

Abstract
Aims
The aim of the present study was to investigate the safety, tolerability and pharmacokinetics of coadministered azithromycin (AZI) and piperaquine (PQ) for treating malaria in pregnant Papua New Guinean women.
Methods
Thirty pregnant women (median age 22 years; 16–32 weeks' gestation) were given three daily doses of 1 g AZI plus 960 mg PQ tetraphosphate with detailed monitoring/blood sampling over 42 days. Plasma AZI and PQ were assayed using liquid chromatography–mass spectrometry and high‐performance liquid chromatography, respectively. Pharmacokinetic analysis was by population‐based compartmental models.
Results
The treatment was well tolerated. The median (interquartile range) increase in the rate‐corrected electrocardiographic QT interval 4 h postdose [12 (6–26) ms0 .5] was similar to that found in previous studies of AZI given in pregnancy with other partner drugs. Six women with asymptomatic malaria cleared their parasitaemias within 72 h. Two apararasitaemic women developed late uncomplicated Plasmodium falciparum infections on Days 42 and 83. Compared with previous pregnancy studies, the area under the concentration–time curve (AUC0–∞) for PQ [38818 (24354–52299) μg h l−1] was similar to published values but there was a 52% increase in relative bioavailability with each dose. The AUC0–∞ for AZI [46799 (43526–49462) μg h l−1] was at least as high as reported for higher‐dose regimens, suggesting saturable absorption and/or concentration‐dependent tissue uptake and clearance from the central compartment.
Conclusions
AZI–PQ appears to be well tolerated and safe in pregnancy. Based on the present/other data, total AZI doses higher than 3 g for the treatment and prevention of malaria may be unnecessary in pregnant women, while clearance of parasitaemia could improve the relative bioavailability of PQ.
Keywords: azithromycin, malaria, pharmacokinetics, piperaquine, pregnancy, safety
What is Already Known about this Subject
Azithromycin has been used as part of combination antimalarial therapy in pregnancy, along with conventional drugs including chloroquine and sulphadoxine–pyrimethamine.
Piperaquine has also been used in pregnancy and has advantages in areas of known chloroquine and sulphadoxine–pyrimethamine parasite resistance.
Both drugs cause nausea and prolong the electrocardiographic QT interval, and it has not been established whether they have a pharmacokinetic interaction.
What this Study Adds
Combination azithromycin–piperaquine was well tolerated and showed promising efficacy in treating malaria in Papua New Guinean women beyond the first trimester of pregnancy
There was no evidence of greater cardiotoxicity with this drug combination than with either drug alone.
Based on relative drug exposure, doses of azithromycin greater than 1 g daily for 3 days may be unnecessary for the treatment of malaria in pregnancy.
Introduction
Malaria in pregnancy remains a major cause of maternal anaemia, low birthweight and increased perinatal mortality 1. One of the major pharmacological strategies used to improve obstetric outcome is intermittent preventive treatment of malaria in pregnancy (IPTp) 2, 3. The World Health Organization (WHO)‐recommended IPTp is sulphadoxine–pyrimethamine (SP) 4 but residual maternal/fetal morbidity (in spite of adherence to SP IPTp regimens), the spread of SP‐resistant Plasmodium falciparum, limited efficacy against P. vivax and the relatively short half‐lives of the component drugs (6–11 days for sulphadoxine and 3–15 days for pyrimethamine 5, 6, 7), have prompted the search for alternatives 8, 9, 10. Prominent among these are combinations that include azithromycin (AZI) 11, 12, 13, 14, 15.
AZI is an azalide antibiotic with unique pharmacokinetic (PK) properties, characterized by prolonged tissue and phagocytic concentrations 16, 17. It has an elimination half‐life (t½) of 12–78 h in pregnant 14, 18 and 54–102 h in non‐pregnant 19, 20, 21, 22, 23 women. It has a slow onset of antimalarial action and modest efficacy as monotherapy 24 but a potentially valuable role as part of combination treatment 11, 24, 25, 26. The advantages of AZI‐based IPTp include its safety 2 and efficacy against other pathogens that can affect obstetric outcomes, such as those causing sexually transmitted diseases 27. In addition, its PK properties show that dose adjustment is probably unnecessary in pregnancy 14. The choice of a partner drug depends on tolerability, safety and PK/pharmacodynamic interactions. Both SP 15 and chloroquine (CQ) 12 added to AZI have proved to be effective in clinical trials but, as with adding artemisinin derivatives to SP or CQ 28, the effectiveness of these combinations is compromised by using failing drugs as partners.
Piperaquine (PQ) combined with dihydroartemisinin (DHA) is effective treatment for uncomplicated malaria 29, 30. No concerns have been raised in animal reproductive toxicity studies of PQ 31, 32 and it appears to be safe and well tolerated in human pregnancy 33, 34, 35, 36, 37, 38. Similarly to other 4‐aminoquinolines, PQ prolongs the electrocardiographic QT interval at therapeutic concentrations 39 and this may have implications for safety. As a result, the manufacturers recommend that DHA–PQ is avoided in patients with a history of arrhythmias, long QT syndrome or who are taking other drugs known to prolong the QT interval 40, 41. It has a long elimination t½ (16–22 days in pregnant and 21–28 days in nonpregnant women 33, 35, 36, 42, 43), which may be advantageous if ensuring regular IPTp dosing is problematic 2. There have, however, been no studies of AZI–PQ in pregnancy.
From available studies, the AZI dose that is both efficacious as part of antimalarial therapy with a longer‐acting drug such as CQ and not associated with tolerability issues, especially nausea, is 1 g daily for 3 days 12. Although higher doses have been used in pregnancy, including 2 g daily for 2 days in an IPTp trial in Papua New Guinea (PNG) 13, gastrointestinal symptoms are comparatively frequent with these regimens 14, 24. Given that PQ is also a drug associated with nausea and vomiting 32, a conservative AZI dose would seem appropriate in initial interaction studies. In addition, both PQ 32 and AZI 44 can prolong the QT interval and, although their individual proarrhythmic effects are unclear 44, 45, it is not known whether they have additive or synergistic cardiotoxicity. This further suggests caution in selecting an AZI dose for human interaction studies.
In view of these considerations, we have assessed the safety, tolerability and PK properties of AZI–PQ in pregnant women from PNG. Given the possibility that some women could have malarial parasitaemia, we used a midrange but previously effective dose of AZI (1 g daily for 3 days) and a conventional adult dose of PQ.
Methods
Study site, approval and participants
The present study was conducted at the Alexishafen Health Centre, Madang Province, PNG, where there is endemic transmission of P. falciparum and P. vivax 46. Pregnant women at 14–32 weeks' gestation and attending their first antenatal visit were eligible if: (i) they had not taken study drugs in the previous 28 days; (ii) they had no history of allergy to study drugs; (iii) there was no significant comorbidity and there were no features of severe malaria; and (iv) they could attend all follow‐up assessments. Witnessed informed consent was obtained from each participant, and verbal consent from their husband/father as culturally appropriate in PNG. Ethics approval was obtained from the PNG Institute of Medical Research Institutional Review Board, the Medical Research Advisory Committee of the PNG Health Department, and the Human Research Ethics Committee of the University of Western Australia.
Baseline assessment and treatment allocation
A detailed medical history, including a standardized symptom questionnaire, was completed, a physical examination (including estimation of gestational age by fundal height) was performed and a 12‐lead electrocardiogram (ECG) was taken. Thick and thin blood smears were prepared. An intravenous cannula was inserted and a 2 ml baseline sample collected for determining haemoglobin and blood glucose levels. The remaining blood was centrifuged within 15 minutes of collection. The separated plasma was placed promptly in a foil‐covered tube, stored immediately at –20 °C and then transferred to –80 °C for long‐term storage prior to drug assay.
All participants received three daily doses (at 0, 24 and 48 h) of 1 g AZI as film‐coated 500 mg tablets (Sandoz, Pyrmont, NSW, Australia), given with three PQ tetraphosphate 320 mg tablets (Sigma‐Tau Industrie Farmaceutiche Riunite S.p.A., Italy) at a dose of 58 mg kg−1 (or 33 mg kg−1 PQ base for a 50 kg woman), consistent with the manufacturer's and WHO recommendations at the time the study was carried out 3. As there was a possibility of hypoglycaemia, especially in the parasitaemic women, participants were fasted for ≥2 h before and after dosing, rather than the ≥3 h recommended by the manufacturer. All treatments were administered with water and observed directly. Women vomiting within 30 min of dosing were to be retreated. After a 48–72 h inpatient stay, each participant was given an insecticide‐treated bed net and instructed on its use for the rest of the pregnancy as an alternative to SP IPTp.
