

Abstract
AIMS
BG00010 is a protein in the glial cell line‐derived neurotrophic factor (GDNF) family. It is a selective ligand for the GDNF family receptor alpha‐3 (GFRα3) co‐receptor that normalizes cellular changes resulting from damage or disease, and potentially alleviates neuropathic pain. The main objectives of this study were to evaluate the pharmacokinetic and safety profiles and to determine the effects on pain of ascending doses of intravenous injections of BG00010 in patients with sciatica.
Methods
This was a randomized, blinded, placebo‐controlled multiple‐dose study in subjects with sciatica. In Part I (16 patients), four IV dose levels were examined (50, 150, 400, 800 μg kg−1) and in Part II (12 patients), three dose levels were examined (400, 600 and 1200 μg kg−1). Safety and efficacy assessments were used as endpoints.
Results
The BG00010 concentration–time data indicated relatively low inter‐patient variability and there was a dose‐dependent (not dose‐proportional) increase in serum exposure from 150 to 1200 μg kg−1. The effective half‐life was between 40 and 60 h. The most frequently occurring adverse events (AEs) reported by patients receiving BG00010 were headache (67–83%), feeling hot (50–100%), and pruritus (42–67%). Most AEs were mild; no serious AEs or AEs leading to discontinuation occurred. Higher dose regimens of BG00010 resulted in greater pain reduction than placebo or lower dose regimens, although a clear dose–response relationship was not seen.
Conclusions
The pharmacokinetic profile of BG00010 was characterized by low intra‐patient variability. These data from a small sample suggest that BG00010 may have a benefit for patients with sciatica.
Keywords: BG00010, neublastin, pharmacodynamics, pharmacokinetics, sciatica
What is Already Known about this Subject
Preclinical data from surgical and chemical nerve‐injury models suggest that BG00010 attenuates pain‐related behaviours and normalizes the neurochemical status of injured small dorsal root ganglion (DRG) neurons without loss of neuronal or axonal function or integrity.
Along with promoting re‐entry of sensory fibres into the spinal cord and re‐establishing synaptic function after crush injury, BG00010 can promote recovery of simple and complex behaviours in preclinical models.
What this Study Adds
Current treatment for sciatica and other neuropathic pain conditions are inadequate, and these data give further support to the concept that compounds targeting neurotrophic factor pathways such as the glial cell‐derived neurotrophic factor (GDNF) family of ligands may provide viable therapies.
These data indicate that initial safety, pharmacokinetic profile and analgesic effects of BG00010 are promising and warrant further evaluation in patients with neuropathic pain.
Introduction
BG00010 (neublastin, artemin) is a glial cell‐derived neurotrophic factor (GDNF) family member 1 that can act as a survival factor for sensory and sympathetic neurons. BG00010 is a selective ligand for the GDNF family receptor alpha‐3 (GFRα3) co‐receptor. The interaction of BG00010 with GFRα3 on nociceptive sensory neurons activates downstream signalling to normalize damage‐ or disease‐induced cellular changes and potentially alleviate neuropathic pain. GFRα3 expression is highly restricted to small dorsal root ganglion (DRG) sensory neurons, reducing the likelihood of unintended side effects 2.
Preclinical data from surgical and chemical nerve‐injury models demonstrate that BG00010 attenuates pain‐related behaviours and normalizes the neurochemical status of injured small DRG neurons without loss of neuronal or axonal function or integrity. Along with promoting re‐entry of sensory fibres into the spinal cord and re‐establishing synaptic function after crush injury, BG00010 can promote recovery of simple and complex behaviours in preclinical models 3, 4, 5. BG00010 is being developed as a first‐in‐class molecule for the treatment of neuropathic pain.
Neuropathic pain results from lesions or disease affecting the peripheral or central somatosensory nervous system. Neuropathic pain is especially problematic because of its severity, chronicity and resistance to simple analgesics. Affecting 2–3% of the population, neuropathic pain is costly to the healthcare system, personally devastating for patients 6, and can substantially impair health‐related quality of life 7.
Sciatica is caused by spinal nerve root compression, often due to lumbar disc prolapse, and is associated with back pain radiating to the leg, occasionally accompanied by neurological deficit 8. Sciatica is common, with reported lifetime incidence of 13–40% 9 and an annual incidence of 1–5%, peaking in the fifth decade of life 10. Most patients with acute sciatica respond to conservative symptom management, with symptom resolution over weeks to months, although some require surgical decompression of the affected nerve root. Nevertheless, 10–40% of patients will develop a chronic pain syndrome 11. Common pharmacotherapies for chronic neuropathic pain include tricyclic antidepressants, serotonin‐norepinephrine reuptake inhibitors, calcium channel α2‐δ ligands, topical lidocaine, opioid agonists and capsaicin. For sciatica, epidural steroid injections have been used for decades despite inconclusive efficacy data [9]. Treatment remains challenging, as many patients do not experience sufficient relief 12. A significant unmet medical need exists for a therapeutic agent with an acceptable safety profile to provide sustained neuropathic pain relief.
In a previous single‐centre study (NCT00961766), BG00010 was administered to 48 patients with sciatica as single intravenous (IV; 0.3–800 μg kg−1) or subcutaneous (50 μg kg−1) doses 13. The results suggested nearly linear pharmacokinetics (PK) over the tested dose range. The most frequently reported adverse events (AEs) were feeling hot, pruritus, headache and rash. In this second study of BG00010 in humans, the main objectives were to evaluate the PK, safety and pharmacodynamics of three IV injections of BG00010 given as two fixed dosing schedules.
