

Abstract
Background
Statins are associated with delayed fibrosis progression and a reduced risk of hepatocellular carcinoma (HCC) in chronic hepatitis C virus (HCV). Limited data exist regarding the most effective type and dose of statin in this population. We sought to determine the impact of statin type and dose upon fibrosis progression and HCC, in patients with HCV.
Methods
Using the Electronically Retrieved Cohort of HCV Infected Veterans (ERCHIVES) database, we identified all subjects initiated on anti-HCV therapy from 2001 to 2014, and all incident cases of cirrhosis and HCC. Statin use was measured using cumulative defined daily dose (cDDD). Multivariable Cox proportional hazard regression models were used to examine the relationship between statin use and the development of cirrhosis and HCC.
Results
Among 9,135 eligible subjects, 1,649 developed cirrhosis, and 239 developed incident HCC. Statin use was associated with a 44% reduction in development of cirrhosis (adjusted HR 0.6, 95% CI 0.53, 0.68). The adjusted HRs (95% CI) of fibrosis progression with statin cDDD 28–89, 89–180, and >180, were 0.74 (0.59,0.93), 0.71 (0.59,0.88), and 0.6 (0.53,0.68), respectively. Mean change in FIB-4 score with atorvastatin (n=944) and fluvastatin (n=34) was −0.17 and −0.13 respectively (p=0.04) after adjustment for baseline FIB-4 score and established predictors of cirrhosis. Statin use was also associated with a 49% reduction in incident HCC (adjusted HR 0.51, 95% CI 0.36, 0.72). A similar dose-response relationship was observed.
Conclusion
In patients with chronic HCV, statin use was associated with a dose-dependent reduction in incident cirrhosis and HCC. Atorvastatin and fluvastatin were associated with the most significant anti-fibrotic effects, compared to other statins.
Keywords: fibrosis, cirrhosis, hepatitis C virus, lipid lowering agent, ERCHIVES
Introduction
Hepatitis C virus (HCV) is one of the most common causes of chronic liver disease and the leading indication for liver transplantation worldwide [1, 2]. Estimates suggest that over a period of twenty to thirty years, cirrhosis will develop in 10% to 25% of patients with chronic hepatitis C (CHC), and hepatocellular carcinoma (HCC) in 1% to 5% [2]. Despite the great success of oral direct-acting antiviral medications, there are still many patients with fibrosis and other clinical complications of HCV, who remain at risk of disease progression despite successful viral clearance [3]. In such patients, reducing the risk of complications related to hepatic fibrosis, including cirrhosis and HCC, are of paramount importance.
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, or statins, exert beneficial effects on circulating lipid levels, and are used for the management and prevention of coronary heart disease and stroke [4]. Recently, statins have garnered attention for their pleiotropic effects. Via both HMG-CoA dependent and independent pathways, statins have been shown to exert antiproliferative, antiangiogenic, proapoptotic and immunomodulatory actions [5–7]. They have inhibit cell growth, decrease proteolysis and block tumor cell spread [8–13], and may offer chemoprevention against many malignancies, including HCC [8, 14–18].
It has been postulated that statins may also exert unique antifibrotic effects in HCV. In animal models, statins have been shown to block activation of hepatic myofibroblasts, inducing apoptosis and preventing both proliferation of hepatic stellate cells (HSCs) and their production of collagens [6, 7, 13, 19–21]. In human studies, use of statins has been associated with a reduced risk of hepatic fibrosis progression [22], and decreased incidence of HCC [23]. In a recent analysis of 7,248 US Veterans infected with chronic HCV, statin use was significantly associated with reduction in fibrosis progression rate, and a lower adjusted risk of developing cirrhosis (hazard ratio [HR] = 0.56; 95% CI = −0.50, 0.63) and HCC (HR=0.51; 95% CI = 0.34, 0.76) [24].
The potency of statin-mediated antiviral effects is thought to depend on the type of statin used, with fluvastatin having the most potent, and pravastatin the least potent, antiviral activity [25–27]. However, data regarding the impact of statin type and dose upon hepatic fibrogenesis and the risk of HCC remain limited. Recently, Yang and colleagues reported the results of a population-based cohort study of Taiwanese patients infected with HCV [28]. The authors observed a significant dose-dependent relationship between increasing statin dose and reduction in risk of cirrhosis [28]. However, the subjects did not have antibody or viral load-confirmed diagnoses of HCV, nor did the study adjust for statin type, baseline disease activity or fibrosis severity. We sought to assess the relationship between statin type and dosage with both hepatic fibrosis progression and the development of HCC, in a large, well-established national cohort of HCV-infected US veterans.
Methods
Study population
Study participants included patients with HCV infection within the Electronically Retrieved Cohort of HCV Infected Veterans (ERCHIVES). ERCHIVES, which has been described in multiple previous publications [24, 29], is a large, established national cohort of HCV-infected veterans along with uninfected controls, created from multiple national databases. Briefly, all HCV-infected veterans observed at any of the nationwide Department of Veterans Affairs (VA) medical facilities who had a positive HCV antibody test between 2001 and 2014, were identified. Demographic, clinical and laboratory data were obtained from the National Patient Care Database and the Corporate Data Warehouse, and pharmacy information including all prescriptions written, doses, duration, number of pills, number of refills and date of refills, were retrieved from the Pharmacy Benefits Management database. Data were then merged based on established algorithms.
Patients were included in the study cohort if they received at least 14 days of treatment for HCV. Patients were excluded if they were coinfected with human immunodeficiency virus (HIV), had a positive hepatitis B surface antigen (HBsAg), baseline cirrhosis or HCC. Patients were also excluded if baseline HCV RNA or FIB-4 score were missing, and if at least one FIB-4 score was not available at least 24 months following completion of anti-HCV therapy.
Definitions
Baseline was defined as the date of HCV treatment initiation. If multiple courses of treatment were given, the date of the most recent course was used for this analysis. Treatment completion was defined according to approved U.S. Food and Drug Administration labeling guidelines [30]. Cirrhosis was defined as a FIB-4 score of > 3.5 based upon previously published work by our group and others [29], and was calculated as follows:
Baseline laboratory values were based on the results of testing performed closest to, but within 365 days prior to, the start date of antiviral therapy. Laboratory data were obtained at yearly intervals and FIB-4 score was re-calculated at each interval. An average of two values closest to the selected time point of interest was used for the calculation of FIB-4 scores. SVR was defined as undetectable HCV RNA in all follow-up HCV RNA tests after the end of treatment, including at least 1 test more than 12 weeks after end of initial treatment [31, 32].