Monitoring, sampling and follow‐up
Detailed assessment, including lying/standing blood pressure and pulse rate, respiratory rate, axillary temperature, haemoglobin and thick/thin blood smears, was carried out at follow‐up visits on Days 1, 2, 3, 7, 14, 28 and 42. Additional monitoring comprised: (i) completion of the standardized side‐effect questionnaire at 6 h and on Days 1, 2, 3 and 7; (ii) blood glucose on Days 1, 2 and 3; (iii) ECG at 4, 12 and 24 h; (iv) assessment of fetal viability (maternally detected movements and fetal heart beat on auscultation) daily on Days 1–4 and then at each subsequent sampling time‐point; and (v) ultrasound scans if required to confirm gestational age and to determine fetal lie and presentation. Clinical review on Days 14, 28 and 42 included assessment of symptoms even though the standardized questionnaire was not administered at these follow‐up visits.
The timing of the ECGs relative to dosing, and the use of Bazett's method to determine the rate‐corrected QT interval in lead II (QTc = QT/√RR) were selected to allow comparison with previous studies in pregnant PNG women 36. Each QT interval was determined by manual measurement from the ECG tracing. All blood smears were examined, and parasitaemia quantified, on site by study staff and subsequently by two skilled independent microscopists in a central laboratory. Discrepancies were adjudicated by a senior microscopist. Parasite densities were calculated from the number of parasites/200 white cells (or /500 white cells if there were <25 parasites/200 white cells) and an assumed total white cell count of 8000 μl−1, with the final density calculated as the geometric mean of the two or three values from expert microscopy 47.
As AZI–PQ is not yet a WHO‐recommended treatment for malaria in pregnancy, all monitoring data were reviewed for each participant, at each study visit, by the on‐site study clinician (JMB). Those women in whom clinical concerns emerged were to be transferred to Modilon Hospital in Madang Town (the provincial referral facility) for further assessment and treatment.
A sparse blood sampling protocol was used, based on previous studies of AZI–CQ 14, AZI–SP 14, PQ–SP 36 and PQ–DHA 36. Participants were randomly assigned to seven of 18 time‐points (1, 2, 3, 6, 12, 24, 32, 40, 48 and 72 h, and Days 4, 5, 7, 10, 14, 21, 28 and 42) at which 3 ml blood samples were drawn either through an intravenous cannula (to 72 h) or by venepuncture. The 24 h and 48 h samples were taken immediately before the second and third doses, respectively.
Participants were returned to usual care after the Day 42 assessment but they were requested to attend Alexishafen Health Centre when they went into labour, or to notify study staff if they delivered in an alternative local healthcare facility or at home. If delivery occurred at the Alexishafen Health Centre, maternal thick/thin blood films were prepared and a maternal haemoglobin concentration was measured. A 3 ml sample of maternal and cord blood was taken for AZI and PQ assay, three thick/thin cord blood smears and a placental smear were prepared, and a section of placenta was collected into 10% neutral buffered formalin for evaluation if cord/placental smears were positive for malaria.
Drug assays
Plasma PQ was measured by a validated high‐performance liquid chromatography assay 48. Briefly, extracted plasma was injected onto a Gemini 5 μm C6‐phenyl column coupled with a SecurityGuard column (Phenomenex®, Lane Cove, NSW, Australia). Analytes were detected at 346 nm and quantified using Chemstation Software (version 9; Agilent Technology, Waldbronn, Germany). For concentrations ranging from 2.5 μg l−1 to 500 μg l−1, intraday relative standard deviations (RSDs) were 2.5–8.1% and interday RSDs were 3.6–9.6%. The limits of quantification (LOQ) and of detection (LOD) were 2 μg l−1 and 1 μg l−1, respectively, with a signal‐to‐noise ratio of 3.0.
Plasma AZI was measured using a triple quadrupole mass spectrometer (LCMS‐8030 plus; Shimadzu, Kyoto, Japan). Plasma samples were extracted as described 14, and 20 μl aliquots were spiked with AZI‐d3 1000 μg l−1 (internal standard), added to 50 μl ammonium hydroxide (40 mM) and 500 μl methanol, mixed for 30 s, and centrifuged at 10 000 × g for 5 min. Supernatant (100 μl) was mixed with 100 μl water, and 5 μl was injected onto an Acquity UPLC BEH C18 column connected to a VanGuard Acquity BEH C18 guard column (Waters Corporation, Wexford, Ireland) at 50 °C. The mobile phase [acetonitrile : methanol: 40 mM ammonium hydroxide (50 : 25 : 25) and acetonitrile : methanol (50 : 50) in equal volumes] was run in isocratic mode at 0.2 ml min−1. Retention times for AZI and AZI‐d3 were both 1.38 min. Quantitation was performed in multiple reaction monitoring mode using an ESI+ ion source. The precursor–product ion pairs were AZI m/z 749.4 → 591.6 and AZI‐d3 m/z 752.5 → 594.5. Optimized mass spectra were acquired with a 4.5 kV interface voltage, a 1.0 kV detector voltage, and heat block and desolvation line temperatures of 400 °C and 250 °C, respectively. Nitrogen was used as the nebulizer and drying gas at flow rates of 3 l min−1 and 10 l min−1, respectively. Argon was used as the collision gas at 230 kPa. Dwell time for both AZI and AZI‐d3 was 100 ms, and their collision energies were −32 V and −33 V, respectively. For concentrations 10–2000 μg l−1, intraday RSDs were 3.2–7.5% and interday RSDs were 4.3–9.1%. The LOQ and LOD were 2.5 μg l−1 and 1.3 μg l−1, respectively, with signal‐to‐noise ratios of ≥10 : 1 and ≥3 : 1, respectively.
PK modelling
The package NONMEM (v 7.2.0, ICON Development Solutions, Ellicott City, MD, USA) with an Intel Visual FORTRAN 10.0 compiler employing nonlinear mixed effects modelling was used to analyse loge plasma concentration–time PQ and AZI data. The first‐order conditional estimation with interaction estimation method was used and a P‐value <0.05 was set as the significance level for a comparison of the nested models (ΔOFV = −6.635, χ2 df = 1). A priori allometric scaling was employed, with volume terms multiplied by (body weight/70)1.0 and clearance terms by (body weight/70)0.75 49. For residual variability (RV), two structures were tested using the log‐transformed data. These were equivalent to exponential and combined RV structures on the normal scale 50. Using final model parameters, post hoc Bayesian prediction in NONMEM was utilized to derive secondary PK parameters, including area under the plasma concentration–time curve (AUC0–∞) and half‐lives (t½s). Simulated plasma concentrations after the final dose were used to obtain estimates of the maximum plasma concentration (Cmax) Base models included the parameters absorption rate constant (ka), central volume of distribution relative to bioavailability (F) (VC/F), clearance (CL/F), and peripheral volumes of distribution and the associated intercompartmental clearances (VP/F and Q/F, respectively).
In the case of PQ, models with two and three compartments (through the NONMEM routines ADVAN 4 and 12) were tested, as were various absorption models, including those with zero‐, first‐ or mixed‐order absorption with and without lag time. There was significant variability in absorption, as reported in other studies 36, 51, which necessitated the assessment of a transit compartment model (the dose passing through a series of compartments before delayed entry into the absorption compartment 36, 51). In this model, there is a single rate constant (ktr) that describes the entry and exit for all transit compartments. The additional continuous variables in this model include the number of transit compartments (NN) and the mean transit time [MTT = (1 + NN)/ktr] 52. Similar models were tested for AZI, including zero‐, first‐ and mixed‐order absorption with and without a lag time linked with two‐ and three‐compartment models. A transit compartment was not included as a mixed‐order model has been shown to describe best the absorption kinetics of AZI in this patient group 14.