Methods
Patients
Eligible patients were aged 18–85 years with a diagnosis of unilateral sciatica, including pain radiating down the leg following a dermatome, suggesting L4, L5 or S1 nerve root involvement, with symptoms present for ≥3 months prior to the screening visit, and pain rated at ≥40 mm on a 100 mm visual analogue scale (VAS) of the Dutch translation of the Short‐Form McGill Pain Questionnaire (SF‐MPQ) 14 at screening and baseline visits.
Key exclusion criteria included: history of severe pain or signs/symptoms of peripheral neuropathy (other than that caused by sciatica) during the 3 months prior to the screening visit; major surgery within the 3 months prior to the screening visit or planned sciatica surgery within 6 months of the screening visit; current generalized myalgia; history of severe allergic or anaphylactic drug‐related reaction; history of malignancy or clinically relevant allergy; and/or cardiac, endocrine, haematologic, hepatic, immunologic, metabolic, urologic, pulmonary, neurologic (not related to sciatica), dermatologic, rheumatic/joint, psychiatric, renal and/or other major disease.
Patients were allowed treatment with a selective serotonin reuptake inhibitor, a serotonin noradrenaline reuptake inhibitor, gabapentin, or a tricyclic antidepressant if doses were stable for 4 weeks prior to the baseline visit, and pregabalin if the dose was stable for 1 week prior to the baseline visit. Doses of other prescription medications and/or over‐the‐counter products were to have been stable for 2 weeks prior to the baseline visit. Previous participation in a study with neurotrophic factors and participation in a study with another investigational drug or approved therapy for investigational use within 3 months prior to the baseline visit was not allowed.
Study design and treatment
This was a single‐centre, randomized, blinded, placebo‐controlled, serial‐cohort, multiple‐dose ascending study that examined two dose schedules (ClinicalTrials.gov NCT01405833). The study was approved by the Medical Ethics Committee (BEBO Foundation, Assen, The Netherlands) and all patients gave written informed consent. ‘BG00010’ is specific for the isoform of the protein used in this study (50:50 mix of 103 and 104 amino acid isoforms). The generic names ‘artemin’ and ‘neublastin’ cover all forms of the protein (104, 113, 125 and full length) and do not accurately describe BG00010. All other drug/target nomenclature is consistent with the British Journal of Pharmacology's ‘Guide to Receptors and Channels’ 15. Since BG000010 is intravenously administered, less frequent dosing is preferable, and unpublished non‐clinical data suggested that less frequent administration with higher doses and more frequent administration at lower doses could be explored clinically, providing the rationale for studying two dosing schedules. In Part I (Cohorts A–D), patients received BG00010 (50, 150, 400 or 800 μg kg−1) or placebo once weekly for 3 weeks. In Part II (Cohorts E–G), patients received BG00010 or placebo dosed every 48 h. The starting dose in Part II was to be no more than 400 μg kg−1 and at least one dose level below the maximum tolerated dose (MTD) established with the once‐weekly schedule.
Patients were enrolled sequentially, cohort by cohort. For each cohort, patients were randomized (three patients to BG00010 and one to placebo) to receive three IV administrations of study treatment. Following the completion of treatment for each cohort, the Data Safety Review Committee (DSRC) determined if it was appropriate to escalate to the next planned dose level. If none of the three BG00010‐treated patients within a cohort experienced a treatment‐related dose‐limiting toxicity (DLT; defined as all BG00010‐related serious AEs [SAEs] and BG00010‐related AEs coded as severe by the Principal Investigator), dose escalation proceeded to the next cohort. If one of three BG00010‐treated patients experienced a treatment‐related DLT, three additional patients were to be enrolled at that dose level and escalation was to continue if there were no other DLTs among the additional BG00010‐treated patients. If two or more BG00010‐treated patients experienced the same or a similar treatment‐related DLT, dosing was to stop, and the previous dose level was to be considered the MTD. If patients experienced DLTs that were not the same or similar in nature, the cohort could be expanded or dosing stopped, as recommended by the DSRC.
Study assessments
The primary objective of the study was to evaluate the PK and safety of three IV injections of BG00010 in two fixed dosing schedules: weekly and as frequently as every 48 h (no more than three times in 1 week). Secondary objectives were to explore the potential of BG00010 to reduce pain following multiple‐dose administration (as measured by a numerical rating scale (NRS) and the SF‐MPQ VAS) and to explore the repeated‐dose immunogenicity of BG00010 (as measured by the incidence of anti‐BG00010 antibodies).
In Parts I and II of the study, blood samples for PK analysis were taken: 30 min pre‐dose and then 15 min and 1, 2.5, 4, 6, 9, 12, 18, 24 and 48 h following the first dose of BG00010; 30 min pre‐dose and then 15 min and 4, 24 and 48 h following the second dose of BG00010; 30 min pre‐dose and then 15 min and 1, 2.5, 4, 6, 9, 12, 18, 24, 48, 72 and 120 h following the third dose of BG00010. The concentration of BG00010 in serum was determined using a chemiluminescent enzyme‐linked immunosorbent assay (quantification range 0.1–10 ng/ml) based on the binding of BG00010 to immobilized anti‐BG00010 antibody (P3B3) using streptavidin conjugated with horseradish peroxidase, which upon addition of luminol substrate produced a chemiluminescent signal. At the lower limit of quantification, assay precision and bias were 12.5% and 13.6% respectively. Assay performance was fully validated in accordance with regulatory guidance and industry best practices.
