of nonfunctioning pNET is about 5 years (Triponez et al. 2006). Both CT and MRI have relatively high sensitivity (81% and 88%, respectively) for detection of pNET, with excellent specificity and positive predictive values. Endoscopic ultrasound (EUS) is the most sensitive (close to 100%) imaging modality for pNET, and can detect lesions as small as 0.3 cm as well as lymph node metastases (Yates et al. 2015). Because of its high sensitivity, some authors recommend the inclusion of EUS in baseline screening and follow-up surveillance of nonfunctioning pNET (Tonelli 2014). Its limitations include poor visualization of distal pancreatic lesions and being highly operator-dependent. Both 111Indium- and 68Gallium-based somatostatin receptor scintigraphy PET/CT can facilitate tumor staging, but have limited availability and radiation exposure risk (Yates et al. 2015). In a recent study of MEN1 patients, 68Ga-DOTATATE PET/CT was reported to show no better sensitivity or specificity in detecting MEN1-related tumors (including pNET) than combined conventional imaging that included invasive endoscopic ultrasound (Lastoria, et al. 2015). However a larger study, comparing only non-invasive imaging modalities, demonstrated a clear superiority of 68Gallium-DOTATATE PET/CT over conventional octreotide scintigraphy and CT for the detection of primary and secondary neuroendocrine tumors, including pNET, in MEN1 patients (Sadowski, et al. 2015).
The utility and accuracy of circulating tumor markers for the early recognition and diagnosis of nonfunctioning pNET is generally poor. For instance, the sensitivity of chromogranin A (CgA) to diagnose pNET was only 0.33 (de Laat, et al. 2013). However, if CgA is elevated at diagnosis, it can be used to evaluate treatment efficacy over time in advanced cases (Kunz, et al. 2013). Interestingly, fasting serum pancreatic polypeptide levels, but not CgA or gastrin, correlated significantly with the number of primary and metastatic lesions detected by 68Gallium-DOTATATE PET/CT in a recent study of neuroendocrine tumors in MEN1 patients, however no pNET-specific analysis was reported (Sadowski et al. 2015). No urinary markers have yet proven useful for the early recognition and diagnosis of nonfunctioning pNET.
Pathological staging is essential prior to initiation of systemic therapy for pNET, because treatment algorithms for well-differentiated and poorly differentiated pNET are different (Kunz et al. 2013). The risk of malignancy significantly increases with tumor size (Tonelli 2014). Besides tumor size, Ki-67 index and mitotic rate are critical determinants for assessing prognosis. The latter two factors are used to assign tumor grade during pathologic examination of pNET. When the mitotic rate and Ki-67 index are discordant, the higher grade is assigned (Kunz et al. 2013). Immunohistochemical staining for CgA and synaptophysin is recommended.
Treatment
Typically nonfunctioning pNETs can be observed in asymptomatic MEN1 patients with low tumor burden or tumor size less than 1 cm. Some authors recommended surgical intervention for nonfunctioning pNET greater than 1 cm due to their potential risk of hepatic metastasis (Imamura 2010; Machado 2012). Other experts however, recognizing the significant mortality and morbidity of pancreatic surgery on one hand and the low risk of metastasis and death in MEN1 patients with nonfunctioning pNETs < 2 cm on the other, recommend reserving surgery for rapidly growing tumors or those tumors > 2 cm (Triponez et al. 2006). Enucleation or surgical resection with adjacent lymph node dissection can be considered for lesions larger than 2 or 2.5 cm (Kunz et al. 2013). Pancreaticoduodenectomy and distal pancreatectomy should be reserved for a small number of patients because of high risk of developing exocrine and endocrine pancreas insufficiency and increased morbidity (Bartsch, et al. 2013; Vezzosi, et al. 2015).
In patients with metastatic nonfunctioning pNET, resection of the primary tumor was associated with better survival (Keutgen, et al. 2016). Resection of single or even multiple surgically amenable hepatic metastatic lesions is recommended by some and can sometimes be curative (Machado 2012). Radiofrequency ablation and trans-arterial chemoembolization can be used to control tumor burden or to treat lesions deep in the hepatic parenchyma (Imamura 2010; Kunz et al. 2013).
Well-differentiated NET in MEN1 usually respond poorly to platinum-based cytotoxic chemotherapy. Streptozocin, 5-flurouracil or temozolomide can be considered in advanced, poorly differentiated pNET for palliative purposes (Kunz et al. 2013). The FDA has approved sunitinib and everolimus for the treatment of progressive metastatic pNET based on positive results from phase III clinical trials. Sunitinib inhibits VEGF receptor signaling. In phase III clinical trials in advanced pNET patients, sunitinib delayed progression of pNET by 6 months and had a favorable adverse events profile (Raymond, et al. 2011). Everolimus inhibits the mTOR pathway signaling. In the RADIANT-3 study, everolimus improved progression-free survival from 4.6 months in the placebo arm to 11 months in the treatment arm (Yao, et al. 2011).
Poorly differentiated pNET may be treated by chemotherapy with platinum based regimen (cisplatin/carboplatin and etoposide for 4–6 cycles). For patients who relapse more than 6 months after the first-line therapy, the same chemotherapy regimen can be repeated; if relapse occurs sooner than 3–6 months, the options for second-line chemotherapy include irinotecan, topotecan, paclitaxel, docetaxel, vinorelbine, gemcitabine, and temozolomide (Kunz et al. 2013).
Despite controversy (Kunz et al. 2013), antitumor effects of somatostatin analogues have been observed in advanced nonfunctioning pNET (Igarashi, et al. 2015; Rinke, et al. 2009). Peptide receptor-conjugated radiotherapy (with 177Lu- or 90Y-labeled somatostatin analogues) is currently under investigation.
Prognosis
The American Joint Committee on Cancer (AJCC) and World Health Organization (WHO) grading systems are useful in predicting pNET disease progression, and tissue immunohistochemistry profile provides additional values (Morin, et al. 2013). Other poor prognostic considerations include a high Ki-67 index/proliferative rate, bone or liver metastasis (Lee, et al. 2015), older age, longer disease history, and associated tumor in the pancreatic head (Keutgen et al. 2016).
It is estimated that at least 50% of pNET eventually recur or metastasize. Their malignant potential correlates with tumor size and extent of lymph node metastasis. Some 50–70% of pNET in the size range of 2 to 3 cm are associated with lymph node metastases, and 25–40% of pNET larger than 4 cm are associated with hepatic metastases (Yates et al. 2015). Patients with nonfunctioning pNET >3 cm have significantly lower survival compared to patients with tumors less than 1 cm or between 1–3 cm (Triponez et al. 2006).
INSULINOMA IN MEN1
In MEN1 patients, insulinoma is the second most common functioning pancreatico-duodenal neuroendocrine tumor after gastrinoma, with a prevalence of 10 to 18% in MEN1 (Bartsch et al. 2013; Goudet et al. 2010). Insulinoma in MEN1 patients has a significantly higher recurrence rate (21%) than sporadic insulinoma (7%) after 20-year follow-up (Service, et al. 1991).
Clinical features
Neuroglycopenic and adrenergic symptoms such as confusion, visual disturbances, and diaphoresis are typical presenting symptoms of insulinoma. Insulinoma can be the first presentation of MEN1, with a median age of 30 at diagnosis (Bartsch et al. 2013). In a Japanese MEN1 cohort, insulinoma was diagnosed at median age of 35, with 24% recognized before age of 20 (Sakurai, et al. 2012). Insulinomas are usually benign and only 10% cases are malignant.
Diagnosis
Diagnosis of insulinoma in patients with typical neuroglycopenic and/or adrenergic symptoms of hypoglycemia requires demonstration of Whipple’s triad, and can be accomplished through supervised fasting (Hirshberg, et al. 2000). Diagnosis of insulinoma in a young patient should prompt genetic testing for MEN1 gene mutation: among MEN1 patients diagnosed before age 21, some 12% presented with insulinoma (Goudet, et al. 2015). MEN1 accounts for about 10% of all insulinoma cases.
Localization of insulinoma in anticipation of surgery should proceed only after biochemical workup has established the diagnosis of insulinoma. Non-invasive localization studies may not prove adequate, since CT and MRI correctly identified insulinoma in fewer than half of cases, and only one-third of cases were detected by trans-abdominal ultrasonography (Taye and Libutti 2015). The successful localization of an insulinoma with 68Gallium-DOTATATE PET/CT was described in a recent case report (Sadowski, et al. 2014). Calcium-stimulated angiography provided correct preoperative localization in nearly 90% of insulinomas (n= 76) in one tertiary referral center (Guettier, et al. 2009). Endoscopic ultrasound also demonstrates high sensitivity for insulinoma localization, especially for pancreatic head lesions (Anderson, et al. 2000). During surgery, the combination of palpation and intra-operative ultrasonography can detect > 90% of insulinomas (Nikfarjam, et al. 2008).
Management
Surgery is the principal treatment modality for insulinoma in MEN1 patients. In MEN1 patients, approximately 50% of insulinomas are located in the tail of the pancreas, with the remainder split roughly evenly between the body and the head (Vezzosi et al. 2015). The choice of operation depends on the presence and location of a dominant lesion. Enucleation or segmental resection can be used for MEN1 patients with a single or dominant lesion (Bartsch et al. 2013). This approach is associated with the fewest post-operative complications but risks a higher recurrence rate. Distal pancreatectomy has the lowest rate of recurrence (Vezzosi et al. 2015). Intraoperative ultrasonography and monitoring serum level glucose/insulin are useful in evaluation of completeness of insulinoma resection (Machado 2012). Diabetes, pancreatic fistula, pancreatitis and sepsis are common complications after surgery (Vezzosi et al. 2015).