When model structures had been determined, variability parameters were added. These included interindividual variability (IIV) and interoccasion variability (IOV) for F and absorption parameters. Correlations between IIV terms were also estimated where possible. Relationships between all model parameters and malaria status, dose number and gestational age were assessed through inspection of scatter plots and box plots of eta (IIV code used in NONMEM) vs. covariate. These associations were then quantified within NONMEM using a stepwise forward inclusion and backward elimination method with P < 0.05 required for inclusion of, and P < 0.01 for retention of, a covariate relationship.
Model evaluation
The median and 2.5th and 97.5th percentiles [95% empirical confidence intervals (CIs)] were obtained from a bootstrap using Perl‐speaks‐NONMEM, using 500 samples to evaluate final model parameter estimates. The predictive performance of the model was evaluated using prediction‐corrected visual predictive checks of 1000 datasets simulated from the final models, from which the simulated 95% CIs for the 10th, 50th and 90th percentiles were plotted together with the corresponding observed data. Numerical predictive checks were also used in the evaluation of the predictive performance of the model.
Results
Subject characteristics
Thirty women (median age 22 years, median gestational age 26 weeks) were recruited, five of whom (16.7%) were aged <20 years (see Table 1). Twelve (40.0%) had been treated with SP between 14 days and 28 days before recruitment but none of the others had taken antimalarial drugs in the previous month. All women were asymptomatic but blood film examination showed that six (20.0%) had an asexual parasitaemia (P. falciparum, n = 4 (13.3%); P. vivax, n = 1 (3.3%); mixed P. falciparum/vivax, n = 1 (3.3%)), and one had P. falciparum gametocytes. The median [interquartile range (IQR)] asexual parasite densities at recruitment were 77 (41–113) μl–1 and 48 (39–56) μl–1 for P. falciparum and P. vivax, respectively. Consistent with PNG policy at the time of recruitment, routine HIV screening was not performed but no participant had clinical evidence of HIV and the prevalence in pregnant PNG women is 1.5% 53.
Table 1.
Baseline characteristics of the 30 pregnant participants. Data are percentages, median [interqutile range] and (range)
| Age (years) | 22 [20–26] | (18 to 32) |
| Weight (kg) | 54 [51–60] | (42 to 74) |
| Height (cm) | 157 [153–158] | (147 to 175) |
| Axillary temperature (°C) | 36.8 [36.4–37.0] | (36.0 to 37.1) |
| Plasmodium falciparum parasitaemia (%) | 16.7 | |
| Gestational age (weeks) | 26 [22–28] | (16 to 32) |
| Gravidity | 2 [1–3] | (1 to 4) |
| Parity | 1 [0–2] | (0 to 3) |
| Respiratory rate ( min −1 ) | 20 [17–22] | (16 to 28) |
| Pulse rate ( min −1 ) | 81 [76–87] | (68 to 108) |
| Systolic blood pressure (mmHg) | 93 [90–104] | (80 to 120) |
| Diastolic blood pressure (mmHg) | 60 [50–60] | (50 to 70) |
| Systolic fall on standing (mmHg) | −4 [−10 to 0] | (‐20 to 20) |
| Diastolic fall standing (mmHg) | −2 [−5 to 0] | (‐10 to 10) |
| Haemoglobin ( g l −1 ) | 97 [85–112] | (62 to 127) |
| Blood glucose ( mmol l −1 ) | 5.4 [4.4–5.8] | (3.1 to 8.3) |
| QT c ( ms 0 .5 ) | 436 [424–445] | (400 to 463) |
Tolerability, safety and efficacy
The treatment was well tolerated and all doses were taken without incident. Side effects during the first 7 days are summarized in Table 2. All were mild (and did not interfere with daily activities) and self‐limiting (resolution within 2 days), with nausea, anorexia and diarrhoea the most frequent. Two women developed skin rashes (on Days 1 and 2, respectively), one in a woman who had contact with a plant to which she had known sensitivity and the other of uncertain aetiology; both rashes resolved within 24 h without requiring treatment. All women were asymptomatic by Day 7. No participant developed a blood glucose <2.5 mmol l−1 during follow‐up. One woman had a baseline haemoglobin of 73 g l−1, which fell to 48 g l−1 on Day 14 without clinical evidence of blood loss. She responded to oral iron/folate (haemoglobin 74 g l−1 on Day 28).
Table 2.
Side effects reported by the 30 participants during the first week after treatment was started. Data are numbers of participants (percentages)
| Fever | 1 (3) |
| Chills | 0 (0) |
| Headache | 1 (3) |
| Nausea | 5 (17) |
| Vomiting | 0 (0) |
| Diarrhoea | 2 (7) |
| Abdominal pain | 1 (3) |
| Anorexia | 2 (7) |
| Insomnia | 0 (0) |
| Dizziness | 2 (7) |
| Rash | 2 (7) |
Changes in QTc after the first dose are summarized in Table 3, together with equivalent data from a previous study of DHA–PQ and CQ–SP in pregnant PNG women 36. There was an overall median increase in QTc from baseline of 12 ms0 .5 at 4 h in the present participants, with a progressive reduction at 12 h and 24 h, but no significant difference between parasitaemic and nonparasitaemic women at any time‐point (P > 0.13 by analysis of variance). These data were similar to those in the previous study of DHA–PQ and CQ–SP. Twenty one of the present 30 women (70.0%) had a 4 h QTc >440 ms0 .5, with a longest recorded QTc of 487 ms0 .5. No woman experienced palpitations or breathlessness during follow‐up. One complained of mild dizziness 6 h after the first dose (simultaneous QTc 453 ms0 .5) which had resolved by 12 h. Postural hypotension (>20 mmHg fall in systolic blood pressure after standing) occurred in seven participants (23.3%) during follow‐up but there were no associated symptoms and there was no clear temporal relationship with the time of drug administration.
Table 3.
Prolongation of the rate‐corrected electrocardiographic QT interval (QTc) in ms0 .5 in the present patients treated with azithromycin–piperaquine (AZI–PQ), including those who were positive or negative for malaria parasites at baseline, and in pregnant Papua New Guinea women recruited to a previous study [36] and treated with dihydroartemisinin–piperaquine (DHA–PQ) or sulphadoxine–pyrimethamine–piperaquine (SP–PQ). Data are median [interquartile range]
| AZI–PQ | DHA–PQ | SP–PQ | |||
|---|---|---|---|---|---|
| All | Malaria | Nonmalaria | All | All | |
| Number | 30 | 6 | 24 | 16 | 16 |
| 4 h | 12 [6 to 26] | 11 [9 to 16] | 16 [5 to 27] | 14 [5 to 26] | 14 [9 to 30] |
| 12 h | 8 [2 to 17] | 3 [−5 to 5] | 12 [3 to 19] | 6 [−1 to 12] | 4 [1 to 16] |
| 24 h | 8 [−1 to 18] | 4 [−1 to 20] | 10 [−1 to 17] | 9 [−1 to 20] | 12 [3 to 19] |
All asexual parasitaemias cleared by Day 3, although one woman developed a gametocytaemia that persisted to Day 7. Two women, both slide‐negative at baseline, developed falciparum malaria during follow‐up, one at Day 42 (with fever) and the other at delivery on Day 83 (asymptomatic but with positive peripheral and placental blood smears). In both cases, the women responded to a 3‐day course of artemether–lumefantrine prescribed under PNG treatment guidelines 54.
Obstetric outcomes
Of the 25 participants (83.3%) followed to parturition, seven successfully delivered at home without medical assistance, two delivered healthy babies at Modilon Hospital and the remaining 16 delivered at the Alexishafen Health Centre (mean ± standard deviation birth weight 2.9 ± 0.5 kg, 48% males). All deliveries were vaginal and two were footling presentations. One involved a posterior presentation accompanied by heavy meconium staining. The baby required respiratory support during transfer to the Modilon Hospital but was discharged well the following day.
PK modelling
None of the 189 PQ and 177 AZI plasma concentrations were below the LOQs. For PQ disposition, a three‐compartment model was superior to a two‐compartment model, with a reduction in the OFV (P < 0.001) and resolution of bias in CWRES plots. The structural model parameters were NN, MTT, VC/FPQ, VP1/FPQ, VP2/FPQ, CL/FPQ, Q1/FPQ and Q2/FPQ. Of these, the IIV of MTT, VC/FPQ and CL/FPQ were estimable. As ka was poorly estimated (high residual standard error, high correlation with MTT) for models using transit compartments, it was set at ktr (1 + NN)/MTT). As correlations between IIV terms for VC/FPQ and CL/FPQ were 0.99, the correlation co‐efficient (r) was fixed at unity to facilitate successful minimization of model estimation. IOV in relative bioavailability was estimated to examine potential differences in between‐dose relative PQ bioavailability (FPQ), as noted previously for PQ and other lipophilic antimalarials 51, 55, 56. There was a significant increase in FPQ for subsequent doses (ΔOFV = −8.414, P < 0.01). No other significant covariate relationships were identified.