Safety assessments included recording AEs and SAEs, measurements of haematology, clinical chemistry, and urinalysis variables; vital signs; physical examinations; neurologic examinations; electrocardiograms (ECGs); numerical pain rating assessments; and longitudinal assessment of quantitative sensory testing (QST) in the unaffected leg (vibratory, cool thermal and heat pain). For vibratory measurements, a Rydel‐Seiffer vibratory tuning fork was used. The Rydel‐Seiffer vibratory tuning fork is an instrument that can determine the vibration extinction threshold. The tuning fork was set to a vibratory frequency of 64 Hz and then placed on the subject's skin. The vibration threshold was the point where the vibration was no longer perceived by the subject 16. Measurements were performed in triplicate on the medial malleolus. Cold and heat stimuli were applied with a thermode device (TSA‐II – NeuroSensory Analyzer, Medoc) which gradually increased or decreased in temperature. The thermode was applied to the inner aspect of the calf muscle of the unaffected leg. Cold sensation, cold pain threshold, heat pain threshold and heat pain tolerance were assessed. For the QST, the change from baseline was noted if there was a change of ≥2 standard deviations (SD) of laboratory normative data from the baseline measurement. As BG00010 is involved in nerve growth, intra‐epidermal nerve fibre density (IENFD) was measured as a safety precaution. A punch biopsy of the distal part (10 cm proximal to the lateral malleolus) of the unaffected leg was performed twice (on the same leg within 1 h) to minimize patient variance.
The presence of anti‐BG00010 antibodies was determined using a tiered assay approach involving a screening assay and a confirmation assay, followed by titration of positive samples. The presence of anti‐BG00010 antibodies in human serum was determined using an electrochemiluminescent assay format. Samples that tested positive for binding antibodies were further evaluated in a neutralizing antibody assay that measured the ability of BG00010 to bind to and activate the extracellular GFRα3 receptor. Assay performance was fully validated in accordance with regulatory guidance and industry best practices.
Plasma and serum samples were also drawn to explore potential pharmacodynamic markers: Substance P, chemokine receptor 2 (CCR2) and norepinephrine.
The efficacy assessments were an 11‐point NRS assessment (general sciatic pain, back pain and leg pain) and pain as measured by the VAS of the SF‐MPQ. Nociceptive testing was performed as exploratory assessment. Electrical, mechanical and cold pressor tests were performed to assess pain detection and pain tolerance thresholds.
Statistical analyses
This was an exploratory study; therefore, the sample size was not based on statistical considerations. PK parameters were calculated using noncompartmental methods, and summary statistics for each PK parameter were calculated by dose. The mean concentration values for each dose group were plotted over time, and dose proportionality was assessed. Calculations were performed using WinNonlin Phoenix version 6.2 (Certara, Princeton, USA).
All patients who were randomized were analysed. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 15.0. The incidences of AEs, SAEs and development of antibodies to BG00010, as well as changes in safety parameters, were summarized by study dose and compared with placebo. Placebo patients were pooled together separately in Parts I and II.
The efficacy analysis population was defined as all patients who received study treatment and had pain data collected post‐dose. Changes in the 11‐point NRS and VAS of the SF‐MPQ were summarized by study dose and compared with placebo.
Results
Study population
Twenty‐eight patients were randomized (see Figure S1 for study flowchart); 16 in Part I of the study to BG00010 (50, 150, 400 or 800 μg kg−1 once weekly) or placebo and 12 in Part II to BG00010 (400, 600 or 1200 μg kg−1 up to every 48 h and no more than 3 times in 1 week) or placebo. The first dose of study treatment was administered on 25 July 2011, and the last study visit was on 20 September 2012.
In Part I, 10 patients (63%) were women; in Part II, 9 patients (75%) were women. The mean age was 53.5 years (range 33–75 years) in Part I and 51.4 years (range 19–74 years) in Part II. The majority of patients were white (22 [79%]). Concomitant medications were taken by 15/16 (94%) patients in Part I and 9/12 (75%) patients in Part II. The most frequent concomitant medication was paracetamol (9/15 [60%] patients in Part I and 3/9 [33%] patients in Part II).
Pharmacokinetics
In all study cohorts, the BG00010 concentration–time data indicated relatively low inter‐patient variability in the time course of BG00010 serum exposure. Serum BG00010 concentrations over time are shown in Figure 1. There was a dose‐dependent increase in serum exposure from 150 to 1200 μg kg−1, but the increase was less than dose‐proportional. The log‐linear BG00010 concentration vs. time course at all dose levels showed a distinct multiphasic disposition in which peak concentrations dropped more than tenfold in the first 2–3 h post‐dose and declined more slowly thereafter. There was no trend toward increasing or decreasing clearance (CL) or steady‐state volume of distribution (V ss) across the body weight range tested (62.6–106.4 kg).
Figure 1.