Patients were defined as having diabetes if they satisfied at least one of the following: (1) ICD-9 coding for Diabetes Mellitus (2) at least 2 outpatient random blood sugars greater than 200 mg/dL, or (3) were being prescribed antidiabetic medications prior to the start of antiviral treatment. History of alcohol and drug abuse or dependence and the diagnosis of HCC were based on the presence of at least one inpatient or two outpatient ICD-9 diagnoses.
Exposure to statin medications
All patients who were prescribed statin medications in any VA pharmacy during the study observation period were identified. Statin prescriptions included simvastatin, lovastatin, fluvastatin, pravastatin, atorvastatin and cerivastatin. We collected the dates of prescriptions ordered, the number of days prescribed, number of pills per prescription and number of refills ordered. With this information the statin “defined daily dose” (DDD) was calculated for each subject. The DDD is a validated unit for measuring a prescribed drug amount, and is defined as the average maintenance dose per day of a drug consumed in an adult [33]. It is calculated as:
The DDD was re-calculated annually for each year of the study observation period.
The “cumulative defined daily dose” (cDDD) was calculated from the DDD. The cDDD is defined as the total sum of dispensed DDDs of a given medication. Both the DDD and cDDD are recommended by the World Health Organization (WHO) and are widely used for comparison of medications, including statins, along a similar standard [33]. Statin use was defined as >28 cDDDs of statin medications prescribed during the study period. Similar information was collected for non-statin lipid lowering agents (cholestyramine, colesevelam, colestipol, ezetimibe, niacin, niacinimide), triglyceride-lowering agents (clofibrate, fenofibrate, gemfibrozil), as well as the antidiabetic agents Metformin, sulfonylureas and thiazolidinedione.
Outcomes
Primary outcome measures were (1) progression of liver fibrosis as measured by FIB-4 score; (2) development of cirrhosis as defined by FIB-4 score ≥ 3.5; and (3) incident HCC, as above.
Statistical analysis
The study cohort was divided into statin users and non-users, with statin use as defined above. Baseline demographic and clinical factors were compared between the two groups using chi-square test or t-test, as appropriate. Predictors of incident cirrhosis and HCC were determined using Cox proportional hazards analysis and generating hazards ratios for each of the predictor variables. Mean change in fibrosis score over the study period was calculated for each of the statin medication by subtracting the baseline score from last available score. Mean fibrosis scores were also plotted over time by the cDDD of statins used. Kaplan-Meier curves were generated to demonstrate time to development of cirrhosis and hepatocellular carcinoma, by statin cDDD. SAS (SAS Institute Inc., Cary, NC) and Stata software (version 11; Stata Corp LP, College Station, TX) were used for statistical analyses.
Regulatory approval
This study was approved by the Institutional Review Board at the VA Pittsburgh Healthcare System. Appropriate permissions were obtained from the sources that provided data for ERCHIVES.
Results
Baseline clinical data
Within the ERCHIVES database, we identified 47,549 subjects with confirmed HCV infection, who were initiated on anti-HCV therapy during the study period. We excluded those who received both Boceprevir and Telaprevir (n=32), and those with HIV coinfection (n=766), as well as those who had positive HBsAg (n=6353), baseline cirrhosis (n=2867) or HCC (n=941), or missing or undetectable HCV RNA at baseline (n=3571 and 2235, respectively), or those without follow-up HCV RNA to measure SVR (n=18,007). We also excluded those without sufficient baseline or follow-up labs to calculate FIB-4 scores (n=1301 and n=2341, respectively) (Figure 1).
Figure 1.
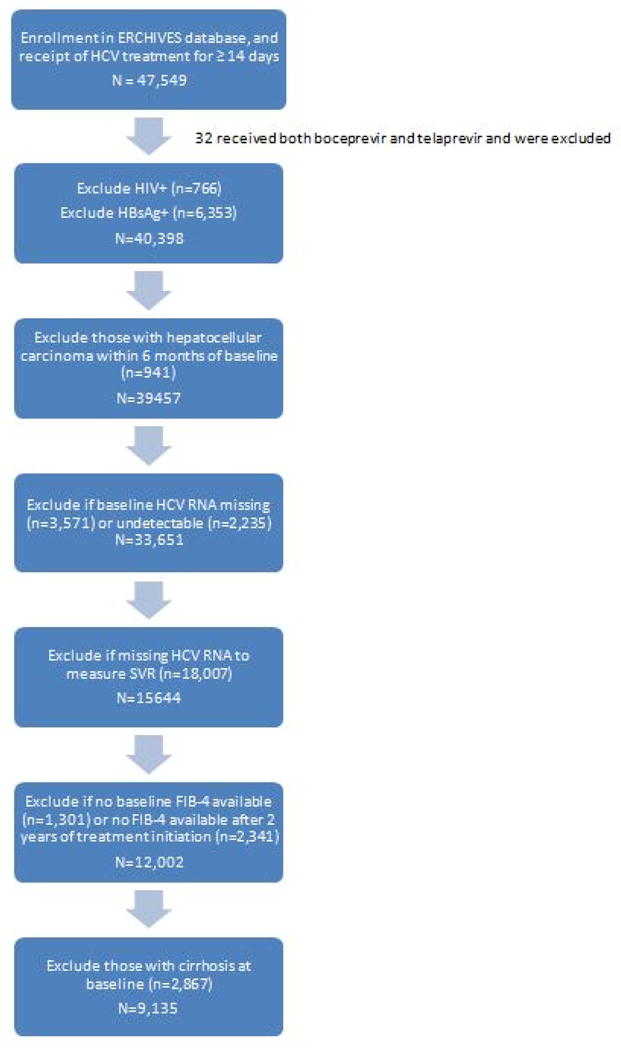
Study flow diagram
Among the 9,135 remaining subjects, 4,165 (45.6%) were statin users, and 4,970 (54.4%) were non-users. A comparison of baseline demographic and clinical characteristics is shown in Table 1. Mean age was 53 among both statin users and non-users (standard deviation [SD] 6.88 and 5.85, respectively; p<0.0001); A similar percentage of both groups (65%) were white, and there was no significant difference in median HCV-RNA levels between groups. Median length of follow up among statin users was 97.9 months (IQR 66.2, 126.6), compared to 81.6 months (IQR 52.9, 113.6) among non-users (p<0.0001).