A three‐compartment model for AZI was also chosen given a significant reduction in OFV (P < 0.001) and no identified bias on conditional weighted residual plots. Mixed zero‐ and first‐order absorption without a lag time described the absorption phase adequately and more complex models were not required. The structural model parameters were, therefore, ka, duration of zero‐order absorption (DUR), VC/FAZI, VP1/FAZI, VP2/FAZI, CL/FAZI, Q1/FAZI and Q2/FAZI. IIV was estimable for DUR, VC/FAZI and CL/FAZI, along with the correlation between IIV terms for VC/FAZI and CL/FAZI. The addition of an IOV term for relative bioavailability did not result in a significant fall in the OFV (P > 0.05). No significant covariate relationships were identified.
The final model parameter estimates and the bootstrap results for PQ and AZI are summarized in Tables 4, 5, respectively. Bias was <12% for all structural and random model parameters for both models. Figures 1, 2 show goodness‐of‐fit plots, and Figures 3, 4 show prediction‐corrected visual predictive checks for PQ and AZI, respectively. Secondary PK parameters for PQ and AZI are shown in Table 6.
Table 4.
Final population pharmacokinetic estimates and bootstrap results for piperaquine in pregnant Papua New Guinean women
| Parameter | Mean | RSE% | Bootstrap median [95% CI ] |
|---|---|---|---|
| Objective function value | −73.222 | −89.430 [−141.288, 34.582] | |
| Structural model parameters | |||
| MTT (h) | 1.42 | 14 | 1.37 [1.11, 2.01] |
| NN | 3.29 | 45 | 3.71 [0.87, 9.88] |
| CL/F PQ ( l h −1 70 kg −1 ) | 89.2 | 15 | 88.7 [68.5, 115.4] |
| V C /F PQ ( l h −1 70 kg −1 ) | 1920 | 23 | 1918 [1123, 3067] |
| Q 1 /F PQ ( l h −1 70 kg −1 ) | 69.2 | 22 | 67.2 [44.2, 98.8] |
| V P1 /F PQ ( l h −1 70 kg −1 ) | 17800 | 17 | 17 838 [12 465, 25 335] |
| Q 2 /F PQ ( l h −1 70 kg −1 ) | 275 | 24 | 269 [134, 429] |
| V P2 /F PQ ( l h −1 70 kg −1 ) | 3720 | 15 | 3707 [2622, 5264] |
| % increase in relative bioavailability with subsequent doses | 51.7 | 30 | 52.0 [25.4, 83.5] |
| Variable model parameters [shrinkage %] | |||
| IOV in F PQ (%) | 62 [8,15,30] | 9 | 60 [47. 70] |
| IIV in MTT (%) | 32 [36] | 24 | 29 [11, 50] |
| IIV in CL/F PQ (%) | 14 [49] | 62 | 15 [2, 27] |
| IIV in V C /F PQ (%) | 21 [49] | 40 | 20 [4, 38] |
| r(CL/F PQ ,V P1 /F PQ ) | 1 | FIX | FIX |
| RV for PQ (%) | 31 [24] | 12 | 29 [23, 37] |
CI, confidence interval; CL/FPQ, clearance relative to bioavailability; FPQ, relative bioavailability of piperaquine; IIV, interindividual variability; IOV, interoccasion variability; MTT, mean transit time; NN, number of transit compartments; Q1/FPQ, intercompartmental clearance for VP1/FPQ; Q2/FPQ, intercompartmental clearance for VP2/FPQ; RSE, residual standard error; RV, residual variability; VC/FPQ, central volume of distribution relative to bioavailability; VP1/FPQ, first peripheral volume of distribution relative to bioavailability; VP2/FPQ, second peripheral volume of distribution relative to bioavailability. IOV and IIV are presented as 100% × √(variability estimate).
Table 5.
Final population pharmacokinetic estimates and bootstrap results for azithromycin in pregnant Papua New Guinean women
| Parameter | Mean | RSE% | Bootstrap median [95% CI ] |
|---|---|---|---|
| Objective function value | −122.715 | −133.692 [−201.308, –73.288] | |
| Structural model parameters | |||
| k a ( h −1 ) | 0.89 | 12 | 0.92 [0.64, 1.44] |
| DUR (h) | 1.67 | 4 | 1.65 [0.98, 2.06] |
| CL/F AZI ( l h −1 70 kg −1 ) | 76.0 | 5 | 75.7 [67.6, 84.5] |
| V C /F AZI ( l h −1 70 kg −1 ) | 365 | 25 | 393 [198, 859] |
| Q 1 /F AZI ( l h −1 70 kg −1 ) | 228 | 14 | 223 [151, 297] |
| V P1 /F AZI ( l h −1 70 kg −1 ) | 2670 | 20 | 2660 [1457, 3883] |
| Q 2 /F AZI ( l h −1 70 kg −1 ) | 59 | 23 | 58.0 [32.9, 93.4] |
| V P2 /F AZI ( l h −1 70 kg −1 ) | 3980 | 13 | 3940 [2809, 4943] |
| Variable model parameters [shrinkage %] | |||
| IIV in DUR (%) | 60 [45] | 23 | 57 [9, 116] |
| IIV in CL/F AZI (%) | 25 [8] | 10 | 24 [13, 33] |
| IIV in V C /F AZI (%) | 127 [12] | 18 | 119 [51, 179] |
| r(CL/F AZI ,V P1 /F AZI ) | −0.80 | 10 | −0.86 [−1.00, –0.11] |
| RV for AZI (%) | 33 [12] | 10 | 29 [27, 32] |
CI, confidence interval; CL/FAZI, clearance relative to bioavailability; DUR, duration of zero‐order absorption; ka, rate for first‐order absorption; IIV, interindividual variability; Q1/FAZI, intercompartmental clearance for VP1/FAZI; Q2/FAZI, intercompartmental clearance for VP2/FAZI; RSE, residual standard error; RV, residual variability; VC/FAZI, central volume of distribution relative to bioavailability; VP1/FAZI, first peripheral volume of distribution relative to bioavailability; VP2/FAZI, second peripheral volume of distribution relative to bioavailability. IIV is presented as 100% × √(variability estimate).
Figure 1.
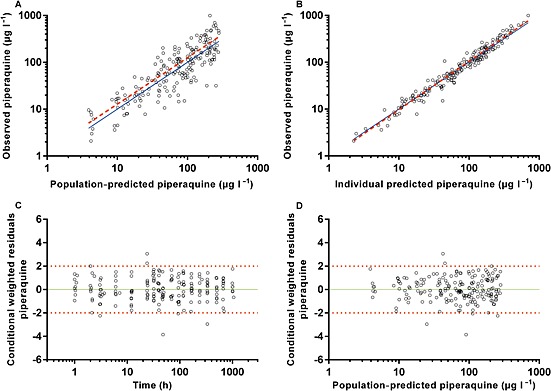
Diagnostic plots of the final plasma piperaquine population pharmacokinetic model. (A) observed vs. population‐predicted plasma concentration. (B) Observed vs. individual predicted plasma concentrations. (C) Conditional weighted residuals (CWRES) vs. time. (D) CWRES vs. population‐predicted concentrations. Solid lines represent lines of identity in A and B, and zero conditional residuals in C and D. Dashed lines represent lines of best fit in A and B, and dotted lines represent ±2 standard deviations in C and D
Figure 2.