Medical therapy may be used for patients who are not surgical candidates, or as a bridging strategy before surgery. Insulinoma patients can be treated with frequent carbohydrate diet, diazoxide, or even corticosteroids (Kunz et al. 2013). Somatostatin analogues, including lanreotide and octreotide, have been used to alleviate symptoms caused by excessive hormone secretion. This approach can be considered in patients with clinically advanced disease and positive somatostatin receptor scintigraphy (Jawiarczyk, et al. 2012).
GASTRINOMA IN MEN1
Approximately 30% of MEN1 patients have gastrinoma, which nearly always manifests as Zollinger-Ellison syndrome (ZES), the constellation of gastric acid hypersecretion with severe acid-related peptic disease and/or diarrhea (Goudet, et al. 2004). Gastrinomas in MEN1 most commonly arise in the duodenum (> 70 %), especially in the first part of the duodenum including the bulb, but can also originate in the pancreas or biliary tree (Tonelli, et al. 2013). MEN1 accounts for about 25% of all ZES.
Clinical features
Clinical findings include epigastric or right upper quadrant pain, diarrhea, esophageal reflux, heartburn or weight loss, which are all related to excessive production of gastric acid. Vomiting may indicate partial or complete gastric outlet obstruction, and hematemesis and/or melena indicate gastrointestinal bleeding. Ulcer perforation may cause acute abdomen with significant complications or even death if not promptly recognized and treated. The clinical symptoms of gastrinoma usually manifest before age 40–50 in MEN1 patients (Gibril, et al. 2004).
Diagnosis
Characteristic clinical symptoms, like severe heartburn or diarrhea, associated with elevated fasting gastrin levels are suggestive but not diagnostic of ZES. The formal diagnosis of ZES usually requires the demonstration of hypergastrinemia with the exclusion of achlorhydria by demonstration of an acidic gastric pH. Somatostatin receptor scintigraphy is the single most sensitive method for imaging primary or metastatic liver lesions in patients with gastrinoma, and is as sensitive as CT, MRI, ultrasound and selective angiography combined (Gibril, et al. 1996). Biopsy through upper GI endoscopy frequently establishes the diagnosis. The majority of gastrinomas in MEN1 arise in the duodenum, whereas sporadic gastrinomas are more commonly located in the pancreas (Jensen, et al. 2006). Although local lymph node involvement can frequently be documented upon initial surgery for gastrinoma in MEN1, the malignant potential of sporadic gastrinomas is greater than those in the context of MEN1 (Jensen et al. 2006; MacFarlane, et al. 1995).
Management
There are proponents of either surgical or non-surgical treatment options for MEN1-associated gastrinoma. Many experts recommend the use of an escalating medical regimen with proton pump inhibitors such as omeprazole and pantoprazole to control symptoms and acid (Ito, et al. 2013b), pointing to the difficulty of establishing a biochemical cure via surgery in MEN1 patients (MacFarlane et al. 1995). Other centers advocate for surgical intervention for duodenal and pancreatic gastrinoma in MEN1, citing significant biochemical cure rates with aggressive surgery (Imamura, et al. 2011; Lopez, et al. 2013). Since correction of primary hyperparathyroidism significantly decreases gastrin level and improves basal acid output (Norton, et al. 2008), surgical treatment for primary hyperparathyroidism is generally recommended prior to any surgery for gastrinoma (Krampitz and Norton 2013; Machado 2012).
BRONCHIAL AND THYMIC NEUROENDOCRINE TUMORS IN MEN1
Thymic NET (sometimes referred to as “carcinoid tumors”), occur in 2–8% of individuals with MEN1. These tumors have a male predominance and a high mortality rate, with a 10-year survival rate between 25–36.1%. On the contrary, bronchial NET are more common (13%) in MEN1, with less gender bias and a more favorable 10-year survival rate (71.1%) (de Laat, et al. 2014; Giusti, et al. 1993; Jensen et al. 2008; Singh Ospina, et al. 2015). Risk factor and mortality analysis of a large cohort of European patients with MEN1 confirmed that thymic NET carries a significantly higher mortality with a hazard ratio of 4.29, while bronchial NET usually follows an indolent course and is not associated with an increased risk of death (Goudet et al. 2010).
Clinical features
Patients with thymic or bronchial NET can be asymptomatic or have chest discomfort or cough. Although most thymic and bronchial NET in MEN1 are hormonally nonfunctioning (Singh Ospina et al. 2015), functioning NET can rarely produce symptoms related to the excessive hormone release. Carcinoid syndrome resulting from primary thymic or bronchial NET is rare. Adrenocorticotropic hormone (ACTH) secreting and growth hormone-releasing hormone (GHRH) secreting thymic tumors have been reported in MEN1 patients resulting in ectopic Cushing’s syndrome and acromegaly respectively (Boix, et al. 2002; Li, et al. 2014; Takagi, et al. 2006). In patients with advanced disease, symptoms and signs can develop from local invasion, mediastinal lymphadenopathy, pulmonary and bone metastases (Otake, et al. 2010; Thomas de Montpreville, et al. 2013). No genotype-phenotype correlation has been observed between MEN1 mutation and the presence of thymic or bronchial NET (Goudet, et al. 2009).
Diagnosis
No circulating tumor marker is sensitive enough to be used for diagnosis of thymic or bronchial NET (Goudet et al. 2009). For tumors that are greater than 1cm size, CT and MRI have sensitivity close to 100% to identify the thymic or bronchial NET. The typical finding on CT scan is an irregular mass enhanced with intravenous contrast that may also contain calcification (Figure 2 and 3). Conventional somatostatin receptor scintigraphy has a lower sensitivity (around 75%) to detect thymic NET (Gibril, et al. 2003). Imaging by 18F-fluorodeoxyglucose (FDG) PET/CT may be useful to detect locally invasive lesions and distant metastases (Abe, et al. 2008; Recuero Diaz, et al. 2013). CT-guided needle biopsy of thymic NET or bronchoscopic biopsy of bronchial NET, followed by pathological examination of the specimens, help confirm diagnosis after clinical evaluation and radiological workup.
Figure 2.
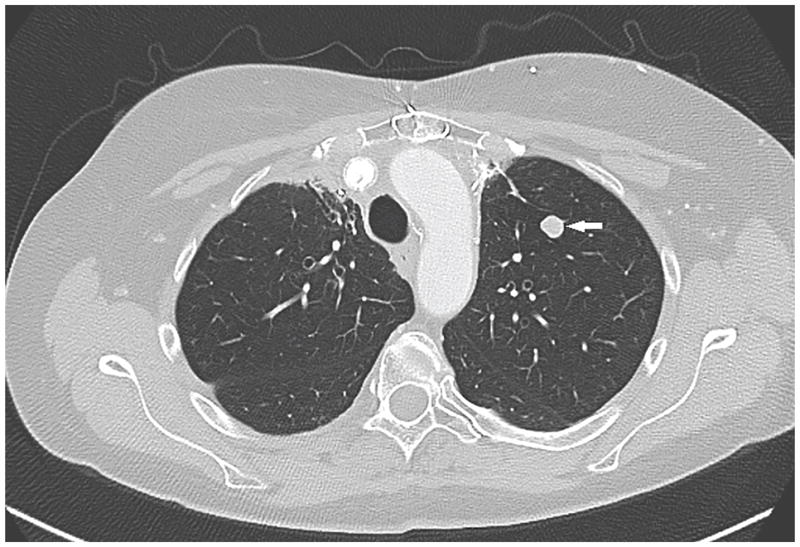
CT of the chest showed a lung nodule of 1.1 cm diameter located in the left upper lobe (marked by arrow) in an MEN1 patient. It was surgically resected and proven to be a well-differentiated pulmonary neuroendocrine tumor (“bronchial carcinoid”).
Figure 3.
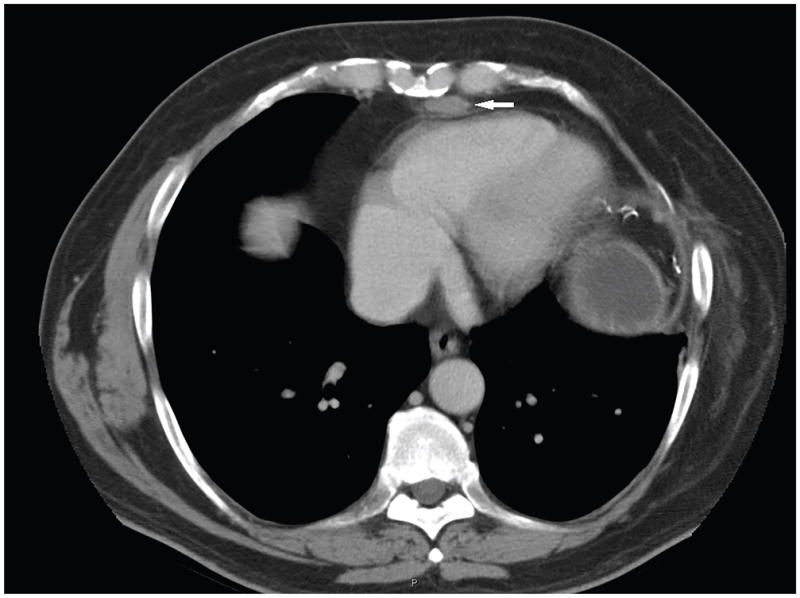
CT of the chest of an MEN1 patient with a history of thymic neuroendocrine tumor and prior surgical resection, showed a 2.5 × 1.2 cm suspicious mediastinal lesion (marked by arrow). It was resected and proven to be a thymic neuroendocrine tumor by surgical pathology.
