

Abstract
Post-translational modifications of core histones play an important role in regulating fundamental biological processes such as DNA repair, transcription and replication. In this paper, we describe a novel assay that allows sequential targeting of distinct histone modifying enzymes to immobilized nucleosomal templates using recombinant chimeric targeting molecules. The assay can be used to study the histone substrate specificity of chromatin modifying enzymes as well as whether and how certain enzymes affect each other's histone modifying activities. As such the assay can help to understand how a certain histone code is established and interpreted.
Keywords: Chromatin, Histone code
Introduction
In a eukaryotic cell, genetic information is stored in a highly conserved and complex structural polymer called chromatin. The nucleosome forms the fundamental repetitive unit of chromatin and consists of 146 base pairs of DNA wrapped around an octamer of histone proteins containing two copies of each of the four core histones, H2A, H2B H3 and H4. DNA packed in nucleosomes and higher order chromatin structures is generally thought to be repressive to processes that require access of proteins to DNA such as transcription initiation, replication or DNA repair. Thus, structural reorganizations that “open” and/or “close” the chromatin template during different genetic processes are required to facilitate proper functioning of the genome. This is largely achieved by two distinct mechanisms: ATP dependent chromatin remodelling (1) and post-translational modifications of core histones (2).
Each of the four core histones is subject to several post-translational modifications. These include: acetylation, methylation, phosphorylation, ubiquitination, poly-ADP ribosylation and biotinylation. During the last decade, a large number of proteins and protein complexes from different organisms have been identified and characterized that catalyse these modifications and contribute, either negatively or positively, to the accessibility of DNA. In order to increase our understanding of fundamental biological processes such as transcription and replication it is of crucial importance to address the biochemical characteristics of these chromatin modifying enzymes. Evidence is accumulating that modified histones serve as a binding scaffold for regulatory proteins. As such, histone modifications and certain combinations of modifications play a major role in dictating the accessibility status of the chromatin. This hypothesis is known as the histone code theory (3).
Of the currently known histone modifications, lysine acetylation has been studied most extensively. Acetylation of histones is a reversible and highly dynamic event catalysed by histone acetyltransferases (HATs) and histone deacetylases (HDACs). All four core histones contain several lysine residues, mostly located in their N-terminus, that are modified by HATs and HDACs. To understand the functioning of these enzymes, in vitro assays are needed that can be used to address how they are targeted to chromatin and to dissect the histone substrate specificity of different HATs and HDACs. Although substantial progress has been made in deciphering the histone substrate specificity and targeting mechanisms of HAT complexes (4, 5) such systems have been lacking for HDACs. Here, we present an assay that allows studying the recruitment and the enzymatic activities of distinct HDAC containing complexes. In addition to analysing histone acetylation and deacetylation, this assay can be used to study the interplay between different histone modifying enzymes and as such can be used to understand how the histone code is established and interpreted.
Materials and Methods
Purification of proteins
The Sin3/HDAC and N-CoR/SMRT complexes were purified from HeLa nuclear extract. Briefly, approximately 5 g of protein was loaded on a P11 phosphocellulose column (Whatman) as described (6). Bound proteins were eluted stepwise with 300 mM, 500 mM and 1000 mM KCl. The majority of the Sin3/HDAC complex was found in the 500mM fraction, whereas the majority of the N-CoR protein was found in the 300mM fraction, as determined by western blotting using antibodies against Sin3a (Santa Cruz) and N-CoR (Santa Cruz). The Sin3/HDAC complex was subsequently enriched on a Phenyl sepharose (AP biotech), DEAE sepharose (AP biotech) and Superose 6 column (AP biotech) whereas the N-CoR/SMRT complex was further purified on a DEAE sepharose and Superose 6 column (7). The enzymatic activity of the obtained fractions was determined by performing in vitro HDAC assays (8).
Constructs encoding LexA-Mad(5-24) and LexA-TR(DE) were cloned in plasmid pET28a (Novagen) and purified as histidine-tagged proteins from BL21DE3 plysS bacteria (Novagen) as described (4). SAGA and NuA4 were purified from S. cerevisiae on Ni2+-nitrilotriacetic acid-agarose (Qiagen), MonoQ (AP biotech) and Superose 6 (AP biotech) columns as described previously (9). A strain was used that contains HA-tagged Ada2, Flag-tagged Tra1 and HA- tagged Esa1 (10) to facilitate detection of these subunits of the SAGA and NuA4 complexes during the purication of the HAT complexes.
