

Abstract
Aim
The aim of the present study was to provide an insight into the characteristics and follow‐up of postmarketing studies of medicines that were conditionally authorized in the European Union (EU).
Methods
We compiled a list of all postmarketing studies attached as specific obligations to the licence of medicines that were granted conditional marketing authorization from January 2006 to April 2014. Studies were characterized based on their objective, design, status upon marketing authorization (MA) and due data set by authorities. They were linked to online study registrations (Clinicaltrials.gov, ENCePP) to determine completion date. We described and associated characteristics of studies and medicines, and determined whether studies were completed on time.
Results
A total of 59 postmarketing studies were requested for 21 conditionally authorized medicines. Most studies had an interventional study design (73%), were ongoing upon MA (61%) and aimed to provide additional data on efficacy (45%). Interventional studies were more often ongoing and providing efficacy data, while observational and other studies were more often new and providing safety data. Frequent grounds for requesting postmarketing studies were ‘long‐term follow‐up’ and ‘increase data on subpopulations’. Of the 34 studies eligible for follow‐up analysis, 26 (76%) were completed and 17 (50%) completed on time. Actual completion time took a median (interquartile range) of 274 (−121 to 556) days longer than expected.
Conclusions
Our results indicated that most postmarketing studies attached to a conditional marketing authorization were eventually completed but that half were completed with a substantial delay. The observations suggest caution when broadening the use of postmarketing studies for resolving uncertainties about benefits and risks after MA.
Keywords: Benefit–risk assessment, drug, drug approval, legislation, postmarketing, product surveillance
What is Already Known about this Subject
The conditional marketing authorization pathway in the EU facilitates early‐access to medicines, provided that postmarketing studies are conducted to reduce uncertainties about benefits and risks.
We know little about the characteristics of these postmarketing studies, the rationales for requesting them and whether they are completed according to the timelines established upon marketing authorization.
What this Study Adds
We found that most requested postmarketing studies are started and eventually completed but that half of all studies are completed with a substantial delay.
Introduction
Postmarketing studies that are requested by drug regulatory authorities upon marketing authorization and conducted by marketing authorization holders have become an increasingly salient instrument in medicines regulation. In spite of extensive testing of medicines before authorization, knowledge of their benefits and risks is inherently limited at the time of marketing authorization 1. Postmarketing studies aim to increase this knowledge by reducing uncertainties about the effectiveness of medicines under real‐world circumstances and identifying or quantifying adverse drug reactions that could influence the benefit–risk balance 2, 3.
The importance of postmarketing studies has markedly increased in recent decades owing to an evolution of medicines regulatory frameworks around the world towards a lifecycle approach 4, 5. Typically, in a lifecycle approach, evidentiary standards for marketing authorization are initially eased, provided that further studies and monitoring activities are conducted to obtain comprehensive data on benefits and risks. Regulators continue to be involved in the evaluation of these data through ongoing assessments of the benefit–risk balance and taking appropriate action when incoming data affect the balance.
In the European Union (EU), conditional marketing authorization is a key example of a regulatory pathway that takes a lifecycle approach. Conditional marketing authorization provides the possibility to grant early access to medicines that treat diseases with unmet medical need in case ‘the benefits to public health of immediate availability outweigh the risks inherent in the fact that additional data are still required’ 6. Marketing authorization applicants that are granted a conditional marketing authorization are ‘required to complete or initiate certain studies with a view to confirming that the risk–benefit balance is positive and resolving any questions relating to the quality, safety and efficacy of the product’ 6. These so‐called ‘specific obligations’ are agreed upon between regulators and marketing authorization applicants on a case‐by‐case basis, depending on the medicine‐specific uncertainties that need to be addressed. They only constitute a part of all postmarketing activities as further studies and activities may be imposed upon marketing authorization applicants by European regulators through ‘obligations in Annex II’, ‘additional pharmacovigilance activities in the risk management plan’ or other ‘legally binding measures’ 7.
The progress in fulfilling specific obligations is evaluated on an annual basis by regulators. When all specific obligations are fulfilled, the medicine is granted a marketing authorization not subject to specific obligations. Although the fulfilment of specific obligations is legally binding, no medicine can be withdrawn from the market purely because the obligation was not fulfilled 8. However, any modification to the obligation with regard to design or due date has to be discussed with and agreed upon by regulators. Moreover, in the case of infringement of specific obligations, regulators can apply a financial penalty to the marketing authorization holder which may amount to a total of 5% of the turnover of the marketing authorization holder in the EU in the preceding year 9.
Little is known about the characteristics and follow‐up of specific obligations attached to conditional marketing authorizations in the EU. Analyses of fulfilment of postmarketing studies in the USA and Canada showed that studies are frequently not conducted or are completed with substantial delays 10, 11, 12, 13. However, the US studies did not focus specifically on postmarketing studies attached to early‐access pathways similar to conditional marketing authorization in the EU, while the means for legal enforcement of study fulfilment also seem to be less specified in the USA compared with the EU. Therefore, it might be expected that the rate of fulfilment of specific obligations in the EU will be relatively high. Nevertheless, a previous analysis of specific obligations in the EU did observe delays and discrepancies in fulfilment 14, although this study did not characterize obligations or quantify the degree to which the individual studies were completed on time. Moreover, one European study on the fulfilment of postauthorization safety studies (PASSs) suggested that fulfilment was generally good, with most studies progressing from protocol to data collection 15. However, at the time of study conduct, most PASSs were not yet completed, limiting the conclusions that could be drawn from this analysis. Further analysis on the characteristics and follow‐up of postmarketing studies in the EU is therefore warranted, especially as confirmation of benefits and risks through postmarketing studies is envisaged to become a major cornerstone for the novel adaptive pathways procedure in the EU 16.
The aim of the present study was therefore to examine the characteristics and follow‐up of postmarketing studies attached as specific obligations to the licence of conditionally authorized medicines in the EU.
Methods
Data collection
We identified all medicines that were granted a conditional marketing authorization in the EU from first use of this pathway in 2006 up until April 2014, based on information from annual reports of the European Medicines Agency (EMA). We excluded two vaccines that were intended for use in emergency situations only.
