

Abstract
Aims
Pharmacokinetic (PK) similarity was assessed among PF‐05280586 (a proposed biosimilar) vs. rituximab sourced from the European Union (rituximab‐EU) and the United States (rituximab‐US). Pharmacodynamics (PD), overall safety and immunogenicity were also evaluated.
Methods
Patients with active rheumatoid arthritis on a background of methotrexate and inadequate response to one or more tumour necrosis factor antagonist therapies were randomized to intravenous PF‐05280586, rituximab‐EU or rituximab‐US 1000 mg doses on study days 1 and 15.
Results
A total of 220 patients were randomized to receive study treatment as assigned. Of these, 198 met per‐protocol population criteria for inclusion in the PK data analysis. PF‐05280586, rituximab‐EU and rituximab‐US exhibited similar PK profiles following administration of assigned study drug on days 1 and 15. The 90% confidence intervals of test‐to‐reference ratios for C max, AUCT, AUC0–∞ and AUC2‐week were within the bioequivalence margin of 80.00–125.00% for comparisons of PF‐05280586 with rituximab‐EU, PF‐05280586 with rituximab‐US, and rituximab‐EU with rituximab‐US. All treatments resulted in a rapid and profound reduction in CD19+ B cells and sustained profound B cell suppression up to week 25. The incidence of antidrug antibody (ADA) response (n = 7, 10 and 9 for PF‐05280586, rituximab‐EU and rituximab‐US, respectively), time to ADA emergence and ADA titres were similar across treatments. None of the ADA‐positive samples was positive for neutralizing activity. No clinically meaningful differences in adverse events were identified.
Conclusions
The study demonstrated PK similarity among PF‐05280586, rituximab‐EU and rituximab‐US. In addition, all treatments showed comparable CD19+ B cell depletion PD responses, as well as safety and immunogenicity profiles.
Keywords: biosimilar, CD19+ B cell counts, immunogenicity, pharmacokinetics, rituximab, safety
What is Already Known about this Subject
Rituximab, a monoclonal antibody directed against the CD20 antigen of B cells, is indicated for non‐Hodgkin's lymphoma, chronic lymphocytic leukaemia (with chemotherapy), rheumatoid arthritis (with methotrexate) and granulomatosis with polyangiitis and microscopic polyangiitis (with glucocorticoids).
What this Study Adds
This is the first clinical report providing evidence of similarity of pharmacokinetics, pharmacodynamics, immunogenicity and safety of PF‐05280586 (a proposed biosimilar) vs. reference products rituximab‐EU and rituximab‐US.
The results provide critical bridging data between the two reference products to allow use of a single reference product in future phase III studies.
Introduction
The term ‘biosimilar’ refers to a biologic product developed to be highly similar to an existing licensed or approved biologic product 1, 2, 3, 4. Biosimilars are expected to be an essential component in enhancing patient access to these important, often lifesaving biologic products. Biologics are large, structurally complex molecules; even minor changes in the manufacturing process could produce differences that can affect the safety, immunogenicity and potency of the molecule 3, 4. Regulatory decisions for approval are based on the totality of the evidence generated from a stepwise approach that generally includes analytical, clinical pharmacokinetic (PK), and efficacy and safety studies intended to support the demonstration of biosimilarity 1, 2, 3, 4.
Rituximab is a genetically engineered chimeric murine/human monoclonal immunoglobulin (Ig) G1κ antibody directed against the CD20 antigen of B cells 5, 6. It is indicated for non‐Hodgkin's lymphoma, chronic lymphocytic leukaemia (in combination with chemotherapy), rheumatoid arthritis (RA; in combination with methotrexate) and granulomatosis with polyangiitis and microscopic polyangiitis (in combination with glucocorticoids) 5, 6. The pathophysiology of RA is unknown, although it is thought to involve activation of an innate immune response, including antigen presentation and production of autoantibodies and cytokines by B cells, and involvement of other key effector cells and inflammatory modulators 7. Thus, B cells are an important and appropriate target for the treatment of RA, as confirmed by the efficacy of rituximab since its approval in 2006. PF‐05280586, which is under development as a potential biosimilar to rituximab, has the same primary amino acid sequence, and similar physicochemical and in vitro functional properties as rituximab 8, 9.
A multicentre, multinational, randomized, double‐blind, controlled trial (REFLECTIONS B328‐01), reported herein, was designed to demonstrate the PK similarity of PF‐05280586 to rituximab sourced from the European Union (rituximab‐EU; MabThera®, F. Hoffmann‐La Roche, Basel, Switzerland 6) and United States (rituximab‐US; Rituxan®, Genentech Inc., South San Francisco, CA, USA 5), and between rituximab‐EU and rituximab‐US. The present study also evaluated the pharmacodynamics (PD) and overall safety, tolerability and immunogenicity of the study drugs.
Methods
This study was registered (ClinicalTrials.gov identifier: NCT01526057) and conducted in compliance with the Declaration of Helsinki and with all International Conference on Harmonisation Good Clinical Practice guidelines. In addition, all local regulatory requirements were followed and, in particular, those affording greater protection to the safety of trial participants. The final protocol, amendments and informed consent documentation were reviewed and approved by institutional review boards and/or independent ethics committees at each investigational centre participating in the study. A signed and dated informed consent was required from each patient before any screening procedures were conducted.
Study population and design
This was a phase I, double‐blind, randomized, parallel‐group, three‐arm trial (Figure S1). The primary objective was to demonstrate PK similarity of PF‐05280586, rituximab‐EU and rituximab‐US in patients with active RA on a background treatment of methotrexate and who had an inadequate response to one or more tumour necrosis factor (TNF) antagonist therapies. Secondary objectives included assessing PD, safety, tolerability and immunogenicity.