Immobilized template pull-down assays
A 520 basepair biotinylated DNA fragment containing 8 LexA binding sites and a 5S nucleosome positioning element was obtained as described (7). This DNA was reconstituted into a nucleosomal template with recombinant Xenopus octamers using a step dilution protocol and immobilized on streptavidin conjugated Dynabeads (Dynal) (11).
Timecourse experiment
Each reaction contained approximately 200 ng of reconstituted immobilized templates. These templates were acetylated with native SAGA complex purified from S. cerevisiae for one hour at 30°C in 45 μl buffer F (50 mM KCl, 10 mM Hepes pH 7.8, 5% v/v glycerol, 2mM MgCl2, 0.05% NP40, 1mM PMSF and 5mM DTT). Templates were extensively washed with buffer F containing 300 mM KCl and 0.5% v/v Triton X-100 to remove the SAGA complex from the beads. Stripping of the HATs from the templates was monitored using western blotting with an HA antibody (Covance) that recognizes the HA tagged Ada2 (for SAGA) or Esa1 (for NuA4) of these HAT complexes (data not shown). The templates were then resuspended in 50 μl buffer F containing 75 mM KCl and 5 μl of the superose 6 fraction containing the Sin3/HDAC compex (7). Reactions were incubated for 5, 10, 20, 40 or 60 minutes at 37°C in a thermoshaker. Beads were washed extensively with buffer F containing 300mM KCl and 0.25% NP40, after which, western blotting analysis was performed. Blots were probed with antibodies against acetylated histone H3 lysine 9,14 (Upstate) and HDAC2 (Santa Cruz).
Competitor titration experiment
Templates were acetylated and subsequently stripped of HATs as described above. Beads were then incubated for 60 minutes at 37°C with 5 μl of the Superose 6 fractions containing the Sin3/HDAC complex with increasing amounts of non-immobilized competitor nucleosome arrays purified from chicken erythrocytes (12). The amount of competitor nucleosomes needed to prevent untargeted HDAC2 association and deacetylation of the acetylated immobilized nucleosomes was subsequently determined by Western blotting using antibodies against acetylated histone H3 lysine 9,14 and HDAC2.
Recruitment of the Sin3/HDAC complex to a long immobilized nucleosomal array
Plasmid L8G5E4T (7) was digested with EcoRI after which a Klenow fill-in was performed using bio-dATP (Gibco). The linearised plasmid was subsequently digested with Asp718, gel-purified, reconstituted with recombinant Xenopus octamers and bound to streptavidin-conjugated Dynabeads (Dynal). Subsequently, targeting experiments were performed as described (7).
Results and Discussion
Immobilized template assay
To study the recruitment and enzymatic activities of different chromatin modifying enzymes, we have established an in vitro reconstituted system containing purified components (Fig. 1A). A relatively short piece of biotinylated DNA, 520 base pairs, is mixed in a 1:1 molar ratio (1 nucleosome per 175 basepairs of DNA) with purified recombinant Xenopus histone octamers (13) and reconstituted into a nucleosomal array using a step dilution protocol (11). The reconstitution can subsequently be analysed using a partial micrococcal nuclease digestion (7). Alternatively, the efficiency of the reconstitution can be analysed on conventional agarose gels (Fig. 1B). Due to the deposition of nucleosomes, the nucleosomal template migrates slower as compared to naked DNA. The templates are subsequently immobilized on streptavidin conjugated Dynabeads. An important feature of these immobilized nucleosomal templates is that they can be washed and resuspended in the buffer of choice after each incubation step. This means that different enzymatic reactions can be performed sequentially, with intervening stripping steps. To mimic the in vivo situation, we aimed to recruit chromatin modifying complexes to in vitro reconstituted immobilized nucleosomal templates in a competitive setting. This means that the recruitment step is performed in the presence of a large molar excess of competitor nucleosomes not bound to beads (Fig. 1A).
Fig. 1. A: Schematic representation of the immobilized template assay.
