

Abstract
Aim
The present study evaluated the pharmacodynamics and pharmacokinetics of nebivolol enantiomers in patients with chronic kidney disease (CKD) and in patients undergoing haemodialysis.
Methods
Forty‐three adult patients were distributed into three groups: healthy volunteers and hypertensive patients with normal kidney function (n = 22); patients with stage 3 and 4 CKD (n = 11); and patients with stage 5 CKD undergoing haemodialysis (n = 10). The subjects received a single oral dose of 10 mg racemic nebivolol. Serial blood samples were collected up to 48 h after administration of the drug and heart rate variation was measured over the same interval during the isometric handgrip test. The nebivolol enantiomers in plasma were analysed by liquid chromatography–tandem mass spectrometry.
Results
The pharmacokinetics of nebivolol is enantioselective, with a greater plasma proportion of l‐nebivolol. CKD increased the area under the concentration–time curve (AUC) of l‐nebivolol (6.83 ng.h ml–1 vs. 9.94 ng.h ml–1) and d‐nebivolol (4.15 ng.h ml–1 vs. 7.30 ng.h ml–1) when compared with the control group. However, the AUC values of l‐nebivolol (6.41 ng.h ml–1) and d‐nebivolol (4.95 ng.h ml–1) did not differ between the haemodialysis and control groups. The administration of a single dose of 10 mg nebivolol did not alter the heart rate variation induced by isometric exercise in the investigated patients.
Conclusions
Stage 3 and 4 CKD increases the plasma concentrations of both nebivolol enantiomers, while haemodialysis restores the pharmacokinetic parameters to values similar to those observed in the control group. No significant difference in heart rate variation induced by isometric exercise was observed between the investigated groups after the administration of a single oral dose of 10 mg nebivolol.
Keywords: CYP2D6, chronic kidney disease, nebivolol, pharmacokinetics, phenotype
What is Already Known about this Subject
Chronic kidney disease decreases the nonrenal clearance of drugs as a result of the accumulation of uraemic toxins, which reduce the activity of cytochrome P 450 (CYP) isoforms and transporters.
Haemodialysis eliminates uraemic toxins and restores the activity of CYP isoforms.
What this Study Adds
The pharmacokinetics of nebivolol is enantioselective, with the plasma accumulation of l‐nebivolol.
Stage 3 and 4 CKD reduces the oral clearance of the two nebivolol enantiomers.
The oral clearance of the nebivolol enantiomers in haemodialysis patients does not differ from that of subjects with normal kidney function.
Introduction
Chronic kidney disease (CKD), which is characterized by a progressive decline in kidney function and the accumulation of uraemic toxins, reduces in parallel the renal clearance of drugs. Velenosi et al. have also shown in animals and patients with CKD a reduction in nonrenal drug clearance due to changes in the expression and activity of metabolizing enzymes and drug transporters 1. Patients with CKD exhibit a plasma accumulation of parathyroid hormone, proinflammatory cytokines [interleukin (IL)1‐β, IL‐6, tumour necrosis factor‐α] and uraemic toxins such as indoxyl sulfate, which can alter gene transcription and protein translation or are involved directly in the inhibition of cytochrome P 450 (CYP) isoforms and drug transporters 2, 3, 4.
The oxidative metabolism dependent on CYP2C9, CYP3A, CYP2C19 and CYP2B6, as well as N‐acetyltransferase 2 and uridine 5′‐diphospho‐glucuronosyltransferase (UGT2B7), is reduced in patients with CKD 3, 5, 6. CKD is also associated with changes in the expression and/or activity of drug transporters such as P‐glycoprotein, multidrug resistance protein (MRP) and organic anion transporting polypeptide (OATP) 7, 8. However, an increase in the hepatic clearance of drugs can be observed in patients with end‐stage renal disease or stage 5 CKD in haemodialysis treatment, which partially eliminates uraemic toxins, restores the activity of CYP isoforms and, in some situations, also contributes to the elimination of some drugs 6, 9.
Activation of the sympathetic nervous system, which is prevalent in CKD patients, plays an important role both in the genesis of hypertension and the progression of CKD, and is associated with increased cardiovascular morbidity and mortality. The magnitude of the increase in sympathetic activity depends on the stage of CKD – i.e. the higher the stage of CKD, the more pronounced the process 10, 11.
Previous studies have suggested that β‐adrenoceptor antagonists such as nebivolol slow the progression of CKD 12. Nebivolol is available in clinical practice as a racemate of the d‐nebivolol (SRRR) and l‐nebivolol (RSSS) enantiomers. Antihypertensive activity is mainly attributed to d‐nebivolol 13. Nebivolol is a highly lipophilic drug [octanol/water partition coefficient (log P) = 4.03 at pH 11.8) and its elimination depends mainly on metabolism. Renal and fecal excretion of the unchanged drug is less than 0.5% of the dose 14. Nebivolol is eliminated mainly by glucuronidation and aromatic hydroxylation mediated by the polymorphic CYP2D6 15, 16. N‐deacylation is a minor metabolic route in extensive (EM) and poor (PM) metabolizers of CYP2D6 14.
As the use of β‐adrenoceptor antagonists for the treatment of hypertension slows the progression of CKD 12 and is associated with reduced cardiovascular mortality in CKD patients undergoing haemodialysis 17, and as CKD can alter nonrenal drug clearance 3, 5, 6, 8, 18, 19, the present study evaluated the influence of CKD and haemodialysis on the pharmacokinetics (PK) of nebivolol enantiomers. Additionally, we investigated the PK–pharmacodynamic (PD) relationship between the plasma concentration of d‐nebivolol and heart rate variation (a PD parameter) during isometric handgrip exercise.
Methods
Patient selection and inclusion criteria
The clinical protocol was approved by the Research Ethics Committee of the University Hospital of the School of Medicine of Ribeirão Preto, University of São Paulo, Brazil. The patients were included in the study after they had signed the free informed consent form.
