

Abstract
Aims
To characterize the potential effect of daclizumab high‐yield process (DAC HYP), a monoclonal antibody that blocks the high‐affinity interleukin‐2 receptors for treatment of multiple sclerosis, on activity of cytochrome P450 (CYP) enzymes.
Methods
Twenty patients with multiple sclerosis received an oral cocktail of probe substrates of CYP1A2 (caffeine 200 mg), CYP2C9 (warfarin 10 mg/vitamin K 10 mg), CYP2C19 (omeprazole 40 mg), CYP2D6 (dextromethorphan 30 mg) and CYP3A (midazolam 5 mg) on two sequential occasions: 7 days before and 7 days after subcutaneous administration of DAC HYP 150 mg every 4 weeks for three doses. Serial pharmacokinetic blood samples up to 96 h post dose and 12‐h urine samples were collected on both occasions. Area under the curve (AUC) for caffeine, S‐warfarin, omeprazole and midazolam, and urine dextromethorphan to dextrorphan ratio were calculated. Statistical analyses were conducted on log‐transformed parameters using a linear mixed‐effects model.
Results
The 90% confidence intervals (CIs) for the geometric mean ratio (probe substrate with DAC HYP/probe substrate alone) for caffeine AUC from 0–12 h (0.93–1.15), S‐warfarin AUC from 0 to infinity (AUC[0–inf]) (0.95–1.06), omeprazole AUC(0–inf) (0.88–1.13) and midazolam AUC(0–inf) (0.89–1.15) were within the no‐effect boundary of 0.80–1.25. The geometric mean ratio for urine dextromethorphan to dextrorphan ratio was 1.01, with the 90% CI (0.76–1.34) extending slightly outside the no‐effect boundary, likely due to high variability with urine collections and CYP2D6 activity.
Conclusions
DAC HYP treatment in patients with multiple sclerosis had no effect on CYP 1A2, 2C9, 2C19, 2D6 and 3A activity.
Keywords: CYP, daclizumab high‐yield process, drug cocktail, therapeutic protein–drug interaction
What is Already Known about this Subject
Therapeutic proteins that modulate cytokine activities can indirectly influence expression of cytochrome P450 (CYP) isoenzymes by affecting cytokine concentrations, and consequently may alter CYP‐mediated metabolism of small molecule drugs when given in combination. Interleukin‐2 (IL‐2) and its modulators have been implicated in CYP‐mediated drug interactions.
Daclizumab high‐yield process (HYP) is a humanized monoclonal antibody that selectively blocks the alpha subunit (CD25) of the high‐affinity IL‐2 receptors and is being developed for treatment of multiple sclerosis.
Daclizumab HYP treatment increases systemic IL‐2 levels.
What this Study Adds
The study demonstrates that repeated administration of daclizumab HYP to patients with multiple sclerosis has no effect on the metabolic activity of the evaluated CYP enzymes.
Results provide important clinical information on impact of cytokine modulators on CYP activities, contributing to the clinical database to guide future recommendation on therapeutic protein–drug interaction assessment.
Introduction
Therapeutic protein–drug interactions (TP‐DIs) are an evolving clinical pharmacology area that has recently garnered significant scientific and regulatory interest. Therapeutic monoclonal antibodies (mAbs) that modulate cytokine activities can indirectly influence the expression of cytochrome P450 (CYP) isoenzymes by affecting cytokine concentrations, and consequently may alter the CYP‐mediated metabolism of small molecule drugs when given in combination 1, 2. The US Food and Drug Administration draft guidance for industry provides perspectives on TP‐DIs and recommendations on evaluating them 3. For therapeutic proteins that are cytokine modulators with known effects on CYPs or transporters, a clinical TP‐DI study should be conducted.
Daclizumab high‐yield process (DAC HYP) is a monoclonal immunoglobulin (Ig) of the human IgG1 isotype that binds specifically to CD25, the alpha subunit of the human high‐affinity interleukin (IL)‐2 receptor (IL‐2R) that is expressed on the surface of activated lymphocytes after interaction with a foreign antigen or in response to IL‐2 4. It is an investigational product being reviewed by health authorities for the treatment of relapsing forms of multiple sclerosis. The pharmacodynamic (PD) effects of DAC HYP include saturation of CD25 and an approximately two‐fold increase in serum IL‐2 levels 5.
