

Abstract
Aims
Free fatty acids (FFA) can act as direct signalling molecules through activation of several membrane‐bound G‐protein coupled receptors. The FFA2 receptor (known as GPR43) is activated by short chain fatty acids (SCFA) such as acetate and has been shown to play a major role in SCFA‐induced neutrophil activation and migration and to contribute in the development and control of inflammation. GLPG0974 is a potent and selective antagonist of the human FFA2. The main objectives of the two phase 1 trials were to characterize the safety, tolerability, pharmacokinetics and pharmacodynamics of GLPG0974.
Methods
Two consecutive randomized, double‐blind, placebo‐controlled, single centre trials in healthy subjects were performed. In the first, GLPG0974 was administered as single doses up to 250 mg and in the second, multiple daily doses up to 400 mg for 14 days were evaluated. Non‐compartmental analysis was used to determine GLPG0974 pharmacokinetics while target engagement was investigated through the inhibition of neutrophils in acetate‐simulated whole blood samples using surface expression of CD11b activated epitope as a marker of neutrophil activation.
Results
The investigation of safety/tolerability and pharmacokinetics in the early development phase showed that GLPG0974 was safe and well tolerated up to a daily dose of 400 mg. GLPG0974 showed good and dose proportional exposure up to 400 mg daily as well as a substantial and sustained inhibition of acetate‐stimulated neutrophil activation.
Conclusion
Based on these results, a proof‐of‐concept study was initiated to evaluate the safety, tolerability and efficacy of GLPG0974 in patients with mild to moderate ulcerative colitis.
Keywords: FFA2, GPR43, GLPG0974, healthy subjects, safety, PK, PD
What is Already Known about this Subject
This is the first report of a clinical trial with a potent and selective antagonist of the human FFA2 receptor.
What this Study Adds
GLPG0974 is the first human FFA2 antagonist to be clinically evaluated. The study presents the molecule's safety, including the absence of metabolic effects on glucose and insulin, and demonstrates good oral pharmacokinetics.
The study presents the practical implementation of a neutrophil activation assay to demonstrate pharmacodynamics through target engagement ex vivo.
The study establishes a dose range from minimal effect to maximal achievable pharmacodynamic response in the early clinical development phase.
Introduction
Free fatty acids, in particular short chain fatty acids (SCFAs) such acetate, propionate and butyrate, are well known to be a direct source of energy. In addition they have been recognized to play an important role in the regulation of metabolic and inflammatory processes. In the human body, SCFAs are mainly produced by anaerobic bacterial fermentation of non‐digestible carbohydrates in the gut. The principal SCFA in human blood is acetate, since butyrate is mainly utilized by enterocytes as metabolic fuel and propionate is mostly converted to glucose in the liver 1, 2. Various studies have shown that treatment with SCFAs of polymorphonuclear cells 3, leukocytes 4 or neutrophils 5 induces a rise in intracellular Ca2 + and subsequent chemotaxis. Thus, immune cells are activated and migrate towards areas with elevated concentrations of SCFAs. This mechanism is likely to offer an evolutionary advantage. Cells such as neutrophils that present the first line of immunological defence are attracted towards areas of bacterial activity.
In recent years, receptors have been identified for which free fatty acids act as ligands. Several G protein‐coupled receptors (GPCRs) including FFA1, FFA2, FFA3, GRP84, GRP119 and GRP120 are activated by free fatty acids of different carbon chain length (short, mid or long chain). The free fatty acid receptor 2 (FFA2), also known as G‐protein‐coupled receptor 43 (GPR43), is activated by SCFAs 3, 6. FFA2 expression has been shown in immune cells like polymorphonuclear leukocytes, neutrophils and eosinophils, peripheral blood mononuclear cells, monocytes and B lymphocytes 3, 4, 7. FFA2 is also expressed on enterocytes and some enteroendocrine cells 8, 9 as well as in adipocytes 10. In human neutrophils in vitro, FFA2 mRNA expression is down regulated by SCFAs, but strongly up‐regulated under conditions of experimental inflammation by bacterial endotoxin lipopolysaccharide 11. SCFAs have been shown to induce neutrophil migration both in vitro 1, 2, 3 and in vivo 5. Intravital microscopy of rat mesenteric venules revealed that SCFAs stimulate neutrophil rolling and adherence to endothelium, which are the key steps in neutrophil transmigration from the blood stream into tissues 5. Neutrophils isolated from FFA2 knock‐out (KO) mice lose the ability to migrate in response to SCFA, indicating a key function for FFA2 in SCFA‐induced neutrophil chemotaxis and tissue accumulation12
Neutrophils play a key role in a number of chronic inflammatory diseases such as inflammatory bowel disease (IBD), chronic obstructive pulmonary disease, cystic fibrosis, periodontitis, vasculitis and others 14. Mechanisms that inhibit neutrophil chemotaxis may attenuate neutrophil‐driven inflammation and resulting tissue destruction. GLPG0974 is a potent and selective FFA2 antagonist that inhibits SCFA‐induced neutrophil activation and migration 15. GLPG0974 is only active against human and monkey FFA2, but not rodent FFA2 (data not shown). For this reason, GLPG0974 could not be evaluated in the classical rodent in vivo models of inflammation. Appropriate primate models are not readily available, so evaluation of this compound in animal models was not possible. GLPG0974 may offer an innovative treatment opportunity for a variety of neutrophil‐dependent pathologies, especially those where SCFA‐induced chemotaxis and activation may play a significant role such as in IBD 16.
Studies in FFA2 KO mice suggest an important contribution of FFA2 in the development and control of inflammation in the gut 13, 17, with conflicting results. While Maslowski et al. 13 and Masui et al. 18 reported that FFA2 KO mice exhibited an exacerbated inflammatory response in models of colitis, arthritis and asthma, Sina et al. 14 and Kim et al. 19 found FFA2 KO mice were protected against chronic dextran sodium sulfate (DSS)‐induced colitis. It remains unclear whether FFA2 deficiency attenuates or exacerbates the inflammatory response. Controversial data between studies may be explained by different genetic backgrounds across FFA2 KO mice studies 20 and microbiota differences between mice strains.