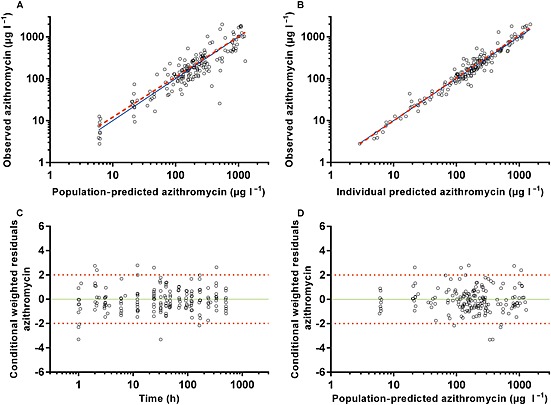
Diagnostic plots of the final azithromycin population pharmacokinetic model. (A) Observed vs. population‐predicted plasma concentration. (B) Observed vs. individual predicted plasma concentrations. (C) Conditional weighted residuals (CWRES) vs. time. (D) CWRES vs. population‐predicted concentrations. Solid lines represent lines of identity in A and B, and zero conditional residuals in C and D. Dashed lines represent lines of best fit in A and B, and dotted lines represent ±2 standard deviations in C and D
Figure 3.
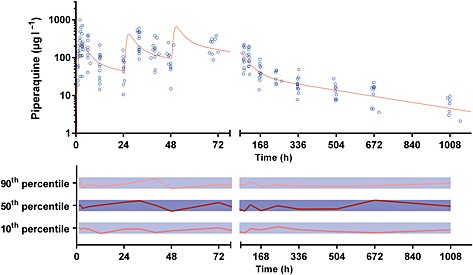
Plasma concentration vs. time data (upper panel) and normalized prediction‐corrected visual predictive check for plasma piperaquine (lower panel). The solid line in the upper panel represents the average concentration vs. time curve for the present patients overlying individual data points, represented as open circles. The solid and dotted lines in the lower panel represent the normalized observed 50th percentile and its 80% prediction interval (i.e. 10th and 90th percentiles), respectively, within their normalized simulated 95% confidence intervals, represented as grey shaded areas
Figure 4.
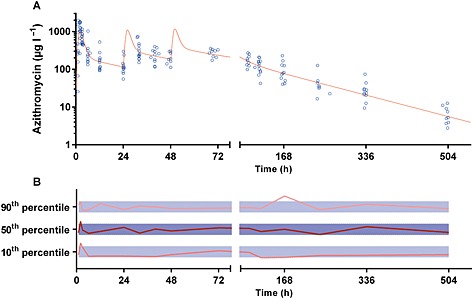
Plasma concentration vs. time data (upper panel) and normalized prediction‐corrected visual predictive check for plasma azithromycin (lower panel). The solid line in the upper panel represents the average concentration vs. time curve for the present patients overlying individual data points, represented as open circles. The solid and dotted lines in the lower panel represent the normalized observed 50th percentile and its 80% prediction interval (i.e. 10th and 90th percentiles), respectively, within their normalized simulated 95 % confidence intervals, represented as grey shaded areas
Table 6.
Post hoc Bayesian estimates of pharmacokinetic parameters and derived secondary parameters for piperaquine and azithromycin in pregnant Papua New Guinean women. Data are median [interquartile range]
| Parameter | Piperaquine | Azithromycin |
|---|---|---|
| MTT (h) | 3.29 [3.29–3.29] | − |
| NN | 1.425 [1.331–1.610] | − |
| k a | − | 0.93 [0.93–0.93] |
| DUR | − | 1.60 [1.29–1.93] |
| CL/F ( l h −1 ) | 75.9 [65.9–83.3] | 64.1 [60.7–68.9] |
| V C /F (l) | 1457 [1122–2013] | 354 [152–628] |
| Q 1 /F ( l h −1 ) | 56.9 [54.5–61.4] | 199 [191–215] |
| V P1 /F (l) | 13 758 [12 994–15 223] | 1837 [1735–2033] |
| Q 2 /F ( l h −1 ) | 227 [217–244] | 56.9 [54.5–61.4] |
| V P2 /F (l) | 2868 [2709–3174] | 3242 [3062–3587] |
| t ½ α (h) | 2.33 [1.88–2.77] | 0.738 [0.316–1.210] |
| t ½ β (h) | 28 [27.5–28.8] | 15.5 [14.9–16.1] |
| t ½ γ (h) | 315 [303–331] | 89.2 [86.1–93.0] |
| C max (μg l −1 ) | 731 [445–900] | 1080 [839–1,439] |
| AUC 0‐∞ (μg h l −1 ) | 38 818 [24 354–52 299] | 46 799 [43 526–49 462] |
| AUC 0‐∞ (μg h l −1 ) per mg kg −1 dose | 1264 [846–1873] | 846 [828–876] |
AUC0–∞, area under the plasma concentration–time curve; CL/F, clearance; Cmax, maximum plasma concentration; MTT, mean transit time; NN, number of transit compartments; Q1/F, intercompartmental clearance for VP1/F; Q2/F, intercompartmental clearance for VP2/F; t½ α, t½ β and t½ γ, first and second distribution, and elimination half‐lives, respectively; VC/F, central volume of distribution; VP1/F, first peripheral volume of distribution; VP2/F, second peripheral volume of distribution.
Discussion
The present study was the first to investigate the potential of AZI–PQ as IPTp or for pregnant women with malarial parasitaemia. The regimen selected, which involved a lower total AZI dose than in some previous studies 13, 14 in view of adverse effect concerns, was well tolerated, with no evidence of greater cardiotoxicity than with PQ in combination with other antimalarial drugs in pregnant PNG women 36. In the relatively few women who were parasitaemic, parasite clearance was prompt. Malaria during follow‐up was infrequent and, reflecting the relatively long elimination t½ of PQ 32, occurred ≥6 weeks after treatment. Consistent with this observation, drug exposure assessed from individual AUC0–∞s was at least that in other PK studies of AZI 14 and PQ 36 in pregnancy.
Nausea affected 17% of the present participants, a percentage within the range (7–24%) seen in previous studies of pregnant PNG women treated AZI or PQ combined with other drugs, including CQ 14, SP 14, 36 or DHA 36. There was no vomiting, and other side effects were mild and infrequent (≤6.7%). The median QTc prolongation 4 h after the first dose (12 ms0 .5) was similar to that in a previous study from our group (14 ms0 .5) in which the same mg kg−1 PQ dose was given to pregnant PNG women in combination with SP or DHA 36. In the latter 36 and present studies, QTc prolongation was resolving by 12 h postdose, no symptoms suggestive of arrhythmia were recorded during follow‐up, and symptomatic postural hypotension was not observed. Although we did not have QTc data beyond 24 h, we interpret these findings as evidence that the addition of AZI to PQ in the present doses is safe, in spite of the fact that both drugs affect ventricular repolarization 32, 44. The lack of adverse outcomes in the majority of women who were followed to parturition is also reassuring.
The AZI PK parameters in the present study differed from those in a previous study in pregnant PNG women 14. The present total AZI dose was lower (3 g over 3 days vs. 4 g over 2 days) but the AUC0–∞ was greater (46 799 vs. 28 713 μg h l−1 14). Although there were some minor differences in characteristics (the present women were younger and later in pregnancy than those in the previous study 14), the most likely explanation for the AUC0–∞ difference is that there are dose‐dependent effects on AZI absorption (saturation), in combination with more extensive tissue distribution, at higher doses. Indeed, Q1/F and Q2/F in the three‐compartment model were >50% higher with the 2 g daily 14 vs. the present 1 g daily dose, consistent with a substantial increase in CL/F. Protein binding diminishes as plasma AZI concentrations increase 21, perhaps facilitating greater tissue uptake and clearance from the central compartment, with a consequent reduction in AUC‐‐∞. Although dose‐dependent effects have not been observed previously 16, 21, the lower individual doses (0.5 g vs, 1.5 g and 0.25 g vs. 0.5 g, compared with 1 g vs. 2 g in our present and previous 14 studies) may have masked an effect. In any case, the present AUC0–∞ data suggest that 1 g AZI daily for 3 days should be as efficacious as higher‐dose regimens 13, 14.
Although CL/F and AUC0–∞ for PQ were consistent with those in our previous study of PQ–SP and PQ–DHA in pregnant PNG women 36, there was a 52% increase in the relative bioavailability of PQ with each subsequent dose in the present study. A study of Thai women with malaria treated with PQ–DHA also showed a time‐dependent increase in bioavailability 33 which we did not find in our previous study 36. Of the potential explanations we postulated previously as an explanation for this latter difference (different sample sizes and durations of sampling, ingestion of fat at the time of PQ dosing, the effect of malaria, and/or racial differences in PQ disposition 36), the present data suggest that the effect of malaria may be most influential.