IL‐2 and its modulators have been implicated in CYP‐mediated drug interactions. Administration of high doses of IL‐2 (9–12 million units m−2) in patients with cancer decreased CYP1A2, CYP2C, CYP2E1 and CYP3A activity by 37%, 45%, 60% and 39%, respectively 6. Basiliximab, an anti‐IL‐2R alpha mAb, has been linked to CYP3A‐mediated drug interactions. Higher concentrations of cyclosporin, a CYP3A substrate, were observed in 24 paediatric renal transplant patients who received an induction dose of basiliximab compared with patients who received placebo 7. Trough concentrations of tacrolimus, a CYP3A substrate, increased by 63% in 12 adult renal transplant patients 2 days after basiliximab administration 8. Considering the effects of DAC HYP on systemic IL‐2 levels and the reported implications of IL‐2‐dependent pathways and IL‐2 modulators on CYP‐mediated drug interactions, this clinical study was conducted to evaluate the effect of DAC HYP on CYP activities using a probe drug cocktail.
A cocktail approach is an effective way to evaluate in vivo TP‐DIs in the target population because it involves administration of a mixture of specific probe substrates of multiple CYP enzymes and/or transporters in a single TP‐DI study to evaluate the effect of a therapeutic protein's inhibition or induction potential 3. A validated and commonly used modified ‘Cooperstown 5 + 1 cocktail’ consisting of oral midazolam (CYP3A), caffeine (CYP1A2), warfarin + vitamin K (CYP2C9), omeprazole (CYP2C19) and dextromethorphan (CYP2D6) has been shown to be safe and well tolerated in patients and healthy volunteers 9, 10, 11, 12, 13.
Methods
This TP‐DI study was conducted as part of a sub‐study within the phase 2 multicentre, open‐label study to assess the immunogenicity, pharmacokinetics (PK), PD and tolerability of DAC HYP in patients with relapsing–remitting multiple sclerosis (main study). The TP‐DI sub‐study was registered (ClinicalTrials.gov identifier NCT01462318 and EudraCT no. 2010‐023856‐97) and conducted at seven clinics in three countries (four clinics in Hungary [DRC Gyógyszervizsgáló Központ Kft, Balatonfüred, Hungary; Kenézy Gyula Kórház és Rendelőintézet, Debrecen, Hungary; Vaszary Kolos Kórház, Ideggyógyászat, Esztergom, Hungary; and Fejer Megyei Szent Gyorgy Egyetemi Oktato Korhaz, Székesfehérvár, Hungary], one clinic in Katowice, Poland [NEURO‐CARE SMO Gabriela Klodowska‐Duda] and two clinics in the United States [Neurology Specialists Inc., Dayton, Ohio, and Associates in Neurology, PSC, Lexington, Kentucky]). The study followed the Declaration of Helsinki and was approved by ethics committees (Egészségügyi Tudományos Tanács Klinikai Farmakológiai Etikai Bizottsága, Budapest, Hungary; Komisja Bioetyczna przy Slaskiej Izbie Lekarskiej w Katowicach, ul., Katowice, Poland; Quorum Review IRB, Seattle, Washington, USA). All patients provided written informed consent before study entry.
Patients and study design
Patients were eligible to be enrolled in the TP‐DI study if they were between 18 and 65 years of age, inclusive; had a confirmed diagnosis of relapsing–remitting multiple sclerosis according to McDonald criteria, and previous cranial magnetic resonance imaging demonstrating lesion(s) consistent with multiple sclerosis; had a baseline Expanded Disability Status Scale score between 0.0 and 5.0, inclusive; and had one or more clinical relapse(s) within the previous 2 years. Patients were excluded if they were on a regimen of therapeutic anticoagulation (low molecular weight heparin or oral anticoagulant agents); used any medication (prescription or nonprescription) or herbal supplement that inhibits or induces CYP1A2, CYP2C9, CYP2C19, CYP2D6 or CYP3A activity within 14 days or five half‐lives (whichever is longer) before the first dose of probe drug cocktail; consumed grapefruit or grapefruit‐containing products or caffeine‐containing products within 3 days before the first dose of probe drug cocktail; had not discontinued interferon (IFN) beta ≥ 1 week before the first dose of probe drug cocktail; or had a history of hypersensitivity to midazolam, caffeine, warfarin, vitamin K, dextromethorphan or omeprazole. The use of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A inhibitors or inducers was not allowed during the study period.