Management
Because of the malignant potential of thymic NET, prophylactic transcervical thymectomy at the time of initial parathyroidectomy is frequently recommended (Altemir Trallero, et al. 2012; Gibril et al. 2003; Goudet et al. 2009; Recuero Diaz et al. 2013). However, such surgical prophylaxis may not always prevent the occurrence of thymic NET in MEN1 patients (Burgess, et al. 2001; Goudet et al. 2009). Prophylactic thymectomy may have the additional benefit of preventing recurrent or persistent hyperparathyroidism, since in one study of 66 MEN1 patients undergoing initial parathyroid surgery, nearly 30% of patients demonstrated intra-thymic parathyroid tissue following such prophylactic thymectomy (Powell, et al. 2008). Screening for thymic or bronchial NET by chest CT or MRI should be implemented at baseline and every one to two years thereafter (Miller, et al. 2008; Thakker, et al. 2012). It must be recognized however that lifelong surveillance for a low-prevalence condition also carries potential risks, including over-detection and overtreatment, increased anxiety, exposure to radiation and medical cost.
For thymic NET in MEN1 patients, en bloc resection of thymus and adjacent compromised structures is recommended (Recuero Diaz et al. 2013). Observation of bronchial NET with surgical resection of any hormonally functional tumors, tumors causing compressive symptoms, or tumors demonstrating significant interval growth is the main management options.
There is only limited data on medical therapy for non-resectable thymic or bronchial NET in MEN1 patients. There is one case report that a partial response was achieved in a MEN1 patient with thymic NET after cisplatin and etoposide chemotherapy (Amano, et al. 2010). Radiation therapy can be utilized for bone metastases.
MEN2A-ASSOCIATED MEDULLARY THYROID CANCER
Classical MEN2A is a heritable syndrome characterized by medullary thyroid cancer (MTC), pheochromocytoma, and primary hyperparathyroidism (virtually always due to benign disease). MEN2A results from germline mutations in the RET protooncogene and is transmitted in an autosomal dominant fashion. Three other non-classical variants of MEN2A are now recognized: MEN2A with cutaneous lichen amyloidosis, MEN2A with Hirschsprung’s disease, and familial MTC (individuals with RET germline mutations from families who have MTC but not pheochromocytoma or primary hyperparathyroidism). MTC arises from parafollicular cells of the thyroid (C-cells) and often develops in early life in MEN2A with an overall penetrance approaching 100% (Wells, et al. 2015). There is a genotype-phenotype association between different RET mutations and clinical manifestation/prognosis of MTC. Current guidelines recommend prophylactic thyroidectomy depending on the risk associated with the particular RET gene mutation (Wells et al. 2015).
Clinical features
Unlike sporadic MTC that most commonly presents in the fourth to sixth decade of life, the incidence of MEN2-associated MTC peaks in the second decade of life. Index cases usually present with a solitary thyroid nodule with or without cervical lymphadenopathy. In rare cases, MTC can cause Cushing’s syndrome through ectopic production of ACTH or CRH.
Molecular pathophysiology
MEN2A is caused by gain-of-function mutation in the RET protooncogene. RET encodes a transmembrane receptor tyrosine kinase which, in conjunction with glial derived neurotrophic factor (GDNF)-family α co-receptors, binds GDNF family ligands (Wells and Santoro 2009). In 95% of patients, classical MEN2A is associated with germline missense mutations in RET codons 609, 611, 618, or 620 of exon 10 or codon 634 of exon 11, that map to the receptor’s extracellular cysteine-rich domain (Frank-Raue and Raue 2015). MEN2A with cutaneous lichen amyloidosis is nearly always associated with mutation of codon 634, while patients with MEN2A and Hirschsprung’s disease typically harbor mutations involving RET exon 10 (Frank-Raue and Raue 2015).
Diagnosis
Neoplastic C-cells of thyroid gland have the potential to secrete several hormones or biogenic amines, including ACTH, β-melanocyte stimulating hormone, calcitonin, carcinoembryonic antigen (CEA), and chromogranin A. Circulating calcitonin and CEA levels are related to C-cell mass and are valuable tumor markers in patients with MTC (Wells et al. 2015). Serum calcitonin level has a high sensitivity but a low specificity for the diagnosis of MTC. It can be elevated in many other medical conditions, including hypercalcemia and hypergastrinemia, and can also be abnormal after the use of certain medications, such as proton pump inhibitors, beta-blockers and glucocorticoids (Toledo, et al. 2009). Higher basal levels and stimulated calcitonin levels increase its predicative value for MTC (Karges 2010).
A higher level of calcitonin may also be indicative of lymph node metastases and can be used to stratify disease and guide neck dissection. In one study, when lymph node metastases were present in the ipsilateral central and lateral neck, contralateral central neck, contralateral lateral neck and upper mediastinum, basal calcitonin levels were found to be greater than 20, 50, 200, and 500 pg/ml, respectively (Machens and Dralle 2010). In patients with calcitonin levels greater than 200 pg/ml, bilateral compartment neck surgery should be considered (Machens and Dralle 2010).
Determination of procalcitonin, the precursor peptide of calcitonin, is comparable to calcitonin in diagnosing primary tumor/extrathyroidal extension and confirming biochemical cure. Because it is more stable and has a longer half-life, procalcitonin has been recommended for use in the community setting (Machens, et al. 2014).
CEA is not a specific marker for MTC, and is not useful for diagnosis. Nevertheless, concurrent measurement of CEA and calcitonin has some benefit in recognizing poorly differentiated or invasive MTC (Wells et al. 2015). It can be elevated in presence of positive heterophilic antibody, tobacco smoking, gastrointestinal tract inflammatory disease, benign lung disease, or in non-thyroid malignancies (Wells et al. 2015).
Commonly used imaging tests to detect and follow primary and secondary MTC include neck US, chest CT, liver MRI, bone scintigraphy and axial skeleton MRI. FDG-PET scan is less sensitive and has low prognostic value (Giraudet, et al. 2007).
Genetic testing is a critical step in diagnosing MEN2A, and also has the advantage of identifying patients at risk for MTC to allow for prophylactic thyroidectomy. If a family member of an index case with a known MEN2A-associated RET mutation is asymptomatic and has negative genetic test, biochemical and image testing can be avoided. American Thyroid Association guidelines recommend starting genetic testing by analyzing RET mutations in the exons 8, 10, 11, 13, 14, 15 and 16. If there is a high clinical suspicion for MEN2A in a patient or family without a known RET mutation, a negative initial genetic testing result warrants sequencing of entire coding region of the RET gene because it is exceedingly rare that a patient diagnosed with MEN2A has no RET germline mutation (Wells et al. 2015).
Management
The timing of prophylactic thyroidectomy is based on the specific RET mutation, and in some cases, serum calcitonin level. Bilateral central neck dissection can be considered for patients with elevated calcitonin level. Patients assigned American Thyroid Association risk stratification levels A to C, with normal basal and simulated calcitonin level, may be operated without central neck dissection (Niederle, et al. 2014).
Surgical cure of MTC might be achieved in patients with negative nodal spread or only a few lymph node metastases (Machens and Dralle 2015). There are different opinions regarding neck lymph node dissection. Some authors propose that central lymph node dissection should be considered for all MTC patients, but lateral lymph node dissection be determined by tumor size, location and extent of lymph node disease (Kunz et al. 2013). Other authors recommend the use of radiological/clinical evaluation or calcitonin level to guide lymph node compartment dissection (Wells et al. 2015).
In advanced MTC that has invaded surrounding structures, extensive debulking surgery involving possible laryngectomy, esophagectomy, or laryngopharyngectomy may be indicated based on patient’s life expectancy and other medical comorbidities, in order to maintain maximal functionality of speech and deglutition. Cytoreductive surgery, external beam radiotherapy, embolization, systemic medical therapy, and other nonsurgical therapies can be considered in order to maintain local tumor control or improve life quality (Wells et al. 2015).
In advanced MTC, somatostatin analogues can be used for refractory diarrhea, which is the major clinical symptom of metastatic disease. Many EGFR targeting agents also inhibit the RET tyrosine kinase, and can be considered for systemic therapy (Wells et al. 2015). Two tyrosine kinase inhibitors, vandetanib (Caprelsa) and cabozantinib (Cometriq), are approved by the FDA to be used as first line therapy in progressive metastatic MTC with significant tumor burden. In randomized, double blind, Phase III trials, both vandetanib and cabozantinib showed statistically significant improvement of progression free survival compared to placebo (Elisei, et al. 2013; Wells, et al. 2012).
MEN2A-ASSOCIATED PHEOCHROMOCYTOMA
Pheochromocytomas are neuroendocrine tumors that originate from the catecholamine-synthesizing chromaffin cells of the adrenal medulla or from extra-adrenal chromaffin tissue. MEN2A patients have a 50% lifetime risk to develop pheochromocytoma, which nearly always arise in the adrenal glands (King and Pacak 2014). MEN2A patients have 50–67% chance of developing bilateral disease (Castinetti, et al. 2014; Castinetti, et al. 2016; Rajan, et al. 2015; Wells et al. 2015), compared to 10% in general pheochromocytoma population. Pheochromocytoma in MEN2A is rarely malignant, and is not associated with a shortened survival (Thosani, et al. 2013).
Clinical features
Pheochromocytoma can be the presenting manifestation of MEN2A (Kinlaw, et al. 2005). Pheochromocytomas in MEN2A typically hypersecrete epinephrine and can result in elevated blood pressure, rapid or forceful heartbeat, profound sweating or even hypertensive crisis and cardiac arrest (Zwolak, et al. 2015). MEN2A patients with unilateral pheochromocytoma can develop a contralateral lesion within the next 10 years (Rajan et al. 2015; Wells et al. 2015), underscoring the need for proper follow up surveillance screening.