The subjects (pregnant PNG women) and protocols (including no food for ≥2 h either side of dosing) were the same for the present and our previous study 36 but the percentage of women with malaria in the present study was greater (20.0% vs. 9.4%) and in the Thai study it was 100% 33. Data relating to the effect of malaria on the disposition of other slowly eliminated 4‐aminoquinoline drugs appear to be inconsistent 57, 58 and malaria at study entry was not a significant covariate in the present PQ PK model, but it is plausible that parasite clearance during PQ dosing progressively improves bioavailability, an effect not observed in our previous study, involving a sample of pregnant women in whom <10% were parasitaemic 36. The Vc/F and t½ estimates also tended to be lower, and the Cmax higher, than in our original study 36 as well as in other published studies 33, 42, 43. For example, the median elimination t½ was 13.1 days in our patients compared with 16–22 days in other studies 33, 36, 42, 43. This could reflect between‐study differences in variables such as maternal age, gestational age and parasitaemia, or the present sparse sampling protocol, with few samples taken late in an inadequately characterized elimination phase 59. An effect of AZI on PQ disposition is also possible but previous studies of AZI and CQ did not show an interaction 60. Indeed, there are relatively few recorded AZI interactions for a drug, reflecting the fact that it is largely excreted unchanged rather than being metabolized 61.
The present study had limitations. It was designed as a tolerability, safety and PK study but a larger AZI–PQ IPTp efficacy study is in progress (ClinicalTrial.gov NCT02575755). We employed a pragmatic blood sampling regimen and, as discussed, could not exclude an effect of this on some PQ PK parameters. Although we had complete follow‐up data to Day 28, three women could not be contacted subsequently, and this also applied to two women after their Day 42 assessment. However, obstetric and other outcomes were available for the remaining 25 women and were reassuring, given the relatively high rate of perinatal morbidity and mortality observed in PNG 62. An important effect of the combination of AZI and PQ accumulation on QTc prolongation beyond 24 h, especially at the PQ Cmax after the third dose, may have been missed. However, there was no evidence from the present and previous 36 data to 24 h that AZI amplified the QTc effects of PQ (see Table 3). In addition, in the case of the 4‐aminoquinoline naphthoquine which, like PQ, has a long t½, studies in PNG children with malaria suggest that repeated daily doses do not lead consistently to significant further QTc prolongation after the first dose 63, 64, while data from animals suggest that that combination of AZI–CQ is not proarrhythmic 65. Nevertheless, given persistent concerns regarding the adverse cardiovascular effects of AZI 66, we recommend that future trials of AZI and 4‐aminoquinoline combinations continue to assess this potential safety issue.
Given the need for novel IPTp regimens, AZI‐based therapies have been recommended for development 2, 11, 12. They have advantages over other candidate therapies, including the longer t½ of AZI compared with the artemisinin derivatives administered with partner drugs such as PQ, and proven safety as well as efficacy against bacterial and other infections that can add to the burden of malaria in pregnancy. The present data suggest that AZI 1 g daily for 3 days in combination with 3 days of conventional adult PQ doses, most likely administered monthly at scheduled antenatal visits if used as IPTp 13, 33, 36, is a suitable regimen for further assessment in clinical trials.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: IM, SJR and TMED received a Malaria in Pregnancy Consortium Grant funded through Bill and Melinda Gates Foundation (#46099) for the submitted work. BRM (Early Career Fellowship #1036951), IM (Senior Research Fellowship #1043345) and TMED (Practitioner Fellowship #1058260) had support from National Health and Medical Research Council of Australia for the submitted work. JBM, SOA, SS, GY, SG, MP‐S, KTB and PMS had no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years. There were no other relationships or activities that could appear to have influenced the submitted work.
We thank the mothers who participated and Sigma‐Tau Industrie Farmaceutiche Riunite S.p.A., Italy, for the manufacturer and provision of all PQ used in the present study. We are grateful to Sister Maria Christina and the staff of the Alexishafen Health Centre and labour ward for their kind assistance and cooperation during the study. We also acknowledge the staff of the Papua New Guinea Institute of Medical Research for their clinical and logistic assistance.
Contributors
BRM coordinated the study, analysed data and drafted the manuscript. JMB collected data and reviewed/edited the manuscript. SOA performed piperaquine assays, contributed to data analysis and reviewed/edited the manuscript. SS performed pharmacokinetic analyses and reviewed/edited the manuscript. GY collected data and reviewed/edited the manuscript. SG collected data and reviewed/edited the manuscript. MP‐S performed drug assays and reviewed/edited the manuscript. KTB supervised drug assays and reviewed/edited the manuscript. PMS facilitated the approval and conduct of the study and reviewed/edited the manuscript. IM assisted with study design and reviewed/edited the manuscript. SJR assisted with study design and reviewed/edited the manuscript. TMED designed the study, assisted with data analysis and produced the final version of the manuscript.
Moore, B. R. , Benjamin, J. M. , Auyeung, S. O. , Salman, S. , Yadi, G. , Griffin, S. , Page‐Sharp, M. , Batty, K. T. , Siba, P. M. , Mueller, I. , Rogerson, S. J. , and Davis, T. ME. (2016) Safety, tolerability and pharmacokinetic properties of coadministered azithromycin and piperaquine in pregnant Papua New Guinean women. Br J Clin Pharmacol, 82: 199–212. doi: 10.1111/bcp.12910.
References
- 1. Desai M, ter Kuile FO, Nosten F, McGready R, Asamoa K, Brabin B, Newman RD. Epidemiology and burden of malaria in pregnancy. Lancet Infect Dis 2007; 7: 93–104. [DOI] [PubMed] [Google Scholar]
- 2. Davis TME, Mueller I, Rogerson SJ. Prevention and treatment of malaria in pregnancy. Future Microbiol 2010; 5: 1599–613. [DOI] [PubMed] [Google Scholar]
- 3. World Health Organization . WHO Global Malaria Programme In: Guidelines for the treatment of malaria. Report. Geneva: World Health Organization, 2010. [Google Scholar]
- 4. World Health Organization, Global Malaria Programme . Updated WHO Policy Recommendation (October 2012): intermittent preventive treatment of malaria in pregnancy using sulfadoxine–pyrimethamine (IPTp–SP). Geneva: World Health Organization, 2012. [Google Scholar]
- 5. Green MD, van Eijk AM, van Ter Kuile FO, Ayisi JG, Parise ME, Kager PA, Nahlen BL, Steketee R, Nettey H. Pharmacokinetics of sulfadoxine–pyrimethamine in HIV‐infected and uninfected pregnant women in Western Kenya. J Infect Dis 2007; 196: 1403–8. [DOI] [PubMed] [Google Scholar]
- 6. Karunajeewa HA, Salman S, Mueller I, Baiwog F, Gomorrai S, Law I, Page‐Sharp M, Rogerson S, Siba P, Ilett KF, Davis TM. Pharmacokinetic properties of sulfadoxine‐pyrimethamine in pregnant women. Antimicrob Agents Chemother 2009; 53: 4368–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nyunt MM, Adam I, Kayentao K, van Dijk J, Thuma P, Mauff K, Little F, Cassam Y, Guirou E, Traore B, Doumbo O, Sullivan D, Smith P, Barnes KI. Pharmacokinetics of sulfadoxine and pyrimethamine in intermittent preventive treatment of malaria in pregnancy. Clin Pharmacol Ther 2010; 87: 226–34. [DOI] [PubMed] [Google Scholar]
- 8. Gosling RD. Intermittent preventive treatment against malaria: an update. Expert Rev Anti Infect Ther 2010; 8: 589–606. [DOI] [PubMed] [Google Scholar]
- 9. Bardaji A, Bassat Q, Alonso PL, Menendez C. Intermittent preventive treatment of malaria in pregnant women and infants: making best use of the available evidence. Expert Opin Pharmacother 2012; 13: 1719–36. [DOI] [PubMed] [Google Scholar]