Twenty patients were enrolled into the TP‐DI sub‐study and received probe drug cocktail in the two sequential treatment periods, 7 days before the first dose of DAC HYP (period 1) and 7 days after the third dose of DAC HYP 150 mg subcutaneous (SC) every 4 weeks for three doses (period 2). The probe drug cocktail consisted of oral midazolam 5 mg (oral syrup or parenteral formulations), caffeine 200 mg (tablet), warfarin 10 mg (tablet), vitamin K 10 mg (tablet), omeprazole 40 mg (capsule) and dextromethorphan 30 mg (oral syrup). Oral vitamin K 10 mg was used to prophylactically counteract warfarin's anticoagulant effect. Midazolam oral syrup (2 mg ml−1) was used at the study sites in the United States and midazolam parenteral formulation was used for oral administration at the study sites in Hungary and Poland due to commercial availability. Using a parenteral formulation with a concentration of 5 mg ml−1, 1 ml of the formulation was diluted with 30 ml of water and the entire preparation was administered 14, 15. The dose of midazolam used in this study (5 mg) was based on the standard dosing range (2–5 mg) oral dosing used in the drug cocktail study 10. Furthermore, the higher dose of midazolam was used to ensure measurable concentrations in period 2 in the event that DAC HYP decreased midazolam concentrations. To detect any potential effects of DAC HYP on the activities of CYP isoenzymes, the probe drug cocktail was administered 7 days after the third dose of DAC HYP 150 mg SC every 4 weeks (the clinical dose investigated in phase 3 studies) based on the following PK and PD considerations: ~90% of steady‐state exposure would be achieved after three doses of DAC HYP based on the terminal half‐life of ~2–3 weeks; time to maximum concentration (Cmax) following SC dosing of DAC HYP is ~7 days; and the increase of serum IL‐2 reached plateau after 4 weeks of DAC HYP dosing. The sample size for this study is similar to that commonly used in cocktail drug interaction studies (n = 15–21) 9, 10, 11, 12, 13.
Patients reported to the study clinic on the morning of the probe drug cocktail administration in both study periods. The probe drug cocktail was administered following an overnight fast for ≥8 h and with ~240 ml of water. A standard lunch was provided 4 h following administration of the probe drug cocktail. After receiving the probe drug cocktail, patients stayed in the clinic for ≥12 h ± 30 min under supervision for safety and PK assessments. Patients were not allowed to leave the clinic until they had completely recovered from the potential sedative effects of midazolam. Patients reported back to the clinic at 24 ± 1, 48 ± 2, 72 ± 2 and 96 ± 2 h after probe drug cocktail administration for collection of PK samples.
PK sampling schedule
Blood samples for the determination of plasma concentrations of midazolam, caffeine, S‐warfarin, omeprazole and 5‐hydroxyomeprazole were collected before probe drug cocktail administration (0 h) and at 0.5, 1.0, 2.0, 3.0, 4.0, 6.0, 8.0, 10, 24, 48, 72 and 96 h after probe drug cocktail administration. Urine samples for the measurement of dextromethorphan and dextrorphan were collected over the 12‐h period immediately after probe drug cocktail administration. The PK sampling schedules were deemed adequate because they have been used in other probe drug cocktail studies 9, 10, 11, 12, 13. Blood samples for the measurement of serum DAC HYP concentrations were collected pre‐dose in period 2.
DNA preparation and genotyping
A single whole‐blood sample for genotyping of polymorphic CYP2D6 and CYP2C19 enzymes was collected at baseline for all patients. Whole‐blood samples were screened for the presence of 27 star alleles (*1, *1XN, *2, *2XN, *3, *4, *4XN, *5, *6, *7, *8, *9, *10, *10XN, *11, *15, *17, *17XN, *19, *20, *29, *35, *35XN, *36, *40, *41 and *41XN) in the CYP2D6 gene, and three star alleles (*1, *2 and *3) in the CYP2C19 gene using the AmpliChip P450 2D6/2C19 assay (Roche Diagnostics, Indianapolis, Indiana, USA).