Table 1.
Baseline† clinical, laboratory and histological characteristics of HCV-infected patients without baseline cirrhosis (n=9135) according to statin medication use.
| Variable* | No Statin use (N=4970) | Statin use1 (N=4165) | P-value |
|---|---|---|---|
| Age, years | 52.5(6.88) | 53.5(5.85) | <.0001 |
| Male, % | 95.37% | 96.16% | 0.06 |
Race
|
65.01% 16.84% 5.77% 12.37% |
65.31% 19.54% 5.35% 9.80% |
<.0001 |
| Diabetes, % | 8.87% | 24.03% | <.0001 |
| Alcohol abuse history | 38.79% | 34.81% | <.0001 |
| Past smoking history, % | 90.44% | 92.65% | 0.0002 |
| BMI | 28.16(5.79) | 29.57(5.83) | <.0001 |
| HCV genotype, % 1 2 3 4 Mix Missing |
31.65% 8.19% 5.84% 0.36% 0.12% 53.84% |
25.55% 8.91% 4.66% 0.31% 0.07% 60.50% |
<.0001 |
| Log10 HCV RNA (IU/mL) | 6.31(1.95) | 6.17(1.99) | 0.002 |
| ALT, U/L | 77.44(64.56) | 73.73(63.16) | 0.01 |
| AST, U/L | 54.62(35.09) | 50.49(32.94) | <.0001 |
| Baseline lipid levels, mean [SD] | |||
| Total Cholesterol | 163.54(33.06) | 179.25(39.08) | <.0001 |
| Low-density lipoprotein (LDL) | 97.83(29.49) | 110.41(34.5) | <.0001 |
| High-density lipoprotein (HDL) | 42.74(14.82) | 39.56(13.14) | <.0001 |
| Triglyceride (TG) | 123.85(85.99) | 155.22(106.72) | <.0001 |
| Non-HDL cholesterol | 121.31(31.67) | 139.81(37.6) | <.0001 |
| Platelets ×1000/mm3 | 212.21(62.93) | 217.02(64.31) | 0.0003 |
| FIB-4 Score | 1.7(0.73) | 1.61(0.69) | <.0001 |
| Metformin use, % | 10.91% | 34.19% | <.0001 |
| Other lipid-lowering agent use2, % | 4.02% | 15.53% | <.0001 |
| ACE-inhibitor use, % | 38.11% | 65.76% | <.0001 |
| Treatment regimen (most recent) | <.0001 | ||
| PEG/RBV only | 88.79% | 93.18% | |
| PEG/RBV/BOC | 10.06% | 6.36% | |
| PEG/RBV/TPV | 1.15% | 0.46% | |
| Completed course of HCV treatment3, % | 63.54% | 68.43% | <.0001 |
| Attainment of sustained virologic response (SVR), % | 47.53% | 55.1% | <.0001 |
| Incident cirrhosis (FIB-4 score > 3.5) | 21.43% | 14.02% | <.0001 |
| Hepatocellular carcinoma (HCC) | 3.22% | 1.75% | <.0001 |
| Median length of follow-up, months (IQR) | 97.9 (66.2, 126.6) | 81.6 (52.9, 113.6) | <.0001 |
Variables expressed as Mean (SD) unless indicated otherwise.
Values obtained at baseline or closest value obtained within 12 months of baseline. The exception was Log10 HCV RNA, for which the closest value to baseline that was obtained within 24 months of baseline, was used.
Duration of infection defined as time from first positive HCV antibody to baseline, in years
Statin use was defined as ≥28 cDDDs per year; nonuse was defined as <28 cDDDs per year.
Other lipid-lowering agents include: fibrates (clofibrate, fenofibrate, gemfibrozil), niacin, ezetimibe, bile acid sequestrants (cholestyramine, colesevelam, colestipol)
A full course of treatment was defined as per the labelling guidelines for the particular regimen, taking into account previous treatment and presence of cirrhosis.
Statin users were more likely to be diabetic (24% vs. 9%; p<0.0001), and to be prescribed metformin, other lipid-lowering medications or angiotensin-converting enzyme (ACE) inhibitors (all p<0.0001). Compared to non-users, statin users had higher baseline total cholesterol (TC), low-density lipoprotein (LDL) and triglyceride (TG) levels, and lower high-density lipoprotein (HDL) (all p<0.0001). In bivariate analysis, patients who were prescribed statins were more likely to achieve SVR (55.1% vs. 47.5%, p<0.0001), less likely to develop cirrhosis (14% vs. 21.4%, p<0.0001), and less likely to be diagnosed with HCC (1.75% vs. 3.22%, p<0.0001), than non-users.
Fibrosis progression
Statin use was associated with reduced risk of fibrosis progression (HR 0.66, 95% CI 0.6, 0.73; p<0.001). In multivariate Cox regression analysis, treatment with statins was associated with a significantly reduced risk of developing cirrhosis (adjusted HR 0.64, 95% CI 0.57, 0.72; p <0.0001 after adjusting for baseline FIB-4 score and univariate predictors of cirrhosis). Over 10 years of follow up, statin use was also significantly correlated with a reduced rate of fibrosis progression compared to non-users, at all doses (p<0.01) and at all time-points, with the exception of two p-values of 0.02 and 0.04 at two time intervals for the higher cDDD (Figure 2).
Figure 2.