Rituximab produces profound and prolonged B cell depletion 5, 6, precluding the conduct of PK studies in healthy subjects. Its use in the present clinical trial population was consistent with the labelled indication for rituximab‐EU and rituximab‐US 5, 6. Eligible participants were adults (aged ≥18 years) with a confirmed diagnosis of RA based on 2010 American College of Rheumatology (ACR)/European League Against Rheumatism classification criteria 10. Patients also had to meet class I, II or III of the ACR 1991 revised criteria for global functional status in rheumatoid arthritis 11; have RA seropositivity, as documented by a screening assessment for rheumatoid factor and/or anticyclic citrullinated peptide antibodies; have active disease, as defined by the following: at least six tender/painful joints (of 68 assessed) and six or more swollen joints (of 66 assessed) at screening and baseline, high‐sensitivity C‐reactive protein (CRP) greater than the upper limit of normal or a Patient's Global Assessment of Arthritis score ≥50 at screening, and Disease Activity Score in 28 joints–CRP >3.2 at screening; be on a stable dose of oral or parenteral methotrexate 10–25 mg per week (or as low as 7.5 mg per week, in the case of prior poor tolerance) for at least 3 months and receiving the stable dose for at least 4 weeks prior to the first dose of the study drug; and have an inadequate response, in the opinion of the investigator, to one or more approved TNF antagonist therapies, defined as failure to achieve an adequate clinical response during prior TNF antagonist therapy, relapse following a clinical response to TNF antagonist therapy or an adverse event (AE) resulting in discontinuation of a TNF antagonist.
Key exclusion criteria were: any prior treatment with lymphocyte‐depleting therapies; pregnancy, lactation or unwillingness to use highly effective contraception for at least 12 months after the last dose of the investigational product; inadequate bone marrow, liver, renal or immune system functions at screening; evidence of untreated, inadequately treated latent, or inadequately treated or active infection with tuberculosis; known or screen‐test positive for HIV, hepatitis B or C, herpes zoster or herpes simplex; primary or secondary immunodeficiency; history of recurrent inflammatory joint disease other than RA; lymphoproliferative disorder; infection requiring hospitalization, parenteral antimicrobial therapy within 3 months prior to the first dose of the study drug or judged clinically significant by the investigator; vaccination with live or attenuated vaccines within the 6 weeks prior to the first dose of the study drug, or planned administration during study participation or within 4 weeks following discontinuation of study drug; current or recent history of uncontrolled clinically significant renal, hepatic, haematological, gastrointestinal, endocrine, metabolic, pulmonary, severe cardiac or neurological disease; addiction or dependence on nonprescribed substances; history of a malignancy, except for nonmelanoma skin cancer, in situ carcinoma or those having nonhaematological tumours treated with curative intent, with no evidence of disease for 5 years; significant trauma or a surgical procedure within 4 weeks prior to the first dose of the study drug; requirement for treatment during the study with prohibited concomitant medications; history of corticosteroid‐induced psychosis or significant hyperglycaemia; known allergy or hypersensitivity to the active drug substance or excipients, or to murine, chimeric, human or humanized proteins; or any other severe acute or chronic medical or psychiatric condition or laboratory abnormality that increased risk associated with study participation or investigational product administration or interfered with interpretation of the study results and, in the judgement of the investigators, made the patient inappropriate for entry into the study.
Eligible patients were randomized 1 : 1 : 1 via a computer‐generated randomization schedule to one course of PF‐05280586, rituximab‐EU or rituximab‐US, administered as two 1000 mg intravenous (i.v.) doses on study days 1 and 15, in accordance with the treatment regimen in the rituximab‐EU and rituximab‐US product labelling for RA 5, 6. All patients received premedication with 100 mg i.v. methylprednisolone (or its equivalent), an antipyretic agent and an antihistaminic (e.g. acetaminophen and diphenhydramine), in accordance with the accepted infusion protocols for rituximab‐EU and rituximab‐US, prior to study drug infusions to decrease the incidence and severity of acute infusion‐related reactions 5, 6. Patients also continued the stable background regimen of methotrexate.
PK
The PK of PF‐05280586, rituximab‐EU and rituximab‐US were determined using serum samples collected predose (day 1, within 1 h prior to the start of the first infusion), and at 3 h (day 1, during the first infusion), 4.25 h (day 1, immediately prior to the end of the first infusion), 72 h (day 4), 168 h (day 8), 335 h (day 15, within 1.5 h prior to the start of the second infusion), 337.5 h (day 15, during the second infusion), 339.25 h (day 15, immediately prior to the end of the second infusion), 408 h (day 18), 504 h (day 22), 672 h (day 29), 1344 h (day 57) and 2016 h (day 85) after the start of the first infusion on day 1. An additional drug concentration sample was collected on day 169 to facilitate immunogenicity assessment at low drug concentrations.
Samples were analysed using a validated, sensitive, specific enzyme‐linked immunosorbent assay (ELISA) with a lower limit of quantification (LLOQ) of 100 ng ml–1. During sample analysis, the precision (expressed as the coefficient of variation [CV%] of quality control samples) was 5.5% to 8.0% and the accuracy (expressed as the percentage relative error [%RE] of quality control samples) was –1.8% to 7.0%. Samples with serum rituximab concentrations below LLOQ were set to zero for PK data analysis. Standard noncompartmental PK data analysis was conducted to estimate PK parameters using Phoenix® WinNonlin® (version 6.3; Pharsight Corporation, Cary, NC, USA).