As FFA2 is also expressed in adipocytes, enterocytes and entero‐endocrine cells, a role in glucose homeostasis, obesity, dyslipidaemia and metabolic processes has been suggested. However, again contradictory results in FFA2 KO mice are described. Some studies have indicated that GPR43 promotes leptin secretion, adipogenesis and inhibition of lipolysis in adipose tissue and adipocytes, thereby regulating energy metabolism 21, 22. Furthermore, in FFA2 KO mice, impaired glucose tolerance with reduced insulin and glucagon‐like peptide (GLP)‐1 levels, as well as decreased insulin sensitivity and obesity are described 23, 24. On the other hand, FFA2 deficiency protected against high fat diet (HFD)‐induced obesity and dyslipidaemia and improved glucose tolerance 25.
Here the first clinical data of GLPG0974 are presented. The main objectives of these first phase I clinical trials were to characterize the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of GLPG0974 in healthy volunteers. As a PD biomarker demonstrating target engagement, the inhibition of activated CD11b (CD11b Active epitope) expression on neutrophils following ex vivo blood stimulation with sodium acetate was used.
Methods
Studies were conducted in accordance with accepted standards for the protection of subject safety and welfare and the principles of the Declaration of Helsinski and its amendments, and were in compliance with Good Clinical Practice. Protocols and informed consents were approved by the Ziekenhuis Netwerk Antwerpen (ZNA) Institutional Review Board (Belgium). All healthy subjects gave written informed consent prior to study initiation.
Study designs
Studies were phase 1, randomized, double‐blind, placebo‐controlled, single centre studies evaluating the safety, tolerability, PK and PD of single and multiple ascending oral doses of GLPG0974 in healthy subjects (NCT01496937 and NCT01721980). Eligible subjects (aged 18–50 years, body mass index [BMI] 18–30 kg m−2) were in good health with no clinically significant deviation from normal ranges in terms of medical history, physical examinations, ECGs or clinical laboratory determinations. Subjects were excluded from the study if they had a medical history of abnormal platelet function or a history of a current immunosuppressive condition.
Thirty‐two subjects in four panels of eight subjects received single ascending doses of 10, 30, 90 and 250 mg or placebo, in such a way that in each panel, six subjects received GLPG0974 and two placebo. In a subsequent study, four cohorts of eight subjects (six on active and two on placebo) received once daily (50 mg once daily, 100 mg once daily, 200 mg once daily) and twice daily doses (200 mg twice daily) of GLPG0974 or placebo for 14 days. Dose selection for the single ascending doses was based on preclinical observations and dose predictions from animal studies, whereas the multiple ascending doses were chosen based on the preclinical data and PK and PD results from the single dose study. Single doses were administered as an oral solution (10 and 50 mg ml−1 L− ‐arginine‐buffered aqueous oral solution and matching placebo) while for repeated doses GLPG0974 and matching placebo were formulated using standard excipients (such as microcrystalline cellulose, croscarmellose sodium, colloidal anhydrous silica and magnesium stearate) in hard gelatin capsules. A volume of 240 ml water was given to each subject immediately at the time of dosing. GLPG0974 is poorly soluble at acidic pH such as human gastric pH (1–3). As food induces a rise in gastric pH, dosing with food would increase the solubility of GLPG0974 and hence its absorption and bioavailability. Thus, treatments were administered after a standard breakfast (and evening meal for b.i.d regimen). Drinks were standardized to at least 1000 ml of water day.
Blood samples for PK were collected at regular intervals over 48 h (single escalating doses) or over 13 days post‐dose (multiple ascending doses). Additionally morning predose samples to determine steady‐state concentrations for GLPG0974 were taken on days 2, 3, 4, 6, 8, and 14 (24 h after dose on day 13 or 12 h in case of twice daily dosing). Blood was collected in tubes containing lithium heparin as an anticoagulant in order to obtain plasma for the analysis of concentrations of GLPG0974. Within 30 min after blood collection the plasma was separated in a refrigerated centrifuge (4–8 °C) for 10 min at circa 1500 g and transferred into two polypropylene tubes with at least 400 μl of plasma per tube. Plasma samples were stored at −20 °C until analysis. Three urine fractions were collected over a 24 h period during the multiple ascending doses to determine the amount of GLPG0974 excreted in urine. After homogenization and recording of the total weight, two 10 ml samples were stored at −20 °C until analysis.
Blood samples for PD analysis were collected at predose and at 1 (or 2), 4 and 8 h post‐dose after single doses and at predose and at 2, 4, 8, 12 and 24 h on day 1 and day 13 during the multiple ascending doses. Blood was obtained by venepuncture in tubes containing sodium citrate as anticoagulant and used subsequently for CD11b activated epitope measurement in neutrophils.
Safety assessments
General safety was evaluated by the incidence of adverse events (AEs) through non‐leading questioning, vital signs, 12‐lead electrocardiograms (ECG), and physical examinations, clinical laboratory parameters (haematology, biochemistry and urinalysis), as well as glucose and insulin monitoring after an oral glucose tolerance test (OGTT) performed prior to and after 14 days dosing with GLPG0974. Exposures under the glucose or insulin plasma concentration–time curve (AUC) were calculated on both occasions.
Bioanalytical and PK methods
Plasma concentrations of GLPG0974 were determined using a validated liquid‐chromatography‐mass spectrometry/mass spectrometry (LC–MS/MS) assay. In brief, the internal standard (deuterated GLPG0974) was added to plasma samples and then processed by liquid/liquid extraction. The evaporated and reconstituted samples were injected into a SCIEX API4000 LC–MS/MS equipped with a XBridge™ C18 chromatography column. GLPG0974 was detected with multiple reaction monitoring. Quantification was performed using peak area ratios and standard curves (with 1/x2 linear regression) prepared from calibration standards. The lower limit of quantification was 1.00 ng ml−1. The overall precision and repeatability precision for quality controls expressed as coefficient of variation (CV%) were not greater than 2.8 and 2.3%, respectively, with deviation from nominal concentrations of no more than −1.9%. At this early development stage, an adapted method from the plasma bioanalytical method was used to determine the concentrations of GLPG0974 in urine.