Forty‐three patients aged 18–65 years, seen at the nephrology division of the local hospital or at the nephrology service of Ribeirão Preto (São Paulo, Brazil) were investigated. The patients were divided into three groups according to CKD stage 20: a control group, consisting of 13 healthy volunteers and nine patients with systemic arterial hypertension and kidney function within the normal range (characterized according to the criteria of the Sixth Brazilian Guidelines on Arterial Hypertension, 2010, and a creatinine clearance >90 ml min–1 1.73 m– 2); a CKD group, consisting of 11 patients with stage 3 and 4 CKD and a creatinine clearance <60 ml min–1 1.73 m– 2; and a haemodialysis group (4‐h sessions given three times per week), consisting of ten patients with stage 5 CKD undergoing haemodialysis and a creatinine clearance <15 ml min–1 1.73 m–2.
The sample size was calculated with the Power and Sample Size Calculation program 21, using the PK parameters of nebivolol in healthy volunteers. A level of significance of P < 0.05, a test power of 80% and a difference in mean nebivolol AUC of 50% between control and CKD groups (AUC 7.76 ng.h ml−1; standard deviation 3.07 ng.h ml−1) were adopted for the sample size calculation 22.
PK sampling and bioanalysis
The morning after a 12‐h fast, the patients received a single oral dose of two tablets of 5 mg racemic nebivolol (Nebilet®, Biolab, Brazil) with 200 ml water. In the haemodialysis group, nebivolol was given after the dialysis session and all blood samples were collected before the next section. The standard diet of the hospital was served 3 h after the administration of nebivolol. Blood samples (10 ml) were collected into heparinized syringes (5000 IU Liquemine®, Roche) at 0 h, 0.5 h, 1 h, 1.5 h, 2 h, 2.5 h, 3 h, 4 h, 6 h, 8 h, 10 h, 12 h, 16 h, 20 h, 24 h, 30 h, 36 h, 42 h and 48 h after administration of the drug. Plasma aliquots for chromatographic analysis were obtained by centrifugation of the blood samples (2800 g for 15 min) and stored at –70 °C until the time of analysis.
The CYP2D6 phenotype was evaluated 48 h after the administration of nebivolol. The patients received a single oral dose of 100 mg metoprolol tartrate (one tablet, Seloken, AstraZeneca, Cotia, SP, Brazil) with 200 ml water. A single 5 ml blood sample was collected after 3 h with a heparinized syringe (5000 IU Liquemin®, Roche, São Paulo, SP, Brazil) and plasma was stored at –70 °C until the time of analysis.
PD evaluation
The sympathetic response to the isometric handgrip exercise (heart rate variation) was evaluated using a Jamar® handgrip dynamometer (Lafayette, IN, USA) over the same interval as the blood collections. Before the administration of nebivolol, the patient was asked to hold the handgrip dynamometer with the dominant hand in the supine position and to perform a maximal voluntary contraction. Only some of the patients in the haemodialysis group, who had an arteriovenous fistula placed on the dominant arm, used the nondominant hand for the handgrip test. The mean of three maximal voluntary contractions was obtained. The patients were asked to perform the isometric handgrip exercise at 30% of maximal voluntary contraction throughout the period of blood collection (0–48 h) 23. The sympathetic response triggered by the handgrip manoeuvre was analysed by calculating the variation in heart rate between the end of the 2‐min isometric exercise and 1 min before the beginning of the exercise. Blood pressure was measured with a noninvasive blood pressure module and heart rate was recorded with a heart rate monitor (Dixtal DX2021, Manaus, AM, Brazil).
The blood pressure and heart rate‐lowering effects of nebivolol and its effect on reduce heart rate were evaluated, comparing the values obtained at baseline (without drug administration) with those measured at the time of maximum concentration (t max) of nebivolol. Both data were not used to construct the PK‐PD model.
Analytical assays
Analysis of metoprolol and α‐hydroxymetoprolol in plasma
Metoprolol and its metabolite α‐hydroxymetoprolol were analysed in the plasma by high‐performance liquid chromatography with fluorescence detection according to a method developed and validated in a previous study from our group (unpublished data). Briefly, 1 ml plasma aliquots spiked with a solution of tramadol as internal standard (1.25 μg) were extracted in basic medium with dichloromethane : diisopropyl ether (1 : 1, v/v). The drugs were separated on a Select B Lichrospher 60 RP column (Merck, Darmstadt, Germany) using 0.05 mol l–1 phosphate buffer (pH 3.5) : acetonitrile (9 : 1, v/v) as the mobile phase. The calibration curves for metoprolol and α‐hydroxymetoprolol were constructed over the concentration range 10–800 ng ml–1 plasma. The coefficients of variation in the study of precision and percentage of inaccuracy inter‐ and intra‐assay were less than 15%, guaranteeing the reproducibility and repeatability of the results.
Analysis of nebivolol enantiomers in plasma
The nebivolol enantiomers were analysed in plasma by a method developed and published in a previous study from our group 24 using liquid chromatography–tandem mass spectrometry coupled to a Chirobiotic V chiral‐phase column. The calibration curves were constructed over the range 25–2500 pg of each nebivolol enantiomer per ml of plasma. The quantification limit was 25 pg of each isomer per ml of plasma. No racemization was detected. The coefficients of variation obtained in the study of inter‐ and intra‐assay precision and accuracy were less than 15%, guaranteeing the reproducibility and repeatability of the results.
PK analysis
The metabolic ratio of metoprolol to α‐hydroxymetoprolol (MET/OHM) and the log metabolic ratio of MET/OHM were calculated by dividing the plasma concentration of metoprolol by the plasma concentration of α‐hydroxymetoprolol obtained 3 h after the administration of a single oral dose of 100 mg racemic metoprolol tartrate 16. Patients presenting a log metabolic ratio of less than 1.5 were classified as PM and those with a log metabolic ratio higher than 1.5 were classified as EM 25.