The primary PK endpoints were maximum serum concentration (C max) and area under the serum concentration–time profile (AUC) from time 0 extrapolated to infinite time (AUC0–∞). Secondary PK endpoints were AUC from time 0 to the last time point with a measurable concentration (AUCT), AUC from time 0 to 2 weeks (AUC2‐week), clearance (CL), volume at steady‐state (V ss) and terminal half‐life (t 1/2). The per‐protocol population, defined as all subjects who were randomized to and received the full dose of the planned study treatment and had no major protocol violations that affected the PK analysis, was used for the PK analyses.
PD
PD was evaluated using circulating CD19+ B cell counts (as a surrogate marker for CD20+ B cells) and serum IgM. Blood samples for CD19+ B cell counts were collected predose (day 1, within 1 h prior to the start of the first infusion), and then at 72 h (day 4), 168 h (day 8), 335 h (day 15, within 1.5 h before the second infusion), 504 h (day 22), 672 h (day 29), 1344 h (day 57), 2016 h (day 85), 2688 h (day 113), 3360 h (day 141) and 4032 h (day 169). As a lowering of IgM levels has been reported in patients treated with B cell‐depleting therapies, serum samples for evaluation of IgM levels were collected at the same time points.
Blood samples were analysed for CD19+ B cell counts using a laser scanning cytometry procedure (ApoCell Inc., Houston, TX, USA). CD19+ B cell count vs.time data were analysed by standard noncompartmental methods using Phoenix WinNonlin and used to calculate the mean minimum CD19+ B cell count (C min,B cell) and median time to C min,B cell (t min B cell). The AUC for CD19+ B cell count vs.time (AUCT, B cell) was estimated using linear trapezoidal linear interpolation. The duration of B cell depletion (τ B cell) was calculated as the time interval over which CD19+ B cell counts were below the quantitation limit. CD19+ B cell counts for those samples without corresponding absolute white blood cell counts could not be calculated and were not included in the data analysis.
Serum samples were assayed for IgM concentrations at Covance Central Laboratory Services Inc. (Indianapolis, IN, USA).
Immunogenicity
Serum samples for detecting antidrug antibodies (ADAs) and neutralizing antibodies (Nabs) were collected on days 1 (predose), 15, 29, 57, 85 and 169. Immunogenicity sample analyses followed a tiered approach of screening, confirmation and endpoint titre determination using two validated electrochemiluminescent assays – one developed to detect antibodies against PF‐05280586 and the other to detect antibodies against rituximab‐EU and rituximab‐US (QPS, LLC, Newark, DE, USA). The two ADA assays demonstrated similar performance characteristics during assay validation. The ADA samples were first analysed using the ADA assay specific for the dosed product. Samples confirmed positive for antibodies to the dosed product were then assessed for cross‐reactivity using the alternative assay. Any samples confirmed positive for ADA were further tested for Nab using one of two (one for PF‐05280586 and one for rituximab‐EU and rituximab‐US) cell‐based Nab assays. Nab assays were based on the functionality of Nab to inhibit the complement‐dependent cytotoxicity of PF‐05280586, rituximab‐EU and rituximab‐US on CD20+ WIL2‐S cells (a human B lymphoma cell line).
Safety
The safety analysis was performed in all enrolled patients who received the study drug (the modified intent‐to‐treat population) and included AEs, electrocardiogram readings, vital signs and clinical laboratory data. The type, incidence and severity (mild, moderate or severe) of AEs, and the investigator's opinion of the relationship to the study treatment of any AEs, including adverse drug reactions, illnesses with onset during the study, exacerbation of previous illnesses or any clinically significant changes in physical examination or abnormal objective laboratory findings, were investigated. Treatment‐emergent AEs were defined as any AE that occurred during or after study drug administration of the first dose of the study drug or any pre‐existing events that worsened in severity after dosing.
Any AEs occurring postinfusion were reviewed as potential infusion‐related reactions and verified by the reporting investigator. All serious AEs (SAEs; i.e. events that resulted in death, were life threatening, required inpatient hospitalization or prolongation of existing hospitalization, resulted in a persistent or significant disability/incapacity, resulted in congenital anomaly/birth defect, or jeopardized the patient or required medical or surgical intervention to prevent one of these outcomes) were to be reported immediately to the sponsor, regardless of treatment or suspected relationship to study drug. If an SAE occurred, the sponsor was to be notified within 24 h of investigator awareness of the event.
Statistical analysis
Statistical analysis consisted of one‐way analyses of variance comparing the natural log‐transformed PK parameters (C max, AUC0–∞, AUC2‐week and AUCT) for each pair‐wise comparison (PF‐05280586 vs. rituximab‐US, PF‐05280586 vs. rituximab‐EU, and rituximab‐EU vs. rituximab‐US). Estimates of adjusted mean differences and corresponding 90% confidence intervals (CIs) were obtained from the model and were exponentiated to provide estimates of the ratio of adjusted geometric means and 90% CIs for the ratios. PK similarity was considered to have been demonstrated for a given test‐to‐reference comparison if the 90% CI of the ratio was within the 80.00–125.00% bioequivalence acceptance window.
A sample size of 65 patients per group (195 patients total) was required to provide approximately 89% power for showing bioequivalence simultaneously for all three AUC0–∞ comparisons, and approximately 97% power for showing bioequivalence simultaneously for all three C max comparisons. Consequently, the study was designed to have at least 86% power overall to demonstrate PK similarity of PF‐05280586 to rituximab‐EU and rituximab‐US. This estimate was based on the assumption that the true ratio of PF‐05280586 to rituximab‐EU and rituximab‐US for both AUC0–∞ and C max was 1.00, and interpatient standard deviations (SDs) were 0.34 for loge AUC0–∞ and 0.30 for loge C max. Approximately 210 patients were to be randomized to ensure that at least 195 patients completed PK‐related procedures per protocol.