The plasma GLPG0974 concentrations were analyzed by non‐compartmental methods. The peak plasma concentrations (C max) and time to reach the C max (t max) were directly observed from the data. The terminal elimination rate constant (λz) was determined by log‐linear regression analysis of the elimination phase. The apparent terminal half‐life calculated from t 1/2,λz = ln2/λz was reported only if more than three data points were used for linear regression to determine λz with an adjusted r 2 value ≥0.900. The area under the curve (AUC) was calculated by the linear‐trapezoidal rule over the dosing interval (AUC(0,τ)) or up to the last quantifiable concentration (AUC(0,t)) using standard non‐compartmental methods (WinNonLin®, version 5.3; Pharsight corporation, Mountain View, CA, USA). Exposures extrapolated to infinity (AUC0‐∞) were calculated from AUC(0,t) + (C t/λz), where C t is the last observed quantifiable concentration. Urine parameters were determined after multiple dosing. The amount of GLPG0974 excreted over the dosing interval period (i.e. 12 or 24 h) expressed in percentage of the dose (Ae%) and the renal clearance (CLR) calculated as Ae(0,τ)/AUC(0,τ), with Ae(0,τ) and AUC(0,τ) corresponding to the amount excreted in urine and the plasma exposure over the dosing interval, respectively.
PD method: measurement of neutrophil CD11b activated epitope
Measurements of the CD11b activated epitope expression on human neutrophils in whole blood were performed as described in Polancec et al. 26. Fresh blood was diluted twice in RPMI medium containing TNFα (2 ng ml−1) and cytochalasin B (20 μg ml−1) and incubated for 15 min at 37 °C for neutrophil priming. After stimulation of diluted blood for 2 h at 37 °C with 10 mm acetate, cells were pelleted with washing/staining buffer (W/S buffer: phosphate‐buffered saline, 2% heat inactivated fetal bovine serum, 0.02% EDTA) and blocked with normal mouse IgG (Invitrogen/molecular probe) for 10 min at 4 °C. Then, cells were labelled for 30 min at 4 °C with anti‐CD45, anti‐CD16 and anti‐CD11b activated epitope antibodies (eBioscience). Red blood cell lysis and white blood cell fixation were performed by incubating the tubes for 10 min at 37 °C in 10 volumes of 1X FACS™ lysing solution (BD Bioscience). After two washing steps, cells were re‐suspended in 400 μl of W/S buffer and the CD11b [AE] marker was quantified on CD45+/CD16high+ neutrophils by flow cytometry (FACSCanto, BD Bioscience). Data are calculated as the percentage of inhibition of CD11b activated epitope expression on neutrophils with regard to data obtained predose on day 1. Maximum percentage of inhibition observed (Emax) on day 1 and day 13, were determined from individual effect–time profiles.
The same assay was used for determination of GLPG0974 in vitro activity on day −1. In this setting, GLPG0974 was added to blood samples in a concentration ranging from 0 to 3 μm, and processed as described above.
Statistical analyses
Clinical safety was addressed by assessing AEs, physical examinations, laboratory assessments (including glucose and insulin monitoring after OGTT), ECG and vital sign results in a descriptive manner. Descriptive statistics and shift tables (according to normal ranges) were calculated for each parameter at every time point and in each treatment group. A treatment‐emergent AE (TEAE) analysis was performed. The change in exposure (AUC) from day −1 at day 14 was compared between the treatment groups (placebo subjects pooled) using an ancova model with treatment and day −1 AUC as fixed effects.
For the PK analyses, the descriptive statistics analysis included the arithmetic means and CVs for C max, AUC, t 1/2,λz, Ae% and CLR and the medians and range for t max.
At this early stage of development, the study was not powered to assess dose proportionality using a 90% CI approach, but rather to get a first estimate of human PK characteristics. Consequently, dose proportionality over the entire dosage range was tested on ln‐transformed GLPG0974 (single dose: C max/dose, AUC(0,24 h)/dose, AUC(0,∞)/dose and t 1/2,λz; repeated dose: Cmax/dose, AUC(0,τ)/dose, Ae(0,τ)/dose, t 1/2,λz and CLR) by means of a mixed‐effect anova. In case of a significant dose effect observed on these parameters, comparison between doses was performed using Tukey's test. For the 200 mg twice daily dose, values of AUC(0,τ) and Ae(0,τ) were multiplied by two to get an estimate of the exposure over the same period as the once daily dosing regimen (i.e. 24 h) and dose‐normalized to the total daily dose (i.e. 400 mg). Wilcoxon–Mann–Whitney non‐parametric test was used to assess the dose proportionality of t max.
Time to reach steady‐state after repeated dosing was assessed by visual inspection of the C trough plasma concentrations of GLPG0974 determined on days 2, 3, 4, 6, 8, 13 and 14 (24 h after dose of day 13 or 12 h in case of twice daily dosing) for each dose group as well by means of a mixed‐effect anova on natural ln‐transformed GLPG0974 trough plasma concentrations.
For PD, mean percentage of inhibition (±SE) vs. time plots were presented after single and repeated administrations at each dose level. The relation between the percentage of inhibition and the plasma concentrations of GLPG0974 in the multiple dose study was explored with an Emax model. After visual inspection of individual plots no real hysteresis was observed and the following Emax model was used:
with Emax and ED 50 corresponding to the maximum inhibition and 50% inhibition, respectively.
Statistical inferential analyses were conducted using SAS® version 9.1 (SAS institute Inc. Cary, NC, USA) at the 0.05 level of significance.
Results
Safety/tolerability
No subjects reported a treatment‐emergent AE that led to treatment discontinuation. All TEAEs were mild in severity and transient in nature. No relevant differences in incidence or severity of AEs were observed between the treatment groups (see Tables 1, 2).
Table 1.