For each medicine, we retrieved the European public assessment report (EPAR) from the Agency's website and extracted information from different components of this report. The authorization details of the EPAR provided the source of information for the authorization date, therapeutic indication, whether the product was indicated for an orphan disease and whether a conditional marketing authorization was requested proactively by the marketing authorization applicant. The assessment history was examined to determine whether and when all specific obligations were considered fulfilled and the medicine converted to a standard marketing authorization.
Annex II of the EPAR was used to retrieve a list of all requested specific obligations, including the text description of the obligation and the due date for completion set by the EMA upon authorization. Obligations were included when they were mentioned under the heading ‘Specific obligation to complete postauthorization measures for the conditional marketing authorization’. We examined all obligation texts to determine whether multiple studies were mentioned in a single obligation text or a single study in multiple obligation texts. Each study was included as a separate observation in our dataset, rendering the number of studies different from the number of obligations. We also excluded one obligation because it was not a request for a study but for the development of a diagnostic test kit.
All studies were characterized by design, status upon marketing authorization, expected duration and objective. Study design was categorized as interventional, observational or other, based on the obligation text. We also determined for each study its status upon marketing authorization as either ongoing or supposed to start postmarketing, based on the obligation text and information from the scientific assessment report. Expected duration was characterized as the difference between the set due date for completion and the marketing authorization date. In case multiple obligations referred to the same study, the last due date was used.
To determine the objectives of the postmarketing studies, we used information from the scientific assessment report, particularly the ‘discussion on clinical efficacy and clinical safety’, which often included information on ‘additional data needed in the context of a conditional marketing authorization’. We also retrieved information from sections on ‘uncertainty in the knowledge about beneficial or unfavourable effects’ or the ‘grounds for re‐examination’ in case the medicine was approved in a re‐examination procedure.
We first categorized objectives in a general way as the need to provide additional efficacy data, additional safety data, or both efficacy and safety data. We subsequently developed a more granular categorization based on the specific grounds for requesting each postmarketing study. Studies were categorized in seven nonmutually exclusive categories: ‘long‐term follow‐up’, ‘additional endpoints’, ‘(additional) comparator’, ‘increase size of study population’, ‘quantification of risk’, ‘understanding posology and drug–drug interactions’ and ‘increase data on subpopulations’. Each study was subsequently categorized into one or more groups depending on which grounds were mentioned in the scientific assessment report.
To provide an insight into study follow‐up, we searched for registrations of all interventional and observational studies in the online public register Clinicaltrials.gov. For observational studies, we also searched the register of the European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP, http://www.encepp.eu). Search terms included the name of the medicine, the study name (if provided) and study description items (e.g. comparator). We merely tried to link interventional and observational studies, as other studies such as bioequivalence studies, pharmacokinetic–pharmacodynamic studies or pooled analysis are generally not registered in a public database while results of these studies as reported in publications often do not mention a completion date. Moreover, we only considered studies with an expected completion date before August 1, 2015 which was the last follow‐up date of this analysis.
For all linked studies, we extracted information on (estimated) completion date. In case studies were completed, we retrieved the actual study completion date. In case studies were still ongoing, we noted the estimated study completion date. Dates listed as month and year were noted as last of the month. Searches for registrations in Clinicaltrials.gov and ENCePP were performed independently by two researchers (JH, TK) and in cases of disagreement, a consensus was sought. All other data were first collected by TK or JH and subsequently reviewed by JH or MB. Disagreement was resolved by consensus. The last follow‐up for data collection was 31 July 2015.
Data analysis
To characterize studies, we first examined associations between study design and their general objective (safety, efficacy, both), status upon marketing authorization and expected duration. We subsequently examined the detailed grounds for the specific obligations and visualized these grounds by treatment indication (cancer vs. noncancer indications), whether or not the medicine was indicated for an orphan disease, whether or not conditional marketing authorization was requested proactively by the applicant and whether or not the postmarketing study was already ongoing at the time of marketing authorization.
To examine fulfilment of the studies, we computed the difference in days between the actual completion date and the set due date for completion. For studies that were not yet completed, in spite of a due date before 1 August 2015, we computed the difference between the expected completion date as listed in the register and the due date. Studies were deemed completed on time when they were completed within a year after the due date. Moreover, medicines were considered to be converted on time when they converted to a standard marketing authorization within a year after the last due date of all postmarketing studies (and hence all obligations).
The analysis to determine study completion was performed on all registered interventional and observational studies as well as on a subset of studies for which there was an explicit request in the obligation text for study completion or a final study report. We conducted this sensitivity analysis as in some cases the specific obligation might have been fulfilled based on an evaluation by the EMA of data from an interim analysis, while the study was still ongoing.
Results
Characteristics
From January 2006 until April 2014, 23 medicines were granted a conditional marketing authorization in the EU, including two vaccines for emergency use. Of the 21 medicines included in the present analysis, 13 were indicated for cancer, three for HIV/AIDS, two for tuberculosis and one each for cystic fibrosis, epilepsy and multiple sclerosis. Almost half (48%) had an orphan designation upon marketing authorization, while a proactive request for a conditional marketing authorization was made for 38% of medicines (Table 1).
Table 1.
Characteristics of medicines and postmarketing studies
| Medicines (n = 21) | |
|---|---|
| Number (%) | |
| Therapeutic indication | |
| Cancer | 13 (62) |
| HIV/AIDS | 3 (14) |
| Epilepsy | 2 (9) |
| Multiple sclerosis | 1 (5) |
| Cystic fibrosis | 1 (5) |
| Tuberculosis | 1 (5) |
| Orphan indication | 10 (48) |
| Proactive request for CMA by MAA | 8 (38) |
| Post‐marketing studies (n = 59) | |
| Design | |
| Interventional studies | 44 (75) |
| Observational studies | 5 (8) |
| Other obligations | 10 (17) |
| General objective | |
| Additional efficacy data | 25 (42) |
| Additional safety data | 9 (15) |
| Additional efficacy/safety data | 25 (42) |
| Status upon MA | |
| New studies | 23 (39%) |
| Expected duration | |
| Expected duration, median (IQR) | 575 (204–1287) |
CMA, conditional marketing authorization; IQR, interquartile range; MA, marketing authorization; MAA, marketing authorization application.