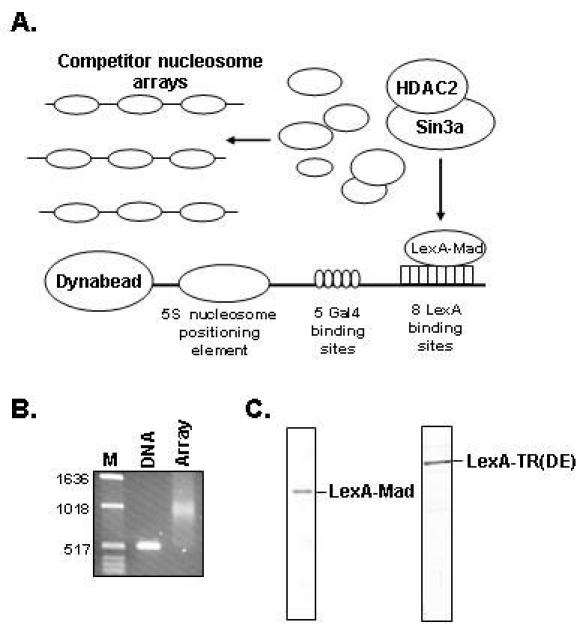
B: DNA reconstituted into a nucleosomal array was loaded on a 1.2% agarose gel and stained with ethidiumbromide afterwards. Naked DNA was loaded as a control. C: The purified LexA-Mad and LexA-TR(DE) proteins were loaded on an SDS-polyacrylamide (12%) gel and stained with Commassie brilliant blue.
In this competitive setting, “targeting” molecules are needed that recruit chromatin modifying enzymes to the immobilized nucleosomal templates. Since we are using a nucleosomal template containing both LexA and Gal4 binding sites, chimeric proteins containing a LexA or Gal4 DNA binding domain can be used and fused to distinct peptides or protein domains that are known to interact with a particular chromatin modifying enzyme. The obvious advantage of using a template containing both LexA and Gal4 binding sites is that co-activator molecules can for example be recruited via Gal4 fusion proteins, such as the commonly used Gal4-VP16 molecule, whereas LexA fusion proteins, such as LexA-Mad, can be used to recruit co-repressor molecules. Thus, this immobilized template assay has the potential to study activating versus repressing activities concomitantly.
Expression and purification of chimeric repessor molecules
We expressed and purified chimeric repressor molecules containing the LexA DBD fused to the N-terminus (amino acids 5-24) of the Mad repressor (LexA-Mad) as well as containing the LexA DBD fused to the DE domain of the thyroid hormone receptor (LexA-TR(DE)). These two molecules have the ability to specifically recruit distinct histone deacetylase containing co-repressor complexes to immobilized nucleosomal templates. The LexA-Mad molecule can recruit the Sin3/HDAC complex whereas the LexA-TR(DE) protein can recruit the N-CoR/SMRT complex to chromatin. The interaction between the N-terminus of Mad and Sin3a as well as the interaction between the DE domain of the unliganded thyroid hormone receptor and N-CoR is well characterized (14-16). LexA-Mad and LexA-TR(DE) are purified from E. coli as histidine-tagged proteins on Ni2+-nitrilotriacetic acid agarose and MonoQ columns. Following these purification steps the purity is estimated to be at least 95% (Fig. 1C). EMSA assays on LexA binding site oligonucleotides (17) can be performed to monitor the DNA binding capacity of the recombinant LexA fusion proteins.
Setting conditions for recruitment experiments
Prior to the recruitment experiments described above and in our previous work, several parameters need to be determined in titration experiments. The deacetylation activity of the Sin3/HDAC complex is determined in a non-competitive setting in a timecourse experiment. As shown in Figure 2A, deacetylation of the immobilized template was completed after approximately 60 minutes for this particular preparation (lane 6). The amount of competitor nucleosomes that needs to be added to prevent non-specific deacetylation of the immobilized templates should be determined in a titration experiment (Fig. 2B) and is influenced by the amount of histone deacetylases added in the reaction as well as the ratio between immobilized templates and competitor nucleosomes. If the concentration of HDACs added in the reaction is too high, even a 50 fold molar excess of competitor nucleosomes versus immobilized nucleosomes may not be sufficient to prevent deacetylation of the immobilized nucleosomes. In the experiment shown in Figure 2B, a 100 fold molar excess of competitor nucleosomes suffices to prevent deacetylation of the immobilized nucleosomes (lane 6). This amount of competitor should subsequently be used in targeting experiments in which the ability of the LexA-Mad or LexA-TR(DE) protein to recruit the Sin3/HDAC or N-CoR/SMRT complex and their enzymatic activity to the immobilized templates are tested (7). An additional advantage of using competitor nucleosomes is that contaminating chromatin modifying enzymes present in the input fraction that are used for the recruitment reactions are diverted away from the immobilized templates to the non-immobilized competitor nucleosomes such that their enzymatic activities do not disturb the analyses and blur any conclusions. This means that relatively crude protein fractions can be used to perform these experiments. The fractions we used are enriched approximately 500-1000 fold for the Sin3/HDAC and N-CoR/SMRT complexes.