Treatment‐emergent adverse events (TEAEs) reported by more than one subject at any single dose
| System organ class adverse event | Pooled placebo n = 8 | GLPG0974 | |||
|---|---|---|---|---|---|
| 10 mg n = 6 | 30 mg n = 6 | 90 mg n = 6 | 250 mg n = 6 | ||
| Any AE | 1 | 1 | 1 | 2 | 0 |
| Gastrointestinal disorders | 0 | 0 | 0 | 1 | 0 |
| • Nausea | 0 | 0 | 0 | 1 | 0 |
| Infections and infestations | 1 | 0 | 0 | 0 | 0 |
| Urethritis chlamydial | 1 | 0 | 0 | 0 | 0 |
| Renal and urinary disorders | 0 | 0 | 0 | 1 | 0 |
| • Polyuria | 0 | 0 | 0 | 1 | 0 |
| Respiratory, thoracic and mediastinal disorders | 0 | 1 | 1 | 1 | 0 |
| • Cough | 0 | 0 | 0 | 1 | 0 |
| • Oropharyngeal pain | 0 | 1 | 1 | 0 | 0 |
n = number of subjects with that observation.
Table 2.
Treatment‐emergent adverse events (TEAEs) reported by more than one subject in any of the repeated dose administration periods
| System organ class adverse event | Pooled placebo n = 8 | GLPG0974 | |||
|---|---|---|---|---|---|
| 50 mg once daily n = 6 | 100 mg once daily n = 6 | 200 mg once daily n = 6 | 200 mg twice daily n = 6 | ||
| Any AE | 4 | 3 | 3 | 4 | 2 |
| Infections and infestations | 2 | 2 | 1 | 3 | 0 |
| • Nasopharyngitis | 2 | 2 | 1 | 2 | 0 |
| • Oral herpes | 0 | 0 | 0 | 1 | 0 |
| Gastrointestinal disorders | 1 | 1 | 2 | 2 | 0 |
| • Diarrhea | 1 | 1 | 1 | 1 | 0 |
| • Abdominal discomfort | 1 | 0 | 0 | 0 | 0 |
| • Abdominal pain | 0 | 0 | 1 | 0 | 0 |
| • Dry mouth | 0 | 0 | 1 | 0 | 0 |
| • Gastro‐oesophageal reflux disease | 0 | 0 | 0 | 1 | 0 |
| • Nausea | 1 | 0 | 0 | 0 | 0 |
| Nervous system disorders | 1 | 0 | 1 | 0 | 2 |
| • Headache | 1 | 0 | 1 | 0 | 0 |
| • Presyncope | 0 | 0 | 0 | 0 | 2 |
n = number of subjects with this observation.
No clinically relevant changes in mean or median laboratory values were observed over time. One subject in the 200 mg twice daily group had an AE related to laboratory abnormalities. This subject had abnormal liver function test values (ALT and AST near to 2‐fold the upper limit of normal) by the end of the treatment period, which were reported as an AE considered possibly related to the study drug by the investigator. The liver function test returned to normal within 14 days after discontinuation of GLPG0974 intake.
Over all groups, no noteworthy effects were seen on vital signs, on ECGs or on physical examinations.
There were no statistically significant differences between the GLPG0974 treatment groups and the pooled placebo group in the glucose and insulin AUC values on day 14 relative to day −1. There were also no statistically significant differences in the glucose and insulin AUC values between the different GLPG0974 treatment groups.
Pharmacokinetics
Mean plasma concentration–time profiles of GLPG0974 are depicted in Figure 1 and PK parameters are summarized in Tables 3, 4.
Figure 1.
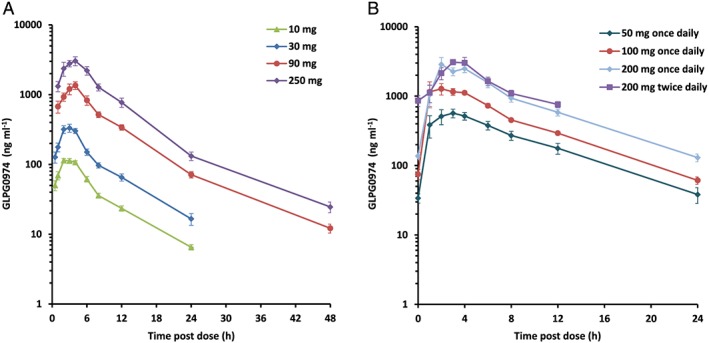
Mean (±SE) plasma concentrations of GLPG0974 after the single (A) and repeated (B) administration of GLPG0974 given as oral solution (A) or capsules (B) to fed healthy male subjects (n = 6 per dose group)
Table 3.
Pharmacokinetic parameters of GLP0974 after a single oral GLPG0974 dose as an oral solution to fed healthy subjects (n = 6 per dose group)
| Dose (mg) | C max (μg ml −1 ) | t max (h) | AUC(0,24 h) (μg ml −1 h) | AUC(0,∞) (μg ml −1 h) | t 1/2,λz (h) |
|---|---|---|---|---|---|
| 10 | 0.122 (13.5) | 3 (2–4) | 0.921 (12.2) | 0.984 (11.5) | 6.62 (17.2) |
| 30 | 0.362 (29.3) | 2.5 (2–4) | 2.51 (18.7) | 2.67 (20.7) | 6.03 (14.1) |
| 90 | 1.45 (34.8) | 4 (3–4) | 11.2 (21.3) | 12.3 (20.2) | 7.37 (9.20) |
| 250 | 3.56 (32.3) | 4 (2–6) | 26.3 (17.5) | 28.4 (18.2) n=5 | 7.28 (11.9) n=5 |
| anova * (P value) Tukey's test | P = 0.0090 | P = 0.0047 | P = 0.0451 | ||
| P = 0.4480 | P = 0.1459 | 30 10 250 90 | 30 10 250 90 | 30 10 250 90 | |
| ————– | ————– |
Estimates are expressed as arithmetic means (CV%) except median (range) for t max.
AUC(0,24 h), AUC up to 24 h; AUC(0,∞), AUC extrapolated up to infinity; C max, maximal plasma concentration; t max, time to reach C max; t 1/2,λz, apparent terminal half‐life.
Dose effect: anova performed on dose normalized parameters, except for t max and t 1/2,λz ‐ Tukey's test (pair comparison): means are sorted in ascending order, doses underlined with the same line are not statistically different.
Table 4.