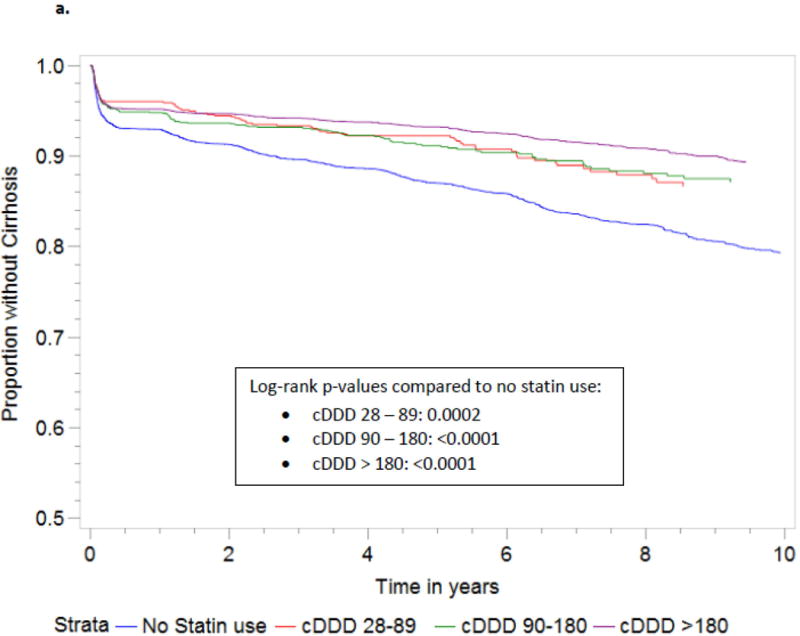
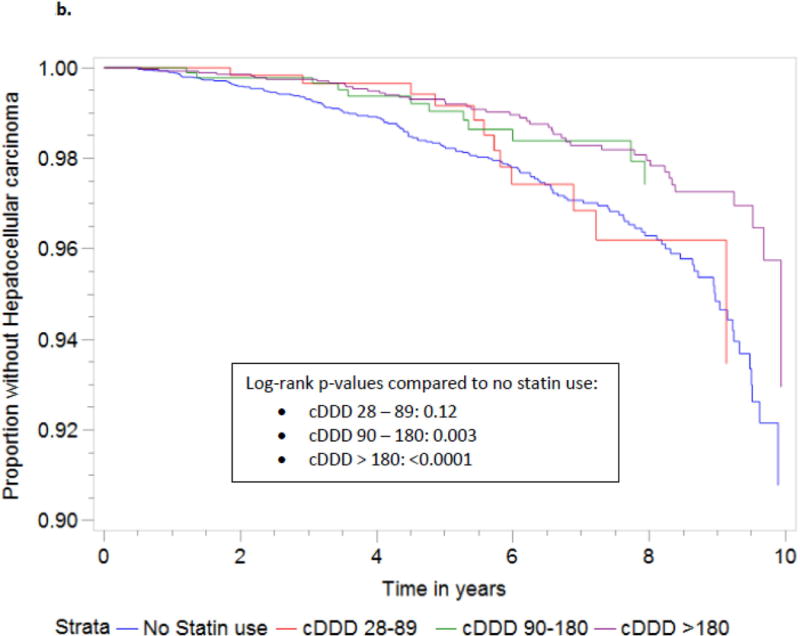
1Development of cirrhosis defined as any follow-up FIB-4 score > 3.5 in patients with baseline FIB-4 score < 3.5.
Abbreviations: HCC, hepatocellular carcinoma; cDDD, cumulative defined daily dose
There was also a significant dose-response relationship between statin use and reduction in fibrosis progression (Table 2). To further define this relationship, statin users were divided into three groups based upon yearly statin cDDD. The mean change in FIB-4 scores according to statin cDDD is shown in Supplementary Figure 1. For patients with cDDDs of 28–89, 90–180 and >180, the adjusted HR of fibrosis progression among statin users was 0.74 (95% CI 0.59, 0.93, p=0.01), 0.71 (95% CI 0.59–0.86, p=0.0006) and 0.6 (95% CI 0.53, 0.68, p<0.0001), respectively, compared to non-users (Table 2).
Table 2.
Complementary log-log regression analysis of statin use according to cumulative defined daily dose (cDDD) and progression of liver fibrosis1, among subjects with baseline FIB-4 score < 3.5 (n=9135)
| Unadjusted HR | Adjusted HR* | ||||||
|---|---|---|---|---|---|---|---|
| Variable, (N) | Fibrosis progression1 N (%) | β coefficient | HR (95% CI) | P-value | β coefficient | HR (95% CI) | P-value |
| Statin use2, (4165) | |||||||
|
81 (14.92%) 122 (14.97%) 381 (13.57%) |
−0.44 −0.42 −0.58 |
0.65(0.52,0.81) 0.66(0.54,0.79) 0.56(0.5,0.63) |
0.0002 <.0001 <.0001 |
−0.31 −0.34 −0.51 |
0.74(0.59,0.93) 0.71(0.59,0.86) 0.6(0.53,0.68) |
0.01 0.0006 <.0001 |
| No Statin use, (4970) | 1065 (21.43%) | 1 | 1 | ||||
HR, hazard ratio; HR adjusted for age, sex, race, smoking history, alcohol abuse history, body mass index (BMI), Diabetes, baseline FIB-4 score, Metformin use, Angiotensin converting enzyme (ACE) inhibitor use, other lipid-lowering agent use, prior completed anti-HCV treatment, attainment of SVR and daily caffeine intake
Progression of fibrosis: defined as any follow-up FIB-4 score ≥ 3.5 during study observation period, in patients with baseline FIB-4 score < 3.5.
Statin use defined as ≥28 cDDDs of statin medications, over study observation period
The mean change in FIB-4 score according to statin type Table 3. Overall, the greatest reduction in FIB-4 scores was observed with atorvastatin and fluvastatin, compared with other statins and compared with the change from baseline (mean change in FIB-4 score of −0.17 and −0.13, respectively; p=0.04 after adjustment for baseline FIB-4 scores and other univariate predictors of cirrhosis).
Table 3.
Mean overall change‡ in FIB-4 scores over the study period, according to type of statin medication used.
| Statin type (N) | Mean Change (SD) | p-value1 |
|---|---|---|
| FIB-4 Score | ||
| ATORVASTATIN (944) | −0.17 (1.01) | 0.04 |
| FLUVASTATIN (34) | −0.13 (0.91) | |
| LOVASTATIN (86) | 0.4 (1.9) | |
| PRAVASTATIN (609) | −0.03 (2.2) | |
| ROSUVASTATIN (187) | −0.07 (0.98) | |
| SIMVASTATIN (2305) | 0.11 (4.92) | |
| No Statin Use (4969) | 0.26 (2.12) | |
Overall change defined as baseline FIB-4 score subtracted from the last FIB-4 score at end of study period
P values obtained using analysis of covariance adjusted for baseline FIB-4 scores, as well as age, sex, race, smoking history, alcohol abuse, body mass index (BMI), Diabetes, Metformin use, other lipid-lowering agent use, prior HCV treatment and attainment of sustained virologic response (SVR).