Fig. 2. Determining the deacetylase capacity of the Sin3/HDAC complex and the amount of competitor nucleosome arrays needed to prevent non-targeted deacetylation.

A: Immobilized nucleosomal templates acetylated with the yeast SAGA complex were incubated with the purified Sin3/HDAC complex using various incubation times. The amount of HDAC2 binding to the templates and the amount of histone H3 acetylation was detemined by western blotting using an antibody against HDAC2 and an antibody that recognizes diacetylated histone H3 Lys 9, 14. B: Immobilized nucleosomal templates acetylated with the yeast SAGA complex were incubated for 1 hour at 37°C with the purified Sin3/HDAC complex in the presence of varying amounts of non-immobilized competitor nucleosomal arrays. The amount of histone H3 acetylation and HDAC2 association was subsequently determined as described in A.
Template requirements
Initially targeting experiments were performed on fairly large nucleosomal templates containing 3500 basepairs of plasmid DNA. However, we were never able to obtain substantial LexA-Mad or LexA-TR(DE) mediated targeted deacetylation of these templates in a competitive setting, whereas complete deacetylation of the template could be observed in the absence of competitor nucleosomes (Fig. 3, compare lane 2 and 4). This is likely due to the fact that HDAC containing complexes only deacetylate locally, i.e. a few nucleosomes surrounding the LexA DNA binding sites. Chromatin immunoprecipitation experiments by other labs support this hypothesis (18). Local deacetylation is difficult to detect on western blot, which assesses the acetylation state of all the nucleosomes on the template. When using a relatively short DNA fragment, the nucleosomes on the immobilized templates are located close to the LexA binding sites and targeted deacetylation can be observed (7).
Fig. 3. Targeted recruitment of the Sin3/HDAC to long nucleosomal templates pre-acetylated with the yeast NuA4 complex.

Immobilized nucleosomal templates acetylated with the yeast NuA4 complex were incubated with or wihout LexA-Mad and subsequently incubated with the Sin3/HDAC complex in the presence or absence of competitor nucleosomal arrays. The amount of histone H4 acetylation was determined with western blotting with an antibody that recognizes tetra-acetylated histone H4.
Acknowledgments
We thank Coen Campsteijn for purifying recombinant Xenopus histone octamers and Martijn van Aart for assistance. We thank Jerry L. Workman and Michael J. Carrozza for providing yeast strain YJW526. Furthermore, we thank Colin Logie for critically reading the manuscript. The research of Michiel Vermeulen is financially supported by the Netherlands Organisation for Scientific Research (NWO).
Appendix
Protocols
Preparation of the DNA template
Digest 500 μg of L8G515S plasmid with 100 units of BamH1 (New England Biolabs) for 2 hours at 37°C in 500 μl in 1 X New England Biolabs (NEB) buffer 2. Check whether digestion is complete by loading a small aliquot on agarose gel
Add: 75 μl of 0.4 mM biotinylated dATP, 75 μl of 1mM dCTP, dTTP and dGTP, 25μl NEB buffer 2, 100 units Klenow polymerase (Invitrogen) and H2O to a final volume of 750 μl. Incubate for 45 minutes at room temperature.
Add 75 μl 3 M NaAc pH 5.2 and 1875 μl ice cold 100% ethanol. Precipitate DNA at -20°C for 15 minutes.
Centrifuge for 10 minutes at 13000 rpm. Wash pellet with ice cold 70% ethanol, air dry the pellet.