Diagnosis
Several studies have shown that penetrance of pheochromocytoma is correlated with the ATA (American Thyroid Association) risk classification of RET gene mutations (Machens, et al. 2005; Machens, et al. 2013; Quayle, et al. 2007; Rowland, et al. 2013). Based on these findings, the ATA issued guidelines in 2015 that recommend MEN2A patients carrying high-risk RET germline mutations (such as A883F, C634F/G/R/S/W/Y; as well as M918T in MEN2B) should start screening for pheochromocytoma at age 11, while MEN2A patients with other moderate-risk RET germline mutations should start screening at age of 16 (Wells et al. 2015). Screening for pheochromocytoma in the context of MEN2A consists of measuring free plasma epinephrine or metanephrine and/or 24-hour urinary metanephrines. Once biochemical evidence of pheochromocytoma is established, the recommended imaging modalities include CT, MRI, 123I-metaiodobenzylguanindine (MIBG) scintigraphy, 18F-fluorodopamine (18F-FDA)-PET and 18F-dihydroxyphenylalanine (18F-DOPA)-PET (Taieb, et al. 2014).
Although fewer than 5% of pheochromocytomas in MEN2A are malignant (Ilias and Pacak 2009), the diagnosis can be made based on documentation of extra-adrenal involvement, nodal or distant metastases, or histopathologic features including high Ki-67/mitotic rate and/or abnormal nuclear pleiomorphism.
Management
Biochemically established pheochromocytoma should be surgically removed prior to the surgery for MTC or HPTH, and blood pressure should be controlled with alpha-blockade prior to surgery.
Laparoscopic resection is appropriate for localized disease. In a retrospective study that compared 438 adrenalectomy and 114 adrenal-sparing surgeries in MEN2 patients with pheochromocytoma, no difference was seen in recurrence rate or disease free survival. About half of bilateral pheochromocytoma patients (57%) had adrenal sparing surgery and didn’t become steroid-dependent (Castinetti et al. 2014). Due to the high prevalence of bilateral disease and the relatively low malignant potential of pheochromocytoma in MEN2A, cortical sparing surgery or subtotal adrenalectomy should be considered as an alternative procedure to preserve adrenal cortical function (Asari, et al. 2006; Taieb et al. 2014; Wells et al. 2015).
Open resection is reserved for malignant or invasive pheochromocytoma, with cytoreductive resection for unresectable disease (Kunz et al. 2013). Progressive disease can be treated with external beam radiation, 131I-MIBG treatment in 123I-MIBG imaging-positive cases, or systemic chemotherapy with cyclophosphamide, vincristine, and dacarbazine (CVD).
CONCLUSIONS AND FUTURE DIRECTIONS
Endocrine and non-endocrine tumors are common in familial syndromes of hyperparathyroidism due to germline mutation of tumor suppressors or proto-oncogenes. These tumors often have different malignant potentials and distinct disease courses, compared to their sporadic counterparts in general population. Our recognition of germline RET activation as the originating molecular defect in MEN2A has accelerated the identification of several tyrosine kinase inhibitors as rational and promising therapeutic agents for MEN2A-associated malignancies. On the other hand, mutations in MEN1 and CDC73 genes are loss-of-function mutations, similar to the findings in other tumor suppressor genes, such as APC. It has proven to be very difficult to restore or compensate for the lost function of these mutated tumor suppressor genes through a pharmacologic approach. The experience from other hereditary tumors may be helpful to guide development of new therapies in the future. For instance, in the field of ovarian cancer, there has been success in using poly (ADP-ribose) polymerase (PARP) inhibitors to treat patients with germline BRCA mutation by causing synthetic lethality (Farmer, et al. 2005; Ledermann 2016). Although disease-specific surveillance and treatment strategies are preferred, there is a paucity of randomized clinical trial data to guide management because of the rarity of these familial syndromes. Advances in tumor imaging modalities, such as 68gallium-based somatostatin receptor scintigraphy, and the greater understanding that is emerging from patient registries that support large-scale cooperative clinical studies in Europe and North America on the natural history and therapeutic outcomes of these rare diseases are quite encouraging. To continue these promising developments, it is important that patients with familial syndromes of hyperparathyroidism and rare endocrine malignancies be referred to specialized research-oriented centers that participate in cooperative clinical studies. This approach will allow critical research to be performed at a limited number of cooperating institutes and ensure an adequate volume of patients. Such a strategy, in conjunction with advances in basic and translational science, should allow the thorough and conclusive exploration of novel and promising initiatives to improve the diagnosis, imaging, surveillance, and surgical and medical therapies of these highly morbid endocrine malignancies.
Acknowledgments
The authors thank Drs. Stephen Marx, Lee Weinstein, Monica Skarulis, and Michael Collins for many helpful discussions, as well as their encouragement and support. We also thank Mr. Craig Cochran and the patients, fellows and nursing staff of 5NW and OP9 in the NIH Clinical Center. The Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases supported this research.
Footnotes
Declaration of interest: there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Abe T, Sato M, Okumura T, Shioyama Y, Kiyoshima M, Asato Y, Saito H, Iijima T, Amemiya R, Nagai H. FDG PET/CT findings of thymic carcinoid and bronchial carcinoid in a patient with multiple neuroendocrine neoplasia type1. Clin Nucl Med. 2008;33:778–779. doi: 10.1097/RLU.0b013e318187efef. [DOI] [PubMed] [Google Scholar]
- Altemir Trallero J, Armengod Grao L, Aguillo Gutierrez E, Cabrejas Gomez C, Ocon Breton J, Garcia Garcia B. Thymic carcinoid in the setting of a multiple endocrine neoplasia syndrome (MEN 1). Prophylactic thymectomy? Endocrinol Nutr. 2012;59:142–144. doi: 10.1016/j.endonu.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Amano H, Yamada T, Jujoh T, Kuroda F, Sakao S, Tada Y, Kurosu K, Kasahara Y, Tanabe N, Takiguchi Y, et al. Case of thymic carcinoid associated with multiple endocrine neoplasia type I treated effectively with chemotherapy. Nihon Kokyuki Gakkai Zasshi. 2010;48:855–859. [PubMed] [Google Scholar]
- Anderson MA, Carpenter S, Thompson NW, Nostrant TT, Elta GH, Scheiman JM. Endoscopic ultrasound is highly accurate and directs management in patients with neuroendocrine tumors of the pancreas. Am J Gastroenterol. 2000;95:2271–2277. doi: 10.1111/j.1572-0241.2000.02480.x. [DOI] [PubMed] [Google Scholar]
- Asare EA, Sturgeon C, Winchester DJ, Liu L, Palis B, Perrier ND, Evans DB, Winchester DP, Wang TS. Parathyroid Carcinoma: An Update on Treatment Outcomes and Prognostic Factors from the National Cancer Data Base (NCDB) Ann Surg Oncol. 2015;22:3990–3995. doi: 10.1245/s10434-015-4672-3. [DOI] [PubMed] [Google Scholar]
- Asari R, Scheuba C, Kaczirek K, Niederle B. Estimated risk of pheochromocytoma recurrence after adrenal-sparing surgery in patients with multiple endocrine neoplasia type 2A. Arch Surg. 2006;141:1199–1205. doi: 10.1001/archsurg.141.12.1199. discussion 1205. [DOI] [PubMed] [Google Scholar]
- Bartsch DK, Albers M, Knoop R, Kann PH, Fendrich V, Waldmann J. Enucleation and limited pancreatic resection provide long-term cure for insulinoma in multiple endocrine neoplasia type 1. Neuroendocrinology. 2013;98:290–298. doi: 10.1159/000357779. [DOI] [PubMed] [Google Scholar]
- Betea D, Bradwell AR, Harvey TC, Mead GP, Schmidt-Gayk H, Ghaye B, Daly AF, Beckers A. Hormonal and biochemical normalization and tumor shrinkage induced by anti-parathyroid hormone immunotherapy in a patient with metastatic parathyroid carcinoma. J Clin Endocrinol Metab. 2004;89:3413–3420. doi: 10.1210/jc.2003-031911. [DOI] [PubMed] [Google Scholar]
- Boix E, Pico A, Pinedo R, Aranda I, Kovacs K. Ectopic growth hormone-releasing hormone secretion by thymic carcinoid tumour. Clin Endocrinol (Oxf) 2002;57:131–134. doi: 10.1046/j.1365-2265.2002.01535.x. [DOI] [PubMed] [Google Scholar]
- Bowyer SE, White AM, Ransom DT, Davidson JA. Resistant hypercalcaemia in metastatic parathyroid carcinoma. Med J Aust. 2013;198:559–561. doi: 10.5694/mja12.11243. [DOI] [PubMed] [Google Scholar]