Measurement of plasma and urine concentrations of probe drugs and serum concentration of DAC HYP
Plasma samples were analysed for caffeine, midazolam, omeprazole, 5‐hydroxyomeprazole and S‐warfarin using validated liquid chromatography (LC) with tandem mass spectrometric detection (MS/MS) methods developed by Covance Inc. (Madison, Wisconsin, USA).
Regarding caffeine, a plasma sample volume of 0.100 ml was combined with the internal standard (caffeine‐d9) before solid‐phase extraction employing a Waters HLB cartridge (Waters Corporation, Milford, Massachusetts, USA). Extracts were evaporated to dryness under a stream of nitrogen and the resulting residue was reconstituted in methanol:water (50:50, v/v) before submission for analysis. Chromatographic separation was achieved using a Genesis C18 column (50 × 4.6 mm, 3 μm) and a gradient of 1% acetic acid in water as mobile phase A and methanol as mobile phase B. An API 4000 mass spectrometer (AB Sciex, Framingham, Massachusetts, USA) employing electrospray ionization in positive ion mode was used to monitor the analyte. Multiple reaction monitoring (MRM) transitions were m/z 194.9 → 138.2 for caffeine and 204.2 → 144.1 for the internal standard (caffeine‐d9). The lower limit of quantification (calibration range), interassay precision and accuracy/bias were 25.0 (25.0–20 000) ng ml−1, 3.4–8.6% and −2.3 to −0.1%, respectively.
Regarding midazolam, a plasma sample volume of 0.200 ml was combined with the internal standard (alpha‐hydroxytriazolam) and extracted from human plasma by automated liquid–liquid extraction. Following extraction, samples were evaporated to dryness under a stream of nitrogen. The resulting residue was reconstituted in methanol:water (50:50, v/v) and the samples were submitted for analysis. Chromatographic separation was achieved using a Merck Chromolith SpeedROD RP‐18e column (50 × 4.6 mm; Merck KGaA, Darmstadt, Germany) and gradient conditions with mobile phase A consisting of 0.15% trifluoroacetic acid in 20 mM ammonium formate and pure methanol as mobile phase B. An API 4000 mass spectrometer (AB Sciex, Framingham, Massachusetts, USA) employing electrospray ionization in positive ion mode was used to monitor the analyte. MRM transitions were m/z 326 → 291 for midazolam and 359 → 176 for the internal standard (alpha‐hydroxytriazolam). The lower limit of quantification (calibration range), interassay precision and accuracy/bias were 0.05000 (0.0500–50.0) ng ml−1, 4.2–11.9% and −4.3 to 4.4%, respectively.
Regarding omeprazole, a plasma sample volume of 50.0 μl was basified using ammonia, combined with the internal standard (omeprazole‐d3) and then underwent liquid–liquid extraction using ethyl acetate. After centrifugation, the organic layer was transferred to a clean plate and diluted with an equal volume of acetonitrile before analysis. Chromatographic separation was achieved using a Thermo Scientific BetaSil Silica‐100 (50 × 3.0 mm, 5 μm; Thermo Fisher Scientific, Waltham, Massachusetts, USA) and a gradient using mobile phases consisting of acetonitrile:water:formic acid (5:95:0.1, v:v:v) and acetonitrile:water:formic acid (95:5:0.1, v:v:v). An API 4000 mass spectrometer (AB Sciex, Framingham, Massachusetts, USA) in positive ion mode was used to monitor the analyte. MRM transitions were m/z 346.0 → 198.3 for omeprazole and 349.0 → 198.3 for the internal standard (omeprazole‐d3). The lower limit of quantification (calibration range), interassay precision and accuracy/bias were 0.500 (0.500–1000) ng ml−1, 3.8–6.6% and −6.5 to 0.6%, respectively.