PK analysis of the nebivolol enantiomers was carried out using the WinNonlin 4.0 program (Pharsight Corporation, Mountain View, CA, USA). The PK parameters were calculated based on the plasma concentrations of the nebivolol enantiomers obtained experimentally with a bicompartmental model.
PK‐PD analysis
PK‐PD analysis using the WinNonlin program was carried out using the plasma concentrations of d‐nebivolol obtained experimentally as the PK parameter (C), and the variation in heart rate between the end of the 2‐min isometric exercise and 1 min before the beginning of the exercise (E max) as the PD parameter 23, 26. The PK‐PD relationship was described by the inhibitory maximum effect model [E = E max*(1 – (C/C+EC50)] (where the EC50 is the plasma concentration of d‐nebivolol that results in 50% E max), which assumes that an increase in plasma d‐nebivolol concentration reduces the variation in heart rate induced by isometric exercise.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism® program (GraphPad Software, La Jolla, CA, USA) for the calculation of the mean, geometric mean, standard error of the mean (SEM) and 95% confidence interval (CI). The two‐tailed Wilcoxon test for paired data was used to evaluate the isomer ratios of nebivolol different from unity. The two‐tailed paired t‐test was used to compare heart rate and blood pressure between baseline and at nebivolol t max. Analysis of variance and Tukey's post‐test for unpaired data were applied to compare the results between the control and CKD groups, control and haemodialysis groups, and CKD and haemodialysis groups. A level of significance of 5% was adopted in all tests.
Results
The demographic data for the patients and the effects of nebivolol on heart rate and blood pressure are shown in Table 1 as the mean and 95% CI. All patients were phenotyped as EM for CYP2D6.
Table 1.
Anthropometric characteristics of the investigated patients (n = 43). Data are expressed as mean (95% confidence interval)
| Control group (n = 22) | CKD group (n = 11) | Haemodialysis group (n = 10) | |
|---|---|---|---|
| Gender (M/F) | 12/10 | 5/6 | 5/5 |
| Age (years) | 36 (32, 41) | 51 (46, 56) | 41 (35, 47) |
| Weight (kg) | 72.0 (68.3, 75.7) | 70.8 (64.8, 76.7) | 63.8 (55.9, 71.7) |
| Height (m) | 1.69 (1.66, 1.72) | 1.61 (1.56, 1.65) | 1.66 (1.61, 1.72) |
| BMI ( kg m – 2 ) | 25.1 (24.1, 26.1) | 27.8 (26.0, 28.7) | 22.8 (21.2, 24.4) |
| CL creatinine (ml min – 1 1. 73 m – 2 ) | 108 (98, 118) | 33 (26, 40) | 9 (7, 11) |
| CYP2D6 phenotype | EM | EM | EM |
| Basal HR | 67 (64, 71) | 69 (63, 75) | 77 (70, 84)‡ |
| HR (d‐nebivolol, t max ) | 63 (60, 67)* | 65 (59, 72)* | 74 (68, 80)*, ‡ |
| Baseline SBP (mmHg) | 123 (117, 130) | 141 (127, 155)† | 133 (122, 145) |
| SBP (d‐nebivolol, t max ) | 117 (111, 122)* | 134 (120, 149)* | 130 (122, 138) |
| Baseline DBP (mmHg) | 72 (68, 77) | 84 (72, 96) | 83 (76, 86) |
| DBP (d‐nebivolol, t max ) | 67 (63, 71)* | 82 (72, 92) | 82 (78, 89) |
| Associated drugs | 5, 6, 9, 11, 18, 19, 22, 23, 24, 25 | 1, 5, 6, 9, 10, 11, 14, 15, 17, 18, 19 | 1, 2, 3, 4, 5, 6, 7, 8, 10, 13, 14, 15, 16, 17, 18, 19, 20 |
BMI, body mass index; CKD, chronic kidney disease; CL, clearance; CYP, cytochrome P450; DBP, diastolic blood pressure, EM, extensive metabolizers of cytochrome P450 2D6; F, female; HR, heart rate; M, male; SBP, systolic blood pressure; t max, maximum concentration. 1 = folic acid; 2 = complex B; 3 = acetylsalicylic acid; 4 = omeprazole; 5 = enalapril; 6 = furosemide; 7 = simvastatin; 8= calcitriol; 9 = captopril; 10 = amlodipine; 11 = hydrochlorothiazide; 12 = diazepam; 13 = sevelamer; 14 = allopurinol; 15 = erythropoietin; 16 = vitamin C; 17 = ferrous sulfate; 18 = losartan; 19 = atorvastatin; 20 = cyclobenzaprine; 21 = indapamide; 22 = levothyroxine; 23 = spironolactone; 24 = olmesartan.
P < 0.05 baseline vs. t max;
P < 0.05 CKD vs. control group;
P < 0.05 haemodialysis vs. control group.
Table 2 shows the kinetic disposition of the l‐nebivolol and d‐nebivolol enantiomers in plasma for the control group (n = 22), CKD group (n = 11) and haemodialysis group (n = 10). The plasma concentration vs. time curves (Figure 1) show plasma accumulation of l‐nebivolol in the three groups investigated.
Table 2.