- Bradley KJ, Bowl MR, Williams SE, Ahmad BN, Partridge CJ, Patmanidi AL, Kennedy AM, Loh NY, Thakker RV. Parafibromin is a nuclear protein with a functional monopartite nuclear localization signal. Oncogene. 2007;26:1213–1221. doi: 10.1038/sj.onc.1209893. [DOI] [PubMed] [Google Scholar]
- Bradley KJ, Cavaco BM, Bowl MR, Harding B, Cranston T, Fratter C, Besser GM, Conceicao Pereira M, Davie MW, Dudley N, et al. Parafibromin mutations in hereditary hyperparathyroidism syndromes and parathyroid tumours. Clin Endocrinol (Oxf) 2006;64:299–306. doi: 10.1111/j.1365-2265.2006.02460.x. [DOI] [PubMed] [Google Scholar]
- Bricaire L, Odou MF, Cardot-Bauters C, Delemer B, North MO, Salenave S, Vezzosi D, Kuhn JM, Murat A, Caron P, et al. Frequent large germline HRPT2 deletions in a French National cohort of patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2013;98:E403–408. doi: 10.1210/jc.2012-2789. [DOI] [PubMed] [Google Scholar]
- Burgess JR, Giles N, Shepherd JJ. Malignant thymic carcinoid is not prevented by transcervical thymectomy in multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf) 2001;55:689–693. doi: 10.1046/j.1365-2265.2001.01348.x. [DOI] [PubMed] [Google Scholar]
- Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, Simonds WF, Gillanders EM, Kennedy AM, Chen JD, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002;32:676–680. doi: 10.1038/ng1048. [DOI] [PubMed] [Google Scholar]
- Castinetti F, Qi XP, Walz MK, Maia AL, Sanso G, Peczkowska M, Hasse-Lazar K, Links TP, Dvorakova S, Toledo RA, et al. Outcomes of adrenal-sparing surgery or total adrenalectomy in phaeochromocytoma associated with multiple endocrine neoplasia type 2: an international retrospective population-based study. Lancet Oncol. 2014;15:648–655. doi: 10.1016/S1470-2045(14)70154-8. [DOI] [PubMed] [Google Scholar]
- Castinetti F, Taieb D, Henry JF, Walz M, Guerin C, Brue T, Conte-Devolx B, Neumann HP, Sebag F. MANAGEMENT OF ENDOCRINE DISEASE: Outcome of adrenal sparing surgery in heritable pheochromocytoma. Eur J Endocrinol. 2016;174:R9–r18. doi: 10.1530/EJE-15-0549. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Chiofalo MG, Sparaneo A, Chetta M, Franco R, Baorda F, Cinque L, Granatiero M, D’Agruma L, Pezzullo L, Scillitani A, et al. A novel CDC73 gene mutation in an Italian family with hyperparathyroidism-jaw tumour (HPT-JT) syndrome. Cellular oncology. 2014;37:281–288. doi: 10.1007/s13402-014-0187-3. [DOI] [PubMed] [Google Scholar]
- de Laat JM, Pieterman CR, van den Broek MF, Twisk JW, Hermus AR, Dekkers OM, de Herder WW, van der Horst-Schrivers AN, Drent ML, Bisschop PH, et al. Natural course and survival of neuroendocrine tumors of thymus and lung in MEN1 patients. J Clin Endocrinol Metab. 2014;99:3325–3333. doi: 10.1210/jc.2014-1560. [DOI] [PubMed] [Google Scholar]
- de Laat JM, Pieterman CR, Weijmans M, Hermus AR, Dekkers OM, de Herder WW, van der Horst-Schrivers AN, Drent ML, Bisschop PH, Havekes B, et al. Low accuracy of tumor markers for diagnosing pancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1 patients. J Clin Endocrinol Metab. 2013;98:4143–4151. doi: 10.1210/jc.2013-1800. [DOI] [PubMed] [Google Scholar]
- Dehghani H, Reith C, Hahnel AC. Subcellular localization of protein kinase C delta and epsilon affects transcriptional and post-transcriptional processes in four-cell mouse embryos. Reproduction. 2005;130:453–465. doi: 10.1530/rep.1.00572. [DOI] [PubMed] [Google Scholar]
- Do Cao C, Aubert S, Trinel C, Odou MF, Bayaram M, Patey M. Parathyroid carcinoma: Diagnostic criteria, classification, evaluation. Ann Endocrinol (Paris) 2015;76:165–168. doi: 10.1016/j.ando.2015.03.016. [DOI] [PubMed] [Google Scholar]
- Elisei R, Schlumberger MJ, Muller SP, Schoffski P, Brose MS, Shah MH, Licitra L, Jarzab B, Medvedev V, Kreissl MC, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31:3639–3646. doi: 10.1200/JCO.2012.48.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Frank-Raue K, Raue F. Hereditary Medullary Thyroid Cancer Genotype-Phenotype Correlation. Recent Results Cancer Res. 2015;204:139–156. doi: 10.1007/978-3-319-22542-5_6. [DOI] [PubMed] [Google Scholar]
- Gao WC, Ruan CP, Zhang JC, Liu HM, Xu XY, Sun YP, Wang Q. Nonfunctional parathyroid carcinoma. J Cancer Res Clin Oncol. 2010;136:969–974. doi: 10.1007/s00432-009-0740-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibril F, Chen YJ, Schrump DS, Vortmeyer A, Zhuang Z, Lubensky IA, Reynolds JC, Louie A, Entsuah LK, Huang K, et al. Prospective study of thymic carcinoids in patients with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. 2003;88:1066–1081. doi: 10.1210/jc.2002-021314. [DOI] [PubMed] [Google Scholar]
- Gibril F, Reynolds JC, Doppman JL, Chen CC, Venzon DJ, Termanini B, Weber HC, Stewart CA, Jensen RT. Somatostatin receptor scintigraphy: its sensitivity compared with that of other imaging methods in detecting primary and metastatic gastrinomas. A prospective study. Ann Intern Med. 1996;125:26–34. doi: 10.7326/0003-4819-125-1-199607010-00005. [DOI] [PubMed] [Google Scholar]
- Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore) 2004;83:43–83. doi: 10.1097/01.md.0000112297.72510.32. [DOI] [PubMed] [Google Scholar]
- Giraudet AL, Vanel D, Leboulleux S, Auperin A, Dromain C, Chami L, Ny Tovo N, Lumbroso J, Lassau N, Bonniaud G, et al. Imaging medullary thyroid carcinoma with persistent elevated calcitonin levels. J Clin Endocrinol Metab. 2007;92:4185–4190. doi: 10.1210/jc.2007-1211. [DOI] [PubMed] [Google Scholar]
- Giusti F, Marini F, Brandi ML. Multiple Endocrine Neoplasia Type 1. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, et al., editors. GeneReviews(R) Seattle (WA): University of Washington, Seattle; 1993. University of Washington, Seattle. All rights reserved. [Google Scholar]
- Goudet P, Dalac A, Le Bras M, Cardot-Bauters C, Niccoli P, Levy-Bohbot N, du Boullay H, Bertagna X, Ruszniewski P, Borson-Chazot F, et al. MEN1 disease occurring before 21 years old: a 160-patient cohort study from the Groupe d’etude des Tumeurs Endocrines. J Clin Endocrinol Metab. 2015;100:1568–1577. doi: 10.1210/jc.2014-3659. [DOI] [PubMed] [Google Scholar]
- Goudet P, Murat A, Binquet C, Cardot-Bauters C, Costa A, Ruszniewski P, Niccoli P, Menegaux F, Chabrier G, Borson-Chazot F, et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d’Etude des Tumeurs Endocrines) cohort study among 758 patients. World J Surg. 2010;34:249–255. doi: 10.1007/s00268-009-0290-1. [DOI] [PubMed] [Google Scholar]
- Goudet P, Murat A, Cardot-Bauters C, Emy P, Baudin E, du Boullay Choplin H, Chapuis Y, Kraimps JL, Sadoul JL, Tabarin A, et al. Thymic neuroendocrine tumors in multiple endocrine neoplasia type 1: a comparative study on 21 cases among a series of 761 MEN1 from the GTE (Groupe des Tumeurs Endocrines) World J Surg. 2009;33:1197–1207. doi: 10.1007/s00268-009-9980-y. [DOI] [PubMed] [Google Scholar]
- Goudet P, Peschaud F, Mignon M, Nicoli-Sire P, Cadiot G, Ruszniewski P, Calender A, Murat A, Sarfati E, Peix JL, et al. Gastrinomas in multiple endocrine neoplasia type-1. A 127-case cohort study from the endocrine tumor group (ETG) Ann Chir. 2004;129:149–155. doi: 10.1016/j.anchir.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Guarnieri V, Scillitani A, Muscarella LA, Battista C, Bonfitto N, Bisceglia M, Minisola S, Mascia ML, D’Agruma L, Cole DE. Diagnosis of parathyroid tumors in familial isolated hyperparathyroidism with HRPT2 mutation: implications for cancer surveillance. J Clin Endocrinol Metab. 2006;91:2827–2832. doi: 10.1210/jc.2005-1239. [DOI] [PubMed] [Google Scholar]
- Guettier JM, Kam A, Chang R, Skarulis MC, Cochran C, Alexander HR, Libutti SK, Pingpank JF, Gorden P. Localization of insulinomas to regions of the pancreas by intraarterial calcium stimulation: the NIH experience. J Clin Endocrinol Metab. 2009;94:1074–1080. doi: 10.1210/jc.2008-1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haciyanli M, Oruk G, Ucarsoy AA, Gur O, Genc H. Multiglandular parathyroid carcinoma: case report and review of the literature. Endocr Pract. 2011;17:e79–83. doi: 10.4158/EP11037.CRR. [DOI] [PubMed] [Google Scholar]
- Hahn MA, Marsh DJ. Identification of a functional bipartite nuclear localization signal in the tumor suppressor parafibromin. Oncogene. 2005;24:6241–6248. doi: 10.1038/sj.onc.1208778. [DOI] [PubMed] [Google Scholar]
- Hahn MA, Marsh DJ. Nucleolar localization of parafibromin is mediated by three nucleolar localization signals. FEBS Lett. 2007;581:5070–5074. doi: 10.1016/j.febslet.2007.09.050. [DOI] [PubMed] [Google Scholar]
- Hara H, Igarashi A, Yano Y, Yashiro T, Ueno E, Aiyoshi Y, Ito K, Obara T. Ultrasonographic features of parathyroid carcinoma. Endocr J. 2001;48:213–217. doi: 10.1507/endocrj.48.213. [DOI] [PubMed] [Google Scholar]