The EMA requested a total of 61 specific obligations for the 21 medicines. We excluded one obligation and observed requests for 59 studies in the 60 obligation texts. Original obligation texts of these 59 studies as retrieved from the respective EPARs are provided in the Appendix. A median [interquartile range (IQR)] of two (1–4) studies per medicine were requested. For 25 studies (42%), the objective of the obligation was to provide additional efficacy data, for nine (16%) additional safety data and for 25 (42%) additional data on both safety and efficacy.
Table 1 shows that there were 44 (75%) requests for interventional studies, five (8%) for observational studies and 10 (17%) for other studies, the latter being mainly reviews of safety or efficacy data, pharmacokinetic–pharmacodynamic studies and post hoc analyses. There were 23 (39%) requests for new studies to be started postmarketing, while 36 (61%) studies were already ongoing at time of marketing authorization (e.g. extension of phase III trials).
Table 2 shows associations between the design of studies and their general objective, status and expected duration. Half of all interventional studies aimed to provide additional efficacy data, while this proportion was lower for observational and other studies (40% and 10%, respectively). Conversely, compared with interventional studies, observational studies and other studies more often aimed to provide additional safety data (9% vs. 40% and 30%, respectively). Only 23% of all interventional studies were expected to start postmarketing, while this proportion was higher for observational studies (100%) and other studies (80%).
Table 2.
Associations between design and characteristics of postmarketing studies
| Interventional (n = 44) | Observational (n = 5) | Other (n = 10) | |
|---|---|---|---|
| General objective | |||
| Additional efficacy data | 22 (50%) | 2 (40%) | 1 (10%) |
| Additional safety data | 4 (9%) | 2 (40%) | 3 (30%) |
| Additional efficacy/safety data | 18 (41%) | 1 (20%) | 6 (60%) |
| Status upon MA | |||
| New study | 10 (23%) | 5 (100%) | 8 (80%) |
| Ongoing study | 34 (77%) | 0 (0%) | 2 (20%) |
| Expected duration | |||
| Duration in days, median (IQR) | 586 (261–1279) | 1402 (1168–1413) | 307 (125–374) |
IQR, interquartile range; MA, marketing authorization.
We found 104 different rationales for the 59 requested postmarketing studies (Figure 1). The most prevalent grounds for requesting these studies were ‘long‐term follow‐up’ (n = 24, 23%) and ‘increase data on subpopulations’ (n = 19, 18%). Figure 1 also indicates differences in the grounds for postmarketing studies by indication type, proactive request for conditional marketing authorization and status of postmarketing study upon authorization. Postmarketing studies for cancer indications were more often motivated by a need to obtain more data on subpopulations (30% vs. 9%) and additional endpoints (19% vs. 4%), while they were less often requested to better understand posology or drug–drug interactions (4% vs. 24%). When a conditional marketing authorization was requested proactively by the applicant, subsequent post‐marketing studies were more often requested to better understand posology or drug–drug interactions (22% vs. 4%) and less often to obtain more data on subpopulations (7% vs. 31%). Ongoing studies were more often used to provide long‐term follow‐up (30% vs. 16%). No pronounced differences were observed between medicines with and without an orphan indication.
Figure 1.
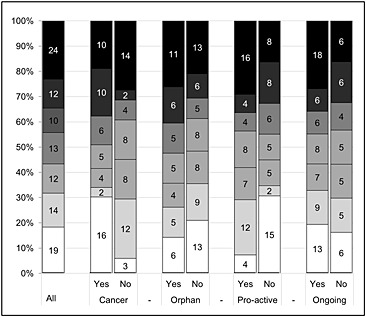
Grounds for requesting post‐marketing studies (from top to bottom). ( ) Long‐term follow‐up, (
) Long‐term follow‐up, ( ) Additional endpoints, (
) Additional endpoints, ( ) (Additional) comparator, (
) (Additional) comparator, ( ) Increase size of study population, (
) Increase size of study population, ( ) Quantification of specific risks, (
) Quantification of specific risks, ( ) Understanding posology and drug‐drug interaction, (□) Increase data on subpopulation(s)
) Understanding posology and drug‐drug interaction, (□) Increase data on subpopulation(s)
Follow‐up
There were 37 interventional and observational studies with a due date before the last follow‐up date. We were able to link 34 (92%) of these studies with a registration on Clinicaltrials.gov or ENCePP (Figure 2). Out of these 34 studies, 26 (76%) were completed before the last follow‐up date. Time to completion took a median (IQR) of 275 (−121 to 773) days longer than expected upon marketing authorization. For eight uncompleted studies, the expected time to completion took a median (IQR) of 913 (853–1248) days longer than expected upon marketing authorization. There was one study that was already completed, in spite of an expected completion date after the last follow‐up date.
Figure 2.
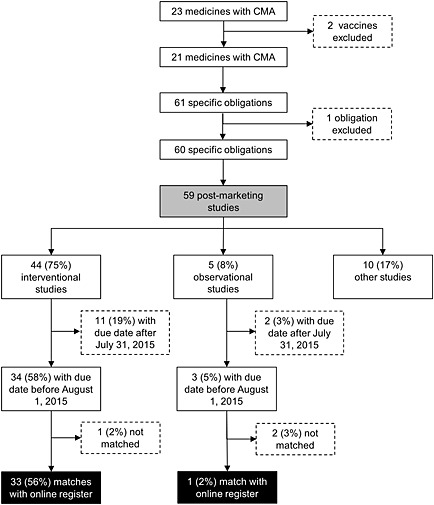
Flowchart describing the identification and matching of postmarketing studies. CMA, conditional marketing authorization. ( ) Analysis of characteristics (n = 59), (
) Analysis of characteristics (n = 59), ( ) Analysis of follow‐up (n = 34)
) Analysis of follow‐up (n = 34)
Seventeen out of 34 (50%) studies were completed within 1 year after the due date. Seven of these 17 studies were not completed before the due date but within 1 year after the due date (Figure 3A). None of the eight uncompleted studies were expected to be completed within 1 year of the due date. When considering only the 15 studies in the follow‐up sample for which there was an explicit request for a final study report, we observed that 12 (80%) of these studies were completed at last follow‐up date and eight (67%) within 1 year after the due date.