Kinetic disposition of the l‐ and d‐ nebivolol enantiomers in the investigated patients (n = 43) following a single oral dose of 10 mg racemic nebivolol. Data are expressed as geometric mean (coefficient of variation)
| l‐nebivolol | d‐nebivolol | |||||
|---|---|---|---|---|---|---|
| Parameters | Control (n = 22) | CKD (n = 12) | Haemodialysis (n = 10) | Control (n = 22) | CKD (n = 12) | Haemodialysis (n = 10) |
| C max (ng ml –1 ) | 1.31 (47) | 1.98 (47) | 1.38 (39) | 0.69 (45)* | 1.34 (54)* | 0.80 (44)* |
| t max (h) † | 1.01 (0.18–2.10) | 1.04 (0.68–2.80) | 1.15 (0.27–1.54) | 1.03 (0.43–2.14) | 0.98 (0.61–3.34) | 1.13 (0.40–1.86) |
| AUC 0–∞ (ng.h ml –1 ) | 6.83 (39) | 9.94 (44) | 6.41 (35) | 4.15 (41)* | 7.30 (51)* | 4.95 (36)* |
| t½ (h) | 13.79 (35) | 12.43 (35) | 12.87 (27) | 13.19 (47) | 11.57 (30) | 16.10 (31) |
| Vd/F (l kg –1 ) | 100.19 (66) | 59.38 (65) | 113.70 (70) | 157.60 (83)* | 71.95 (64)* | 197.49 (65)* |
| CL/F (l h –1 kg –1 ) | 10.24 (47) | 7.18 (32) | 12.45 (35) | 16.84 (41)* | 9.77 (51)* | 15.86 (33)* |
AUC0–∞, area under the concentration–time curve from time zero extrapolated to infinity; CKD, chronic kidney disease; C max, maximal plasma concentration; CV, coefficient of variation; CL/F, oral clearance; t½, terminal elimination half‐life; t max, time to reach C max; Vd/F, apparent volume of distribution.
Paired t‐test, P < 0.05 (l‐nebivolol vs. d‐nebivolol);
median (range).
Figure 1.
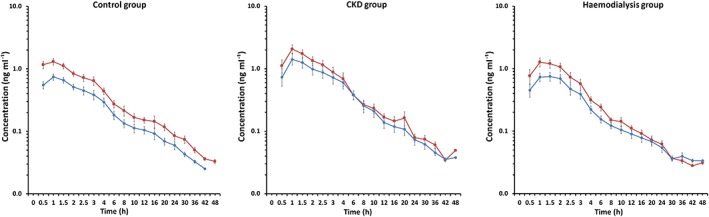
Plasma concentration vs. time curves of the nebivolol enantiomers in patients in the control (n = 22), chronic kidney disease (CKD) (n = 11) and haemodialysis (n = 10) groups treated with a single oral dose of 10 mg racemic nebivolol. Values are expressed as the mean ± standard error of the mean
The comparison of the geometric mean ratios of the PK parameters for l‐nebivolol and d‐nebivolol between the control (n = 22) and CKD (n = 11) groups and between the control and haemodialysis (n = 10) groups is shown in Table 3. A difference in maximum concentration (C max), AUC and oral clearance (CL/F, where CL is clearance and F is bioavailability) of l‐nebivolol and d‐nebivolol was observed between patients in the control and CKD groups, but not between the control and haemodialysis groups, showing that haemodialysis restores normal PK parameters. Figure 2 illustrates the differences in the PK parameters (C max, AUC and oral clearance) of the l‐nebivolol and d‐nebivolol enantiomers between the control vs. CKD and control vs. haemodialysis groups, using box plots.
Table 3.
Pharmacokinetic parameter ratios between control (n = 22) and CKD (n = 11) groups and between control and haemodialysis (n = 10) groups following a single oral dose of 10 mg racemic nebivolol. Ratios are expressed as geometric mean
| Control/CKD | Control/Haemodialysis | |||
|---|---|---|---|---|
| Parameters | l‐nebivolol | d‐nebivolol | l‐nebivolol | d‐nebivolol |
| C max (ng ml –1 ) | 0.66* | 0.51* | 0.95 | 0.86 |
| AUC 0–∞ (ng.h ml –1 ) | 0.69* | 0.57* | 1.07 | 0.84 |
| CL/F (l h –1 kg –1 ) | 1.43* | 1.72* | 0.82 | 1.06 |
AUC0–∞, area under the concentration–time curve from time zero extrapolated to infinity; CKD, chronic kidney disease; CL/F, oral clearance; C max, maximal plasma concentration.
One‐way analysis of variance and Tukey's post‐test, P < 0.05 (control vs. CKD) and (control vs. haemodialysis).
Figure 2.
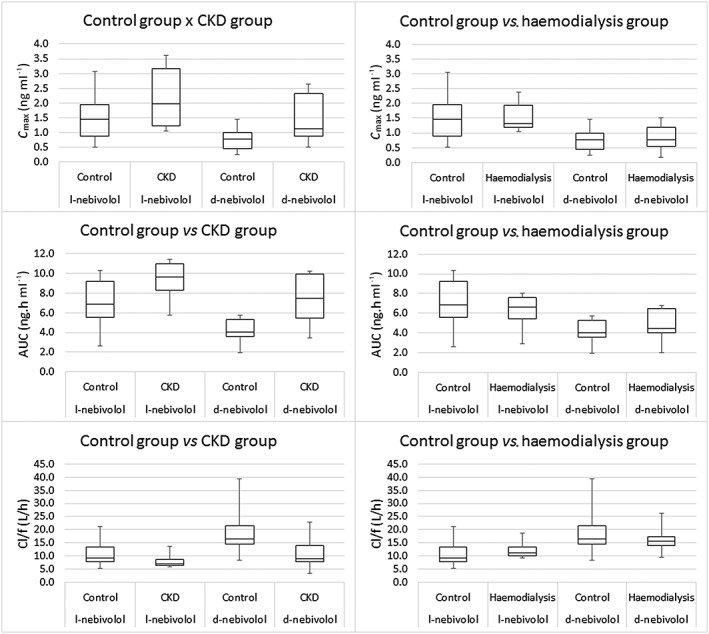
Box plot comparing pharmacokinetic parameters (C max, AUC and oral clearance) of the l‐nebivolol and d‐nebivolol enantiomers between the control and CKD groups and between the control and haemodialysis groups. AUC, area under the concentration–time curve; CKD, chronic kidney disease; C max, maximal plasma concentration
Figure 3 shows the PK‐PD modelling (inhibitory E max model) of the heart rate variation between the end of the 2‐min isometric exercise and 1 min before the beginning of the exercise as a function of the plasma concentrations of d‐nebivolol for all patients included in the study and classified as EM (n = 43). The data show high variability in the PD parameter. Table 4 shows the E max and EC50 parameters obtained using the inhibitory E max model.