- Harari A, Waring A, Fernandez-Ranvier G, Hwang J, Suh I, Mitmaker E, Shen W, Gosnell J, Duh QY, Clark O. Parathyroid carcinoma: a 43-year outcome and survival analysis. J Clin Endocrinol Metab. 2011;96:3679–3686. doi: 10.1210/jc.2011-1571. [DOI] [PubMed] [Google Scholar]
- Harari A, Zarnegar R, Lee J, Kazam E, Inabnet WB, 3rd, Fahey TJ., 3rd Computed tomography can guide focused exploration in select patients with primary hyperparathyroidism and negative sestamibi scanning. Surgery. 2008;144:970–976. doi: 10.1016/j.surg.2008.08.029. discussion 976–979. [DOI] [PubMed] [Google Scholar]
- Hirshberg B, Livi A, Bartlett DL, Libutti SK, Alexander HR, Doppman JL, Skarulis MC, Gorden P. Forty-eight-hour fast: the diagnostic test for insulinoma. J Clin Endocrinol Metab. 2000;85:3222–3226. doi: 10.1210/jcem.85.9.6807. [DOI] [PubMed] [Google Scholar]
- Horie I, Ando T, Inokuchi N, Mihara Y, Miura S, Imaizumi M, Usa T, Kinoshita N, Sekine I, Kamihara S, et al. First Japanese patient treated with parathyroid hormone peptide immunization for refractory hypercalcemia caused by metastatic parathyroid carcinoma. Endocr J. 2010;57:287–292. doi: 10.1507/endocrj.k09e-283. [DOI] [PubMed] [Google Scholar]
- Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, Chen J, Fleuren GJ, Robinson BG, Delbridge LW, Philips J, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003;40:657–663. doi: 10.1136/jmg.40.9.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Gurung B, Wan B, Matkar S, Veniaminova NA, Wan K, Merchant JL, Hua X, Lei M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature. 2012;482:542–546. doi: 10.1038/nature10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundahl SA, Fleming ID, Fremgen AM, Menck HR. Two hundred eighty-six cases of parathyroid carcinoma treated in the U.S. between 1985–1995: a National Cancer Data Base Report. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer. 1999;86:538–544. doi: 10.1002/(sici)1097-0142(19990801)86:3<538::aid-cncr25>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Igarashi H, Hijioka M, Lee L, Ito T. Biotherapy of pancreatic neuroendocrine tumors using somatostatin analogs. J Hepatobiliary Pancreat Sci. 2015;22:618–622. doi: 10.1002/jhbp.227. [DOI] [PubMed] [Google Scholar]
- Ilias I, Pacak K. Diagnosis, localization and treatment of pheochromocytoma in MEN 2 syndrome. Endocr Regul. 2009;43:89–93. [PubMed] [Google Scholar]
- Imamura M. Recent standardization of treatment strategy for pancreatic neuroendocrine tumors. World J Gastroenterol. 2010;16:4519–4525. doi: 10.3748/wjg.v16.i36.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura M, Komoto I, Ota S, Hiratsuka T, Kosugi S, Doi R, Awane M, Inoue N. Biochemically curative surgery for gastrinoma in multiple endocrine neoplasia type 1 patients. World J Gastroenterol. 2011;17:1343–1353. doi: 10.3748/wjg.v17.i10.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Igarashi H, Uehara H, Berna MJ, Jensen RT. Causes of death and prognostic factors in multiple endocrine neoplasia type 1: a prospective study: comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Medicine (Baltimore) 2013a;92:135–181. doi: 10.1097/MD.0b013e3182954af1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Igarashi H, Uehara H, Jensen RT. Pharmacotherapy of Zollinger-Ellison syndrome. Expert Opin Pharmacother. 2013b;14:307–321. doi: 10.1517/14656566.2013.767332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MART, Hu MI, et al. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 2015. CDC73-Related Disorders. 1993–2015. [Google Scholar]
- Jaehning JA. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim Biophys Acta. 2010;1799:379–388. doi: 10.1016/j.bbagrm.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenicke LA, von Eyss B, Carstensen A, Wolf E, Xu W, Greifenberg AK, Geyer M, Eilers M, Popov N. Ubiquitin-Dependent Turnover of MYC Antagonizes MYC/PAF1C Complex Accumulation to Drive Transcriptional Elongation. Mol Cell. 2016;61:54–67. doi: 10.1016/j.molcel.2015.11.007. [DOI] [PubMed] [Google Scholar]
- Jawiarczyk A, Bolanowski M, Syrycka J, Bednarek-Tupikowska G, Kaluzny M, Kolodziejczyk A, Domoslawski P. Effective therapy of insulinoma by using long-acting somatostatin analogue. A case report and literature review. Exp Clin Endocrinol Diabetes. 2012;120:68–72. doi: 10.1055/s-0031-1287792. [DOI] [PubMed] [Google Scholar]
- Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer. 2008;113:1807–1843. doi: 10.1002/cncr.23648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RT, Niederle B, Mitry E, Ramage JK, Steinmuller T, Lewington V, Scarpa A, Sundin A, Perren A, Gross D, et al. Gastrinoma (duodenal and pancreatic) Neuroendocrinology. 2006;84:173–182. doi: 10.1159/000098009. [DOI] [PubMed] [Google Scholar]
- Jumpertz von Schwartzenberg R, Elbelt U, Ventz M, Mai K, Kienitz T, Maurer L, Rose T, Ruckert JC, Strasburger CJ, Spranger J. Palliative treatment of uncontrollable hypercalcemia due to parathyrotoxicosis: denosumab as rescue therapy. Endocrinol Diabetes Metab Case Rep. 2015;2015:150082. doi: 10.1530/EDM-15-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karges W. Calcitonin determination for early diagnosis of medullary thyroid cancer. Chirurg. 2010;81:620, 622–626. doi: 10.1007/s00104-009-1883-9. [DOI] [PubMed] [Google Scholar]
- Karuppiah D, Thanabalasingham G, Shine B, Wang LM, Sadler GP, Karavitaki N, Grossman AB. Refractory hypercalcaemia secondary to parathyroid carcinoma: response to high-dose denosumab. Eur J Endocrinol. 2014;171:K1–5. doi: 10.1530/EJE-14-0166. [DOI] [PubMed] [Google Scholar]
- Kebebew E, Arici C, Duh QY, Clark OH. Localization and reoperation results for persistent and recurrent parathyroid carcinoma. Arch Surg. 2001;136:878–885. doi: 10.1001/archsurg.136.8.878. [DOI] [PubMed] [Google Scholar]
- Keutgen XM, Nilubol N, Glanville J, Sadowski SM, Liewehr DJ, Venzon DJ, Steinberg SM, Kebebew E. Resection of primary tumor site is associated with prolonged survival in metastatic nonfunctioning pancreatic neuroendocrine tumors. Surgery. 2016;159:311–319. doi: 10.1016/j.surg.2015.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khadilkar KS, Budyal SR, Kasliwal R, Lila AR, Bandgar T, Shah NS. HRPT2- (CDC73) RELATED HEREDITARY HYPERPARATHYROIDISM: A CASE SERIES FROM WESTERN INDIA. Endocr Pract. 2015;21:1010–1016. doi: 10.4158/EP15648.OR. [DOI] [PubMed] [Google Scholar]
- King KS, Pacak K. Familial pheochromocytomas and paragangliomas. Mol Cell Endocrinol. 2014;386:92–100. doi: 10.1016/j.mce.2013.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinlaw WB, Scott SM, Maue RA, Memoli VA, Harris RD, Daniels GH, Porter DM, Belloni DR, Spooner ET, Ernesti MM, et al. Multiple endocrine neoplasia 2A due to a unique C609S RET mutation presents with pheochromocytoma and reduced penetrance of medullary thyroid carcinoma. Clin Endocrinol (Oxf) 2005;63:676–682. doi: 10.1111/j.1365-2265.2005.02400.x. [DOI] [PubMed] [Google Scholar]
- Kong J, Wang O, Nie M, Shi J, Hu Y, Jiang Y, Li M, Xia W, Meng X, Xing X. Familial isolated primary hyperparathyroidism/hyperparathyroidism-jaw tumour syndrome caused by germline gross deletion or point mutations of CDC73 gene in Chinese. Clin Endocrinol (Oxf) 2014;81:222–230. doi: 10.1111/cen.12461. [DOI] [PubMed] [Google Scholar]
- Korpi-Hyovalti E, Cranston T, Ryhanen E, Arola J, Aittomaki K, Sane T, Thakker RV, Schalin-Jantti C. CDC73 intragenic deletion in familial primary hyperparathyroidism associated with parathyroid carcinoma. J Clin Endocrinol Metab. 2014;99:3044–3048. doi: 10.1210/jc.2014-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouvaraki MA, Shapiro SE, Cote GJ, Lee JE, Yao JC, Waguespack SG, Gagel RF, Evans DB, Perrier ND. Management of pancreatic endocrine tumors in multiple endocrine neoplasia type 1. World J Surg. 2006;30:643–653. doi: 10.1007/s00268-006-0360-y. [DOI] [PubMed] [Google Scholar]
- Krampitz GW, Norton JA. Current management of the Zollinger-Ellison syndrome. Adv Surg. 2013;47:59–79. doi: 10.1016/j.yasu.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Kunz PL, Reidy-Lagunes D, Anthony LB, Bertino EM, Brendtro K, Chan JA, Chen H, Jensen RT, Kim MK, Klimstra DS, et al. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas. 2013;42:557–577. doi: 10.1097/MPA.0b013e31828e34a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutcher MR, Rigby MH, Bullock M, Trites J, Taylor SM, Hart RD. Hyperparathyroidism-jaw tumor syndrome. Head Neck. 2013;35:E175–177. doi: 10.1002/hed.22918. [DOI] [PubMed] [Google Scholar]
- Lastoria S, Marciello F, Faggiano A, Aloj L, Caraco C, Aurilio M, D’Ambrosio L, Di Gennaro F, Ramundo V, Camera L, et al. Role of Ga-DOTATATE PET/CT in patients with multiple endocrine neoplasia type 1 (MEN1) Endocrine. 2015 doi: 10.1007/s12020-015-0702-y. [DOI] [PubMed] [Google Scholar]