Regarding S‐warfarin, a plasma sample volume of 0.250 ml was combined with the internal standard (S‐warfarin‐d6) and extracted from human plasma by liquid–liquid extraction using ethyl acetate in hexane. The organic layer was evaporated to dryness under a stream of nitrogen and the resulting residue was reconstituted in acetonitrile before analysis. Chromatographic separation was achieved using an Astec β‐Cyclodextrin Cyclobond I column (250 × 4.6 mm, 5 μm, Phenomenex Inc., Torrance, California, USA) and isocratic conditions with a mobile phase of acetonitrile:acetic acid:triethylamine (1000:3:2.5, v:v:v). An API 4000 mass spectrometer (AB Sciex, Framingham, Massachusetts, USA) employing atmospheric pressure chemical ionization in negative ion mode was used to monitor the analyte. MRM transitions were m/z 307.1 → 161.0 for S‐warfarin and 313.1.1 → 161.0 for the internal standard (S‐warfarin‐d6). The lower limit of quantification (calibration range), interassay precision and accuracy/bias were 5.00 (5.00–1500) ng ml−1, 12.8–18.6% and −2.7 to 2.6%, respectively.
Urine samples were analysed for dextromethorphan and dextrorphan using a validated assay developed by Pharmaceutical Product Development, LLC (Middleton, Wisconsin, USA) 10. Dextromethorphan and dextrorphan were extracted from 50 μl of urine incubated with beta‐glucuronidase solution and internal standards (dextromethorphan‐D3/dextrorphan‐D3/3‐methoxymorphinan‐D3). Extracts were analysed by LC with MS/MS detection. The lower limit of quantification (calibration range), interassay precision and accuracy/bias were, respectively, 0.00100 (0.00100–1.00) μg ml−1, 3.59–6.71% and −12.5 to −6.84% for dextromethorphan and 0.0200 (0.0200–20.0) μg ml−1, 0.895–2.31% and −5.88 to 2.89% for dextrorphan.
A validated sandwich enzyme‐linked immunosorbent assay method was used to determine DAC HYP concentrations in human serum. In this assay format, microtiter plates were coated with anti‐idiotype DAC HYP antibody, followed by blocking, washing and incubation with 1/100 diluted calibrators, controls and samples. A biotin‐conjugated anti‐human IgG was then added to detect bound DAC HYP. After another plate washing, a horseradish peroxidase conjugated to streptavidin was added and bound horseradish peroxidase conjugate was detected with a tetramethyl benzidine substrate, which was read colorimetrically on a plate reader. The standard curve range is from 500–7500 ng ml−1 for DAC HYP in human serum. Samples measuring outside assay limits of quantitation were re‐analysed at an appropriate dilution. If the sample result fell below the lower limit of quantitation of 500 ng ml−1 and the sample dilution was at the minimum required dilution (1/100), the result for this sample was reported as below the limit of quantitation.
PK and statistical analysis
Plasma PK parameters for the probe drug cocktail were determined using the actual sample collection times and non‐compartmental method (Phoenix® WinNonlin® 6.2.1; Pharsight Corporation, Sunnyvale, California, USA). Pre‐dose concentrations higher than 5% of Cmax were excluded from all PK evaluations. The 12 h urinary ratio of dextromethorphan to dextrorphan was calculated. Pre‐specified primary PK endpoints were area under the curve (AUC) from 0 to infinity (AUC[0–inf]) for midazolam, caffeine, S‐warfarin and omeprazole, and 12 h urine dextromethorphan to dextrorphan ratio. Considering the objective of this study was to determine the effect of DAC HYP on CYP activities using sensitive probe substrates, evaluation of plasma AUC of the probe substrates provides a better and more accurate phenotypic measure of the CYP activities than Cmax or plasma ratio at a given time point. Caffeine metabolites were not analysed because the impact of DAC HYP on CYP1A2 can be determined based on caffeine exposure.
Statistical analyses were conducted on log‐transformed PK parameters and urinary ratio using a linear mixed‐effects model with treatment as a fixed effect and subject as a random effect using SAS version 9.3 (SAS Institute, Cary, North Carolina, USA). To evaluate the effect of DAC HYP on probe drug PK, the adjusted geometric mean ratio between treatment in period 2 (probe drug cocktail + DAC HYP) and treatment in period 1 (probe drug cocktail alone) was calculated, along with the associated 90% confidence interval (CI).