Redissolve the pellet in 400 μl H2O. Add 50 μl NEB buffer 2, 100 units of HinDIII (New England Biolabs) and H2O to a final volume of 500 μl. Incubate for 2 hours at 37°C. Check whether digestion is complete on agarose gel. A 520 basepair fragment should be observed.
Load sample on an agarose gel. Divide the sample over a number of slots. Run the DNA long enough for the short fragment (520 basepairs) to be separated from the long fragment (3000 basepairs).
Gel purify the short DNA fragment using a gel-purification kit (Qiagen). The amount of short DNA fragment should be around 70 μg starting from 500 μg of plasmid. Use 8 Qiagen spin columns (capacity 10 μg of DNA per column).
After elution of the DNA from the columns according to the manufacturer's instructions, pool and reprecipitate the purified DNA, wash the pellet with 70% ethanol, air dry the pellet and finally redissolve the DNA in 10 μl of H2O. A final yield of approximately 30 μg of DNA is usually obtained.
Nucleosome reconstitution
Add together: 2 μg of purified recombinant Xenopus octamers, 2.6 μg of purified DNA, 1 μl of 1 mg/ml BSA, 2.79 μl 5M NaCl and H2O to a final volume of 10 μl. Using these amounts of DNA and histones results in a 1:1 molar ratio, meaning approximately 1 nucleosome per 175 basepairs of DNA.
Incubate at 37°C for 15 minutes.
Dilute the reaction by adding 3.3, 6.7, 5, 3.6, 4.7, 6.7, 10, 30, and 20 μl, respectively, of 50 mM Hepes-KOH (pH 7.5), 1 mM EDTA, 5 mM DTT, and 0.5 mM PMSF, with 15 minutes incubations at 30°C for each incubation step. Finally, add 100 μl of 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, 0.1% NP40, 5 mM DTT, 0.5 mM PMSF, 20% glycerol, and 0.1 mg/ml BSA and incubate the reaction for 15 minutes at 30°C. Reconstitutions can be stored at 4°C up to 4 months.
Analysing histone deaceylase activity
Add 22 μl 3 M KCl to the reconstitution (final concentration KCl 300 mM).
Take 60 μl of streptavidin conjugated dynabeads (Dynal) and wash them twice with 200 μl buffer F (50 mM KCl, 10 mM Hepes-KOH pH 7.8, 5% glycerol, 2mM MgCl2, 5 mM DTT, 0.25 mg/ml BSA and 0.5 mM PMSF) according to the manufacturer's instructions.
Add the reconstitution to the beads and incubate at for 1 hour at 37°C in a thermoshaker.
Wash beads twice with 200 μl buffer F containing 300 mM KCl, then twice with 200 μl buffer F. Finally, resuspend beads in 200 μl buffer F. Immobilized nucleosomal templates can be stored at 4°C for about a week prior to usage.
Use 20 μl of immobilized nucleosomal template per reaction. Wash beads twice with 50 μl buffer F, then resuspend the beads in 45 μl buffer F. Add 1 μM AcetylCoA (Sigma), 10 mM sodium butyrate (Invitrogen) and purified HAT complex. The amount of HAT complex added in the reaction should produce at least 6000 dpm in an in vitro HAT assay. Incubate beads for 1 hour at 30°C in a thermoshaker.
Wash beads twice with 100 μl buffer F containing 300 mM KCl and then wash and resuspend the beads in 45 μl Buffer F containing 75 mM KCl, 0.1% NP40 and complete protease inhibitors (Roche biochemicals).
Add purified HDAC complex to the reactions. Incubate the samples at 37°C for 5, 10, 20, 40 or 60 minutes in a thermoshaker. Wash beads three times with buffer F containing 300 mM KCl. Add Laemmli buffer to the beads, incubate the beads for 5 minutes at 95°C and load the samples on a 15% SDS PAGE gel.
Perform western blotting. Cut the membrane in two parts. Incubate the upper part with an antibody against the HDAC molecule or co-repressor of interest. Incubate the lower part of the blot with an antibody against acetylated histone H3 or H4 or any histone antibody of choice.