Figure 3.
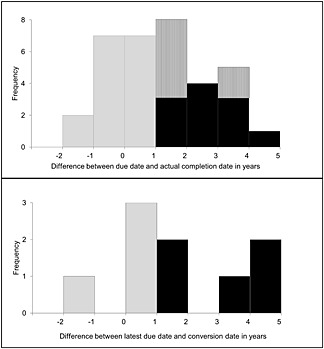
Distribution of time to completion for postmarketing studies (upper panel) and time to conversion for medicines (lower panel). ( ) Completion in time, (
) Completion in time, ( ) Completion delayed, (
) Completion delayed, ( ) Expected completion delayed, (
) Expected completion delayed, ( ) Conversion in time, (
) Conversion in time, ( ) Conversion not in time
) Conversion not in time
When assessing conversion of the licence of medicines to a standard marketing authorization, there were 14 medicines that had a due date for all obligations before the last follow‐up date. Nine (64%) of these medicines converted to a standard marketing authorization and four (29%) were converted within 1 year after the last due date (Figure 3B). There were no other medicines that were converted. Conversion occurred a median (IQR) of 470 (114–1295) days later than expected based on the last due date as agreed at the time of market authorization. None of the 21 included medicines were withdrawn from the market owing to obligation outcomes that affected the benefit–risk balance of the product.
Conclusions
The aim of the present study was to examine the characteristics and follow‐up of postmarketing studies that were attached as specific obligations to the licence of conditionally authorized medicines in the EU. We observed that during the study period 2006–2014, a median of two specific obligations were requested for conditionally authorized medicines, with most requests for additional efficacy data from interventional studies that were already ongoing upon marketing authorization. Moreover, although most studies were started as judged from information in a publicly accessible registry, completion of half of all studies was substantially delayed and only four out of 14 medicines were converted to a standard marketing authorization in time.
Regarding study characteristics, we observed mainly requests for interventional studies, with about three‐quarter of these studies already ongoing at time of marketing authorization. It seems that these medicines were authorized relatively often at a stage when early data from ongoing pivotal studies were available but collection of longer‐term follow‐up data from these studies was still deemed necessary by regulators. This observation is supported by our finding that relatively many postmarketing studies were requested in order to provide long‐term follow‐up data.
Prior research focusing specifically on oncology medicines showed that conditional marketing authorization is not always used in a proactive manner to bring the most promising and transformative therapies to the market 17. These authors concluded that, in some cases, the rationale for granting a conditional marketing authorization to oncology medicines was not a general lack of data but rather a lack of strong enough data to warrant a standard marketing authorization. In these cases, the conditions for authorization were generally less well planned and authorization was relatively often accompanied by a narrowing of the indication to a specific subpopulation 17. The present study indicated that the grounds for requesting postmarketing studies were different for developer‐initiated compared with regulator‐initiated conditional marketing authorizations. More specifically, when conditional marketing authorization was not proactively applied for, regulators tended to be more likely to request longer‐term follow‐up data from ongoing trials and/or additional data on the safety/efficacy of these medicines in subpopulations. When conditional marketing authorization was applied for proactively, there tended to be relatively more requests for further data on posology and drug–drug interactions.
A mixed picture emerges from our results with regard to the follow‐up of specific obligations. We showed that three‐quarter of studies are started and eventually completed by marketing authorization holders, yet also demonstrated that half of all interventional and observational studies are completed with a substantial delay. Apart from the fact that many studies were already ongoing upon marketing authorization, there are a number of other factors rooted in European legislation, as well as regulatory practice, that may contribute to eventual study start and completion.
First, regulators consider the likelihood that a marketing authorization holder is in the position to conduct studies as a formal evaluation criterion when deciding to grant a conditional marketing authorization 6. In doing so, they may take into account factors such as the resources of marketing authorization holders to conduct studies, the complexity of the study and the possibility that, upon marketing authorization, a window of opportunity for conducting a study is closed because of ethical or logistical reasons. When regulators expect that timely completion of studies will be challenging, marketing authorisation can be denied on this ground 18. Second, marketing authorization holders need to apply for a renewal of a conditional marketing authorization on a yearly basis. In preparation for this procedure, marketing authorization holders are expected to draft a report on progress in fulfilling the requested obligations. This provides regulatory authorities with an opportunity to monitor study completion and requires a substantial effort on the part of marketing authorization holders annually, which may incentivize study conduct.
At the same time, our findings raise concerns over the timely completion of studies, given that only half of all studies were completed within 1 year after the due date set by authorities. There may be several explanations for this observation. Marketing authorization holders may face critical ethical and logistical challenges in conducting studies. Once a drug is on the market, patients may not be willing to participate in an interventional study in case they are randomized to a control group 19. Moreover, physicians and academic researchers may have limited interests in contributing to studies that have the sole purpose of confirming earlier findings and ask no novel scientific questions 20. Furthermore, upon marketing authorization, regulators may have been too optimistic about study completion. The risk of an inaccurate prediction of study completion might be especially high when a conditional marketing authorization is not requested proactively by the marketing authorization applicant and agreement on specific obligations needs to be reached within a short timeframe at the end of the marketing authorization procedure 17. Although the present study did not substantiate this claim directly, it showed that the grounds for imposing specific obligations were different when conditional marketing authorization was requested proactively.