Results
Patient demographics
Twenty patients with relapsing–remitting multiple sclerosis (13 women and seven men; 16 white, three black and one other race; nine patients from three clinics in Hungary, seven patients from one clinic in Poland and four patients from two clinics in the United States) were enrolled. The mean (range) age, body weight and body mass index were 36 (19–52) years, 72.9 (48.5–118) kg and 24.5 (17.5–40.8), respectively. Ten (50%) patients had a history of receiving prior medications for multiple sclerosis (IFN beta‐1a, IFN beta‐1b, glatiramer acetate, fumaric acid, laquinimod and natalizumab). The mean ± standard deviation (SD) number of gadolinium‐enhancing (Gd+) lesions was 2.3 ± 3.11 at baseline. Ten (50%) patients had one or more Gd+ lesion(s) at baseline, of which five (25%) patients had four or more Gd+ lesions. All patients completed the study. However, one patient did not have blood PK samples collected during period 2 due to study site error. No patients received any prohibited medications during the study.
Pharmacogenetics
Pharmacogenetic analysis identified three patients with predicted poor metabolizer (PM) status for CYP2D6 and one patient with predicted PM status for CYP2C19. Urine concentration ratios of dextromethorphan to dextrorphan were consistent with the CYP2D6 genotype, with the highest ratios observed in the three PMs of CYP2D6 (data not shown). However, omeprazole exposure from the PM of CYP2C19 was within the range of exposures from the other extensive metabolizers of CYP2C19 (data not shown). Statistical analysis for the effect of DAC HYP on CYP probe substrates included all data, because excluding data from the patients with predicted PM status did not influence the results.
Pharmacokinetics
Pre‐dose serum DAC HYP concentrations
The mean (SD) pre‐dose serum concentration of DAC HYP at the third dose was 16.5 (8.46) μg ml−1, which is consistent with the previously characterized mean steady‐state trough concentration of 14.9 (6.33) μg ml−1 following repeated dosing of DAC HYP 150 mg SC in patients with multiple sclerosis 16.
Effects of DAC HYP on exposure of the CYP probes
The mean (SD) plasma concentration–time profiles for midazolam, S‐warfarin, omeprazole and caffeine following administration of the probe drug cocktail before or after multiple doses of DAC HYP are illustrated in Figure 1 and are almost superimposable. The mean plasma concentration–time profiles for caffeine are shown over the 24 h post dose because caffeine concentrations in most patients rebounded at or after 24 h post dose, which was likely due to ingestion of caffeinated drinks or food because the patients were discharged from the clinic ~12 h after dosing. This observation has also been reported in another drug cocktail study where caffeine drug concentrations rebounded after the patients were discharged from the clinic 24 h post dose 17.
Figure 1.
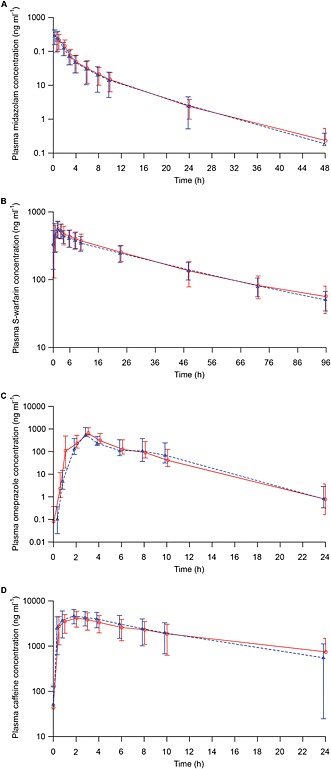
Plasma concentration–time profiles for (A) midazolam, (B) S‐warfarin, (C) omeprazole and (D) caffeine following administration of a probe drug cocktail alone or with daclizumab high‐yield process (DAC HYP) in patients with multiple sclerosis. Mean and standard deviation for midazolam (n = 19), S‐warfarin (n = 19), omeprazole (n = 18) and caffeine (n = 12). Treatment: ( ) Cocktail alone, (