- 10. Alifrangis M, Nag S, Schousboe ML, Ishengoma D, Lusingu J, Pota H, Kavishe RA, Pearce R, Ord R, Lynch C, Dejene S, Cox J, Rwakimari J, Minja DT, Lemnge MM, Roper C. Independent origin of Plasmodium falciparum antifolate super‐resistance, Uganda, Tanzania, and Ethiopia. Emerg Infect Dis 2014; 20: 1280–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chandra RS, Orazem J, Ubben D, Duparc S, Robbins J, Vandenbroucke P. Creative solutions to extraordinary challenges in clinical trials: methodology of a phase III trial of azithromycin and chloroquine fixed‐dose combination in pregnant women in Africa. Malar J 2013; 12: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chico RM, Chandramohan D. Azithromycin plus chloroquine: combination therapy for protection against malaria and sexually transmitted infections in pregnancy. Expert Opin Drug Metab Toxicol 2011; 7: 1153–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Unger HW, Ome‐Kaius M, Wangnapi RA, Umbers AJ, Hanieh S, Suen CS, Robinson LJ, Rosanas‐Urgell A, Wapling J, Lufele E, Kongs C, Samol P, Sui D, Singirok D, Bardaji A, Schofield L, Menendez C, Betuela I, Siba P, Mueller I, Rogerson SJ. Sulphadoxine–pyrimethamine plus azithromycin for the prevention of low birthweight in Papua New Guinea: a randomised controlled trial. BMC Med 2015; 13: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salman S, Rogerson SJ, Kose K, Griffin S, Gomorai S, Baiwog F, Winmai J, Kandai J, Karunajeewa HA, O'Halloran SJ, Siba P, Ilett KF, Mueller I, Davis TM. Pharmacokinetic properties of azithromycin in pregnancy. Antimicrob Agents Chemother 2010; 54: 360–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luntamo M, Kulmala T, Mbewe B, Cheung YB, Maleta K, Ashorn P. Effect of repeated treatment of pregnant women with sulfadoxine–pyrimethamine and azithromycin on preterm delivery in Malawi: a randomized controlled trial. Am J Trop Med Hyg 2010; 83: 1212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Amsden GW, Gray CL. Serum and WBC pharmacokinetics of 1500 mg of azithromycin when given either as a single dose or over a 3 day period in healthy volunteers. J Antimicrob Chemother 2001; 47: 61–6. [DOI] [PubMed] [Google Scholar]
- 17. Amsden GW, Nafziger AN, Foulds G, Cabelus LJ. A study of the pharmacokinetics of azithromycin and nelfinavir when coadministered in healthy volunteers. J Clin Pharmacol 2000; 40: 1522–7. [PubMed] [Google Scholar]
- 18. Ramsey PS, Vaules MB, Vasdev GM, Andrews WW, Ramin KD. Maternal and transplacental pharmacokinetics of azithromycin. Am J Obstet Gynecol 2003; 188: 714–8. [DOI] [PubMed] [Google Scholar]
- 19. Ballow C, Amsden G, Highet V, Forrest A. Pharmacokinetics of oral azithromycin in serum, urine, polymorphonuclear leucocytes and inflammatory vs. non‐inflammatory skin blisters in healthy volunteers. Clin Drug Investig 1998; 15: 159–67. [DOI] [PubMed] [Google Scholar]
- 20. Boonleang J, Panrat K, Tantana C, Krittathanmakul S, Jintapakorn W. Bioavailability and pharmacokinetic comparison between generic and branded azithromycin capsule: a randomized, double‐blind, 2‐way crossover in healthy male Thai volunteers. Clin Ther 2007; 29: 703–10. [DOI] [PubMed] [Google Scholar]
- 21. Foulds G, Shepard RM, Johnson RB. The pharmacokinetics of azithromycin in human serum and tissues. J Antimicrob Chemother 1990; 25: 73–82. [DOI] [PubMed] [Google Scholar]
- 22. Setiawati E, Deniati SH, Yunaidi DA, Handayani LR, Harinanto G, Santoso ID, Sari AP, Rimainar A. Bioequivalence study of two azithromycin formulations in healthy subjects. Arzneimittelforschung 2009; 59: 471–5. [DOI] [PubMed] [Google Scholar]
- 23. Kent JR, Almond MK, Dhillon S. Azithromycin: an assessment of its pharmacokinetics and therapeutic potential in CAPD. Perit Dial Int 2001; 21: 372–7. [PubMed] [Google Scholar]
- 24. van Eijk AM, Terlouw DJ. Azithromycin for treating uncomplicated malaria. Cochrane Database Syst Rev 2011; 2: : Cd006688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gilliams EA, Jumare J, Claassen CW, Thesing PC, Nyirenda OM, Dzinjalamala FK, Taylor T, Plowe CV, Tracy LA, Laufer MK. Chloroquine–azithromycin combination antimalarial treatment decreases risk of respiratory‐ and gastrointestinal‐tract infections in Malawian children. J Infect Dis 2014; 210: 585–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parnham MJ, Haber VE, Giamarellos‐Bourboulis EJ, Perletti G, Verleden GM, Vos R. Azithromycin: mechanisms of action and their relevance for clinical applications. Pharmacol Ther 2014; 143: 225–45. [DOI] [PubMed] [Google Scholar]
- 27. Centre for Disease Control and Prevention . Sexually transmitted diseases treatment guidelines, 2006: Diseases characterized by urethritis and cervicitis. MMWR Morb Mortal Wkly Rep 2006; 55: 35–49.16424856 [Google Scholar]
- 28. Adjuik M, Babiker A, Garner P, Olliaro P, Taylor W, White N, International Artemisinin Study G . Artesunate combinations for treatment of malaria: meta‐analysis. Lancet 2004; 363: 9–17. [DOI] [PubMed] [Google Scholar]
- 29. Poespoprodjo JR, Fobia W, Kenangalem E, Lampah DA, Sugiarto P, Tjitra E, Anstey NM, Price RN. Dihydroartemisinin–piperaquine treatment of multidrug resistant falciparum and vivax malaria in pregnancy. PLoS One 2014; 9: : e84976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Denis MB, Davis TME, Hewitt S, Incardona S, Nimol K, Fandeur T, Poravuth Y, Lim C, Socheat D. Efficacy and safety of dihydroartemisinin–piperaquine (Artekin) in Cambodian children and adults with uncomplicated falciparum malaria. Clin Infect Dis 2002; 35: 1469–76. [DOI] [PubMed] [Google Scholar]
- 31. Batty KT, Moore BR, Stirling V, Ilett KF, Page‐Sharp M, Shilkin KB, Mueller I, Rogerson SJ, Karunajeewa HA, Davis TM. Investigation of reproductive toxicity of piperaquine in mice. Reprod Toxicol 2010; 29: 206–13. [DOI] [PubMed] [Google Scholar]
- 32. Davis TM, Hung TY, Sim IK, Karunajeewa HA, Ilett KF. Piperaquine: a resurgent antimalarial drug. Drugs 2005; 65: 75–87. [DOI] [PubMed] [Google Scholar]
- 33. Tarning J, Rijken MJ, McGready R, Phyo AP, Hanpithakpong W, Day NP, White NJ, Nosten F, Lindegardh N. Population pharmacokinetics of dihydroartemisinin and piperaquine in pregnant and nonpregnant women with uncomplicated malaria. Antimicrob Agents Chemother 2012; 56: 1997–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rijkin MJ, McGready R, Boel ME, Barends M, Proux S, Pimanpanarak M, Singhasivanon P, Nosten F. Dihydroartemisinin–piperaquine rescue treatment of multidrug‐resistant Plasmodium falciparum malaria in pregnancy: a preliminary report. Am J Trop Med Hyg 2008; 78: 543–5. [PubMed] [Google Scholar]
- 35. Hoglund RM, Adam I, Hanpithakpong W, Ashton M, Lindegardh N, Day NP, White NJ, Nosten F, Tarning J. A population pharmacokinetic model of piperaquine in pregnant and non‐pregnant women with uncomplicated Plasmodium falciparum malaria in Sudan. Malar J 2012; 11: 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benjamin JM, Moore BR, Salman S, Page‐Sharp M, Tawat S, Yadi G, Lorry L, Siba PM, Batty KT, Robinson LJ, Mueller I, Davis TME. Population pharmacokinetics, tolerability and safety of dihydroartemisinin–piperaquine and sulfadoxine–pyrimethamine–piperaquine in pregnant and nonpregnant Papua New Guinean women. Antimicrob Agents Chemother 2015; 59: 4260–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nosten F, McGready R, Mutabingwa T. Case management of malaria in pregnancy. Lancet Infect Dis 2007; 7: 118–25. [DOI] [PubMed] [Google Scholar]
- 38. D'Alessandro U. Progress in the development of piperaquine combinations for the treatment of malaria. Curr Opin Infect Dis 2009; 22: 588–92. [DOI] [PubMed] [Google Scholar]
- 39. White NJ. Cardiotoxicity of antimalarial drugs. Lancet Infect Dis 2007; 7: 549–58. [DOI] [PubMed] [Google Scholar]
- 40. European Medicines Agency (EMA) . Eurartesim. EU Summary of Product Characteristics. 2011. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Summary_for_the_public/human/001199/WC500118117.pdf (last accessed 31 January 2016).