It has also been suggested by several authors that there is little incentive for marketing authorization holders to complete postmarketing studies in a timely manner 8. Postmarketing studies generally offer little financial benefit and may even result in a reduced market share if new safety concerns are identified or the indication is narrowed down following the identification of subpopulations that respond best to therapy. The legal design of the conditional marketing authorization regulation 5 may partly contribute to this, given that regulators will already have agreed that the benefit–risk balance of the product is positive when granting a conditional marketing authorization. Once this decision has been made, there seems to be little chance of revoking it, unless new data from post‐marketing studies dictate otherwise. This concern can be mitigated if regulators would judge that the ‘benefit–risk balance is reasonably likely to be positive’. This would put the ‘burden of proof’ to confirm the likelihood that the positive benefit–risk balance is positive on the marketing authorization holder. Such proof could then be demanded, within a legally defined term‐limited period after which regulators would assess whether the benefit‐risk balance is positive and decide on conversion or revocation of the marketing authorisation.
It is highly likely that regulators are aware of the adjusted timelines of the delayed studies, given that progress is monitored on a yearly basis. It is also likely that they have agreed upon modifications to the study timelines and are aware of the results of interim analyses. However, although there may be good reasons for study delay, it goes without saying that these delays are not in the interests of public health. When remaining questions about safety and/or efficacy are not answered within set time frames, patients may be exposed to unnecessary treatment risks. Moreover, given the limited data availability, it is more challenging for regulators to balance the benefits and risks of medicines in a scientifically sound way. This is particularly important, given the fact that a relatively large number of the medicines that were granted a conditional marketing authorization were authorized without consensus about the positive benefit‐risk balance upon marketing authorization 17.
One other result of the present analysis stands out. Although, eventually, the vast majority of interventional studies could be linked to a registration at Clinicaltrials.gov or ENCePP, establishing a link was a time‐consuming process, especially when only short study descriptions, without a study name, were available from the EPAR. Moreover, this indirect way of assessing study completion was only possible for interventional and observational studies, and not for other studies. Our results therefore also stress the need for more transparency on the part of authorities to provide better information on the design and follow‐up of specific obligations attached to a conditional marketing authorization. Transparency could be increased by publishing the summaries of annual reassessment reports or establishing a register of postmarketing authorization measures, with regular status updates. It is in the interests of patients and healthcare providers to have access to this information, given that patients are exposed to higher treatment risks when comprehensive data on benefits and risks are not available.
There were a number of limitations to the present analysis. A first limitation is that we assessed completion of postmarketing studies instead of completion of specific obligations. Consequently, we do not know how regulators assessed the fulfilment of obligations in light of study progress and whether changes were made to the obligations in the postmarketing phase. It may, for instance, be the case that some studies are not completed but that regulators consider the data to be comprehensive enough for the fulfilment of obligations, or that the study design is changed in response to critical challenges. There may also be cases in which there are requests for additional obligations after the initial study results have become available. To limit the influence of these time‐varying factors, we conducted a sensitivity analysis on studies for which there was an explicit request in the obligation text for a final study report or study completion. We observed similar delays in this sensitivity analysis. A more in‐depth study, focusing specifically on the assessment of incoming data by regulators during annual renewals, may provide more insight into whether the fulfilled obligations solve the issues outstanding at the time of a conditional marketing authorization. A previous study, focusing specifically on the safety concerns listed in the risk management plans (RMP) of a cohort of medicines intended for chronic use, showed in this respect that after 5 years, 20% of the mentioned uncertainties had been resolved but that new uncertainties had been included in the RMP at a similar rate 21. A second limitation is that the follow‐up time of the present analysis was limited specifically the case for medicines that had been recently authorized for use. As a result, the present analysis had limited statistical power to test for associations. For example, we could not discern statistically the factors that contribute to the timely completion of studies. This was remains an area for further research. A third limitation is that we were able to assess compliance only for a subset of interventional and observational studies. We therefore do not know whether our results hold for studies that are smaller and less complex to conduct than interventional and observational studies.
In conclusion, our results indicate that most postmarketing studies attached as specific obligations to conditionally authorized medicines in the EU are started and eventually completed; however, half of all studies are completed with a substantial delay compared with the timelines expected at time of authorization. These observations suggest that caution is necessary when broadening the use of this regulatory instrument for resolving significant uncertainties about the benefits and risks of medicines in the postmarketing phase, especially when designing novel authorization procedures, such as adaptive pathways. To mitigate concerns, such pathways should be used in a prospective manner, including early discussions on design and request for study completion. Moreover, care should be taken further to incentivize the timely conduct of postmarketing studies, to facilitate the balancing of benefits and risks by regulators based on comprehensive data.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work. JH has performed research within the context of Escher Projects. This Dutch public–private partnership resides under the umbrella of Top Institute Pharma (www.ti‐pharma.com) and has received funding from the European Federation of Pharmaceutical Industry & Associations (EFPIA).
Contributors
JH, TK, AKMT, HGML and MLDB designed the research; JH and TK collected and analysed the data; JH and TK wrote the first draft; JH, TK, AMT, HGML and MLDB wrote the final draft.