Figure 3.
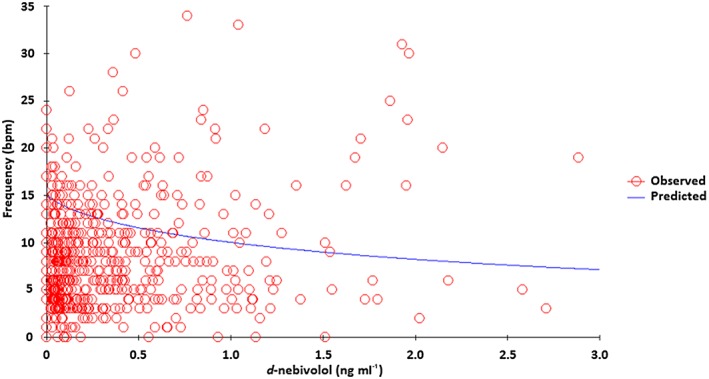
Pharmacokinetic–pharmacodynamic analysis (inhibitory E max model) of the d‐nebivolol isomer as a function of heart rate variation induced by isometric handgrip exercise in all patients classified as extensive metabolizers of cytochrome P450 2D6 (n = 43), E max, maximum heart rate variation
Table 4.
Parameters obtained by pharmacokinetic–pharmacodynamic analysis (inhibitory E max model) relating the plasma concentrations of the d‐nebivolol enantiomer as a function of the heart rate variation induced by isometric handgrip exercise, including all investigated patients classified as extensive metabolizers of cytochrome P450 2D6 (n = 43). Data are expressed as mean (95% confidence interval)
| Parameters | Control group (n = 22) | CKD group (n = 12) | Haemodialysis (n = 10) |
|---|---|---|---|
| E max (bpm) | 11.87 (7.78, 21.09) | 15.53 (11.19, 23.80) | 11.62 (9.16, 17.43) |
| EC 50 (pg ml –1 ) | 1411.29 (1071.22, 2166.66) | 1656.11 (966.98, 3522.85) | 1254.50 (997.67, 1900.62) |
CKD, chronic kidney disease; E max, maximum heart rate variation. *One‐way analysis of variance and Tukey's post‐test, P < 0.05 (control vs. CKD) and (control vs. haemodialysis).
Discussion
The present study investigated the influence of CKD and haemodialysis treatment on the PK and PK‐PD modelling of nebivolol enantiomers in patients phenotyped as EM for CYP2D6.
The PK of nebivolol was found to be enantioselective, with plasma accumulation of the l‐nebivolol enantiomer in all investigated groups (Table 2). In the control group, higher C max (1.31 ng ml–1 vs. 0.69 ng ml–1) and AUC0–∞ (6.83 ng.h ml–1 vs. 4.15 ng.h ml–1) values were observed for l‐nebivolol when compared with d‐nebivolol. The present results agree with those reported by Lindamood et al. 27, who found a C max of 1.30 ng ml–1 for l‐nebivolol and of 0.70 ng ml–1 for d‐nebivolol after the oral administration of 10 mg racemic nebivolol to healthy volunteers. Similar to the control group, patients in the CKD group exhibited higher C max (1.98 vs. 1.34 ng ml–1) and AUC (9.94 ng.h ml–1 vs. 7.30 ng.h ml–1) values for l‐nebivolol. Plasma accumulation of l‐nebivolol was also observed in the haemodialysis group (C max: 1.38 vs. 0.80 ng ml–1 and AUC0–∞: 6.41 ng.h ml–1 vs. 4.95 ng.h ml–1).
The oral clearance values of the two nebivolol enantiomers were high and indicated extensive hepatic metabolism (10.24 l h–1 kg–1 and 16.84 l h–1 kg–1 for l‐nebivolol and d‐nebivolol, respectively; Table 2). The volume of distribution of the two enantiomers was also very high in the control group, with values of 100.19 l kg–1 and 157.60 l kg–1 for l‐nebivolol and d‐nebivolol, respectively (Table 2).
Comparison of the PK parameters between the control and CKD groups allowed us to infer that stage 3 and 4 CKD reduces the oral clearance of l‐nebivolol (7.18 l h–1 kg–1 vs. 10.24 l h–1 kg–1) and d‐nebivolol (9.77 l h–1 kg–1 vs. 16.84 l h–1 kg–1) and increases the C max and AUC values of the two enantiomers [analysis of variance (ANOVA), P < 0.05; Table 3). The oral clearance values observed in the CKD group (508.34 l h–1 for l‐nebivolol and 691.72 l h–1 for d‐nebivolol) agree with those obtained by Shaw et al. 28 for patients with moderate CKD who received nebivolol as a racemic mixture (738 l h–1). Shaw et al. 28 observed a progressive increase in the AUC values of nebivolol as kidney disease progresses. The authors reported AUC values for the isomeric mixture of 6.59 ng.h ml–1, 4.55 ng.h ml–1, 11.28 ng.h ml–1 and 23.36 ng.h ml–1 for healthy volunteers and patients with mild, moderate and severe CKD, respectively.
Several mechanisms have been proposed to explain the reduction in CYP‐mediated hepatic drug clearance in patients with CKD, such as alterations in gene transcription and protein translation, reduced CYP expression due to the inhibition of haemoprotein biosynthesis and/or increased enzyme degradation, depletion of cofactors such as NADPH, and direct inhibition of CYP enzymes by uraemic toxins 2, 29.