- Ledermann JA. PARP inhibitors in ovarian cancer. Ann Oncol. 2016;27:i40–i44. doi: 10.1093/annonc/mdw094. [DOI] [PubMed] [Google Scholar]
- Lee L, Igarashi H, Fujimori N, Hijioka M, Kawabe K, Oda Y, Jensen RT, Ito T. Long-term outcomes and prognostic factors in 78 Japanese patients with advanced pancreatic neuroendocrine neoplasms: a single-center retrospective study. Jpn J Clin Oncol. 2015;45:1131–1138. doi: 10.1093/jjco/hyv143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Su J, Zhao L, Wu J, Ding X, Fang F, Wu Y, Sun H, Peng Y. Familial Cushing syndrome due to thymic carcinoids in a multiple endocrine neoplasia type 1 kindred. Endocrine. 2014;47:183–190. doi: 10.1007/s12020-013-0141-6. [DOI] [PubMed] [Google Scholar]
- Lin L, Zhang JH, Panicker LM, Simonds WF. The parafibromin tumor suppressor protein inhibits cell proliferation by repression of the c-myc proto-oncogene. Proc Natl Acad Sci U S A. 2008;105:17420–17425. doi: 10.1073/pnas.0710725105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez CL, Falconi M, Waldmann J, Boninsegna L, Fendrich V, Goretzki PK, Langer P, Kann PH, Partelli S, Bartsch DK. Partial pancreaticoduodenectomy can provide cure for duodenal gastrinoma associated with multiple endocrine neoplasia type 1. Ann Surg. 2013;257:308–314. doi: 10.1097/SLA.0b013e3182536339. [DOI] [PubMed] [Google Scholar]
- MacFarlane MP, Fraker DL, Alexander HR, Norton JA, Lubensky I, Jensen RT. Prospective study of surgical resection of duodenal and pancreatic gastrinomas in multiple endocrine neoplasia type 1. Surgery. 1995;118:973–979. doi: 10.1016/s0039-6060(05)80102-3. discussion 979–980. [DOI] [PubMed] [Google Scholar]
- Machado MC. Surgical treatment of pancreatic endocrine tumors in multiple endocrine neoplasia type 1. Clinics (Sao Paulo) 2012;67(Suppl 1):145–148. doi: 10.6061/clinics/2012(Sup01)24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machens A, Brauckhoff M, Holzhausen HJ, Thanh PN, Lehnert H, Dralle H. Codon-specific development of pheochromocytoma in multiple endocrine neoplasia type 2. J Clin Endocrinol Metab. 2005;90:3999–4003. doi: 10.1210/jc.2005-0064. [DOI] [PubMed] [Google Scholar]
- Machens A, Dralle H. Biomarker-based risk stratification for previously untreated medullary thyroid cancer. J Clin Endocrinol Metab. 2010;95:2655–2663. doi: 10.1210/jc.2009-2368. [DOI] [PubMed] [Google Scholar]
- Machens A, Dralle H. Surgical Treatment of Medullary Thyroid Cancer. Recent Results Cancer Res. 2015;204:187–205. doi: 10.1007/978-3-319-22542-5_9. [DOI] [PubMed] [Google Scholar]
- Machens A, Lorenz K, Dralle H. Peak incidence of pheochromocytoma and primary hyperparathyroidism in multiple endocrine neoplasia 2: need for age-adjusted biochemical screening. J Clin Endocrinol Metab. 2013;98:E336–345. doi: 10.1210/jc.2012-3192. [DOI] [PubMed] [Google Scholar]
- Machens A, Lorenz K, Dralle H. Utility of serum procalcitonin for screening and risk stratification of medullary thyroid cancer. J Clin Endocrinol Metab. 2014;99:2986–2994. doi: 10.1210/jc.2014-1278. [DOI] [PubMed] [Google Scholar]
- Marx SJ, Agarwal SK, Kester MB, Heppner C, Kim YS, Skarulis MC, James LA, Goldsmith PK, Saggar SK, Park SY, et al. Multiple endocrine neoplasia type 1: clinical and genetic features of the hereditary endocrine neoplasias. Recent Prog Horm Res. 1999;54:397–438. discussion 438–399. [PubMed] [Google Scholar]
- Mathews JW, Winchester R, Alsaygh N, Bartlett AM, Luttrell LM. Hyperparathyroidism-Jaw Tumor Syndrome-An Overlooked Cause of Severe Hypercalcemia. Am J Med Sci. 2015 doi: 10.1016/j.amjms.2016.06.020. [DOI] [PubMed] [Google Scholar]
- Mehta A, Patel D, Rosenberg A, Boufraqech M, Ellis RJ, Nilubol N, Quezado MM, Marx SJ, Simonds WF, Kebebew E. Hyperparathyroidism-jaw tumor syndrome: Results of operative management. Surgery. 2014;156:1315–1324. doi: 10.1016/j.surg.2014.08.004. discussion 1324–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BS, Rusinko RY, Fowler L. Synchronous thymoma and thymic carcinoid in a woman with multiple endocrine neoplasia type 1: case report and review. Endocr Pract. 2008;14:713–716. doi: 10.4158/EP.14.6.713. [DOI] [PubMed] [Google Scholar]
- Morin E, Cheng S, Mete O, Serra S, Araujo PB, Temple S, Cleary S, Gallinger S, Greig PD, McGilvray I, et al. Hormone profiling, WHO 2010 grading, and AJCC/UICC staging in pancreatic neuroendocrine tumor behavior. Cancer Med. 2013;2:701–711. doi: 10.1002/cam4.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newey PJ, Bowl MR, Cranston T, Thakker RV. Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid tumors. Hum Mutat. 2010;31:295–307. doi: 10.1002/humu.21188. [DOI] [PubMed] [Google Scholar]
- Newey PJ, Bowl MR, Thakker RV. Parafibromin--functional insights. J Intern Med. 2009;266:84–98. doi: 10.1111/j.1365-2796.2009.02107.x. [DOI] [PubMed] [Google Scholar]
- Niederle B, Sebag F, Brauckhoff M. Timing and extent of thyroid surgery for gene carriers of hereditary C cell disease--a consensus statement of the European Society of Endocrine Surgeons (ESES) Langenbecks Arch Surg. 2014;399:185–197. doi: 10.1007/s00423-013-1139-5. [DOI] [PubMed] [Google Scholar]
- Nikfarjam M, Warshaw AL, Axelrod L, Deshpande V, Thayer SP, Ferrone CR, Fernandez-del Castillo C. Improved contemporary surgical management of insulinomas: a 25-year experience at the Massachusetts General Hospital. Ann Surg. 2008;247:165–172. doi: 10.1097/SLA.0b013e31815792ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton JA, Krampitz G, Jensen RT. Multiple Endocrine Neoplasia: Genetics and Clinical Management. Surg Oncol Clin N Am. 2015;24:795–832. doi: 10.1016/j.soc.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton JA, Venzon DJ, Berna MJ, Alexander HR, Fraker DL, Libutti SK, Marx SJ, Gibril F, Jensen RT. Prospective study of surgery for primary hyperparathyroidism (HPT) in multiple endocrine neoplasia-type 1 and Zollinger-Ellison syndrome: long-term outcome of a more virulent form of HPT. Ann Surg. 2008;247:501–510. doi: 10.1097/SLA.0b013e31815efda5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otake Y, Aoki M, Nakanishi T, Hashimoto K. Atypical carcinoid of thymus associated with multiple endocrine neoplasia syndrome type 1. Gen Thorac Cardiovasc Surg. 2010;58:534–537. doi: 10.1007/s11748-009-0557-3. [DOI] [PubMed] [Google Scholar]
- Pichardo-Lowden AR, Manni A, Saunders BD, Baker MJ. Familial hyperparathyroidism due to a germline mutation of the CDC73 gene: implications for management and age-appropriate testing of relatives at risk. Endocr Pract. 2011;17:602–609. doi: 10.4158/EP10337.RA. [DOI] [PubMed] [Google Scholar]
- Powell AC, Alexander HR, Pingpank JF, Steinberg SM, Skarulis M, Bartlett DL, Agarwal S, Cochran C, Seidel G, Fraker D, et al. The utility of routine transcervical thymectomy for multiple endocrine neoplasia 1-related hyperparathyroidism. Surgery. 2008;144:878–883. doi: 10.1016/j.surg.2008.08.031. discussion 883–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quayle FJ, Fialkowski EA, Benveniste R, Moley JF. Pheochromocytoma penetrance varies by RET mutation in MEN 2A. Surgery. 2007;142:800–805. doi: 10.1016/j.surg.2007.09.013. discussion 805 e801. [DOI] [PubMed] [Google Scholar]
- Rajan S, Zaidi G, Agarwal G, Mishra A, Agarwal A, Mishra SK, Bhatia E. Genotype-Phenotype Correlation in Indian Patients with MEN2-Associated Pheochromocytoma and Comparison of Clinico-Pathological Attributes with Apparently Sporadic Adrenal Pheochromocytoma. World J Surg. 2015 doi: 10.1007/s00268-015-3255-6. [DOI] [PubMed] [Google Scholar]
- Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- Recuero Diaz JL, Embun Flor R, Menal Munoz P, Hernandez Ferrandez J, Arraras Martinez MJ, Rivas de Andres JJ. Thymic carcinoid associated with multiple endocrine neoplasia syndrome type I. Arch Bronconeumol. 2013;49:122–125. doi: 10.1016/j.arbres.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Rinke A, Muller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, Mayer C, Aminossadati B, Pape UF, Blaker M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27:4656–4663. doi: 10.1200/JCO.2009.22.8510. [DOI] [PubMed] [Google Scholar]