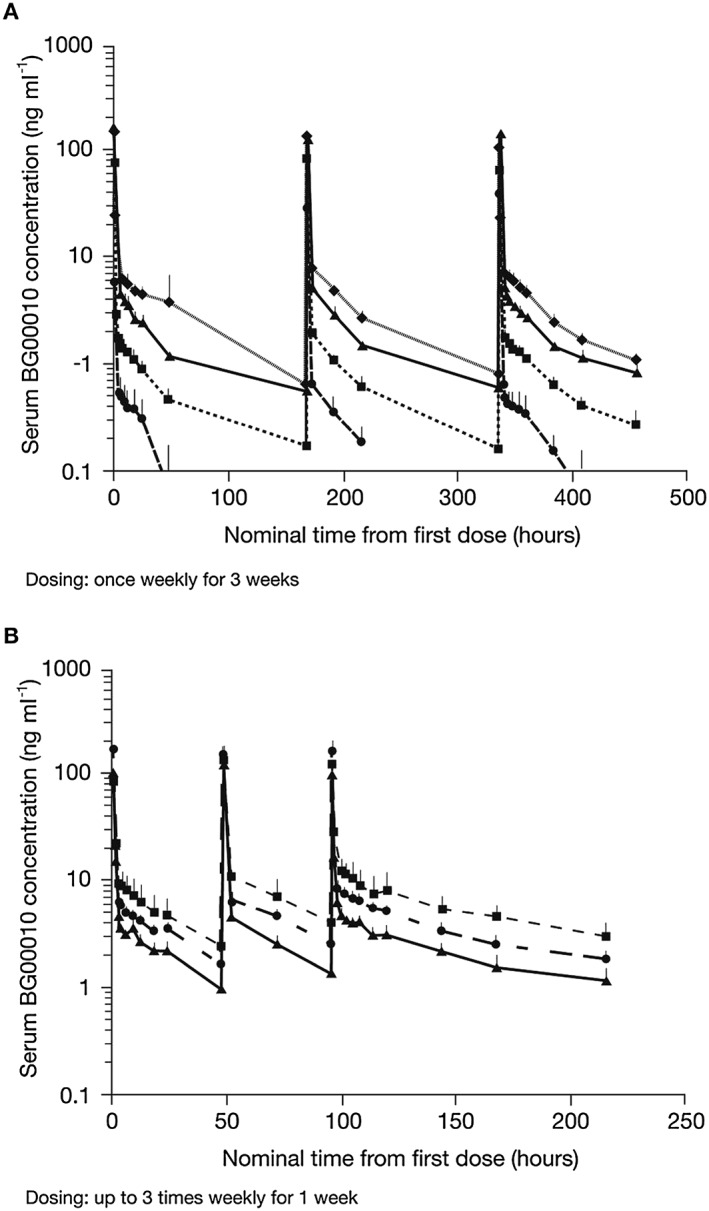
Mean (+ SD) serum BG00010 concentration vs. time by cohort in Part I (A: ( ) 50 µg kg−1 (n = 3), (
) 50 µg kg−1 (n = 3), ( ) 150 µg kg−1 (n = 3), (
) 150 µg kg−1 (n = 3), ( ) 400 µg kg−1 (n = 3), (
) 400 µg kg−1 (n = 3), ( ) 800 µg kg−1 (n = 3)) and Part II (B: (
) 800 µg kg−1 (n = 3)) and Part II (B: ( ) 400 µg kg−1 (n = 3), (
) 400 µg kg−1 (n = 3), (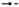 ) 600 µg kg−1 (n = 3), (
) 600 µg kg−1 (n = 3), ( ) 1200 µg kg−1 (n = 3))
) 1200 µg kg−1 (n = 3))
A summary of the PK parameters for Parts I and II of the study is given in Table 1. In Part I of the study, the area under the concentration–time curve from time zero to infinity (AUCinf) increased approximately in proportion to dose from 0 to 400 μg kg−1 and somewhat less than dose proportionally from 400 to 800 μg kg−1 (Table 1). There was no difference in AUCinf between the first (Dose 1) and third (Dose 3) dose within each cohort, indicating little or no BG00010 accumulation. The maximal plasma concentration (C max) also increased with BG00010 dose, but reached a plateau from 400 to 800 μg kg−1 (Table 1). Reaching a plateau in C max between these dose levels was expected due to increasing IV infusion time. CL was relatively constant over the dose range tested for Cohorts A to D. Mean V ss for all cohorts increased as a function of dose. Estimation of V ss at the lower dose levels (Cohorts A and B) was influenced by the limited range of concentration–time data and therefore produced underestimates of the true volume. At the higher dose levels (Cohorts C and D), mean V ss fell within a tighter range (Table 1).
Table 1.
Summary of the PK parameters for Parts I and II of the study
| Part I (weekly dosing) | Part II (dosing every 48 h) | ||||||
|---|---|---|---|---|---|---|---|
| BG00010 50 μg kg −1 (n = 3) | BG00010 150 μg kg −1 (n = 3) | BG00010 400 μg kg −1 (n = 3) | BG00010 800 μg kg −1 (n = 3) | BG00010 400 μg kg −1 (n = 3) | BG00010 600 μg kg −1 (n = 3) | BG00010 1200 μg kg −1 (n = 3) | |
| Injection 1 | |||||||
| AUC (inf) h ng ml −1 | 45.0 (9.1) | 156.7 (36.7) | 399.0 (37.3) | 652.7 (243.2) | |||
| AUC tau * h ng ml −1 | 40.7 (12.7) | 142.3 (28.4) | 347.0 (27.5) | 606.0 (247.7) | 180.7 (5.0) | 294.7 (51.5) | 313.7 (113.6) |
| Clearance (l h −1 kg −1 ) | 1.1 (0.2) | 1.0 (0.2) | 1.0 (0.1) | 1.3 (0.4) | |||
| C max (ng ml −1 ) | 33.2 (2.6) | 76.4 (17.3) | 156.0 (32.9) | 149.3 (29.5) | 100.9 (9.7) | 174.7 (52.3) | 84.0 (18.1) |
| t ½ (h)† | 24.1 (9.6) | 54.9 (8.5) | 63.8 (16.5) | 49.9 (4.0) | |||
| V SS (l kg −1 ) | 18.3 (7.4) | 44.5 (2.5) | 62.3 (11.0) | 71.4 (27.6) | |||
| Injection 2 | |||||||
| C max (ng ml −1 ) | 28.5 (3.6) | 84.1 (12.4) | 118.9 (35.9) | 135.0 (32.9) | 118.3 (25.8) | 154.7 (22.0) | 135.0 (43.6) |
| Injection 3 | |||||||
| AUC (inf) h ng ml −1 | 52.1 (22.6) | 159.7 (20.5) | 435.7 (121.2) | 516.3 (70.5) | |||
| AUC (0–168) h ng ml −1 | 47.6 (20.5) | 149.7 (16.5) | 354.7 (52.5) | 471.3 (73.0) | 232.3 (8.1) | 386.0 (61.1) | 487.7 (167.9) |
| Clearance (l h −1 kg −1 ) | 1.1 (0.4) | 0.9 (0.1) | 1.0 (0.2) | 1.6 (0.2) | |||
| C max (ng ml −1 ) | 38.8 (15.2) | 65.2 (7.8) | 140.3 (47.9) | 106.8 (23.2) | 96.9 (24.4) | 175.0 (47.9) | 121.3 (30.4) |
| t ½ (h)‡ | 41.5 (27.1) | 48.4 (12.7) | 86.6 (53.4) | 53.8 (14.2) | 66.1 (15.5) | 62.5 (15.8) | 99.0 (37.5) |
| V SS (l kg −1 ) | 25.3 (8.5) | 40.5 (9.2) | 74.8 (32.1) | 91.2 (23.5) | |||
All values are mean (±SD). AUC, area under the concentration–time curve; AUCtau, AUC during a dosing interval at steady state; AUC(inf), AUC from time zero to infinity; C max, maximal plasma concentration; T ½, half‐life; V ss, steady‐state volume of distribution.