Michaud et al. 30 showed that the serum of patients with CKD contains uraemic toxins that reduce the CYP content of normal rat hepatocytes. These mediators decrease the expression of CYP isoforms (CYP1A2, CYP2C6, CYP2C11, CYP2D1/2D2, CYP3A2 and CYP4A1/4A3) secondarily to the low levels of mRNA. It should be noted that these authors observed a strong correlation between the severity of CKD and a reduction in hepatic CYP. Furthermore, there was a strong correlation between serum parathyroid hormone concentration and the reduction in CYP expression. Rostami‐Rodjegan et al. reported a decrease in CYP2D6 activity in patients with CKD 31. Nolin found that the CYP2D6‐dependent metabolism of propranolol is not altered in patients with early‐stage CKD but is acutely reduced in patients with stage 5 CKD 32. It is important to highlight that nebivolol is classified as a class 2 drug (poor solubility and high permeability) by the Biopharmaceutical Classification System, so its PK parameters are not linked to drug transporters 33.
The oral clearance values of 12.45 l h–1 kg–1 for l‐nebivolol and 15.86 l h–1 kg–1 for d‐nebivolol obtained for the haemodialysis group (Table 2) did not differ from those in the control group (10.24 l h–1 kg–1 and 16.84 l h–1 kg–1, respectively) but were different from those obtained for the CKD group (7.18 l h–1 kg–1 and 9.77 l h–1 kg–1, respectively).
Considering that the reduction in nonrenal drug clearance depends on the serum concentration of uraemic toxins 6, and that patients undergoing haemodialysis eliminate uraemic toxins at least partially, it is possible that the activity of enzymes and transporters is restored in haemodialysis patients. Momper et al. showed that haemodialysis acutely restores the hepatic clearance of erythromycin, suggesting that the dialysis of uraemic toxins may directly and acutely reverse the inhibition of drug metabolism and transporter activity in patients with stage 5 CKD 6. Nolin et al. reported that the high concentration of uraemic toxins in CKD patients reduces the activity of CYP3A, while haemodialysis acutely restores CYP3A activity by removing uraemic toxins 34. This activity was increased by 27% 2 h after haemodialysis. Hence, the oral clearance values of the two nebivolol enantiomers, as well as the other PK parameters obtained for haemodialysis patients, did not differ from those of the control group (ANOVA; P < 0.05; Table 3).
As nebivolol is a highly cardioselective β‐adrenoceptor antagonist, we evaluated its PD using the heart rate variation induced by isometric handgrip exercise at 30% of maximal voluntary contraction. The heart rate variation (the difference between the value observed after 2 min of isometric exercise and the value observed 1 min before the isometric exercise) was evaluated up to 48 h after the administration of a single oral dose of 10 mg racemic nebivolol 23, 35. As can be seen in Figure 3, there was wide variability in the PD parameter. Isometric handgrip exercise activates the baroreflex control of blood pressure, which results in an increase in blood pressure, accompanied by an increase in heart rate and peripheral vascular resistance in resting skeletal muscle 36. The inhibitory E max model was used for PK‐PD modelling of plasma d‐nebivolol concentrations as a function of heart rate variation (Figure 2) in all patients phenotyped as EM for CYP2D6 (n = 43). However, it should be noted that CYP2D6 activity‐dependent hydroxylated metabolites are equipotent with d‐nebivolol as β‐adrenoceptor antagonists and that the sum of unchanged nebivolol and hydroxylated metabolites is similar in patients phenotyped as EM or PM for CYP2D6 37.
The heart rate variation induced by isometric handgrip exercise was not correlated with the plasma concentrations of d‐nebivolol (Figure 3) but showed wide interindividual variability among all patients studied (n = 43). The E max (11.87 bpm vs. 15.53 bpm vs. 11.62 bpm for the control, CKD and haemodialysis groups, respectively) and EC50 values (1411.29 pg ml–1 vs. 1656.11 pg ml–1 vs. 1254.50 pg ml–1 for the control, CKD and haemodialysis groups, respectively) did not differ between the three groups (ANOVA; P > 0.05; Table 4), suggesting that CKD does not alter the heart rate variation induced by isometric handgrip exercise at 30% of maximal voluntary contraction. In line with these results, all groups showed a significant reduction in heart rate after nebivolol use.
The PD parameter heart rate variation induced by isometric handgrip exercise did not show sufficient sensitivity to detect a greater acute bradycardic response induced by d‐nebivolol in patients with CKD. This fact does not rule out the possibility that CKD patients using nebivolol could have a greater therapeutic response, placing these patients at risk of excessive bradycardia or arterial hypotension, especially during chronic nebivolol use. However, the acute blood pressure‐lowering effect of nebivolol appears to be blunted by CKD. In CKD patients, only systolic blood pressure was reduced, and in haemodialysis patients no reduction in blood pressure was observed (Table 1). These data could be attributed to the presence of marked endothelial dysfunction in haemodialysis patients, as the acute blood pressure effect of nebivolol is mediated by a reduction in peripheral vascular resistance owing to endothelial nitric oxide release 38, 39, 40. Although the PD assessment was a limitation of the present study, the finding of an increase of more than 40% in the exposure of CKD patients to d‐nebivolol suggests that these patients may be at risk of more prominent bradycardia or arterial hypotension, especially in clinical situations of exacerbated sympathetic activity, such as heart failure, which is frequent in this population. It is therefore prudent that patients with CKD initiate treatment with low doses of nebivolol and that more attention be paid to the monitoring of PD effects.
Conclusion
The present study showed that the PK of nebivolol is enantioselective, with plasma accumulation of the l‐nebivolol enantiomer which is responsible for the vasodilating effect of the drug. CKD reduces the oral clearance of the two nebivolol enantiomers. By contrast, haemodialysis, by eliminating uraemic toxins, restores the oral clearance values of the nebivolol enantiomers to those observed in the control group.