- 41. Darpo B, Ferber G, Siegl P, Laurijssens B, Macintyre F, Toovey S, Duparc S. Evaluation of the QT effect of a combination of piperaquine and a novel anti‐malarial drug candidate OZ439, for the treatment of uncomplicated malaria. Br J Clin Pharmacol 2015; 80: 706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adam I, Tarning J, Lindegardh N, Mahgoub H, McGready R, Nosten FH. Pharmacokinetics of piperaquine in pregnant women in Sudan with uncomplicated Plasmodium falciparum malaria. Am J Trop Med Hyg 2012; 87: 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rijken MJ, McGready R, Phyo AP, Lindegardh N, Tarning J, Laochan N, Than HH, Mu O, Win AK, Singhasivanon P, White N, Nosten FH. Pharmacokinetics of dihydroartemisinin and piperaquine in pregnant and nonpregnant women with uncomplicated falciparum malaria. Antimicrob Agents Chemother 2011; 55: 5500–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Howard PA. Azithromycin‐induced proarrhythmia and cardiovascular death. Ann Pharmacother 2013; 47: 1547–51. [DOI] [PubMed] [Google Scholar]
- 45. Borsini F, Crumb W, Pace S, Ubben D, Wible B, Yan GX, Funck‐Brentano C. In vitro cardiovascular effects of dihydroartemisin–piperaquine combination compared with other antimalarials. Antimicrob Agents Chemother 2012; 56: 3261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Michon P, Cole‐Tobian JL, Dabod E, Schoepflin S, Igu J, Susapu M, Tarongka N, Zimmerman PA, Reeder JC, Beeson JG, Schofield L, King CL, Mueller I. The risk of malarial infections and disease in Papua New Guinean children. Am J Trop Med Hyg 2007; 76: 997–1008. [PMC free article] [PubMed] [Google Scholar]
- 47. Laman M, Moore BR, Benjamin J, Padapu N, Tarongka N, Siba P, Betuela I, Mueller I, Robinson LJ, Davis TM. Comparison of an assumed versus measured leucocyte count in parasite density calculations in Papua New Guinean children with uncomplicated malaria. Malar J 2014; 13: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Karunajeewa HA, Ilett KF, Mueller I, Siba P, Law I, Page‐Sharp M, Lin E, Lammey J, Batty KT, Davis TM. Pharmacokinetics and efficacy of piperaquine and chloroquine in Melanesian children with uncomplicated malaria. Antimicrob Agents Chemother 2008; 52: 237–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Anderson BJ, Holford NH. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 2009; 24: 25–36. [DOI] [PubMed] [Google Scholar]
- 50. Karlsson M. [NMusers] When to do transformation of data? 2002. [8th May 2014]. Available at http://www.cognigencorp.com/nonmem/nm/99apr232002.html (last accessed 31 January 2016).
- 51. Salman S, Page‐Sharp M, Batty KT, Kose K, Griffin S, Siba P, Ilett KF, Mueller I, Davis TME. A pharmacokinetic comparison of two piperaquine‐containing artemisinin combination therapies in Papua New Guinean children with uncomplicated malaria. Antimicrob Agents Chemother 2012; 56: 3288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Savic RM, Jonker DM, Kerbusch T, Karlsson MO. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn 2007; 34: 711–26. [DOI] [PubMed] [Google Scholar]
- 53. Coghlan B, Millan J, Malau C, Kaldor J, Toole M. The HIV epidemic in Papua New Guinea. J Acquir Immune Defic Syndr 2011; 58: e48–51. [DOI] [PubMed] [Google Scholar]
- 54. Papua New Guinea National Department of Health . National malaria treatment protocol. Port Moresby: National Department of Health, 2009. [Google Scholar]
- 55. Karunajeewa HA, Salman S, Mueller I, Baiwog F, Gomorrai S, Law I, Page‐Sharp M, Rogerson S, Siba P, Ilett KF, Davis TM. Pharmacokinetics of chloroquine and monodesethylchloroquine in pregnancy. Antimicrob Agents Chemother 2010; 54: 1186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Salman S, Page‐Sharp M, Griffin S, Kose K, Siba PM, Ilett KF, Mueller I, Davis TM. Population pharmacokinetics of artemether, lumefantrine, and their respective metabolites in Papua New Guinean children with uncomplicated malaria. Antimicrob Agents Chemother 2011; 55: 5306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Edwards G, Looareesuwan S, Davies AJ, Wattanagoon Y, Phillips RE, Warrell DA. Pharmacokinetics of chloroquine in Thais: plasma and red‐cell concentrations following an intravenous infusion to healthy subjects and patients with Plasmodium vivax malaria. Br J Clin Pharmacol 1988; 25: 477–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Reuter SE, Upton RN, Evans AM, Navaratnam V, Olliaro PL. Population pharmacokinetics of orally administered mefloquine in healthy volunteers and patients with uncomplicated Plasmodium falciparum malaria. J Antimicrob Chemother 2015; 70: 868–76. [DOI] [PubMed] [Google Scholar]
- 59. Tarning J, Lindegardh N, Annerberg A, Singtoroj T, Day NPJ, Ashton M, White NJ. Pitfalls in estimating piperaquine elimination. Antimicrob Agents Chemother 2005; 39: 5127–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cook JA, Randinitis EJ, Bramson CR, Wesche DL. Lack of a pharmacokinetic interaction between azithromycin and chloroquine. Am J Trop Med Hyg 2006; 74: 407–12. [PubMed] [Google Scholar]
- 61. Jain R, Danziger LH. The macrolide antibiotics: a pharmacokinetic and pharmacodynamic overview. Curr Pharm Des 2004; 10: 3045–53. [DOI] [PubMed] [Google Scholar]
- 62. Jimmy S, Kemiki AD, Vince JD. Neonatal outcome at Modilon Hospital, Madang: a 5‐year review. PNG Med J 2003; 46: 8–15. [PubMed] [Google Scholar]
- 63. Benjamin J, Moore B, Lee ST, Senn M, Griffin S, Lautu D, Salman S, Siba P, Mueller I, Davis TM. Artemisinin–naphthoquine combination therapy for uncomplicated pediatric malaria: a tolerability, safety, and preliminary efficacy study. Antimicrob Agents Chemother 2012; 56: 2465–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Laman M, Moore BR, Benjamin JM, Yadi G, Bona C, Warrel J, Kattenberg JH, Koleala T, Manning L, Kasian B, Robinson LJ, Sambale N, Lorry L, Karl S, Davis WA, Rosanas‐Urgell A, Mueller I, Siba PM, Betuela I, Davis TM. Artemisinin–naphthoquine versus artemether‐lumefantrine for uncomplicated malaria in Papua New Guinean children: an open‐label randomized trial. PLoS Med 2014; 11: : e1001773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fossa AA, Wisialowski T, Duncan JN, Deng S, Dunne M. Azithromycin/chloroquine combination does not increase cardiac instability despite an increase in monophasic action potential duration in the anesthetized guinea pig. Am J Trop Med Hyg 2007; 77: 929–38. [PubMed] [Google Scholar]
- 66. Cheng YJ, Nie XY, Chen XM, Lin XX, Tang K, Zeng WT, Mei WY, Liu LJ, Long M, Yao FJ, Liu J, Liao XX, Du ZM, Dong YG, Ma H, Xiao HP, Wu SH. The role of macrolide antibiotics in increasing cardiovascular risk. J Am Coll Cardiol 2015; 66: 2173–84. [DOI] [PubMed] [Google Scholar]