Overview of all post‐marketing studies included in the analysis
| Trade name (international nonproprietary name) | Study identification | Postmarketing studies | Registration number |
|---|---|---|---|
| Deltyba (delamanid) | 1.1 | Phase III trial comparing delamanid 100 mg b.i.d. for 2 months +200 mg q.d. for 4 months plus OBR for 18–24 months vs. optimized background regimen for 18–24 months with placebo for the first 6 months | NCT01424670 |
| 1.2 | Controlled study of the efficacy, safety and pharmacokinetics of delamanid 100 mg b.i.d. for 2 months, followed by delamanid 200 mg q.d. for 4 months or delamanid 400 mg q.d. for 6 months in adult patients with pulmonary multidrug‐resistant tuberculosis | Registration not found | |
| Cometriq (cabozantinib) | 2.1 | A dose‐comparison study (XL‐184‐401) (140 mg vs. 60 mg) in 112 patients with hereditary or sporadic medullary thyroid cancer | NCT01896479 |
| Sirturo (bedaquiline) | 3.1 | Confirmatory phase III study to evaluate additional efficacy and safety data of bedaquiline in different treatment regimens compared with a regimen that does not include bedaquiline | NCT02409290 |
| Erivedge (vismodegib) | 4.1 | Safety update of the pooled safety population | Not included in follow‐up analysis |
| 4.2 | Final analysis of SHH4476g (pivotal study) | NCT00833417 | |
| 4.3 | Interim analysis of 500 patients with a potential 1‐year follow‐up and final analysis on safety and efficacy data efficacy in patients with symptomatic metastatic BCC in study MO25616 | NCT01367665 | |
| Bosulif (bosutinib) | 5.1 | Single‐arm, open‐label, multicentre efficacy and safety study of bosutinib in patients with Philadelphia chromosome‐positive chronic myelogenous leukaemia (Ph + CML) previously treated with one or more tyrosine kinase inhibitor(s) and for whom imatinib, nilotinib and dasatinib are not considered appropriate treatment options | NCT02228382 |
| Adcetris (brentuximab) | 6.1 | Overall survival follow‐up of the patients included in study SG035–0003, including subanalysis of patients ≥ 100 kg body weight. The data should be presented in the context of historical controls | NCT00848926 |
| 6.2 | Overall survival follow‐up of the patients included in study SG035–0004, including subanalysis of patients ≥ 100 kg body weight. The data should be presented in the context of historical controls | NCT00866047 | |
| 6.3 | A postauthorization safety study (PASS) in both studied HL and sALCL patient populations (n = 500), including a sufficient number of sALCL patients (i.e. at least n = 50; study MA25101). | ENCEPP5744 | |
| 6.4 | A single‐arm study in a similar patient population as the sALCL population investigating response rate, duration of response, rate of (second) ASCT and data in subpopulations (including but not necessarily restricted to ALK status and age) (study C25006). | NCT01909934 | |
| 6.5 | A single‐arm studying r/r HL population not eligible for ASCT, investigating response rate, PFS, OS, proportion of patients proceeding to transplant and safety (n = approximately 60 patients) | NCT01990534 | |
| Xalkori (crizotinib) | 7.1 | Clinical study report of study A8081007, including a detailed analysis of outcome on postprogression treatments as well as efficacy and baseline data according to race (Caucasian/Asian) by treatment groups | NCT00932893 |
| 7.2 | Updated safety (serious adverse events and deaths) and efficacy (PFS, OS) data for study 1001 | NCT00585195 | |
| 7.3 | Updated safety (serious adverse events and deaths) and efficacy (PFS, OS) data for study 1005 | NCT00932451 | |
| 7.4 | Safety review of main (severe) hepatic disorders from all available main studies of crizotinib (including studies 1001, 1005 and 1007) | Not included in follow‐up analysis | |
| Pixuvri (pixantrone) | 8.1 | Randomized controlled phase III study (PIX306) of pixantrone–rituximab vs. gemcitabine–rituximab in patients with aggressive B‐cell NHL, who failed frontline CHOP‐R, who are not eligible for autologous stem cell transplant (ASCT) (2nd line) or failed ASCT (3rd or 4th line) | NCT01321541 |
| Caprelsa (vandetanib) | 9.1 | Open‐label trial based on a CHMP‐approved protocol, comparing RET‐negative and RET‐positive patients with sporadic medullary thyroid cancer treated with vandetanib | NCT01945762 |
| Fampyra (fampridine) | 10.1 | Double‐blind, placebo‐controlled, long‐term efficacy and safety study to investigate a broader primary endpoint clinically meaningful in terms of walking ability and to further evaluate the early identification of responders in order to guide further treatment. A study report is to be submitted | NCT02219932 |
| Votrient (pazopanib) | 11.1 | Study report for VEG108844 (a study of pazopanib vs. sunitinib in the treatment of subjects with locally advanced and/or metastatic renal cell carcinoma) | NCT00720941 |
| 11.2 | Pooled analysis of data from study VEG108844 and VEG113078 (a study to evaluate the efficacy and safety of pazopanib vs. sunitinib for the treatment of Asian subjects with locally advanced and/or metastatic renal cell carcinoma) | Not included in follow‐up analysis | |
| Arzerra (ofatumumab) | 12.1 | Open‐label, multicentre study investigating the safety and efficacy of ofatumumab therapy vs. physician's choice in patients with bulky fludarabine‐refractory chronic lymphocytic leukaemia | NCT01313689 |
| 12.2 | Phase IV observational study to provide further data on the clinical efficacy and safety of ofatumumab | NCT01453062 | |
| Cayston (aztreonam) | 13.1 | Clinical study report of study GS‐US‐205‐0110: ‘Open‐label, randomized phase 3 study to evaluate the efficacy and safety of AZLI versus tobramycin nebulizer solutions in an intermittent aerosolized regimen in patients with CF’ | NCT00757237 |
| 13.2 | Clinical study report of study GS‐205‐0117: ‘Phase 3, double‐blind, multicenter, multinational randomized, placebo controlled trial evaluating AZLI in patients with cystic fibrosis, mild lung disease and PA’ | NCT00712166 | |
| 13.3 | Review of all paediatric data from controlled studies | Not included in follow‐up analysis | |
| 13.4 | Paediatric development plan consisting of well‐controlled trials to support short‐term and long‐term repeated use in this patient group | Not included in follow‐up analysis | |
| Votubia (everolimus) | 14.1 | Long‐term follow‐up on duration of response and time to progression for study C2485 | NCT00411619 |
| 14.2 | Interim and final safety and efficacy results of pivotal clinical study M2301 | NCT00789828 | |