Heart rate variation induced by isometric handgrip exercise at 30% of maximal voluntary contraction, used as the PD parameter, exhibits wide variability among patients, is not altered by CKD and is not correlated with individual plasma concentrations of d‐nebivolol after the administration of a single oral dose of 10 mg racemic nebivolol. The blood pressure‐lowering effect of nebivolol could be blunted in haemodialysis patients.
Competing Interest
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years.
The authors are grateful to Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (CAPES) for financial support and for granting a research fellowship, and to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Apoio ao Ensino, Pesquisa e Assistência do Hospital das Clínicas, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo (FAEPA), for financial support.
Neves, D. V. , Lanchote, V. L. , Moysés Neto, M. , Cardeal da Costa, J. A. , Vieira, C. P. , and Coelho, E. B. (2016) Influence of chronic kidney disease and haemodialysis treatment on pharmacokinetics of nebivolol enantiomers. Br J Clin Pharmacol, 82: 83–91. doi: 10.1111/bcp.12917.
References
- 1. Velenosi TJ, Urquhart BL. Pharmacokinetic considerations in chronic kidney disease and patients requiring dialysis. Expert Opin Drug Metab Toxicol 2014; 10: 1131–43. [DOI] [PubMed] [Google Scholar]
- 2. Nolin TD, Naud J, Leblond FA, Pichette V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin Pharmacol Ther 2008; 83: 898–903. [DOI] [PubMed] [Google Scholar]
- 3. Dreisbach AW. The influence of chronic renal failure on drug metabolism and transport. Clin Pharmacol Ther 2009; 86: 553–6. [DOI] [PubMed] [Google Scholar]
- 4. Sayama H, Takubo H, Komura H, Kogayu M, Iwaki M. Application of a physiologically based pharmacokinetic model informed by a top‐down approach for the prediction of pharmacokinetics in chronic kidney disease patients. AAPS J 2014; 16: 1018–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Simard E, Naud J, Michaud J, Leblond FA, Bonnardeaux A, Guillemette C, Sim E, Pichette V. Downregulation of hepatic acetylation of drugs in chronic renal failure. J Am Soc Nephrol 2008; 19: 1352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Momper JD, Venkataramanan R, Nolin TD. Nonrenal drug clearance in CKD: searching for the path less traveled. Adv Chronic Kidney Dis 2010; 17: 384–91. [DOI] [PubMed] [Google Scholar]
- 7. Naud J, Michaud J, Leblond FA, Lefrancois S, Bonnardeaux A, Pichette V. Effects of chronic renal failure on liver drug transporters. Drug Metab Dispos 2008; 36: 124–8. [DOI] [PubMed] [Google Scholar]
- 8. Naud J, Nolin TD, Leblond FA, Pichette V. Current understanding of drug disposition in kidney disease. J Clin Pharmacol 2012; 52: 10S–22S. [DOI] [PubMed] [Google Scholar]
- 9. Redon J, Martinez F, Cheung AK. Special considerations for antihypertensive agents in dialysis patients. Blood Purif 2010; 29: 93–8. [DOI] [PubMed] [Google Scholar]
- 10. Zilch O, Vos PF, Oey PL, Cramer MJ, Ligtenberg G, Koomans HA, Blankestijn PJ. Sympathetic hyperactivity in haemodialysis patients is reduced by short daily haemodialysis. J Hypertens 2007; 25: 1285–9. [DOI] [PubMed] [Google Scholar]
- 11. Grassi G, Quarti‐Trevano F, Seravalle G, Arenare F, Volpe M, Furiani S, Dell'Oro R, Mancia G. Early sympathetic activation in the initial clinical stages of chronic renal failure. Hypertension 2011; 57: 846–51. [DOI] [PubMed] [Google Scholar]
- 12. Hart PD, Bakris GL. Should beta‐blockers be used to control hypertension in people with chronic kidney disease? Semin Nephrol 2007; 27: 555–64. [DOI] [PubMed] [Google Scholar]
- 13. Pauwels PJ, Van Gompel P, Leysen JE. Human beta 1‐ and beta 2‐adrenergic receptor binding and mediated accumulation of cAMP in transfected Chinese hamster ovary cells. Profile of nebivolol and known beta‐adrenergic blockers. Biochem Pharmacol 1991; 42: 1683–9. [DOI] [PubMed] [Google Scholar]
- 14. Peer A, Snoeck E, Woestenborghs R, Velde V, Mannens G, Meuldermans W, Heykants J. Clinical pharmacokinetics of nebivolol. Drug Investig 1991; 3: 25–30. [Google Scholar]
- 15. Fux R, Mörike K, Pröhmer AMT, Delabar U, Schwab M, Schaeffeler E, Lorenz G, Gleiter CH, Eichelbaum M, Kivistö KT. Impact of CYP2D6 genotype on adverse effects during treatment with metoprolol: a prospective clinical study. Clin Pharmacol Ther 2005; 78: 378–87. [DOI] [PubMed] [Google Scholar]
- 16. Frank D, Jaehde U, Fuhr U. Evaluation of probe drugs and pharmacokinetic metrics for CYP2D6 phenotyping. Eur J Clin Pharmacol 2007; 63: 321–33. [DOI] [PubMed] [Google Scholar]
- 17. Matsue Y, Suzuki M, Nagahori W, Ohno M, Matsumura A, Hashimoto Y. β‐blocker prevents sudden cardiac death in patients with hemodialysis. Int J Cardiol 2013; 165: 519–22. [DOI] [PubMed] [Google Scholar]
- 18. Verbeeck RK, Musuamba FT. Pharmacokinetics and dosage adjustment in patients with renal dysfunction. Eur J Clin Pharmacol 2009; 65: 757–73. [DOI] [PubMed] [Google Scholar]