Steady‐state pharmacokinetic parameters of GLPG0974 after once or twice daily oral doses as capsules to fed healthy subjects (n = 6 per dose group)
| Dose (mg) | Regimen | C max (μg ml −1 ) | t max (h) | AUC(0,τ) (μg ml −1 h) | t 1/2,λz (h) | Ae(0,τ) (%) | CL R (l h −1 ) |
|---|---|---|---|---|---|---|---|
| 50 | Once daily | 0.695 (17.2) | 2.5 (1–6) | 5.48 (31.4) | 5.46 (16.4) | 2.97 (36.9) | 0.289 (45.9) |
| 100 | 1.58 (48.4) | 2.5 (1–4) | 10.8 (21.4) | 5.42 (12.4) | 2.14 (35.4) | 0.209 (40.1) | |
| 200 | 3.34 (42.7) | 3.0 (2–4) | 21.4 (20.9) | 5.68 (13.1) | 1.91 (17.7) | 0.186 (29.3) | |
| 200 | Twice daily | 3.84 (27.3) | 3.0 (2–4) | 19.4 (20.6) | 5.39 (10.6)n=3 | 2.10 (35.8) | 0.217 (33.5) |
| anova * (P value) | P = 0.2767 | P = 0.6605 | P = 0.9088 | P = 0.5531 | P = 0.2239 | P = 0.1860 | |
Estimates are expressed as arithmetic means (CV%) except median (range) for t max.
AUC(0,τ) and Ae(0,τ), AUC and amount excreted in urine over the dosing interval i.e. 12 h (twice daily) or 24 h (once daily); CLR; renal clearance; C max, maximal concentration; t max, time to reach the C max; t 1/2,λz, apparent terminal half‐life.
Dose effect: anova performed on dose normalized parameters, except for t max, t 1/2,λz) and CLR.
After single and repeated oral administration of GLPG0974 to healthy fed male subjects, GLPG0974 was rapidly absorbed with a median t max of 2.5–3 h. Decrease in plasma concentrations of GLPG0974 displayed a multiphasic profile (see Figure 1A,B).
After single dosing, maximal plasma concentration (Cmax) increased dose proportionally over the entire dose range while a statistically significant dose effect was observed on the dose normalized exposure (AUC(0,∞)/dose). However, pairwise comparison performed by Tukey's test did not show ranking between doses. Therefore, it was concluded that AUCs increased dose proportionally within the 10 to 250 mg single dose range (Table 3).
After repeated dosing, steady‐state for GLPG0974 plasma concentrations were attained by day 2, regardless of dose and dose regimen (once daily or twice daily see Figure 2) without accumulation. At steady‐state, the plasma exposure (both C max and AUC over the 24 h time interval) increased dose proportionally over the 50 to 400 mg daily dose range (Table 4). The overall plasma terminal elimination half‐life was 5.3 h.
Figure 2.
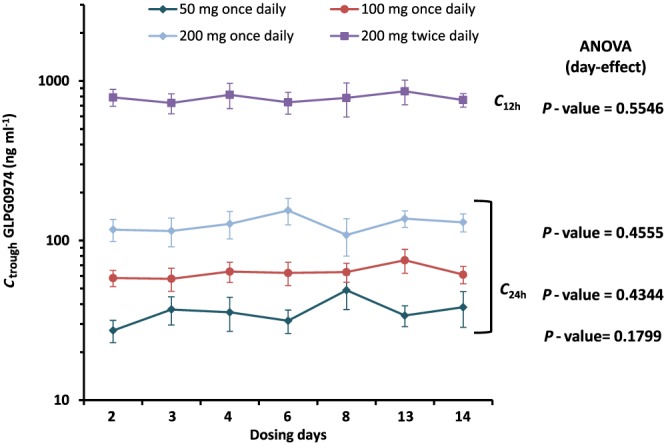
Mean (±SE) trough plasma concentrations of GLPG0974 after repeated dosing (C 24h after once daily and C 12h after twice daily dosing; n = 6 per dose group)
Low amounts of GLPG0974 were excreted unchanged in urine (less than 3.0%). To the extent there was excretion of unchanged GLPG0974, this was rapid with 88% of the amount excreted within the first 12 h (data not shown). The renal clearance of GLPG0974 was about 0.23 l h−1.
Overall, the between‐subject variability of AUC and C max at steady‐state was low to moderate (between‐subject CV% range 17–48%).
At the highest dose tested for the oral solution (250 mg) and the capsule (200 mg), the C max values were 3.56 and 3.34 μg ml−1 and AUC(0,24 h) values were 26.3 and 21.4 μg ml−1 h (Tables 3, 4). Although the doses of GLPG0974 administered were not exactly the same, the bioavailability could be considered as similar for GLPG0974 given as an oral solution or capsule.
Pharmacodynamics
In the single dose study, in vitro activity of GLPG0974 was evaluated in the blood of the subjects before GLPG0974 administration. The compound inhibited the CD11b activated epitope expression on neutrophils concentration‐dependently in all subjects, with an IC 50 of 483 ± 286 nm, corresponding to 234 ± 139 ng ml−1.
In the single dose study, target engagement was explored up to 8 h after administration of the 30, 90 and 250 mg doses, evaluating the inhibitory effect of GLPG0974 on the ex vivo sodium acetate‐induced CD11b activated epitope expression on neutrophils. GLPG0974 dose‐dependently inhibited the PD marker, from 1–2 h post‐dose up to 8 h post‐dosing with maximal percentages of inhibition of 61%, 73% or 89%, respectively, compared with baseline. A clear relationship between GLPG0974 exposure and PD effect was observed.
In the multiple ascending dose study, the PD effects were evaluated more extensively. Figure 3A shows that a single administration of GLPG0974 (day 1) dose‐dependently inhibited sodium acetate‐induced expression of CD11b activated epitope on neutrophils. The average inhibition reached its maximum 4 h post‐dose with corresponding values ranging between 68% and 78% over the 50–400 mg daily dose range. The PD effect was sustained for at least 12 h from the 100 mg dose onwards with inhibition ranging from 59% to 74%, while a decline of about half the maximum inhibition (31%) was observed at 50 mg once daily. Dose–response effect of GLPG0974 clearly appeared 24 h post‐dose, with a marked dose‐dependent decrease in the inhibition of C11b activated epitope expression. Twice daily administration of GLPG0974 resulted in the maintenance of maximum inhibition of CD11b activated epitope expression in circulating neutrophils over 24 h. Average maximal percentages of inhibition were 70%, 76%, 82% and 93% for 50, 100, 200 mg once daily and 200 mg twice daily respectively.