Results
Patient demographics and disposition
A total of 220 patients were randomized to treatment groups and received the study treatment as assigned (Figure 1). Baseline demographics were generally similar among patients (Table 1). The majority of patients were female [n = 170 (77.3%)] and white [n = 171 (77.7%)], with a mean age of 54.4 years, mean weight 83.13 kg and mean body mass index 30.07 kg m–2. Of these 220 patients, 198 met per‐protocol population criteria for inclusion in the PK data analysis. Patient disposition and ineligibility information for inclusion in the per‐protocol population is provided in Figure 1 and Table S1.
Figure 1.
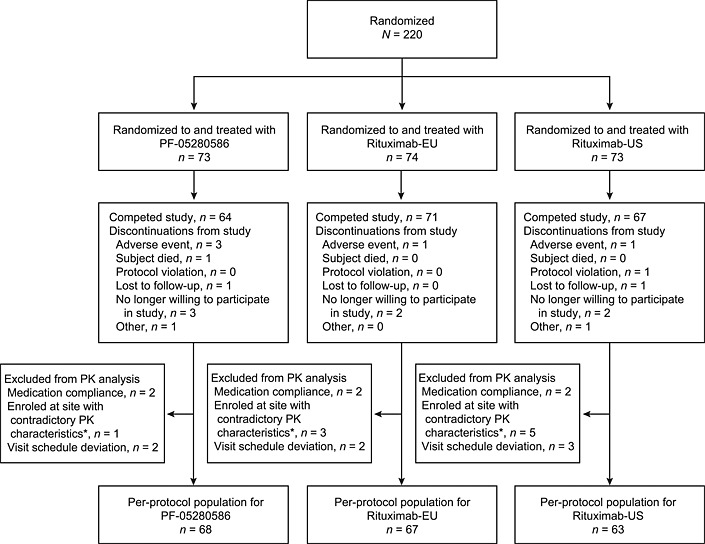
Patient disposition. PK, pharmacokinetics. *Patients enrolled at one site were excluded because the validity of study conduct was not established. (The vast majority of pharmacokinetic profiles from the site were contradictory to the known pharmacokinetic characteristics for rituximab.)
Table 1.
Baseline demographics (mITT population)
| PF‐05280586 | Rituximab‐EU | Rituximab‐US | |
|---|---|---|---|
| n = 73 | n = 74 | n = 73 | |
| Gender, n (%) | |||
| Male | 14 (19.2) | 17 (23.0) | 19 (26.0) |
| Female | 59 (80.8) | 57 (77.0) | 54 (74.0) |
| Age, mean ± SD, years | 54.9 ± 11.52 | 54.9 ± 11.07 | 53.4 ± 11.87 |
| Race, n (%) | |||
| White | 56 (76.7) | 57 (77.0) | 58 (79.5) |
| Black | 2 (2.7) | 6 (8.1) | 5 (6.8) |
| Asian | 3 (4.1) | 0 (0.0) | 1 (1.4) |
| Other | 12 (16.4) | 11 (14.9) | 9 (12.3) |
| Disease duration since first diagnosis, mean ± SD, months | 153.3 ± 99.34 | 140.6 ± 98.91 | 125.0 ± 96.83 |
| Body mass index, mean ± SD, kg m–2 | 31.47 ± 8.133 | 29.76 ± 6.29 | 29.00 ± 6.74 |
mITT, modified intent‐to‐treat; SD, standard deviation.
A total of 18 (8.2%) patients discontinued before completing the study, including nine, three and six treated with PF‐05280586, rituximab‐EU and rituximab‐US, respectively (Figure 1). No notable differences were observed across groups with regard to reason for discontinuation of the study. In the per‐protocol population, there were no significant imbalances in demographic characteristics known to affect the disposition of the study drugs among groups. Mean ± SD body weights in the PF‐05280586, rituximab‐EU and rituximab‐US groups were 86.0 ± 22.2, 82.4 ± 20.4 and 79.9 ± 21.4 kg, respectively.
PK
The study drugs exhibited similar PK profiles, characterized by a rapid increase in serum drug concentration during each infusion on days 1 and 15, followed by a multiphasic decline in drug concentrations after each infusion (Figure 2). Mean peak concentrations ranged from 327 μg ml–1 to 357 μg ml–1 following the first infusion and from 385 μg ml–1 to 421 μg ml–1 following the second infusion. The trough concentration (C trough) levels immediately before the second infusion were between 78.4 μg ml–1 and 91.8 μg ml–1.
Figure 2.
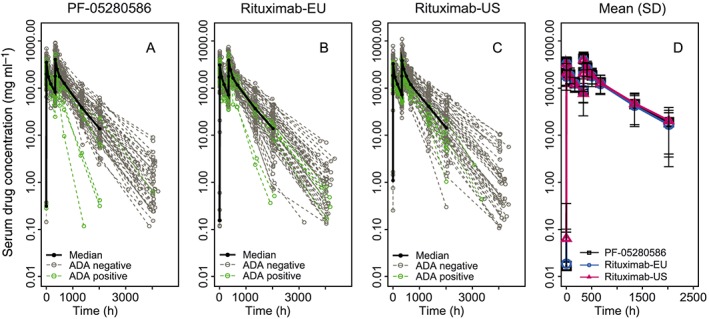
Individual and mean ± SD serum concentration–time profiles in patients with rheumatoid arthritis receiving 1000 mg intravenous doses of study drugs on day 1 and day 15 (per‐protocol population). Panels A–C: Green dashed lines represent patients testing positive for ADAs postdose; grey dashed lines represent patients testing negative for ADAs postdose. ADAs, antidrug antibodies; SD, standard deviation
Mean values and interpatient variability of the PK parameters obtained from the concentration–time data of the 12‐week PK assessment period (C max, AUCT, AUC0–∞, AUC2‐week, CL, V ss and t 1/2) were similar across all groups (Table 2; Figure S2). The CV% was 26–38% for C max, 34–40% for AUCT and 37–45% for AUC0–∞. Adjusted geometric means and 90% CIs for the ratios of C max, AUCT, AUC0–∞ and AUC2‐week were within the acceptance window of 80.00–125.00% for comparisons of PF‐05280586 with rituximab‐EU and rituximab‐US, and rituximab‐EU with rituximab‐US (Table 3).