- 19. Neirynck N, Vanholder R, Schepers E, Eloot S, Pletinck A, Glorieux G. An update on uremic toxins. Int Urol Nephrol 2013; 45: 139–50. [DOI] [PubMed] [Google Scholar]
- 20. K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43: S1–290. [PubMed] [Google Scholar]
- 21. Dupont WD, Plummer WD. PS power and sample size program available for free on the internet. Control Clin Trials 1997; 18: 274. [Google Scholar]
- 22. Kamali F, Howes A, Thomas SH, Ford GA, Snoeck E. A pharmacokinetic and pharmacodynamic interaction study between nebivolol and the H2‐receptor antagonists cimetidine and ranitidine. Br J Clin Pharmacol 1997; 43: 201–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kubo T, Azevedo ER, Newton GE, Parker JD, Floras JS. Lack of evidence for peripheral alpha(1)‐adrenoceptor blockade during long‐term treatment of heart failure with carvedilol. J Am Coll Cardiol 2001; 38: 1463–9. [DOI] [PubMed] [Google Scholar]
- 24. Neves DV, Vieira CP, Coelho EB, Marques MP, Lanchote VL. Stereoselective analysis of nebivolol isomers in human plasma by high‐performance liquid chromatography–tandem mass spectrometry: application in pharmacokinetics. J Chromatogr B Analyt Technol Biomed Life Sci 2013; 940: 47–52. [DOI] [PubMed] [Google Scholar]
- 25. Sohn DR, Kusaka M, Shin SG, Jang IJ, Chiba K, Ishizaki T. Utility of a one‐point (3‐hour postdose) plasma metabolic ratio as a phenotyping test using metoprolol in two east Asian populations. Ther Drug Monit 1992; 14: 184–9. [DOI] [PubMed] [Google Scholar]
- 26. Duprez D, Lefebvre R, De Backer T, De Sutter P, Trouerbach J, Clement DL. Influence of nebivolol on the cardiovascular hemodynamics during postural changes and isometric exercise. Cardiovasc Drugs Ther 1991; 5: 709–17. [DOI] [PubMed] [Google Scholar]
- 27. Lindamood C, Ortiz S, Shaw A, Rackley R, Gorski JC. Effects of commonly administered agents and genetics on nebivolol pharmacokinetics: drug–drug interaction studies. J Clin Pharmacol 2011; 51: 575–85. [DOI] [PubMed] [Google Scholar]
- 28. Shaw A, Liu S, Zachwieja L, Eddy T, Donnelly C, Huang M. Effects of varying degrees of renal impairment on the pharmacokinetic disposition of nebivolol. Clin Pharmacol Ther 2005; 77: P38. [Google Scholar]
- 29. Yeung CK, Shen DD, Thummel KE, Himmelfarb J. Effects of chronic kidney disease and uremia on hepatic drug metabolism and transport. Kidney Int 2014; 85: 522–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Michaud J, Dubé P, Naud J, Leblond FA, Desbiens K, Bonnardeaux A, Pichette V. Effects of serum from patients with chronic renal failure on rat hepatic cytochrome P450. Br J Pharmacol 2005; 144: 1067–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rostami‐Hodjegan A, Kroemer HK, Tucker GT. In‐vivo indices of enzyme activity: the effect of renal impairment on the assessment of CYP2D6 activity. Pharmacogenetics 1999; 9: 277–86. [DOI] [PubMed] [Google Scholar]
- 32. Nolin TD. Altered nonrenal drug clearance in ESRD. Curr Opin Nephrol Hypertens 2008; 17: 555–9. [DOI] [PubMed] [Google Scholar]
- 33. Thadkala K, Sailu C, Aukunuru J. Formulation, optimization and evaluation of oral nanosuspension tablets of nebivolol hydrochloride for enhancement of dissolution rate. Der Pharm Lett 2015; 7: 71–84. [Google Scholar]
- 34. Nolin TD, Appiah K, Kendrick SA, Le P, McMonagle E, Himmelfarb J. Hemodialysis acutely improves hepatic CYP3A4 metabolic activity. J Am Soc Nephrol 2006; 17: 2363–7. [DOI] [PubMed] [Google Scholar]
- 35. Van Nueten L, De Crée J. Nebivolol: comparison of the effects of dl‐nebivolol, d‐nebivolol, l‐nebivolol, atenolol, and placebo on exercise‐induced increases in heart rate and systolic blood pressure. Cardiovasc Drugs Ther 1998; 12: 339–44. [DOI] [PubMed] [Google Scholar]
- 36. Kamiya A, Michikami D, Fu Q, Niimi Y, Iwase S, Mano T, Suzumura A. Static handgrip exercise modifies arterial baroreflex control of vascular sympathetic outflow in humans. Am J Physiol Regul Integr Comp Physiol 2001; 281: R1134–9. [DOI] [PubMed] [Google Scholar]
- 37. Lefebvre J, Poirier L, Poirier P, Turgeon J, Lacourciere Y. The influence of CYP2D6 phenotype on the clinical response of nebivolol in patients with essential hypertension. Br J Clin Pharmacol 2007; 63: 575–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Georgianos PI, Sarafidis PA, Zoccali C. Intradialysis hypertension in end‐stage renal disease patients: clinical epidemiology, pathogenesis, and treatment. Hypertension 2015; 66: 456–63. [DOI] [PubMed] [Google Scholar]
- 39. Van de Water A, Janssens W, Van Neuten J, Xhonneux R, De Cree J, Verhaegen H, Reneman RS, Janssen PA. Pharmacological and hemodynamic profile of nebivolol, a chemically novel, potent, and selective beta 1‐adrenergic antagonist. J Cardiovasc Pharmacol 1988; 11: 552–63. [DOI] [PubMed] [Google Scholar]
- 40. Ritter JM. Nebivolol: endothelium‐mediated vasodilating effect. J Cardiovasc Pharmacol 2001; 38 (Suppl. 3): S13–6. [DOI] [PubMed] [Google Scholar]