AUCtau: Tau =0–168 for Part I; 0–48 for Part II;
For Dose 1, Part I, mean terminal t ½ values are based on concentration data to 168 h post‐dose;
For Dose 3, Part I, mean terminal t ½values are based on concentration data to 120 h post‐dose.
In Part II of the study, the increase in AUC during a dosing interval (0–48 h) at steady state (AUCtau) was less than proportional to dose, and AUCtau for Dose 3 was higher than that for Dose 1 in all cohorts, indicating some degree of accumulation (Table 1). C max for Cohorts E–G increased with dose, but formed a similar plateau as observed in Cohorts A–D. There was little or no BG00010 accumulation with a 168 h dose interval in Cohorts A to D and some accumulation (29–55%) with a 48 h dose interval in Cohorts E–G, indicating an effective half‐life (t ½ (eff)) for BG00010 between 30 and 60 h.
Safety
All patients experienced AEs. The most frequently reported AEs reported by patients receiving BG00010 were headache (10 BG00010‐treated patients [83%] vs. 3 placebo‐treated patients [75%] in Part I and 6 [67%] vs. 1 [33%] in Part II), feeling hot (6 [50%] vs. 0 in Part I and 9 [100%] vs. 0 in Part II), generalized pruritus (8 [67%] vs. 1 [25%] in Part I and 3 [33%] vs. 0 in Part II) and pruritus (5 [42%] vs. 2 [50%] in Part I and 6 [67%] vs. 0 in Part II). The majority of AEs were mild; no severe AEs, SAEs or AEs leading to discontinuation were observed. There was no indication for increased reporting of AEs with increasing BG00010 dose or increasing frequency of BG00010 dosing.
An overview of pruritus‐related AEs is shown in Figure 2. All AEs pertaining to pruritus were considered related to study treatment and resolved by the end of the follow‐up period. Pruritus that lasted >28 days was reported in five patients: this was mild in severity and no modification of study treatment was required in any of the five patients. One patient reported a generalized rash during the study. The patient was in the BG00010 50 μg kg−1 group and the AE was mild in severity and lasted 5 days. One patient in the BG00010 150 μg kg−1 group had a temperature‐related AE of moderate severity (feeling hot) that lasted 3 days. The same event occurred after the second and third dose, but were of mild severity. Five patients in Part I experienced mild temperature‐related AEs that lasted between 1 and 5 days. Nine patients in Part II experienced mild temperature‐related AEs with duration of 1–2 days. No clear trends in response to multiple doses of BG00010 were observed in blood levels of Substance P or CCR2 in Parts I or II of the study.
Figure 2.
Adverse‐event overview in Part I (A) and Part II (B) for pruritus events. *Patient received placebo. Severity: 1 = mild; 2 = moderate. ( ) Pruritus‐related events
) Pruritus‐related events
In Parts I and II, the most frequent concomitant medication was paracetamol. Use of concomitant medications as permitted by the protocol was not expected to affect study results.
No clinically significant changes were found in vital signs, ECG, IENFD, QST or safety laboratory tests. Although some patients, at some points, had QST values outside the normal range, none of these changes were determined to be a reason for concern. Observed changes were considered within the expected variability of QST assessments (data not shown). The change in IENFD from baseline is shown in Figure 3. No trends in IENFD data were observed and no clear dose effect was documented in Parts I or II of the study. There was no correlation between changes in IENFD and clinical findings, and the changes that were seen were considered not clinically significant.
Figure 3.