Table 2.
Mean ± SD PK parameter estimates (per‐protocol population)
| PF‐05280586 | Rituximab‐EU | Rituximab‐US | |
|---|---|---|---|
| n = 68 | n = 67 | n = 63 | |
| C max, μg ml–1 | 453 ± 153 | 422 ± 111 | 430 ± 163 |
| AUC2‐week, μg·h ml–1 | 52, 100 ± 18, 000 | 49, 600 ± 14, 200 | 49, 200 ± 15, 900 |
| AUCT, μg·h ml–1 † | 198, 000 ± 79, 600 | 188, 000 ± 64, 300 | 196, 000 ± 78, 300 |
| AUC0‐∞, μg·h ml–1 ‡ | 213, 000 ± 90, 400 | 200, 000 ± 74,600 | 214, 000 ± 95, 300* |
| CL, ml h–1 kg–1 | 11.2 ± 4.91 | 11.4 ± 4.55 | 11.3 ± 4.87* |
| V ss, ml kg–1 | 5490 ± 1740 | 5590 ± 1320 | 5810 ± 1590* |
| t ½, h | 434 ± 142 | 424 ± 125 | 456 ± 145* |
AUC2‐week, area under the serum concentration–time curve from time 0 to 2 weeks; AUCT, area under the serum concentration–time curve from time 0 to the last time point with a quantifiable concentration; AUC0–∞, area under the serum concentration–time curve from time 0 extrapolated to infinite time; CL, clearance; C max, maximum serum concentration; PK, pharmacokinetic; SD, standard deviation; t ½, terminal elimination half‐life; V ss, volume of distribution at steady‐state.
n = 62 as one patient missed multiple samples, for whom the terminal phase could not be determined adequately.
AUCT was estimated based on the 12‐week concentration–time data. When the additional drug concentration samples collected on day 169 were included in the calculation, the mean (±SD) values of AUCT were 209,000 ± 87, 800 μg·h ml–1, 198, 000 ± 70, 800 μg·h ml–1 and 209, 000 ± 89, 800 μg·h ml–1 for PF‐05280586, rituximab‐EU and rituximab‐US, respectively.
AUC0–∞ was estimated based on the 12‐week concentration–time data. When the additional drug concentration samples collected on day 169 were included in the calculation, the mean (±SD) values of AUC0–∞ were 213, 000 ± 89, 600 μg·h ml–1, 200, 000 ± 72, 500 μg·h ml–1 and 214, 000 ± 94, 000 μg·h ml–1 for PF‐05280586, rituximab‐EU and rituximab‐US, respectively.
Table 3.
Statistical comparison of PK exposure parameters between test and reference products (per‐protocol population)
| Adjusted geometric mean | ||||||
|---|---|---|---|---|---|---|
| Test | Reference | Parameter | Test | Reference | Ratio (%) * | 90% CI (%) |
| PF‐05280586 | Rituximab‐EU | C max, μg ml–1 | 432 | 409 | 105.67 | 96.91–115.21 |
| AUC2‐week, μg·h ml–1 | 49 500 | 47 700 | 103.74 | 95.10–113.15 | ||
| AUCT, μg·h ml–1 † | 184 000 | 178 000 | 103.36 | 92.81–115.12 | ||
| AUC0–∞, μg·h ml–1 ‡ | 196 000 | 188 000 | 104.19 | 92.75–117.06 | ||
| PF‐05280586 | Rituximab‐US | C max, μg ml–1 | 432 | 405 | 106.62 | 97.65–116.41 |
| AUC2‐week, μg·h ml–1 | 49 500 | 46 900 | 105.56 | 96.64–115.30 | ||
| AUCT, μg·h ml–1 † | 184 000 | 181 000 | 101.33 | 90.82–113.04 | ||
| AUC0–∞, μg·h ml–1 ‡ | 196 000 | 195 000 | 100.45 | 89.20–113.11 | ||
| Rituximab‐EU | Rituximab‐US | C max, μg ml–1 | 409 | 405 | 100.90 | 92.38–110.20 |
| AUC2‐week, μg·h ml–1 | 47 700 | 46 900 | 101.76 | 93.13–111.18 | ||
| AUCT, μg·h ml–1 † | 178 000 | 181 000 | 98.03 | 87.83–109.40 | ||
| AUC0–∞, μg·h ml–1 ‡ | 188 000 | 195 000 | 96.40 | 85.57–108.60 | ||
AUC2‐week, area under the serum concentration–time profile from time 0 to 2 weeks; AUCT, area under the serum concentration–time curve from time 0 to the last time point with a quantifiable concentration; AUC0‐∞, area under the serum concentration–time curve from time 0 extrapolated to infinite time; CI, confidence interval; C max, maximum serum concentration; PK, pharmacokinetic.
Test/reference ratio of adjusted geometric means.