References
- Becker PB, Horz W. ATP-dependent nucleosome remodeling. Annual Review of Biochemistry. 2002;71:247–273. doi: 10.1146/annurev.biochem.71.110601.135400. [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Current Opinion in Cell Biology. 2003;15:172–183. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Kundu TK, Palhan VB, Wang Z, An W, Cole PA, Roeder RG. Activator-dependent transcription from chromatin in vitro involving targeted histone acetylation by p300. Molecular Cell. 2000;6:551–561. doi: 10.1016/s1097-2765(00)00054-x. [DOI] [PubMed] [Google Scholar]
- Utley RT, Ikeda K, Grant PA, Cote J, Steger DJ, Eberharter A, John S, Workman JL. Transcriptional activators direct histone acetyltransferase complexes to nucleosomes. Nature. 1998;394:498–502. doi: 10.1038/28886. [DOI] [PubMed] [Google Scholar]
- Valcarcel R, Stunnenberg HG. Retinoid-dependent in vitro transcription. Methods in Enzymology. 1996;274:149–161. doi: 10.1016/s0076-6879(96)74014-4. [DOI] [PubMed] [Google Scholar]
- Vermeulen M, Carroza MJ, Lasonder E, Workman JL, Logie C, Stunnenberg HG. In vitro targeting reveals histone tail specificity of the Sin3/Histone Deacetylase and N-CoR/SMRT co-repressor complexes. Molecular and Cellular Biology. 2004;24:2364–2372. doi: 10.1128/MCB.24.6.2364-2372.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolle D, Brosch G, Lechner T, Lusser A, Loidl P. Biochemical methods for analysis of histone deacetylases. Methods. 1998;15:323–331. doi: 10.1006/meth.1998.0636. [DOI] [PubMed] [Google Scholar]
- Eberharter A, John S, Grant PA, Utley RT, Workman JL. Identification and analysis of yeast nucleosomal histone acetyltransferase complexes. Methods. 1998;15:315–321. doi: 10.1006/meth.1998.0635. [DOI] [PubMed] [Google Scholar]
- Carroza MJ, John S, Sil AK, Hopper JE, Workman JL. Gal80 confers specificity on HAT complex interactions with activators. Journal of Biological Chemistry. 2002;277:24648–24652. doi: 10.1074/jbc.M201965200. [DOI] [PubMed] [Google Scholar]
- Steger DJ, Workman JL. Transcriptional analysis of purified histone acetyltransferase complexes. Methods. 1999;19:410–416. doi: 10.1006/meth.1999.0877. [DOI] [PubMed] [Google Scholar]
- Logie C, Peterson CL. Purification and biochemical properties of yeast SWI/SNF complex. Methods in Enzymology. 1999;304:726–741. doi: 10.1016/s0076-6879(99)04044-6. [DOI] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Glass CK, Rosenfeld MG. The c oregulator exchange in transcriptional functions of nuclear receptors. Genes & Development. 1997;14:121–141. [PubMed] [Google Scholar]
- Spronk CA, Tessari M, Kaan AM, Jansen JF, Vermeulen M, Stunnenberg HG, Vuister GW. The Mad1-Sin3B interaction involves a novel helical fold. Nature Structural Biology. 2000;7:1100–1104. doi: 10.1038/81944. [DOI] [PubMed] [Google Scholar]
- Brubaker K, Cowley SM, Huang K, Loo L, Yochum GS, Ayer DE, Eisenman RN, Radhakrishnan I. Solution structure of the interacting domains of the Mad-Sin3 complex: implications for recruitment of a chromatin-modifying complex. Cell. 2000;103:655–665. doi: 10.1016/S0092-8674(00)00168-9. [DOI] [PubMed] [Google Scholar]
- Mohana-Borges R, Pacheco AB, Sousa FJ, Foguel D, Almeida DF, Silva JL. LexA repressor forms stable dimers in solution. The role of specific dna in tightening protein-protein interactions. Journal of Biological Chemistry. 2000;275:4708–4712. doi: 10.1074/jbc.275.7.4708. [DOI] [PubMed] [Google Scholar]
- Deckert J, Struhl K. Targeted recruitment of Rpd3 histone deacetylase represses transcription by inhibiting recruitment of Swi/Snf, SAGA, and TATA binding protein. Molecular and Cellular Biology. 2002;22:6458–6470. doi: 10.1128/MCB.22.18.6458-6470.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]