- Rowland KJ, Chernock RD, Moley JF. Pheochromocytoma in an 8-year-old patient with multiple endocrine neoplasia type 2A: implications for screening. J Surg Oncol. 2013;108:203–206. doi: 10.1002/jso.23378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski SM, Millo C, Cottle-Delisle C, Merkel R, Yang LA, Herscovitch P, Pacak K, Simonds WF, Marx SJ, Kebebew E. Results of (68)Gallium-DOTATATE PET/CT Scanning in Patients with Multiple Endocrine Neoplasia Type 1. J Am Coll Surg. 2015;221:509–517. doi: 10.1016/j.jamcollsurg.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski SM, Neychev V, Cottle-Delisle C, Merkel R, Yang LA, Quezado MM, Chang R, Kebebew E. Detection of insulinoma using (68)Gallium-DOTATATE PET/CT: a case report. Gland Surg. 2014;3:E1–5. doi: 10.3978/j.issn.2227-684X.2014.10.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai A, Yamazaki M, Suzuki S, Fukushima T, Imai T, Kikumori T, Okamoto T, Horiuchi K, Uchino S, Kosugi S, et al. Clinical features of insulinoma in patients with multiple endocrine neoplasia type 1: analysis of the database of the MEN Consortium of Japan. Endocr J. 2012;59:859–866. doi: 10.1507/endocrj.ej12-0173. [DOI] [PubMed] [Google Scholar]
- Schantz A, Castleman B. Parathyroid carcinoma. A study of 70 cases. Cancer. 1973;31:600–605. doi: 10.1002/1097-0142(197303)31:3<600::aid-cncr2820310316>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Schussheim DH, Skarulis MC, Agarwal SK, Simonds WF, Burns AL, Spiegel AM, Marx SJ. Multiple endocrine neoplasia type 1: new clinical and basic findings. Trends Endocrinol Metab. 2001;12:173–178. doi: 10.1016/s1043-2760(00)00372-6. [DOI] [PubMed] [Google Scholar]
- Service FJ, McMahon MM, O’Brien PC, Ballard DJ. Functioning insulinoma--incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc. 1991;66:711–719. doi: 10.1016/s0025-6196(12)62083-7. [DOI] [PubMed] [Google Scholar]
- Shane E. Clinical review 122: Parathyroid carcinoma. J Clin Endocrinol Metab. 2001;86:485–493. doi: 10.1210/jcem.86.2.7207. [DOI] [PubMed] [Google Scholar]
- Sharretts JM, Simonds WF. Clinical and molecular genetics of parathyroid neoplasms. Best Pract Res Clin Endocrinol Metab. 2010;24:491–502. doi: 10.1016/j.beem.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattuck TM, Valimaki S, Obara T, Gaz RD, Clark OH, Shoback D, Wierman ME, Tojo K, Robbins CM, Carpten JD, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003;349:1722–1729. doi: 10.1056/NEJMoa031237. [DOI] [PubMed] [Google Scholar]
- Shibata Y, Yamazaki M, Takei M, Uchino S, Sakurai A, Komatsu M. Early-onset, severe, and recurrent primary hyperparathyroidism associated with a novel CDC73 mutation. Endocr J. 2015;62:627–632. doi: 10.1507/endocrj.EJ15-0057. [DOI] [PubMed] [Google Scholar]
- Sidhu PS, Talat N, Patel P, Mulholland NJ, Schulte KM. Ultrasound features of malignancy in the preoperative diagnosis of parathyroid cancer: a retrospective analysis of parathyroid tumours larger than 15 mm. Eur Radiol. 2011;21:1865–1873. doi: 10.1007/s00330-011-2141-3. [DOI] [PubMed] [Google Scholar]
- Silverberg SJ, Rubin MR, Faiman C, Peacock M, Shoback DM, Smallridge RC, Schwanauer LE, Olson KA, Klassen P, Bilezikian JP. Cinacalcet hydrochloride reduces the serum calcium concentration in inoperable parathyroid carcinoma. J Clin Endocrinol Metab. 2007;92:3803–3808. doi: 10.1210/jc.2007-0585. [DOI] [PubMed] [Google Scholar]
- Singh Ospina N, Thompson GB, Nichols CFrSDC, Young WF., Jr Thymic and Bronchial Carcinoid Tumors in Multiple Endocrine Neoplasia Type 1: The Mayo Clinic Experience from 1977 to 2013. Horm Cancer. 2015;6:247–253. doi: 10.1007/s12672-015-0228-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu WK, Law CY, Lam CW, Mak CM, Wong GW, Ho AY, Ho KY, Loo KT, Chiu SC, Chow LT, et al. Novel nonsense CDC73 mutations in Chinese patients with parathyroid tumors. Fam Cancer. 2011;10:695–699. doi: 10.1007/s10689-011-9466-6. [DOI] [PubMed] [Google Scholar]
- Sriphrapradang C, Sornmayura P, Chanplakorn N, Trachoo O, Sae-Chew P, Aroonroch R. Fine-needle aspiration cytology of parathyroid carcinoma mimic hurthle cell thyroid neoplasm. Case Rep Endocrinol. 2014;2014:680876. doi: 10.1155/2014/680876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taieb D, Kebebew E, Castinetti F, Chen CC, Henry JF, Pacak K. Diagnosis and preoperative imaging of multiple endocrine neoplasia type 2: current status and future directions. Clin Endocrinol (Oxf) 2014;81:317–328. doi: 10.1111/cen.12513. [DOI] [PubMed] [Google Scholar]
- Takagi J, Otake K, Morishita M, Kato H, Nakao N, Yoshikawa K, Ikeda H, Hirooka Y, Hattori Y, Larsson C, et al. Multiple endocrine neoplasia type I and Cushing’s syndrome due to an aggressive ACTH producing thymic carcinoid. Intern Med. 2006;45:81–86. doi: 10.2169/internalmedicine.45.1427. [DOI] [PubMed] [Google Scholar]
- Taye A, Libutti SK. Diagnosis and management of insulinoma: current best practice and ongoing developments. Research and Reports in Endocrine Disorders. 2015;5:125–133. [Google Scholar]
- Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, Melmed S, Sakurai A, Tonelli F, Brandi ML. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1) J Clin Endocrinol Metab. 2012;97:2990–3011. doi: 10.1210/jc.2012-1230. [DOI] [PubMed] [Google Scholar]
- Thomas de Montpreville V, Ghigna MR, Lacroix L, Besse B, Broet P, Dartevelle P, Fadel E, Dorfmuller P. Thymic carcinomas: clinicopathologic study of 37 cases from a single institution. Virchows Arch. 2013;462:307–313. doi: 10.1007/s00428-013-1371-y. [DOI] [PubMed] [Google Scholar]
- Thosani S, Ayala-Ramirez M, Palmer L, Hu MI, Rich T, Gagel RF, Cote G, Waguespack SG, Habra MA, Jimenez C. The characterization of pheochromocytoma and its impact on overall survival in multiple endocrine neoplasia type 2. J Clin Endocrinol Metab. 2013;98:E1813–1819. doi: 10.1210/jc.2013-1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo SP, Lourenco DM, Jr, Santos MA, Tavares MR, Toledo RA, Correia-Deur JE. Hypercalcitoninemia is not pathognomonic of medullary thyroid carcinoma. Clinics (Sao Paulo) 2009;64:699–706. doi: 10.1590/S1807-59322009000700015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli F. How to follow up and when to operate asymptomatic pancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1? J Clin Gastroenterol. 2014;48:387–389. doi: 10.1097/MCG.0000000000000087. [DOI] [PubMed] [Google Scholar]
- Tonelli F, Giudici F, Nesi G, Batignani G, Brandi ML. Biliary tree gastrinomas in multiple endocrine neoplasia type 1 syndrome. World J Gastroenterol. 2013;19:8312–8320. doi: 10.3748/wjg.v19.i45.8312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triponez F, Dosseh D, Goudet P, Cougard P, Bauters C, Murat A, Cadiot G, Niccoli-Sire P, Chayvialle JA, Calender A, et al. Epidemiology data on 108 MEN 1 patients from the GTE with isolated nonfunctioning tumors of the pancreas. Ann Surg. 2006;243:265–272. doi: 10.1097/01.sla.0000197715.96762.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truran PP, Johnson SJ, Bliss RD, Lennard TW, Aspinall SR. Parafibromin, galectin-3, PGP9.5, Ki67, and cyclin D1: using an immunohistochemical panel to aid in the diagnosis of parathyroid cancer. World J Surg. 2014;38:2845–2854. doi: 10.1007/s00268-014-2700-2. [DOI] [PubMed] [Google Scholar]
- Vezzosi D, Cardot-Bauters C, Bouscaren N, Lebras M, Bertholon-Gregoire M, Niccoli P, Levy-Bohbot N, Groussin L, Bouchard P, Tabarin A, et al. Long-term results of the surgical management of insulinoma patients with MEN1: a Groupe d’etude des Tumeurs Endocrines (GTE) retrospective study. Eur J Endocrinol. 2015;172:309–319. doi: 10.1530/EJE-14-0878. [DOI] [PubMed] [Google Scholar]
- Wei CH, Harari A. Parathyroid carcinoma: update and guidelines for management. Curr Treat Options Oncol. 2012;13:11–23. doi: 10.1007/s11864-011-0171-3. [DOI] [PubMed] [Google Scholar]
- Wells SA, Jr, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25:567–610. doi: 10.1089/thy.2014.0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SA, Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, Baudin E, Elisei R, Jarzab B, Vasselli JR, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134–141. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SA, Jr, Santoro M. Targeting the RET pathway in thyroid cancer. Clin Cancer Res. 2009;15:7119–7123. doi: 10.1158/1078-0432.CCR-08-2742. [DOI] [PubMed] [Google Scholar]
- Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–523. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates CJ, Newey PJ, Thakker RV. Challenges and controversies in management of pancreatic neuroendocrine tumours in patients with MEN1. Lancet Diabetes Endocrinol. 2015;3:895–905. doi: 10.1016/S2213-8587(15)00043-1. [DOI] [PubMed] [Google Scholar]
- Zwolak A, Rudzki G, Swirska J, Dudzinska M, Daniluk J, Tarach J. Catecholamine crisis as a first manifestation of familial bilateral pheochromocytoma caused by RET proto-oncogene mutation in codon C 634R. Endokrynol Pol. 2015;66:462–468. doi: 10.5603/EP.2015.0056. [DOI] [PubMed] [Google Scholar]