Lipophilic statin medications included Atorvastatin, lovastatin, simvastatin
Hydrophilic statin medications included Rosuvastatin, fluvastatin, pravastatin
Development of HCC
Statin use was also associated with a dose-dependent reduction in the incidence of HCC (Table 4). Those subjects who received more than 90 cDDDs demonstrated a significant reduction in HCC risk (p=0.004), and the greatest benefit was observed in those who received >180 cDDD of statins (p<0.0001). For subjects with 90–180 cDDDs and >180 cDDDs, the adjusted HRs of incident HCC were 0.48 (95% CI 0.27, 0.88; p=0.02), and 0.51 (95% CI 0.36, 0.72, p=0.0001). Increasing statin dose was also associated with significantly delayed time to the development of HCC (Figure 2b).
Table 4.
Hazard Ratio (HR) of statin use and reduction of hepatocellular carcinoma (HCC) risk, among included subjects (n=9135)
| Model | Non-users1 | Statin use1 | |||||
|---|---|---|---|---|---|---|---|
| 28 to 89 cDDDs2 | 90 to 180 cDDDs | >180 cDDDs | |||||
HCC status, No. of patients
|
160 4810 |
12 531 |
12 803 |
49 2758 |
|||
HCC status, No. of person-years
|
863.06 33556.21 |
65.84 3936.89 |
54.16 5957.27 |
299.43 22528.85 |
|||
| Incidence rate (per 105 person-years from baseline) | 464.86 (392.83,536.89) | 299.8 (130.17,469.42) | 199.62 (86.67,312.56) | 214.65 (154.55,274.75) | |||
| Absolute risk reduction* | n/a | 165.06 | 265.24 | 250.21 | |||
| HR (95% CI) | HR(95% CI) | p-value | HR(95% CI) | p-value | HR(95% CI) | p-value | |
| Crude HR | 1 | 0.63 (0.35,1.13) | 0.12 | 0.42 (0.23,0.75) | 0.004 | 0.43 (0.31,0.6) | <.0001 |
| Adjusted HR† | 1 | 0.85 (0.47,1.53) | 0.58 | 0.48 (0.27,0.88) | 0.02 | 0.51 (0.36,0.72) | 0.0001 |
Statin use defined as ≥ 28 cumulative defined daily doses (cDDDs) of statin medications, over study observation period
cDDDs, cumulative defined daily doses
Absolute risk reduction per 105 person-years, from baseline
HR, hazard ratio; HR adjusted for age, sex, race, smoking history, alcohol abuse history, caffeine intake, body mass index (BMI, Diabetes, baseline FIB-4 score, Metformin use, Angiotensin converting enzyme (ACE) inhibitor use, other lipid-lowering agent use, non-steroidal anti-inflammatory medication use, prior completed HCV treatment, attainment of sustained virologic response (SVR), and daily caffeine intake
In order to estimate whether the anti-HCC effects of statins were independent of their antiviral and antifibrotic effects, we conducted two sensitivity analyses of HCC risk. The first was stratified by the mean change in FIB-4 score between baseline and year 1 of follow-up. We excluded those subjects who derived an anti-fibrotic benefit from statin use, defined as mean annual reduction in FIB-4 ≥ 0.4, as has been validated in previously-published literature [34]. This adjustment did not significantly impact the relationship between statin use and HCC risk (HR 0.6 among patients who received > 180 cDDDs (95% CI 0.38, 0.94; p=0.03 after adjusting for baseline FIB-4). In a separate sensitivity analysis, we excluded subjects who had attained SVR, and this adjustment also did not diminish the strength of the association between statin cDDD and risk of either fibrosis progression or incident HCC.
Discussion
To our knowledge, this is the first US study to demonstrate a dose-response relationship between statin use and reduction in hepatic fibrosis progression and the development of HCC, after controlling for the potentially confounding effects of age, sex, HCV RNA level, baseline level of fibrosis, alcohol use, smoking history, and the concomitant use of potentially beneficial medications such as metformin or other lipid-lowering agents. It is also the first study to document a relationship between statin type and fibrosis progression. In the final adjusted model, statin use was associated with a 44% overall reduction in risk of fibrosis progression, and a 49% reduction in the incidence of HCC.
Fibrosis progression
When patients were stratified according to cumulative statin use, we observed a statistically significant inverse relationship between increasing statin dose and reduction in the HR of fibrosis progression and HCC, at each time point during the 10-year follow up period. These associations were statin-specific, and remained significant even after adjustment for established predictors of both fibrosis progression and HCC. These results therefore add to the growing body of data showing that statins not only treat dyslipidemia but also delay disease progression, in patients with HCV [22, 24].
Statins are among the most commonly prescribed medications worldwide, and have been shown to offer significant benefit to patients with chronic liver disease [11, 22] through pleiotropic antiproliferative, antiangiogenic, anti-inflammatory and anti-neoplastic actions [6–8]. Despite this, clinical data regarding the antifibrotic effects of statins in CHC remain limited. In a recent analysis of the HALT-C Trial cohort, a significant association between statin use and reduced fibrosis progression was observed [22], and similar results were found in a large national cohort study of US Veterans with chronic HCV [24]. Until recently, however, the impact of dose, duration or statin type upon hepatic fibrosis progression had not been evaluated, and this is the first study to do so in a US population.
Yang and colleagues recently reported a dose-response relationship between statin use and fibrosis progression in a population-based cohort from a national Taiwanese database [28]. However, this study was limited in several important ways. First, the diagnosis of HCV was defined only by reported ICD-9 code, rather than by HCV antibody or confirmatory HCV RNA. Similarly, a diagnosis of cirrhosis was defined only by ICD-9 code, without additional direct or surrogate clinical measurements. It is conceivable that cirrhosis – particularly compensated cirrhosis – may have gone unrecognized in a disproportionate group of patients, resulting in underestimation or misclassification of outcomes. Additionally, fibrosis severity, which is an important predictor of cirrhosis risk, was not assessed at baseline or at follow-up time points, and the study did not adjust for other potential confounders, including baseline HCV viral load, obesity, alcohol use or smoking status.