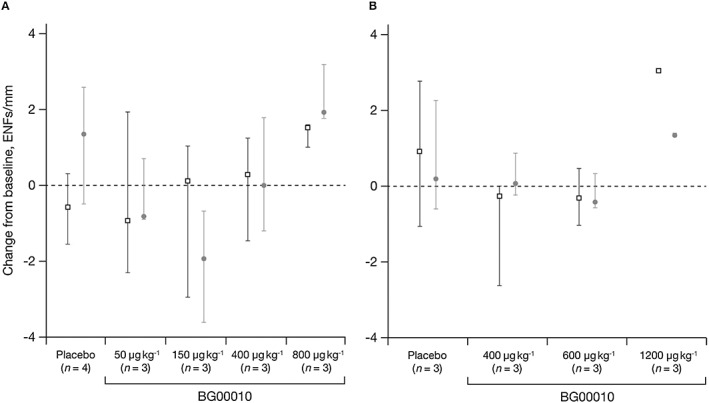
Median (± range) change in IENFD from baseline by treatment and visit in Part I (A: ( ) Week 6, (
) Week 6, ( ) Week 10) and Part II (B: (
) Week 10) and Part II (B: ( ) 28 days post Dose 3, (
) 28 days post Dose 3, ( ) 56 days post Dose 3). ENF, epidermal nerve fibre; IENFD, intra‐epidermal nerve fibre density
) 56 days post Dose 3). ENF, epidermal nerve fibre; IENFD, intra‐epidermal nerve fibre density
The only case of anti‐BG00010 antibodies detected in the study was in a patient receiving placebo. This was a false positive result on Day 20; results were negative on Day 43. No neutralizing antibodies were detected.
Pharmacodynamics
Change from baseline in NRS over time is shown in Figure 4. On the NRS in Part I, there was little differentiation between placebo‐treated patients and patients treated with BG00010 50 or 150 μg kg−1 in mean change in general sciatica pain from baseline. There was more differentiation for patients treated with BG00010 400 or 800 μg kg−1, observed particularly after Dose 2. After Dose 2, patients receiving BG00010 ≥ 400 μg kg−1 reported greater reductions in general sciatica pain and these reductions were maintained after Dose 3. However, overall there was substantial variability in the NRS data. In Part II of the study, mean changes from baseline among placebo‐treated patients remained within a lower range relative to that observed for placebo treatment in Part I and the treatment response was far more evident with the Part II dosing regimen. Results for leg pain and back pain, as measured by the NRS, were similar to those seen for general pain.
Figure 4.
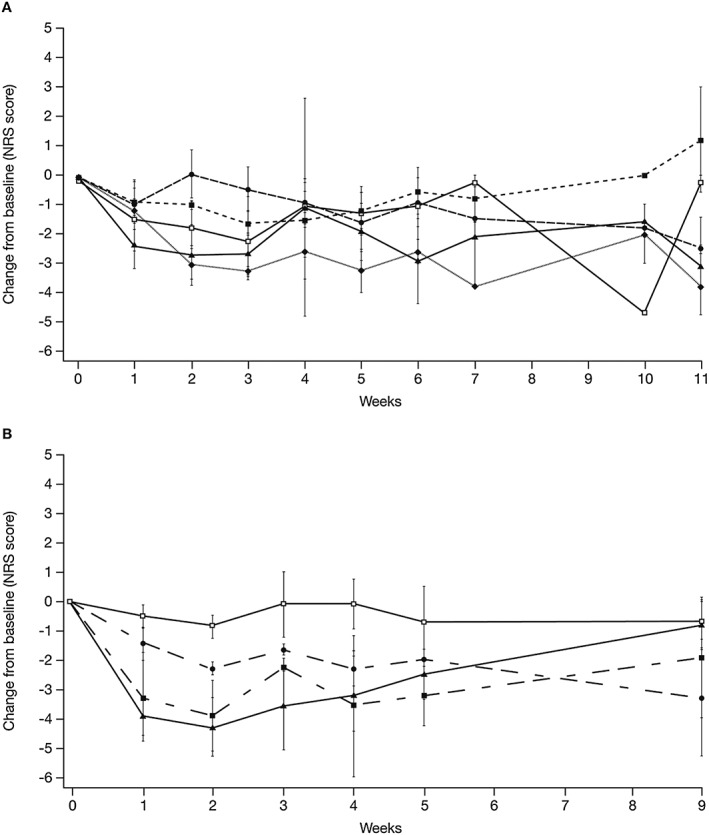
Mean (± SE) NRS pain general assessment change from baseline for Part I (A: ( ) Placebo (n = 4), (
) Placebo (n = 4), ( ) 50 µg kg−1 (n = 3), (
) 50 µg kg−1 (n = 3), ( ) 150 µg kg−1 (n = 3), (
) 150 µg kg−1 (n = 3), ( ) 400 µg kg−1 (n = 3), (
) 400 µg kg−1 (n = 3), ( ) 800 µg kg−1 (n = 3)) and Part II (B: (
) 800 µg kg−1 (n = 3)) and Part II (B: ( ) Placebo (n = 3), (
) Placebo (n = 3), ( ) 400 µg kg−1 (n = 3), (
) 400 µg kg−1 (n = 3), ( ) 600 µg kg−1 (n = 3), (
) 600 µg kg−1 (n = 3), ( ) 1200 µg kg−1 (n = 3)). NRS, numerical rating scale
) 1200 µg kg−1 (n = 3)). NRS, numerical rating scale
In Part I, BG00010 800 μg kg−1 resulted in the greatest mean (SD) decrease from baseline in VAS score (−34 [14] mm at Week 6). The greatest mean decrease from baseline for placebo‐treated patients was seen at Week 10 (−20.67 [31.63] mm). For the other doses tested in Part I, no pain reduction was seen compared with placebo‐treated patients. In Part II of the study, the greatest mean decrease from baseline was seen 56 days after Dose 3 in the 600 μg kg−1 dose group (mean [SD] of −33.33 [33.38] mm vs. −3.67 [20.03] mm with placebo). A trend in pain reduction from baseline as measured by the VAS of the SF‐MPQ was seen in the BG00010 600 and 1200 μg kg−1 dose groups compared with placebo. No notable trends were seen in the nociceptive testing data (Table S1a‐f; Figure S2).