Figure 3.
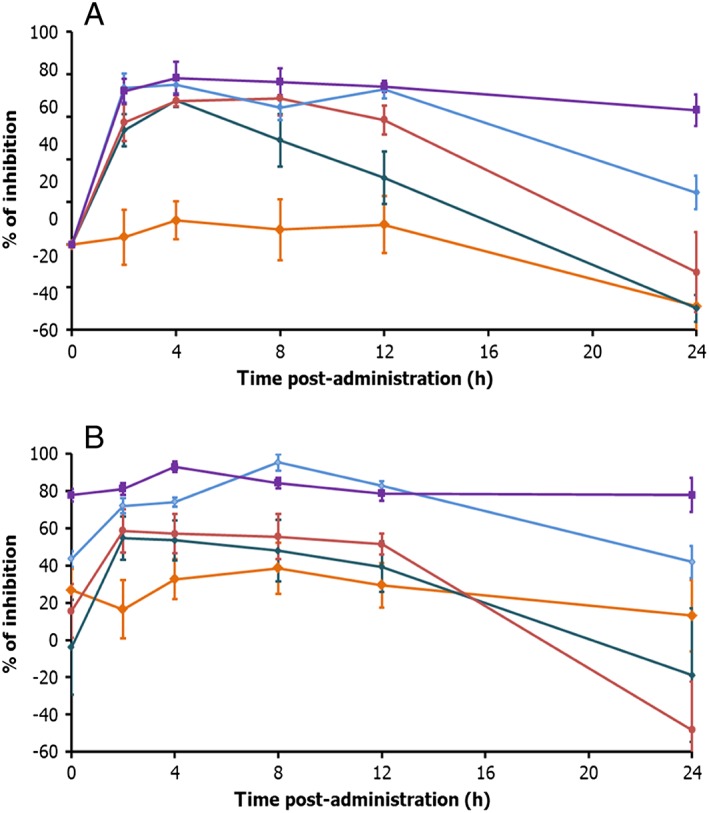
Mean (±SE) percentages of inhibition of sodium acetate‐induced CD11b activated epitope in the whole blood of healthy subjects administered with GLPG0974 at 50 mg once daily (green curve), 100 mg once daily (red curve), 200 mg once daily (blue curve), 200 mg twice daily (purple curve) or with placebo (orange curve) (n = 8 placebo; n = 6 per dose group) A: day 1 B: day 13
At steady‐state (day 13, Figure 3B), a similar PD profile was observed as on day 1, with all doses of GLPG0974 inhibiting the C11b activated epitope expression on neutrophils. The maximal inhibition was obtained with the highest dose (200 mg once daily) showing a sustained PD effect for 12 h. The same dose given twice daily (200 mg twice daily) resulted in sustained inhibition of C11b activated epitope expression for 24 h. At pre‐dose (day 13), the groups receiving 200 mg GLPG0974, either once daily or twice daily, displayed a similar percentage inhibition as 24 h after start of the administration (day 1), indicating the target engagement is maintained at the same level over the 13 days treatment period. Average maximal percentages of inhibition were 66%, 66%, 95% and 94% for 50, 100, 200 mg once daily and 200 mg twice daily, respectively.
Overall a robust and sustained PD effect was observed with 200 mg twice daily GLPG0974, allowing a maximal inhibition of the target over 24 h.
In Figure 4, plasma concentrations of GLPG0974 are plotted against the percentage of CD11b activated epitope inhibition. The PK/PD analysis indicated that 50% inhibition was obtained at a plasma concentration of 238 ng ml−1 (ED50), which was very similar to the IC50 observed in vitro (234 ng ml−1). Inhibition of CD11b activated epitope activation on neutrophils plateaued at a plasma concentration of 1100 ng ml−1, which is close to the plasma concentrations observed 12 h after administration of 200 mg. As such 200 mg twice daily warrants a maximal inhibition of neutrophil activation for 24 h.
Figure 4.
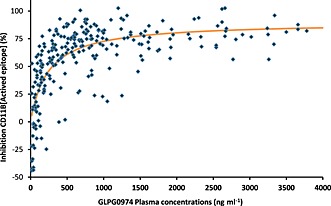
Correlation between the GLPG0974 plasma concentration and the percentage of CD11b activated epitope inhibition measured in whole blood of the same healthy subjects
Discussion
The safety, PK and PD of GLPG0974 were evaluated over a wide dose range (10 to 400 mg) and different dosing regimens (once daily and twice daily) in healthy male subjects.
In healthy male subjects, single doses up to 250 mg and multiple dosing up to 400 mg daily of GLPG0974 for 14 days were generally safe and well tolerated. Overall, no noteworthy effects were seen on laboratory parameters, on vital signs or on ECGs. At the end of the 14 day dosing period one subject in the 200 mg twice daily cohort showed increased liver function test results up to near two‐fold of the upper limit of normal, which returned to normal within a fortnight after the end of GLPG0974 administration.
In the literature there is some debate on the role of FFA2 in the regulation of the energy balance explored in vivo in FFA2 knock‐out mice 23, 24, 25. On the one hand, obesity, impaired systemic insulin sensitivity and glucose tolerance with reduced insulin and (GLP)‐1 levels were reported in FFA2−/− mice 24, 27. On the other hand, FFA2 deficiency improved insulin sensitivity and glucose tolerance and protected against HFD‐induced obesity and dyslipidaemia 25. As such an oral glucose tolerance test was performed to evaluate the potential effect of GLPG0974 on glucose homeostasis in man. Up to the highest dose tested, GLPG0974 had no effect on the glucose and insulin concentrations, suggesting that the compound has no negative impact on glucose homeostasis, which should be further corroborated in larger follow‐up trials.