A number of mechanisms have been proposed to explain the antifibrotic effects of statins, related to their antiviral and immunomodulatory effects [5]. Statins inhibit the formation of lipid rafts and block the formation of geranylgeranylated F-box/leucine-rich repeat protein 2, both of which are necessary for HCV replication [35, 36]. They also block the activation of hepatic myofibroblasts, inducing apoptosis and preventing hepatic stellate cell proliferation and collagen synthesis [6, 7, 13, 19–21]. They have also been shown to upregulate transcription factors that exert vasoprotective effects in the liver and inhibit stellate cells [37]. Finally, and perhaps most importantly, statins have demonstrated clinical efficacy in the treatment of the metabolic syndrome, the components of which are independent risk factors for HCC among patients with chronic HCV [38–40].
Statin type and fibrosis progression
Whether certain types of statins possess greater antifibrotic potential in chronic HCV has been previously unknown. Our study is the first to compare the effects of different statin types on fibrosis progression in this population. We observed the greatest antifibrotic benefit with atorvastatin and fluvastatin, compared to simvastatin, pravastatin, lovastatin, or no statin use. This association remained significant even after adjustment for established predictors of cirrhosis. In clinical studies, fluvastatin has been associated with potent anti-HCV effects and enhanced rates of sustained virological response (SVR) [26, 41], and a recent analysis showed a reduction in NASH-mediated fibrosis through inhibition of paracrine signaling, with fluvastatin use [42]. Future studies with large sample sizes and histological endpoints will be necessary, in order to validate our findings.
Statin use and HCC
This is the first US study to demonstrate a significant dose-response relationship between statin use and reduction in the incidence of HCC. We observed a 47% overall reduction in incident HCC among statin users, which is in accord with previously published values, including a recent meta-analysis which reported a 37% reduced risk of HCC among statin users [18]. As with the analyses of fibrosis, this dose-response relationship was statin-specific, and remained significant after adjustment for other established predictors of HCC. Moreover, in sensitivity analysis, the strength of this association was not diminished by the exclusion of subjects who had attained SVR.
Statins may exert chemoprotective effects through the inhibition of thioredoxin, a hepatic enzyme which is increased in pre-malignant hepatic nodules and plays a role in cell survival [43]. By blocking cyclins and cyclin-dependent kinases [44], statins induce tumor cell apoptosis [45] and microtubule bundling [46], while also promoting cell cycle arrest [45] Statins also interfere with lipid rafts, and inhibit tumor cell adhesion and migration [36]. In vitro, HCC cells treated with statins have been found to decrease the expression of cell adhesion molecules thus preventing cell growth and invasion possibly via a rho-dependent kinase [47].
It has also been postulated that the lipid-lowering action of statins may directly result in chemoprevention [48, 49]. Cancer cells undergo metabolic reprogramming that increases lipid biosynthesis [50] and enhances expression of enzymes within the mevalonate pathway [51]; this upregulation has been associated with mutations in tumor suppressor genes, increased cell spread and the development of HCC [35, 51]. Conversely, the suppression of these pathways results in tumor suppression and apoptosis [35, 51]. It has also been shown that statin-mediated inhibition of mevalonate prevents the generation of multiple end producs, including geranylgeranyl pyrophosphate, which may result in reduced development of HCC [35, 52].
Strengths of this study include the use of a large, unselected national cohort of US subjects, with long length or follow-up and serial measurements of clinical and laboratory parameters. Medication information was obtained from an integrated, comprehensive pharmacy database, which collected all prescription data including dose, quantity and refills. Additionally, to minimize confounding by indication and by severity of liver disease, we excluded patients with baseline cirrhosis, baseline HCC, and adjusted for baseline fibrosis scores in the final multivariable model. Though residual confounding cannot be completely excluded in an observational study, a dose-dependent relationship between statin use and reduced fibrosis progression and incident HCC was seen in all subjects, regardless of baseline severity of disease.
With a non-randomized study design, this analysis was subject to potential selection bias and unmeasured confounding variables. However, with such a large cohort, we were able to control for many observed and well-described confounders, in our adjusted models. A second potential limitation is use of a surrogate clinical score rather than liver biopsy or transient elastography, for the measurement and determination of hepatic fibrosis stage. However, the FIB-4 score is a well-validated and widely used marker of liver fibrosis progression, in the published literature. In addition, use of such a large sample size may minimize the variance otherwise attributable to this index score. Finally, and perhaps most importantly, it must be underscored that prescription data is inherently imprecise, and true patient adherence is often significantly reduced in comparison to prescription pharmacy information.
In conclusion, our findings demonstrate a significant dose-dependent reduction in the risk of both fibrosis progression and incident HCC, among statin users with CHC. These results support the possible role for statins in the prevention of liver disease progression. Future prospective studies with histological and clinical endpoints are needed. Such analyses will need to define the optimal timing of statin initiation, ideal duration of therapy, and the relative antifibrotic potential of different types of statins, in patients with chronic hepatitis C as well as other etiologies of liver disease.
Supplementary Material
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the VA Pittsburgh Healthcare System and the central data repositories maintained by the VA Information Resource Center, including the National Patient Care Database, Decisions Support System Database and Pharmacy Benefits Management Database. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs.
Financial Support: K24 DK078772 (RTC)
Footnotes
Conflict of Interest: Dr. Butt has received investigator initiated grants (to the institution) from Gilead and AbbVie.