| Intelence (etravirine) | 15.1 | Pooled 48‐weeks data from the two pivotal trials C206 and C216 (DUET‐1 and DUET‐2) to substantiate the durability of the virological suppression achieved with etravirine and to assess the safety profile of the compound further | Not included in follow‐up analysis |
| 15.2 | Confirmatory study to provide reassurance on the extrapolation of the study results from the two pivotal studies (DUET‐1 and DUET‐2) to the combined use of etravirine with boosted PIs other than darunavir/ritonavir | Registration not found | |
| Tyverb (lapatinib) | 16.1 | Updated analysis of survival data for study EGF100151 | NCT00078572 |
| 16.2 | Phase III randomized, controlled clinical study to evaluate the incidence of brain metastases as the site of relapse with a lapatinib‐containing therapy compared with an appropriate, trastuzumab‐containing control arm | NCT00820222 | |
| Isentress (raltegravir) | 17.1 | 48‐week safety and efficacy data from the ongoing phase III protocol 018 (‘A multicenter, double‐blind, randomized, placebo‐controlled study to evaluate the safety and antiretroviral activity of MK‐0518 in combination with an optimized background therapy (OBT), versus optimized background therapy alone, in HIV‐infected patients with documented resistance to at least 1 drug in each of the 3 classes of licensed oral antiretroviral therapies’) | NCT00293267 |
| 17.2 | 48‐week safety and efficacy data from the ongoing phase III protocol 019 (‘A multicenter, double‐blind, randomized, placebo‐controlled study to evaluate the safety and antiretroviral activity of MK‐0518 in combination with an optimized background therapy (OBT), versus optimized background therapy alone, in HIV‐infected patients with documented resistance to at least 1 drug in each of the 3 classes of licensed oral antiretroviral therapies’) | NCT00293254 | |
| 17.3 | Specific plans for the monitoring of resistance, with frequent reporting intervals | Not included in follow‐up analysis | |
| 17.4 | Observational postauthorization safety study as specified in the RMP | Registration not found | |
| Vectibix (panitumumab | 18.1 | Study report of 20 050 181 study, including the safety–efficacy analysis in relation to KRAS | NCT00339183 |
| 18.2 | Study report of 20 050 203 study, including the safety–efficacy analysis in relation to KRAS | NCT00364013 | |
| 18.3 | Clinical study report of 20 030 167 study, including the safety–efficacy analysis in relation to KRAS | NCT00083616 | |
| 18.4 | Clinical study report of 20 030 250 study, including the safety–efficacy analysis in relation to KRAS | NCT00089635 | |
| 18.5 | Clinical study report of PACCE study, including the safety–efficacy analysis in relation to KRAS | NCT00115765 | |
| 18.6 | Clinical study report of SPIRITT study, including the safety–efficacy analysis in relation to KRAS | NCT00418938 | |
| 18.7 | Clinical study report of PRECEPT study, including the safety–efficacy analysis in relation to KRAS | NCT00411450 | |
| 18.8 | Clinical study report of STEPP study, including the safety–efficacy analysis in relation with KRAS | NCT00332163 | |
| Prezista (darunavir) | 19.1 | Interaction study TMC114‐C163 (‘A phase I, open‐label, randomized, crossover trial in healthy subjects to investigate the pharmacokinetic interaction between rifabutin and TMC114, coadministered with low‐dose ritonavir, at steady‐state’) | Not included in follow‐up analysis |
| 19.2 | Interaction study TMC114‐C123 (‘A phase I, open label, randomized, crossover trial in healthy subjects to investigate the pharmacokinetic interaction between didanosine and TMC114, coadministered with low‐dose ritonavir, at steady‐state’) should be submitted | Not included in follow‐up analysis | |
| 19.3 | Study report from study TMC114‐C214 (‘A randomized, controlled, open‐label trial to compare the efficacy, safety and tolerability of TMC114/RTV versus LPV/RTV in treatment‐experienced HIV‐1 infected subjects’) | NCT00110877 | |
| 19.4 | Study report from study TMC114‐C202 (‘A phase II randomized, controlled, partially blinded trial to investigate dose response of TMC114/RTV in 3‐class‐experienced HIV‐1 infected subjects, followed by an open‐label period on the recommended dose of TMC114/RTV’) | NCT00071097 | |
| 19.5 | Study report from study TMC114‐C213 (‘A phase II randomized, controlled, partially blinded trial to investigate dose–response of TMC114/RTV in 3‐class‐experienced HIV‐1 infected subjects, followed by an open‐label period on the recommended dose of TMC114/RTV’) | NCT00650832 | |
| 19.6 | Study report from study TMC114‐C215 (‘An open label trial of TMC114/RTV in HIV‐1 infected, treatment experienced subjects’) | NCT00081588 | |
| 19.7 | Study report from study TMC114‐C208 (‘An open label trial of TMC114/RTV in HIV‐1 infected subjects who were randomized in the trials TMC114‐C201, TMC114‐C207 or in sponsor selected phase I trials’) | NCT02187107 | |
| 19.8 | Study report from study TMC114‐C209 (‘Open‐label safety study of TMC114 in combination with low dose RTV and other ARVs in highly experienced HIV‐1 infected patients with limited or no treatment options’) | NCT00115050 | |
| 19.9 | Study report from study TMC125‐C206 (‘A phase III randomized, double‐blinded, placebo‐controlled trial to investigate the efficacy, tolerability and safety of TMC125 as part of an ART including TMC114/RTV and an investigator‐selected OBR in HIV‐1 infected subjects with limited to no treatment options’) | NCT00254046 | |
| 19.10 | Study report from study TMC125‐C216 (‘A phase III randomized, double‐blinded, placebo‐controlled trial to investigate the efficacy, tolerability and safety of TMC125 as part of an ART including TMC114/RTV and an investigator‐selected OBR in HIV‐1 infected subjects with limited to no treatment options’) | NCT00255099 | |
| Diacomit (stiripentol) | 20.1 | Randomized controlled clinical trial with stiripentol in the add‐on therapy using maximally safe doses of clobazam + valproate | Registration not found |
| 20.2 | Bioavailability study in 24 subjects to determine the relative bioavailability of the stiripentol sachet vs. stiripentol capsule by 2007 (STP 166) | Not included in follow‐up analysis | |
| Sutent (sunitinib) | 21.1 | Results of an ongoing study in cytokine‐naïve patients with metastatic renal cell carcinoma | NCT00083889 |
b.i.d., twice daily; q.d., single daily dose
Hoekman, J. , Klamer, T. T. , Mantel‐Teeuwisse, A. K. , Leufkens, H. G. M. , and De Bruin, M. L. (2016) Characteristics and follow‐up of postmarketing studies of conditionally authorized medicines in the EU. Br J Clin Pharmacol, 82: 213–226. doi: 10.1111/bcp.12940.