For AUCT calculated after inclusion of the additional drug concentration samples collected on day 169, the ratio (90% CI for ratio) was 103.26 (92.13–115.73), 100.45 (89.46–112.79) and 97.28 (86.60–109.27) for the comparisons of PF‐05280586 vs. rituximab‐EU, PF‐05280586 vs. rituximab‐US, and rituximab‐EU vs. rituximab‐US, respectively.
For AUC0–∞ calculated after inclusion of the additional drug concentration samples collected on day 169, the ratio (90% CI for ratio) was 104.19 (92.83–116.93), 100.21 (89.12–112.67) and 96.18 (85.51–108.19) for the comparisons of PF‐05280586 vs. rituximab‐EU, PF‐05280586 vs. rituximab‐US, and rituximab‐EU vs. rituximab‐US, respectively.
PD
Mean CD19+ B cell counts at baseline were 84–100 cells μl–1. All treatments resulted in rapid CD19+ B cell depletion following dose administration on day 1 (Figure 3). By week 2, mean CD19+ B cell count values had decreased by 98.2% to 99.8% in all groups. CD19+ B cell counts remained suppressed for the study duration (24 weeks). CD19+ B cell counts recovered to at least 50% of baseline at the end of treatment in three (4.1%) patients treated with PF‐05280586, six (8.1%) treated with rituximab EU and six (8.2%) treated with rituximab‐US.
Figure 3.
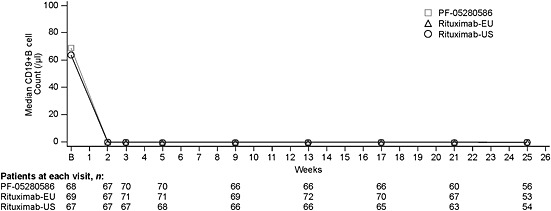
CD19+ B cell count over time in the modified intent‐to‐treat population. B, baseline
In all three groups, C min,B cell values were below the quantitation limit, with a t min,B cell of 1 week. The mean (±SD) area under the CD19+ B cell count vs. time curve (AUCT, B cell) was 13 312 [±13 309 (n = 68)], 14 304 [±13146 (n = 69)] and 12 496 [±13500 (n = 67)] cells·day ml–1 in the PF‐05280586, rituximab‐EU and rituximab‐US groups, respectively. The mean duration of B cell depletion (τB cell) was 120–126 days. Overall, there were no observed differences among groups in the CD19+ B cell count vs. time profile and the parameters determined.
There were no major variations in circulating IgM values over time, with only small differences among the groups (data not shown).
Immunogenicity
Immunogenicity was evaluated in all 220 patients who received at least one dose of study drug for ADA responses (Table S2). Thirteen patients (n = 4, n = 6 and n = 3 receiving PF‐05280586, rituximab‐EU and rituximab‐US, respectively) tested positive for low titres of ADAs in samples collected at baseline. Of these 13 patients, eight (n = 4, n = 3 and n = 1, respectively) tested negative in all subsequent postdose samples. The other five patients who were positive for ADAs at baseline also tested positive for ADAs at one or more time points postdose.
A total of 26 patients (n = 7, n = 10 and n = 9 receiving PF‐05280586, rituximab‐EU and rituximab‐US, respectively) tested positive for ADAs for at least one time point postdose. Of these, 22 patients did not have detectable levels of ADAs until day 85 or later (n = 6, n = 7 and n = 9, respectively). In two patients (one each in the PF‐05280586 and rituximab‐EU groups), ADAs first emerged on day 15 and persisted until the last sample collection on day 169. These patients also had the highest ADA titres among all patients, with peak titres of 4.24 detected on day 15 in one patient (rituximab‐EU) and 4.57 on day 169 in the other (PF‐05280586). In addition, two patients receiving rituximab‐EU had lower titres of ADAs that were first detected on days 29 and 57, and persisted until the last sampling on day 169. On day 169, ADA samples were available from 55, 58 and 52 patients from the rituximab‐US, rituximab‐EU and PF‐05280586 groups, respectively. Among these patients, six (10.9%), eight (13.8%) and six (11.5%) patients from the rituximab‐US, rituximab‐EU and PF‐05280586 groups, respectively, tested positive for ADAs in the day 169 sample. Overall, there were no observed differences among treatment groups in the incidence of ADAs, time of ADA emergence or ADA titres. Most (∼83%) of the 42 postdose samples that tested positive for ADAs also tested positive in the cross‐reactivity assay with similar titres. None of the samples testing positive for ADAs was positive for neutralizing activity on the cell‐based Nab assays.
In general, patients who tested positive for ADAs appeared to have lower AUC0–∞ and higher CL compared with patients who did not have any samples testing positive for ADAs (Figure 2); however, the effect of ADA formation appeared to be comparable among the treatment groups as there were no apparent differences in group mean (±SD) estimates of the PK parameters. Overall, ADA development had no apparent effect on CD19+ B cell depletion/repletion profiles. For the two patients with persistent ADAs and the highest titres, one (administered rituximab‐EU) had CD19+ B cells depleted postdose for the entire study duration – up to day 169 – whereas the other patient (administered PF‐05280586) had CD19+ B cell counts postdose that became detectable again after day 85 and reached approximately 5% of the pretreatment baseline counts on day 141.
Safety
Safety data from all 220 patients enrolled were evaluated. Overall, no clinically meaningful differences in the incidence or severity of AEs were found among groups (Table 4). A total of 136 (61.8%) patients experienced a treatment‐emergent AE [n = 50 (68.5%), n = 41 (55.4%) and n = 45 (61.6%) receiving PF‐05280586, rituximab‐EU and rituximab‐US, respectively]. Across all treatments, 57 (25.9%) patients experienced a treatment‐related AE [n = 22 (30.1%), n = 17 (23.0%) and n = 18 (24.7%), respectively).