Author’s Contributions:
Tracey G. Simon: conception and design, interpretation of data, drafting of the article, critical revision
Hector Bonilla: conception and design, interpretation of data, drafting of the article, critical revision
Peng Yan: acquisition of data, statistical analysis
Raymond T. Chung: conception and design, data interpretation, critical revision, study supervision
Adeel A. Butt: conception and design, acquisition of data, statistical analysis, data interpretation, critical revision, study supervision
References
- 1.Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Annals of internal medicine. 2006;144:705–714. doi: 10.7326/0003-4819-144-10-200605160-00004. [DOI] [PubMed] [Google Scholar]
- 2.Thomas DL, Seeff LB. Natural history of hepatitis C. Clinics in liver disease. 2005;9:383–398, vi. doi: 10.1016/j.cld.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 3.van der Meer AJ, Veldt BJ, Feld JJ, Wedemeyer H, Dufour JF, Lammert F, et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA: the journal of the American Medical Association. 2012;308:2584–2593. doi: 10.1001/jama.2012.144878. [DOI] [PubMed] [Google Scholar]
- 4.Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Journal of the American College of Cardiology. 2014;63:2889–2934. doi: 10.1016/j.jacc.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Sun HY, Singh N. Antimicrobial and immunomodulatory attributes of statins: relevance in solid-organ transplant recipients. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2009;48:745–755. doi: 10.1086/597039. [DOI] [PubMed] [Google Scholar]
- 6.Wu J, Wong WW, Khosravi F, Minden MD, Penn LZ. Blocking the Raf/MEK/ERK pathway sensitizes acute myelogenous leukemia cells to lovastatin-induced apoptosis. Cancer research. 2004;64:6461–6468. doi: 10.1158/0008-5472.CAN-04-0866. [DOI] [PubMed] [Google Scholar]
- 7.Rao S, Porter DC, Chen X, Herliczek T, Lowe M, Keyomarsi K. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7797–7802. doi: 10.1073/pnas.96.14.7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuoppala J, Lamminpaa A, Pukkala E. Statins and cancer: A systematic review and meta-analysis. European journal of cancer. 2008;44:2122–2132. doi: 10.1016/j.ejca.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 9.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. The Journal of clinical investigation. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kisseleva T, Brenner DA. Mechanisms of fibrogenesis. Experimental biology and medicine. 2008;233:109–122. doi: 10.3181/0707-MR-190. [DOI] [PubMed] [Google Scholar]
- 11.Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371:838–851. doi: 10.1016/S0140-6736(08)60383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwaisako K, Brenner DA, Kisseleva T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. Journal of gastroenterology and hepatology. 2012;27(Suppl 2):65–68. doi: 10.1111/j.1440-1746.2011.07002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shirin H, Sharvit E, Aeed H, Gavish D, Bruck R. Atorvastatin and rosuvastatin do not prevent thioacetamide induced liver cirrhosis in rats. World journal of gastroenterology: WJG. 2013;19:241–248. doi: 10.3748/wjg.v19.i2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kisseleva T, Brenner DA. Anti-fibrogenic strategies and the regression of fibrosis. Best practice & research Clinical gastroenterology. 2011;25:305–317. doi: 10.1016/j.bpg.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nature reviews Cancer. 2005;5:930–942. doi: 10.1038/nrc1751. [DOI] [PubMed] [Google Scholar]
- 16.Welzel TM, Graubard BI, Zeuzem S, El-Serag HB, Davila JA, McGlynn KA. Metabolic syndrome increases the risk of primary liver cancer in the United States: a study in the SEER-Medicare database. Hepatology. 2011;54:463–471. doi: 10.1002/hep.24397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonovas S, Filioussi K, Tsavaris N, Sitaras NM. Statins and cancer risk: a literature-based meta-analysis and meta-regression analysis of 35 randomized controlled trials. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24:4808–4817. doi: 10.1200/JCO.2006.06.3560. [DOI] [PubMed] [Google Scholar]
- 18.Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology. 2013;144:323–332. doi: 10.1053/j.gastro.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Trebicka J, Hennenberg M, Odenthal M, Shir K, Klein S, Granzow M, et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. Journal of hepatology. 2010;53:702–712. doi: 10.1016/j.jhep.2010.04.025. [DOI] [PubMed] [Google Scholar]
- 20.Miyaki T, Nojiri S, Shinkai N, Kusakabe A, Matsuura K, Iio E, et al. Pitavastatin inhibits hepatic steatosis and fibrosis in non-alcoholic steatohepatitis model rats. Hepatology research: the official journal of the Japan Society of Hepatology. 2011;41:375–385. doi: 10.1111/j.1872-034X.2010.00769.x. [DOI] [PubMed] [Google Scholar]
- 21.Marcelli M, Cunningham GR, Haidacher SJ, Padayatty SJ, Sturgis L, Kagan C, et al. Caspase-7 is activated during lovastatin-induced apoptosis of the prostate cancer cell line LNCaP. Cancer research. 1998;58:76–83. [PubMed] [Google Scholar]
- 22.Simon TG, King LY, Zheng H, Chung RT. Statin Use is Associated with a Reduced Risk of Fibrosis Progression in Chronic Hepatitis C. Journal of hepatology. 2014 doi: 10.1016/j.jhep.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsan YT, Lee CH, Ho WC, Lin MH, Wang JD, Chen PC. Statins and the risk of hepatocellular carcinoma in patients with hepatitis C virus infection. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:1514–1521. doi: 10.1200/JCO.2012.44.6831. [DOI] [PubMed] [Google Scholar]
- 24.Butt AA, Yan P, Bonilla H, Abou-Samra AB, Shaikh OS, Simon TG, et al. Effect of addition of statins to antiviral therapy in hepatitis C virus-infected persons: Results from ERCHIVES. Hepatology. 2015;62:365–374. doi: 10.1002/hep.27835. [DOI] [PubMed] [Google Scholar]
- 25.Ikeda M, Abe K, Yamada M, Dansako H, Naka K, Kato N. Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology. 2006;44:117–125. doi: 10.1002/hep.21232. [DOI] [PubMed] [Google Scholar]
- 26.Bader T, Fazili J, Madhoun M, Aston C, Hughes D, Rizvi S, et al. Fluvastatin inhibits hepatitis C replication in humans. The American journal of gastroenterology. 2008;103:1383–1389. doi: 10.1111/j.1572-0241.2008.01876.x. [DOI] [PubMed] [Google Scholar]