References
- 1. Duijnhoven RG, Straus SMJM, Raine JM, de Boer A, Hoes AW, De Bruin ML. Number of patients studied prior to approval of new medicines: a database analysis. PLoS Med 2013; 10: e1001407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Giezen TJ, Mantel‐Teeuwisse AK, Straus SMJM, Egberts TCG, Blackburn S, Persson I, et al Evaluation of post‐authorization safety studies in the first cohort of EU risk management plans at time of regulatory approval. Drug Saf 2009; 32: 1175–87. [DOI] [PubMed] [Google Scholar]
- 3. Eichler H‐G, Abadie E, Breckenridge A, Flamion B, Gustafsson LL, Leufkens H, et al Bridging the efficacy–effectiveness gap: a regulator's perspective on addressing variability of drug response. Nat Rev Drug Discov 2011; 10: 495–506. [DOI] [PubMed] [Google Scholar]
- 4. Breckenridge A, Feldschreiber P, Gregor S, Raine J, Mulcahy L‐A. Evolution of regulatory frameworks. Nat Rev Drug Discov 2011; 10: 3–4. [DOI] [PubMed] [Google Scholar]
- 5. Breckenridge A, Mello M, Psaty BM. New horizons in pharmaceutical regulation. Nat Rev Drug Discov 2012; 11: 501–2. [DOI] [PubMed] [Google Scholar]
- 6. European Commission . Commission Regulation (EC) No 507/2006 of 29 March 2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of Regulation (EC) No 726/2004 of the European Parliament and of the Council. Official J Eur Union 2006; L/92: 6–9 [Online]. Available at http://ec.europa.eu/health/files/eudralex/vol‐1/reg_2006_507/reg_2006_507_en.pdf (last accessed 11 February 2016).
- 7. European Medicines Agency . European Medicines Agency post‐authorisation procedural advice for users of the centralised procedure. Doc. Ref. EMEA‐H‐19984/03 Rev. 58, 2016 [Online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500003981.pdf (last accessed 11 February 2016).
- 8. Wagstaff A. Fast and effective? How will EMEA use its new powers of conditional marketing authorisation? Canc World 2007; 1: 22–8. [Google Scholar]
- 9. European Commission . Commission Regulation (EC) No 658/2007. of 14 June 2007 concerning financial penalties for infringement of certain obligations in connection with marketing authorisations granted under Regulation (EC) No 726/2004 of the European Parliament and of the Council. Official J Eur Union 2007; L155/10 [Online]. Available at http://ec.europa.eu/health/files/eudralex/vol‐1/reg_2007_658/reg_2007_658_en.pdf (last accessed 11 February 2006).
- 10. Fain K, Daubresse M. The Food and Drug Administration Amendments Act and postmarketing commitments to the FDA. JAMA 2013; 310: 202–4. [DOI] [PubMed] [Google Scholar]
- 11. Willyard C. FDA's post‐approval studies continue to suffer delays and setbacks. Nat Med 2014; 20: 1224–5. [DOI] [PubMed] [Google Scholar]
- 12. Moore T, Furberg C. Development times, clinical testing, postmarket follow‐up, and safety risks for the new drugs approved by the us food and drug administration: the class of 2008. JAMA Intern Med 2014; 174: 90–5. [DOI] [PubMed] [Google Scholar]
- 13. Law MR. The characteristics and fulfillment of conditional prescription drug approvals in Canada. Health Policy 2014; 116: 154–61. [DOI] [PubMed] [Google Scholar]
- 14. Banzi R, Gerardi C, Bertele’ V, Garattini S. Approvals of drugs with uncertain benefit–risk profiles in Europe. Eur J Intern Med 2015; 26: 572–84. [DOI] [PubMed] [Google Scholar]
- 15. Blake KV, Prilla S, Accadebled S, Guimier M, Biscaro M, Persson I, et al European Medicines Agency review of post‐authorisation studies with implications for the European Network of Centres for Pharmacoepidemiology and Pharmacovigilance. Pharmacoepidemiol Drug Saf 2011; 20: 1021–9. [DOI] [PubMed] [Google Scholar]
- 16. Eichler H‐G, Baird LG, Barker R, Bloechl‐Daum B, Børlum‐Kristensen F, Brown J, et al From adaptive licensing to adaptive pathways: delivering a flexible life‐span approach to bring new drugs to patients. Clin Pharmacol Ther 2014; 97: 234–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoekman J, Boon WPC, Bouvy JC, Ebbers HC, de Jong JP, De Bruin ML. Use of the conditional marketing authorisation pathway for oncology medicines in Europe. Clin Pharmacol Ther 2015; 98: 534–41. [DOI] [PubMed] [Google Scholar]
- 18. European Medicines Agency . Guideline on the scientific application and the practical arrangements necessary to implement Commission Regulation (EC) No. 507/2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of regulation (EC) No 726/2004. Doc. Ref. EMEA/509951/2006, 2006 [Online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/10/WC500004908.pdf (last accessed 20 February 2015).
- 19. Eichler H‐G, Pignatti F, Leufkens H, Breckenridge A. Balancing early market access to new drugs with the need for benefit/risk data: a mounting dilemma. Nat Rev Drug Discov 2008; 7: 818–26. [DOI] [PubMed] [Google Scholar]
- 20. Porter J, Desmond M, O’ Donnell M, McNamara P, Lehnert M, Wang B, et al Challenges of a post‐authorisation safety study (PASS) in an orphan oncology indication. Pharmacoepidemiol Drug Saf 2014; 23: 472. [Google Scholar]
- 21. Vermeer NS, Duijnhoven RG, Straus SMJM, Mantel‐Teeuwisse AK, Arlett PR, Egberts ACG, et al Risk management plans as a tool for proactive pharmacovigilance: a cohort study of newly approved drugs in Europe. Clin Pharmacol Ther 2014; 96: 723–31. [DOI] [PubMed] [Google Scholar]