Table 4.
Summary of treatment‐emergent adverse events (all‐causality; mITT population)
| n (%) | PF‐05280586 | Rituximab‐EU | Rituximab‐US |
|---|---|---|---|
| n = 73 | n = 74 | n = 73 | |
| Patients with AEs | 50 (68.5) | 41 (55.4) | 45 (61.6) |
| Patients with treatment‐related AEs | 22 (30.1) | 17 (23.0) | 18 (24.7) |
| Patients with serious AEs | 5 (6.8) | 1 (1.4) | 4 (5.5) |
| Patients withdrawn from treatment owing to AEs | 2 (2.7) | 1 (1.4) | 1 (1.4) |
| Patients with AEs grade ≥3 | 10 (13.7) | 1 (1.4) | 10 (13.7) |
AE, adverse event; mITT, modified intent‐to‐treat.
One death was reported in a 66‐year‐old female who developed a presumed bone neoplasm (no histological confirmation) 51 days after the first dose of PF‐05280586. The patient was discontinued from the study owing to this AE and subsequently died. The AE was not considered to be related to the study drug.
The overall incidence of SAEs was consistent with what might be expected in an RA population. A total of 12 treatment‐emergent SAEs occurred in 11 patients. Five PF‐05280586‐treated patients had six SAEs: cardiac failure, intentional self‐injury, presumed bone neoplasm, bacterial arthritis and, in one patient, bacterial sepsis and septic shock. In the rituximab‐EU group, two patients reported SAEs of (one each) thrombocytopenic purpura and pericarditis. Four rituximab‐US–treated patients had SAEs (one each): cardiac failure congestive, atrial flutter, pyelonephritis and arthropathy. Two patients had SAEs that were considered to be treatment related: the thrombocytopenic purpura reported by a patient in the rituximab‐EU group and the case of atrial flutter in the rituximab‐US group.
A total of four (1.8%) patients were withdrawn from treatment owing to an AE: two (2.7%) receiving PF‐05280586, one (1.4%) receiving rituximab‐EU and one (1.4%) receiving rituximab‐US. In all, 21 (9.5%) patients experienced an AE of grade 3 or higher. The incidence of grade 3 or higher AEs was lower among patients receiving rituximab‐EU [n = 1 (1.4%)] compared with rituximab‐US [n = 10 (13.7%)] or PF‐05280586 [n = 10 (13.7%)). There were no dose reductions or temporary discontinuations due to AEs during the study. Nine patients (n = 5, n = 2 and n = 2, receiving PF‐05280586, rituximab‐EU and rituximab‐US, respectively) had the infusion rate reduced owing to an AE. There were no AEs related to the ADA response (e.g. infusion related or hypersensitivity reactions), and the incidence and types of AEs observed in those patients who tested positive for ADAs were similar to those in patients testing negative for ADAs. In general, the mean changes from baseline in vital signs were small and no notable differences were observed across groups with regard to 12‐lead electrocardiogram results.
Discussion
The primary objective of the present study was to demonstrate the PK similarity of PF‐05280586, rituximab‐EU and rituximab‐US in patients with active RA on a background of methotrexate who had an inadequate response to one or more TNF antagonist therapies, using the standard bioequivalence acceptance range of 80.00–125.00%. The 90% CIs for test‐to‐reference ratios for the exposure parameters evaluated were within the predefined acceptance range for comparisons of PF‐05280586 with rituximab‐EU and rituximab‐US, and for rituximab‐EU compared with rituximab‐US, demonstrating PK similarity among the three products. The PK characteristics evaluated were consistent with those reported for rituximab in patients with RA 12.
Baseline CD19+ B cell values were comparable among the three groups and were similar to previous reports 12. All three treatments decreased CD19+ B cell counts, reaching a maximum reduction by week 2. These counts remained suppressed up to week 25, consistent with previous reports showing substantial and sustained depletion of CD19+ B cells following administration of the same doses of rituximab 12. There were no major variations in circulating IgM values over time and only small differences were noted among the groups. Taken together, these results demonstrate that PF‐05280586, rituximab‐EU and rituximab‐US had comparable CD19+ B cell depletion profiles in patients with active RA on a background of methotrexate who had an inadequate response to one or more TNF antagonist therapies.
The incidence of ADA responses, time of ADA emergence and ADA titres were similar across treatments. The overall incidence of ADA responses observed was consistent with that reported (11%) in controlled and long‐term studies 5. The majority of samples that tested positive for ADAs also tested positive in the cross‐reactivity assay with similar titres, suggesting that ADAs were likely to have developed against shared epitopes among the study drugs.
Overall, no clinically meaningful differences in safety events were identified, although some numerical differences were observed across treatments. Although numerically more patients receiving PF‐05280586 than in the other groups discontinued the study, no differences were noted in discontinuations due to AEs. Similarly, a numerical difference in the incidence of grade 3 or above AEs was found across groups, with a lower incidence occurring in patients receiving rituximab‐EU compared with rituximab‐US or PF‐05280586. None of the individual (preferred term) AEs of grade 3 or higher occurred in more than two patients treated with any of the three treatments.
In summary, the evidence of PK similarity of PF‐05280586 to rituximab‐EU and rituximab‐US, together with the results of safety, immunogenicity and PD assessments from the present study, support the continued development of PF‐05280586 as a potential biosimilar for rituximab. In addition, the results of PK similarity between rituximab‐EU and rituximab‐US indicated that either of the two products by itself can be used a comparator for future comparative studies with PF‐05280586.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (http://www.icmje.org/coi_disclosure.pdf) (available on request from the corresponding author) and declare the following financial relationships with organizations that might have an interest in the submitted work in the previous 3 years: SC has been a consultant and investigator for Pfizer Inc. PE has undertaken clinical trials and provided expert advice to Abbott/Abbvie, Bristol Myers Squibb, Pfizer, UCB, MSD, Roche, Novartis, Samsung, Takeda and Lilly. MG has an ongoing clinical research grant from Pfizer Inc. DY, LAM, RL, BG, DT and XM are employees of Pfizer Inc. JCB and GSG were employees of Pfizer at the time of study conduct.