After oral administration, GLPG0974 was rapidly absorbed with peak plasma concentrations reached between 2 and 4 h. The apparent elimination half‐life ranged between 5 and 7 h. Given this moderate elimination half‐life, a twice daily dosing regimen may be considered to ensure a sustained and maximal PD effect over a 24 h period. Approximately 3% of GLPG0974 was excreted unchanged in urine, suggesting metabolism to be the primary route of elimination of GLPG0974. At steady‐state, the exposure to GLPG0974 increased in proportion to the dose over the 50–400 mg daily dose range. Although the initial GLPG0974 oral solution did show sufficient bioavailability to trigger a PD effect, an oral solid dosage form (i.e. capsule) was envisaged and tested during the multiple ascending dose study. Identical doses were not tested with the two formulations (90 and 250 mg for the oral solution and 100 and 200 mg for the capsule). However given the absence of a difference in the PK of GLPG0974 at very close doses (90 and 100 mg or 200 and 250 mg), it could be concluded that the bioavailability of GLPG0974 given as a capsule or oral solution to fed healthy subjects was similar. These data suggest that the disposition of GLPG0974 administered in the fed state not to be limited by drug dissolution.
In human whole blood, GLPG0974 potently inhibits the acetate‐induced expression of activated CD11b (CD11b activated epitope) on neutrophils, in vitro as well as ex vivo. CD11b forms together with CD18 the membrane activated complex‐1 (Mac‐1), a cell surface receptor. Through interaction with ICAM‐1, MAC‐1 mediates firm adhesion and arrest of neutrophils on endothelium, an essential step in neutrophil extravasation, leading to neutrophil migration into tissues at foci of inflammation and infection. For interaction with ICAM‐1, a conformational change of CD11b (CD11b activation‐specific epitope) is needed. Different inflammatory stimuli upregulate CD11b activated epitope expression which is considered a more relevant marker of neutrophil activation in blood than the total CD11b level 27, 28. The current studies corroborate further the potential of CD11b activated epitope as a biomarker for neutrophil activation in a clinical setting. Ex vivo measurement of acetate‐induced CD11b activated epitope in whole blood clearly indicates target engagement by GLPG0974 after single as well as multiple oral doses. A rapid and substantial inhibition is observed with all doses, in line with the PK profile. A dose–response effect is observed, best exemplified in the maintenance of inhibition obtained over time (after t max). A dose of 200 mg twice daily results in a robust and maximal inhibition of the biomarker during 24 h. At steady‐state (day 13), inhibition levels at predose are in the same range as 24 h after the first dose (day 1) and the inhibition profiles over time are similar to the profiles at day 1 after t max, indicative for a sustained effect of GLPG0974 on the target receptor, without inducing tachyphylaxis. These data are further corroborated by the good PK/PD relationship observed between GLPG0974 plasma concentrations and inhibition of neutrophil biomarker expression. Inhibition of CD11b activated epitope activation on neutrophils plateaued at a plasma concentration of 1100 ng ml−1, further substantiating that at 200 mg twice daily GLPG0974 maximal target engagement is maintained for 24 h.
The data presented here show that GLPG0974 is safe and well‐tolerated up to a daily dose of 400 mg. Twice daily 200 mg dosing leads to sufficiently high GLPG0974 plasma concentrations to inhibit SCFA‐induced GPR43 activity on neutrophil activation for 24 h. One of the hallmarks of IBD is the invasion of activated neutrophils in the inflamed gut. Due to the high level of SCFA produced in the gut, it is tempting to consider GLPG0974 as a potential candidate molecule to inhibit neutrophil activation and subsequent recruitment in the gut, thus potentially impacting positively pathologies involving gut inflammation, such as IBD. Based on these results, a proof‐of‐concept study is being performed to evaluate the safety, tolerability and efficacy of 200 mg twice daily. GLPG0974 in patients with mild to moderate ulcerative colitis.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
The authors would like to acknowledge Dr M. Petkova and W. Haazen and their study teams from SGS Life Science Services Clinical Pharmacology Unit (Antwerpen, Belgium) for conducting these studies, Mr D. Polancec from Fidelta (Zagreb, Croatia) and D. Van Bockstaele from Laboratory Corporation of America Holdings/LabCorp Clinical Trials (Mechelen, Belgium), for the development and application of the flow cytometry method for the PD assay, Ms L. Allamassey from Galapagos NV for the PK/PD analysis and Mr M. Boterman and A. Muntendam from Analytical Biochemical Laboratory (Assen, The Netherlands) for plasma and urine samples analysis.
Namour, F. , Galien, R. , Van Kaem, T. , Van der Aa, A. , Vanhoutte, F. , Beetens, J. , and van't Klooster, G. (2016) Safety, pharmacokinetics and pharmacodynamics of GLPG0974, a potent and selective FFA2 antagonist, in healthy male subjects. Br J Clin Pharmacol, 82: 139–148. doi: 10.1111/bcp.12900.