) Cocktail alone, ( ) Cocktail + DAC HYP
) Cocktail + DAC HYP
Statistical comparisons (probe drug cocktail with DAC HYP vs. probe drug cocktail alone) of midazolam, S‐warfarin, omeprazole and caffeine AUC and 12 h urine concentration ratio for dextromethorphan to dextrorphan are presented in Table 1. Repeated administration of DAC HYP 150 mg SC every 4 weeks in patients with multiple sclerosis had no effect on the primary PK endpoint for caffeine (CYP1A2), S‐warfarin (CYP2C9), omeprazole (CYP2C19) and midazolam (CYP3A). The 90% CIs for AUC(0–inf) for S‐warfarin, omeprazole and midazolam were within the default no‐effect boundary of 0.80–1.25. Caffeine AUC(0–inf) and terminal elimination half‐life were not reportable because caffeine concentrations in most patients rebounded at or after 24 h post dose. Therefore, AUC from 0–12 h (AUC0–12) for caffeine, which is not expected to be confounded by external caffeine ingestion after end of the 12 h clinic confinement, was calculated and reported. The 90% CI for AUC0–12 for caffeine was within the default no‐effect boundary of 0.80–1.25. The geometric mean ratio for the 12 h urine dextromethorphan to dextrorphan ratio was close to 1; however, 90% CIs for the ratio extended on both sides slightly outside the default no‐effect boundary of 0.80–1.25, most likely due to high variability associated with urine collections and CYP2D6 activity. For the assessment of secondary PK endpoint, the 90% CIs for S‐warfarin and caffeine Cmax were within the boundary of 0.80–1.25. However, the 90% CIs for midazolam and omeprazole Cmax extended slightly outside of the boundary of 0.80–1.25 (with point estimates of 1.01 and 1.08, respectively; Table 1).
Table 1.
Effects of DAC HYP 150 mg SC every 4 weeks on the primary PK parameters of CYP probes: summary of analysis of variance
| Probe drug | Parameter (unit) | n | Mean (%CV) | Ratio ([probe drug + DAC HYP]/probe drug alone) | 90% CI for the ratio | |
|---|---|---|---|---|---|---|
| Probe drug alone | Probe drug + DAC HYP | |||||
| Midazolam | AUC(0–inf) (ng h ml−1) | 19 | 800.8 (41) | 816.9 (49) | 1.01 | 0.89–1.15 |
| Cmax (ng ml−1) | 19 | 278 (38) | 311 (48) | 1.01 | 0.90–1.26 | |
| t1/2 (h) | 19 | 5.68 (25) | 5.57 (25) | NC | NC | |
| S‐warfarin | AUC(0–inf) (ng h ml−1) | 16* | 19 760 (27) | 19 680 (25) | 1.00 | 0.95–1.06 |
| Cmax (ng ml−1) | 19 | 641 (22) | 650 (24) | 1.01 | 0.95–1.07 | |
| t1/2 (h) | 19 | 31.1 (26) | 31.9 (20) | NC | NC | |
| Omeprazole | AUC(0–inf) (ng h ml−1) | 17† | 2040 (127) | 1810 (97) | 1.00 | 0.88–1.13 |
| Cmax (ng ml−1) | 18 | 745 (68) | 785 (70) | 1.08 | 0.82–1.43 | |
| t1/2 (h) | 17† | 1.22 (72) | 1.12 (64) | NC | NC | |
| Caffeine | AUC0–12 (ng h ml−1)‡ | 12§ | 32 350 (37) | 36 110 (43) | 1.03 | 0.93–1.14 |
| Cmax (ng ml−1) | 12§ | 4734 (29) | 5352 (30) | 1.11 | 1.00–1.23 | |
| t1/2 (h)‡ | NC | NC | NC | NC | NC | |
| Dextromethorphan | 12‐h urine dextromethorphan to dextrorphan ratio | 20 | 0.425 (296)¶ | 0.489 (370)¶ | 1.01 | 0.76–1.34 |
%CV, coefficient of variation; AUC0–12, area under the curve from 0–12 h; AUC(0–inf), area under the curve from 0 to infinity; CI, confidence interval; Cmax, maximum concentration; CYP, cytochrome P450; DAC HYP, daclizumab high‐yield process; NC, not calculated; PK, pharmacokinetics; SC, subcutaneous; t1/2, terminal elimination half‐life.