Discussion
Overall, BG00010 dosed weekly or every 48 h demonstrated dose‐dependent PK in this ascending‐dose study in patients with sciatica. There were no DLTs, the MTD was not identified, and BG00010 administration appeared to lead to a reduction in back pain and leg pain in some patients.
When administered as multiple doses by IV infusion at 48‐ or 168‐hour dose intervals, serum BG00010 exposure increased in a dose‐dependent, but less than dose‐proportional, manner with relatively low variability within dose cohorts. At all dose levels, the log‐linear BG00010 concentration–time course showed a distinct multiphasic disposition in which peak concentrations dropped more than tenfold in the first 2–3 h after administration and then declined more slowly over the following days with a t ½ that generally fell between 40 and 60 h. There was no accumulation of BG00010 when dosed at a 168‐hour interval, but there was some accumulation when dosed every 48 h (accumulation ratio: 29–55%), suggesting a t ½ (eff) for BG00010 of approximately 30–60 h. The PK results in the current study are consistent with those observed in the previous single ascending‐dose study with BG00010 13.
All patients experienced at least one AE during the study and AEs were more frequently reported in patients receiving BG00010 versus placebo. There was no clear increase in the incidence of the most frequent AEs for the higher‐ versus lower‐dose cohorts or in the cohorts where BG00010 was administered three times during 1 week versus once weekly for 3 weeks. One of the most frequently reported AEs in this study was mild or moderate pruritus, starting approximately 1–3 days following administration of Dose 1. The exact mechanism of the pruritus is not known, but pruritus was also seen in a single ascending‐dose study where it was more common with higher BG00010 doses (≥100 μg kg−1) 13. In our study, with weekly dosing, patients who received the highest BG00010 dose (800 μg kg−1) experienced pruritus AEs of longer duration than patients in the other treatment groups. However, in Part II of the study, when BG00010 was dosed every 48 h, there was no clear indication of pruritus AEs having a longer duration with increasing BG00010 dose. The single case of anti‐BG00010 antibodies being detected in a patient receiving placebo was a false positive result, not unexpected as the assay cut point incorporates a 5% false positive rate.
The effects of BG00010 on neuropathic pain behaviour have been studied extensively in rats. Three rat models of neuropathic pain have been examined: spinal nerve ligation 3, chronic constriction injury of the sciatic nerve 17 and distal root crush 18. In these models, BG00010 substantially attenuated neuropathic pain behaviour. Exposure to BG00010 at single doses of 400 or 800 μg kg−1 in humans was comparable to exposure in rats at doses that have shown efficacy in these nonclinical pain models. In this study, based on the NRS and VAS of the SF‐MPQ, the Part II multiple‐dose BG00010 regimen resulted in greater pain reduction versus placebo and the lower‐dose regimens, although a clear dose–response relationship was not seen. These limited data suggest that BG00010 may benefit patients with sciatica and that further development of this compound for the treatment of neuropathic pain is warranted.
No formal sample size calculation was performed for the efficacy endpoints; therefore, owing to the small sample size of each cohort, no final conclusions can be made about the efficacy of BG00010 at this point. Nociceptive testing was included as an exploratory measure. With only three subjects per dose group, large variability was observed in baseline values of the different nociceptive tasks. No effects of BG00010 on evoked pain were observed. Likely the group size was too small to detect clear effects on evoked pain.
A recent review 19 concluded that available evidence does not clearly show favourable effects with nonsteroidal anti‐inflammatory drugs, corticosteroids, antidepressants or opioid analgesics for the treatment of sciatica. There has, however, been significant progress over the last 20 years in the understanding of the biology of pain sensory neurons, and the discovery that neurotrophic factors play an important role in neuropathic pain has provided several new therapeutic targets. Although sciatica probably has mixed neuropathic and nociceptive/inflammatory components, neurotrophic factors such as the GDNF family of ligands may address certain aspects of the underlying causes of neuropathic pain.
In conclusion, PK results were consistent with those observed in previous research. There were no DLTs and a MTD was not identified. These data from a small sample suggest that further evaluation of BG00010 in patients with sciatica is warranted. Additionally, a phase II study in patients with radiculopathy has recently been completed.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare that P.O., J.H. and G.J.G. have no conflict of interest. E.V. is a former employee of Biogen and owns shares of Biogen stock. Y.T., B.R., E.A. and G.G. are former employees of Biogen. A.V. and L.W. are employees of Biogen.
The funder of the study was involved in the study design and the data collection and analysis. The funder reviewed and provided feedback on the paper. All authors had full access to all the data in the study, made the decision to submit these data for publication, were involved in writing the manuscript and agreed upon the final content of the paper. The corresponding author had final responsibility for the decision to submit for publication.
This study was sponsored by Biogen. The Centre for Human Drug Research received funding from Biogen. Writing support under the direction of the authors was provided by Sarah Feeny of Complete Medical Communications, and was funded by Biogen.
Supporting information
Table S1a Nociceptive testing data, Part I, cold pressor test, pain tolerance threshold in seconds, mean (±SD)
Table S1b Nociceptive testing data, Part I, electrical pain test, pain tolerance threshold in mA, mean (±SD)
Table S1c Nociceptive testing data, Part I, pressure pain test, pain tolerance threshold in kPa, mean (±SD)
Table S1d Nociceptive testing data, Part II, cold pressor test, pain tolerance threshold in seconds, mean (±SD)
Table S1e Nociceptive testing data, Part II, electrical pain test, pain tolerance threshold in mA, mean (±SD)
Table S1f Nociceptive testing data, Part II, pressure pain test, pain tolerance threshold in kPa, mean (±SD)
Figure S1 Consort E‐flowchart
Figure S2 Nociceptive data. PTT, pain tolerance threshold
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Okkerse, P. , Hay, J. L. , Versage, E. , Tang, Y. , Galluppi, G. , Ravina, B. , Verma, A. , Williams, L. , Aycardi, E. , and Groeneveld, G. J. (2016) Pharmacokinetics and pharmacodynamics of multiple doses of BG00010, a neurotrophic factor with anti‐hyperalgesic effects, in patients with sciatica. Br J Clin Pharmacol, 82: 108–117. doi: 10.1111/bcp.12941.