References
- 1. Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987; 28: 1221–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pouteau E, Nguyen P, Ballèvre O, Krempf M. Production rates and metabolism of short chain fatty acids in the colon and whole body using stable isotopes. Proc Nutr Soc 2003; 62: 87–93. [DOI] [PubMed] [Google Scholar]
- 3. Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, Brezillon S, Dupriez V, Vassart G, Van Damme J, Parmentier M, Detheux M. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem 2003; 278: 25481–9. [DOI] [PubMed] [Google Scholar]
- 4. Cavaglieri CR, Nishiyama A, Fernandes LC, Curi R, Miles EA, Calder PC. Differential effects of short chain fatty acids on proliferation and production of pro‐ and anti‐inflammatory cytokines by cultured lymphocytes. Life Sci 2003; 73: 1683–960. [DOI] [PubMed] [Google Scholar]
- 5. Vinolo MAR, Rodrigues HG, Hatanaka E, Hebeda CB, Farsky SHP, Curi R. Short chain fatty acids stimulate the migration of neutrophils to inflammatory sites. Clin Sci 2009; 117: 331–8. [DOI] [PubMed] [Google Scholar]
- 6. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ, Pike NB, Strum JC, Steplewski KM, Murdock PR, Holder JC, Marshall FH, Szekeres PG, Wilson S, Ignar DM, Foord SM, Wise A, Dowell SJ. The orphan G protein‐coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 2003; 278: 11312–9. [DOI] [PubMed] [Google Scholar]
- 7. Nilsson NE, Kotarsky K, Owman C, Olde B. Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short‐chain fatty acids. Biochem Biophys Res Commun 2003; 303: 1047–52. [DOI] [PubMed] [Google Scholar]
- 8. Tazoe H, Otomo Y, Kaji I, Tanaka R, Karaki SI, Kuwahara A. Roles of short‐chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J Physiol Pharmacol 2008; 59: 251–62. [PubMed] [Google Scholar]
- 9. Karaki S, Tazoe H, Hayashi H, Kashiwabara H, Tooyama K, Suzuki Y, Kuwahara A. Expression of the short‐chain fatty acid receptor, GPR43, in the human colon. J Mol Histol 2008; 39: 135–42. [DOI] [PubMed] [Google Scholar]
- 10. Hong YH, Nishimura Y, Hishikawa D, Tsuzuki H, Miyahara H, Gotoh C, Choi KC, Feng DD, Chen C, Lee HG, Katoh K, Roh SG, Sasaki S. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 2005; 146: 5092–9. [DOI] [PubMed] [Google Scholar]
- 11. Aoyama M, Kotani J, Usami M. Butyrate and propionate induced activated or nonactivated neutrophil apoptosis via HDAC inhibitor activity but without activating GPR‐41/GPR‐43 pathways. Nutrition 2010; 26: 653–61. [DOI] [PubMed] [Google Scholar]
- 12. Vinolo MA, Ferguson GJ, Kulkarni S. SCFAs induce mouse neutrophil chemotaxis through the GPR43 receptor. PLoS One 2011; 6: e21205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM, Mackay CR. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009; 461: 1282–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Witko‐Sarsat V, Rieu P, Descamps‐Latscha B, Lesavre P, Halbwachs‐Mecarelli L. Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest 2000; 80: 617–53. [DOI] [PubMed] [Google Scholar]
- 15. Pizzonero M, Dupont S, Babel M, Beaumont S, Bienvenu N, Blanqué R, Cherel L, Christophe T, Crescenzi B, De Lemos E, Delerive P, Deprez P, De Vos S, Djata F, Fletcher S, Kopiejewski S, L'Ebraly C, Lefrançois JM, Lavazais S, Manioc M, Nelles L, Oste L, Polancec D, Quénéhen V, Soulas F, Triballeau N, van der Aar EM, Vandeghinste N, Wakselman E, Brys R, Saniere L. Discovery and optimization of an azetidine chemical series as a free fatty acid receptor 2 (FFA2) antagonist: from hit to clinic. J Med Chem 2014; 57: 10044–57. [DOI] [PubMed] [Google Scholar]
- 16. Niederman R, Zhang J, Kashket S. Short‐chain carboxylic‐acid‐stimulated, PMN‐mediated gingival inflammation. Crit Rev Oral Biol Med 1997; 8: 269–90. [DOI] [PubMed] [Google Scholar]
- 17. Sina C, Gavrilova O, Förster M, Till A, Derer S, Hildebrand F, Raabe B, Chalaris A, Scheller J, Rehmann A, Franke A, Ott S, Häsler R, Nikolaus S, Fölsch UR, Rose‐John S, Jiang HP, Li J, Schreiber S, Rosenstiel P. G protein‐coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J Immunol 2009; 183: 7514–22. [DOI] [PubMed] [Google Scholar]
- 18. Masui R, Sasaki M, Funaki Y, Ogasawara N, Mizuno M, Iida A, Izawa S, Kondo Y, Ito Y, Tamura Y, Yanamoto K, Noda H, Tanabe A, Okaniwa N, Yamaguchi Y, Iwamoto T, Kasugai K. G protein‐coupled receptor 43 moderates gut inflammation through cytokine regulation from mononuclear cells. Inflamm Bowel Dis 2013; 19: 2848–56. [DOI] [PubMed] [Google Scholar]
- 19. Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH. Short‐chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 2013; 145: 396–406. [DOI] [PubMed] [Google Scholar]
- 20. Blad CC, Tang C, Offermanns S. G protein‐coupled receptors for energy metabolites as new therapeutic targets. Nat Rev Drug Discov 2012; 11: 603–19. [DOI] [PubMed] [Google Scholar]
- 21. Ge H, Li X, Weiszmann J, Wang P, Baribault H, Chen JL, Tian H, Li Y. Activation of G protein‐coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology 2008; 149: 4519–26. [DOI] [PubMed] [Google Scholar]
- 22. Zaibi MS, Stocker CJ, O'Dowd J, Davies A, Bellahcene M, Cawthorne MA, Brown AJ, Smith DM, Arch JR. Roles of GPR41 and GPR43 in leptin secretory responses of murine adipocytes to short chain fatty acids. FEBS Lett 2010; 584: 2381–6. [DOI] [PubMed] [Google Scholar]
- 23. Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, Cameron J, Grosse J, Reimann F, Gribble FM. Short‐chain fatty acids stimulate glucagon‐like peptide‐1 secretion via the G‐protein‐coupled receptor FFAR2. Diabetes 2012; 61: 364–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, Terasawa K, Kashihara D, Hirano K, Tani T, Takahashi T, Miyauchi S, Shioi G, Inoue H, Tsujimoto G. The gut microbiota suppresses insulin‐mediated fat accumulation via the short chain fatty acid receptor GPR43. Nat Commun 2013; 4: 1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bjursell M, Admyre T, Göransson M, Marley AE, Smith DM, Oscarsson J, Bohlooly YM. Improved glucose control and reduced body fat mass in free fatty acid receptor 2‐deficient mice fed a high‐fat diet. Am J Physiol Endocrinol Metab 2011; 00: E211–20. [DOI] [PubMed] [Google Scholar]
- 26. Polancec D, Vrancic M, Dupont S, Oreskovic K, Erakovic Haber V. Flow cytometric measurement of the CD11b Activated Epitope [AE] expression on human neutrophils in whole blood as a tool in drug development. Cyto 2013. [Google Scholar]
- 27. Orr Y, Taylor JM, Cartland S, Bannon PG, Geczy C, Kritharides L. Conformational activation of C11b without shedding of L‐selectin on circulating human neutrophils. J Leukoc Biol 2007; 82: 1115–25. [DOI] [PubMed] [Google Scholar]
- 28. Zarbock A, Ley K. Neutrophil adhesion and activation under flow. Microcirculation 2009; 16: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]