- 27.Wuestenberg A, Kah J, Singethan K, Sirma H, Keller AD, Rosal SR, et al. Matrix conditions and KLF2-dependent induction of heme oxygenase-1 modulate inhibition of HCV replication by fluvastatin. PloS one. 2014;9:e96533. doi: 10.1371/journal.pone.0096533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y-H, Chen WC, Tsan YT, Chen MJ, Shih WT, Tsai YH, Chen PC. Statin use and the risk of cirrhosis development in patients with hepatitis C virus infection. Journal of hepatology. 2015 doi: 10.1016/j.jhep.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 29.Butt AA, Yan P, Lo Re V, 3rd, Rimland D, Goetz MB, Leaf D, et al. Liver fibrosis progression in hepatitis C virus infection after seroconversion. JAMA internal medicine. 2015;175:178–185. doi: 10.1001/jamainternmed.2014.6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Department of Veterans Affairs Hepatitis CRC. Yee HS, Currie SL, Darling JM, Wright TL. Management and treatment of hepatitis C viral infection: recommendations from the Department of Veterans Affairs Hepatitis C Resource Center program and the National Hepatitis C Program office. The American journal of gastroenterology. 2006;101:2360–2378. doi: 10.1111/j.1572-0241.2006.00754.x. [DOI] [PubMed] [Google Scholar]
- 31.Backus LI, Boothroyd DB, Phillips BR, Mole LA. Predictors of response of US veterans to treatment for the hepatitis C virus. Hepatology. 2007;46:37–47. doi: 10.1002/hep.21662. [DOI] [PubMed] [Google Scholar]
- 32.Zeuzem S, Heathcote EJ, Shiffman ML, Wright TL, Bain VG, Sherman M, et al. Twelve weeks of follow-up is sufficient for the determination of sustained virologic response in patients treated with interferon alpha for chronic hepatitis C. Journal of hepatology. 2003;39:106–111. doi: 10.1016/s0168-8278(03)00187-9. [DOI] [PubMed] [Google Scholar]
- 33.World Health Organization Collaborating Center for Drugs Statistical Methodology. ATC Index with Defined Daily Dose. Oslo, Noway: World Health Organization (WHO); 2003. p. 2003. [Google Scholar]
- 34.Tamaki N, Kurosaki M, Tanaka K, Suzuki Y, Hoshioka Y, Kato T, et al. Noninvasive estimation of fibrosis progression overtime using the FIB-4 index in chronic hepatitis C. Journal of viral hepatitis. 2013;20:72–76. doi: 10.1111/j.1365-2893.2012.01635.x. [DOI] [PubMed] [Google Scholar]
- 35.Kapadia SB, Chisari FV. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2561–2566. doi: 10.1073/pnas.0409834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clendening JW, Penn LZ. Targeting tumor cell metabolism with statins. Oncogene. 2012;31:4967–4978. doi: 10.1038/onc.2012.6. [DOI] [PubMed] [Google Scholar]
- 37.Marrone G, Maeso-Diaz R, Garcia-Cardena G, Abraldes JG, Garcia-Pagan JC, Bosch J, et al. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: behind the molecular mechanisms of statins. Gut. 2014 doi: 10.1136/gutjnl-2014-308338. [DOI] [PubMed] [Google Scholar]
- 38.Siegel AB, Zhu AX. Metabolic syndrome and hepatocellular carcinoma: two growing epidemics with a potential link. Cancer. 2009;115:5651–5661. doi: 10.1002/cncr.24687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohki T, Tateishi R, Sato T, Masuzaki R, Imamura J, Goto T, et al. Obesity is an independent risk factor for hepatocellular carcinoma development in chronic hepatitis C patients. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2008;6:459–464. doi: 10.1016/j.cgh.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 40.Konishi I, Hiasa Y, Shigematsu S, Hirooka M, Furukawa S, Abe M, et al. Diabetes pattern on the 75 g oral glucose tolerance test is a risk factor for hepatocellular carcinoma in patients with hepatitis C virus. Liver international: official journal of the International Association for the Study of the Liver. 2009;29:1194–1201. doi: 10.1111/j.1478-3231.2009.02043.x. [DOI] [PubMed] [Google Scholar]
- 41.Bader T, Hughes LD, Fazili J, Frost B, Dunnam M, Gonterman A, et al. A randomized controlled trial adding fluvastatin to peginterferon and ribavirin for naive genotype 1 hepatitis C patients. Journal of viral hepatitis. 2013;20:622–627. doi: 10.1111/jvh.12085. [DOI] [PubMed] [Google Scholar]
- 42.Chong LW, Hsu YC, Lee TF, Lin Y, Chiu YT, Yang KC, et al. Fluvastatin attenuates hepatic steatosis-induced fibrogenesis in rats through inhibiting paracrine effect of hepatocyte on hepatic stellate cells. BMC gastroenterology. 2015;15:22. doi: 10.1186/s12876-015-0248-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skogastierna C, Johansson M, Parini P, Eriksson M, Eriksson LC, Ekstrom L, et al. Statins inhibit expression of thioredoxin reductase 1 in rat and human liver and reduce tumour development. Biochemical and biophysical research communications. 2012;417:1046–1051. doi: 10.1016/j.bbrc.2011.12.091. [DOI] [PubMed] [Google Scholar]
- 44.Relja B, Meder F, Wilhelm K, Henrich D, Marzi I, Lehnert M. Simvastatin inhibits cell growth and induces apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. International journal of molecular medicine. 2010;26:735–741. doi: 10.3892/ijmm_00000520. [DOI] [PubMed] [Google Scholar]
- 45.Zhang W, Wu J, Zhou L, Xie HY, Zheng SS. Fluvastatin, a lipophilic statin, induces apoptosis in human hepatocellular carcinoma cells through mitochondria-operated pathway. Indian journal of experimental biology. 2010;48:1167–1174. [PubMed] [Google Scholar]
- 46.Ali N, Allam H, Bader T, May R, Basalingappa KM, Berry WL, et al. Fluvastatin interferes with hepatitis C virus replication via microtubule bundling and a doublecortin-like kinase-mediated mechanism. PloS one. 2013;8:e80304. doi: 10.1371/journal.pone.0080304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Relja B, Meder F, Wang M, Blaheta R, Henrich D, Marzi I, et al. Simvastatin modulates the adhesion and growth of hepatocellular carcinoma cells via decrease of integrin expression and ROCK. International journal of oncology. 2011;38:879–885. doi: 10.3892/ijo.2010.892. [DOI] [PubMed] [Google Scholar]
- 48.Farwell WR, Scranton RE, Lawler EV, Lew RA, Brophy MT, Fiore LD, et al. The association between statins and cancer incidence in a veterans population. Journal of the National Cancer Institute. 2008;100:134–139. doi: 10.1093/jnci/djm286. [DOI] [PubMed] [Google Scholar]
- 49.Poynter JN, Gruber SB, Higgins PD, Almog R, Bonner JD, Rennert HS, et al. Statins and the risk of colorectal cancer. The New England journal of medicine. 2005;352:2184–2192. doi: 10.1056/NEJMoa043792. [DOI] [PubMed] [Google Scholar]
- 50.Hirsch HA, Iliopoulos D, Joshi A, Zhang Y, Jaeger SA, Bulyk M, et al. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer cell. 2010;17:348–361. doi: 10.1016/j.ccr.2010.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes & development. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karlic H, Thaler R, Gerner C, Grunt T, Proestling K, Haider F, et al. Inhibition of the mevalonate pathway affects epigenetic regulation in cancer cells. Cancer genetics. 2015;208:241–252. doi: 10.1016/j.cancergen.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.