The authors would like to thank Sherry Cai from the Pfizer Clinical Assay Group for managing the analyses of drug concentration samples. This study was funded by Pfizer Inc. Editorial assistance was provided by Christina McManus, PhD, of Engage Scientific Solutions and funded by Pfizer Inc.
Contributors
SC, PE, MG, DY, J‐CB, LAM, RL, BG, DT, GS‐G and XM wrote the manuscript; RL, DY and XM designed the research; SC, MG, DY, LAM, RL, BG and XM performed the research; and DY, LAM, RL, BG and XM analysed the data.
Supporting information
Figure S1 Study design
Figure S2 Individual and mean estimates of PK parameters for (A) Cmax, (B) AUCT and (C) AUC0‐∞
Table S1 Summary of patients ineligible for inclusion in the per‐protocol population
Table S2 Incidence of anti‐drug antibodies by treatment (mITT population)
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Cohen, S. , Emery, P. , Greenwald, M. , Yin, D. , Becker, J. ‐C. , Melia, L. A. , Li, R. , Gumbiner, B. , Thomas, D. , Spencer‐Green, G. , and Meng, X. (2016) A phase I pharmacokinetics trial comparing PF‐05280586 (a potential biosimilar) and rituximab in patients with active rheumatoid arthritis. Br J Clin Pharmacol, 82: 129–138. doi: 10.1111/bcp.12916.
References
- 1. European Medicines Agency . Guideline on similar biological medicinal products containing biotechnology‐derived proteins as active substance: non‐clinical and clinical Issues. Draft [online]. London, UK: European Medicines Agency (EMEA); 2013. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/06/WC500144124.pdf (last accessed 16 September 2015).
- 2. Health Canada . Guidance for sponsors: information and submission requirements for subsequent entry biologics (SEBs) [online]. Ottawa, Ontario: Health Canada Health Products and Food Branch, Minister of Public Works and Government Services; 2008. Available at http://www.hc‐sc.gc.ca/dhp‐mps/alt_formats/pdf/brgtherap/applic‐demande/guides/seb‐pbu/seb‐pbu‐2010‐eng.pdf (last accessed 16 September 2015).
- 3. US Food and Drug Administration , US Department of Health and Human Services, Center for Drug Evaluation and Research (CDER) , Center for Biologics Evaluation and Research (CBER) . Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product. Draft guidelines [online]. Rockville, MD: (CBER); 2012. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf (last accessed 16 September 2015).
- 4. World Health Organization . Guidelines on evaluation of similar biotherapeutic products (SBPs) [online]. Geneva, Switzerland: World Health Organization; 2009. Available at http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf (last accessed 16 September 2015).
- 5. Genentech Inc . Rituxan® (rituximab) prescribing information [online]. South San Francisco, CA, 2013. Available at http://www.gene.com/download/pdf/rituxan_prescribing.pdf (last accessed 7 October 2014).
- 6. F. Hoffmann‐La Roche Ltd . MabThera (rituximab) Summary of product characteristics [online]. Basel, Switzerland; 2008. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/000165/WC500025821.pdf (last accessed 7 October 2014).
- 7. Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2012; 51 (Suppl. 5): v3–11. [DOI] [PubMed] [Google Scholar]
- 8. Karnik S, Thompson MS, DeGruttola H, Ferrari S, Gu L, Aggarwal P, Porter TJ, Rouse JC, Ng C‐K. Characterization and comparison of PF‐05280586, a proposed rituximab biosimilar, to the licensed product. Proceedings of the American Association of Pharmaceutical Scientists – 2013 National Biotechnology Conference (APS‐NBC 2013); 20–22 May, 2013; San Diego, CA, USA.
- 9. Yin D, Becker J‐C, Melia L, Li R, Gumbiner B, Thomas D, Spencer‐Green G, Meng X. A phase I pharmacokinetics trial comparing PF‐05280586 (a potential biosimilar) and rituximab in subjects with active rheumatoid arthritis (REFLECTIONS B328‐01). Ann Rheum Dis 2014; 73: FRI0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Menard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska‐Biernat E, Symmons D, Tak PP, Upchurch KS, Vencovsky J, Wolfe F, Hawker G. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010; 62: 2569–81. [DOI] [PubMed] [Google Scholar]
- 11. Hochberg MC, Chang RW, Dwosh I, Lindsey S, Pincus T, Wolfe F. The American College of Rheumatology 1991 revised criteria for the classification of global functional status in rheumatoid arthritis. Arthritis Rheum 1992; 35: 498–502. [DOI] [PubMed] [Google Scholar]
- 12. Breedveld F, Agarwal S, Yin M, Ren S, Li NF, Shaw TM, Davies BE. Rituximab pharmacokinetics in patients with rheumatoid arthritis: B‐cell levels do not correlate with clinical response. J Clin Pharmacol 2007; 47: 1119–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Study design
Figure S2 Individual and mean estimates of PK parameters for (A) Cmax, (B) AUCT and (C) AUC0‐∞
Table S1 Summary of patients ineligible for inclusion in the per‐protocol population
Table S2 Incidence of anti‐drug antibodies by treatment (mITT population)
Supporting info item
Supporting info item
Supporting info item
Supporting info item