References
- 1. Baloh RH, Tansey MG, Lampe PA, Fahrner TJ, Enomoto H, Simburger KS, et al. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRalpha3‐RET receptor complex. Neuron 1998; 21: 1291–302. [DOI] [PubMed] [Google Scholar]
- 2. Ossipov MH. Growth factors and neuropathic pain. Curr Pain Headache Rep 2011; 15: 185–92. [DOI] [PubMed] [Google Scholar]
- 3. Gardell LR, Wang R, Ehrenfels C, Ossipov MH, Rossomando AJ, Miller S, et al. Multiple actions of systemic artemin in experimental neuropathy. Nat Med 2003; 9: 1383–9. [DOI] [PubMed] [Google Scholar]
- 4. Wang R, King T, Ossipov MH, Rossomando AJ, Vanderah TW, Harvey P, et al. Persistent restoration of sensory function by immediate or delayed systemic artemin after dorsal root injury. Nat Neurosci 2008; 11: 488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wong LE, Gibson ME, Arnold HM, Pepinsky B, Frank E. Artemin promotes functional long‐distance axonal regeneration to the brainstem after dorsal root crush. Proc Natl Acad Sci U S A 2015; 112: 6170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schmader KE. Epidemiology and impact on quality of life of postherpetic neuralgia and painful diabetic neuropathy. Clin J Pain 2002; 18: 350–4. [DOI] [PubMed] [Google Scholar]
- 7. O'Connor AB. Neuropathic pain: quality‐of‐life impact, costs and cost effectiveness of therapy. Pharmacoeconomics 2009; 27: 95–112. [DOI] [PubMed] [Google Scholar]
- 8. El Barzouhi A, Vleggeert‐Lankamp CL, Lycklama ANG, Van der Kallen BF, van den Hout WB, Verwoerd AJ, et al. Magnetic resonance imaging interpretation in patients with sciatica who are potential candidates for lumbar disc surgery. PLoS One 2013; 8: e68411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stafford MA, Peng P, Hill DA. Sciatica: a review of history, epidemiology, pathogenesis, and the role of epidural steroid injection in management. Br J Anaesth 2007; 99: 461–73. [DOI] [PubMed] [Google Scholar]
- 10. Frymoyer JW. Back pain and sciatica. N Engl J Med 1988; 318: 291–300. [DOI] [PubMed] [Google Scholar]
- 11. Weber H, Holme I, Amlie E. The natural course of acute sciatica with nerve root symptoms in a double‐blind placebo‐controlled trial evaluating the effect of piroxicam. Spine (Phila Pa 1976) 1993; 18: 1433–8. [PubMed] [Google Scholar]
- 12. Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol 2010; 9: 807–19. [DOI] [PubMed] [Google Scholar]
- 13. Rolan PE, O'Neill G, Versage E, Rana J, Tang Y, Galluppi G, et al. First‐in‐human, double‐blind, placebo‐controlled, randomized, dose‐escalation study of BG00010, a glial cell line‐derived neurotrophic factor family member, in subjects with unilateral sciatica. PLoS One 2015; 10: e0125034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Melzack R. The short‐form McGill Pain Questionnaire. Pain 1987; 30: 191–7. [DOI] [PubMed] [Google Scholar]
- 15. Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol 2011; 164: S1–S324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martina IS, van Koningsveld R, Schmitz PI, van der Meche FG, van Doorn PA. Measuring vibration threshold with a graduated tuning fork in normal aging and in patients with polyneuropathy. J Neurol Neurosurg Psychiatry 1998; 65: 743–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arnold HM, Huang C, Gong B, Rossomando AJ, Engber T, Pepinsky B. Neublastin (artemin) alleviates neuropathic pain in rats produced by chronic constriction injury. Paper presented at the IASP: 14th World Congress on Pain, Milan, Italy, August 2012.
- 18. Wang R, Rossomando A, Sah DW, Ossipov MH, King T, Porreca F. Artemin induced functional recovery and reinnervation after partial nerve injury. Pain 2014; 155: 476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pinto RZ, Maher CG, Ferreira ML, Ferreira PH, Hancock M, Oliveira VC, et al. Drugs for relief of pain in patients with sciatica: systematic review and meta‐analysis. BMJ 2012; 344: e497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1a Nociceptive testing data, Part I, cold pressor test, pain tolerance threshold in seconds, mean (±SD)
Table S1b Nociceptive testing data, Part I, electrical pain test, pain tolerance threshold in mA, mean (±SD)
Table S1c Nociceptive testing data, Part I, pressure pain test, pain tolerance threshold in kPa, mean (±SD)
Table S1d Nociceptive testing data, Part II, cold pressor test, pain tolerance threshold in seconds, mean (±SD)
Table S1e Nociceptive testing data, Part II, electrical pain test, pain tolerance threshold in mA, mean (±SD)
Table S1f Nociceptive testing data, Part II, pressure pain test, pain tolerance threshold in kPa, mean (±SD)
Figure S1 Consort E‐flowchart
Figure S2 Nociceptive data. PTT, pain tolerance threshold
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item