AUC(0–inf) values extrapolated by >20% were excluded from the analysis.
Two patients with uncharacterizable terminal elimination phase and hence no AUC(0–inf) and t1/2 determined.
Caffeine AUC(0–inf) and t1/2 not reportable due to caffeine concentrations in most patients rebounded at or after 24 h post dose, which is probably due to ingestion of caffeinated drinks or food as patients were discharged 12 h post dose.
Seven patients in periods 1 and 2 had predose caffeine concentrations >5% of Cmax and data from these patients were excluded from all PK and statistical analysis.
High %CV due to three patients with poor metabolizer status for CYP2D6.
Safety and tolerability
One (5%) patient in period 1 (probe drug cocktail alone) and three (15%) patients in period 2 (probe drug cocktail + DAC HYP) experienced one or more treatment‐emergent adverse event(s) (e.g. diarrhoea, nasopharyngitis, sensation of heaviness). All adverse events were mild in severity and were not considered related to DAC HYP therapy. No clinically relevant changes in vital signs or electrocardiogram were observed during the TP‐DI sub‐study.
Discussion
To our knowledge, this is first comprehensive clinical study to assess potential effects of a mAb that blocks the high‐affinity IL‐2R on activity of CYP enzymes, and one of the few robust therapeutic protein–small molecule drug interaction studies conducted to date. Results from this study show that repeated administration of the DAC HYP dosing regimen evaluated in pivotal studies in patients with multiple sclerosis (i.e. 150 mg SC every 4 weeks) did not affect the systemic exposure of concomitantly administered sensitive probe substrates for CYP1A2 (caffeine), CYP2C9 (S‐warfarin), CYP2C19 (omeprazole), CYP2D6 (dextromethorphan) and CYP3A (oral midazolam). While meeting strict bioequivalence criteria is not generally required to declare no clinically relevant drug interaction, the 90% CIs for plasma AUC for caffeine, S‐warfarin, omeprazole and midazolam were within the default no‐effect boundary of 0.80–1.25, with point estimates ranging from 1.00–1.03. The 90% CIs for S‐warfarin and caffeine Cmax were within the bioequivalence criteria, while the 90% CIs for midazolam and omeprazole Cmax extended slightly outside the upper boundary of 1.25 (with point estimates of the ratios close to 1). Considering the objective of this study was to determine the effect of DAC HYP on CYP activities using sensitive probe substrates, evaluation of AUC of the probe substrates provides a more robust phenotypic measure of the CYP activities than Cmax of the probe substrates. The 90% CI for the urine dextromethorphan to dextrorphan ratio (a phenotypic measure of CYP2D6 activity) extended slightly outside the default no‐effect boundary on both sides (0.76–1.34), with a point estimate of 1.01. The wider CI for the urine dextromethorphan to dextrorphan ratio is most likely due to the high intra‐individual variability associated with urine collections and CYP2D6 activity, which has been reported to be ~50% 18. Another study with a comparable sample size (21 patients with cancer) showed a comparable CI (90% CI, 0.70–1.30) for the 12 h urine dextromethorphan to dextrorphan ratio with sorafenib, a drug with no clinically relevant interaction with CYP2D6 13.
The potential impact of the IL‐2 modulator on CYP3A‐mediated metabolism has been suggested based on two reports for basiliximab, an anti‐IL‐2R alpha mAb. In both reports, a retrospective analysis or review of medical records suggests that concentrations of CYP3A substrates (i.e. cyclosporine, tacrolimus) were decreased in paediatric renal transplant patients after basiliximab administration or were lower in transplant patients who received basiliximab compared with patients who received placebo 7, 8. The authors hypothesized that binding of basiliximab to the IL‐2R on activated T cells allows circulating IL‐2 to bind to IL‐2Rs on hepatic and intestinal cells, resulting in downregulation of CYP3A activity 8. The major limitations of these previous reports include the retrospective nature of these analyses, limited sample size, variability in monitoring and measuring drug concentrations, high PK variability for the evaluated drug and cross‐patient comparison. In contrast, the present study for DAC HYP was a well‐designed prospective clinical TP‐DI study using recommended sensitive probe substrates of CYP activities. Another difference between our study and the retrospective reports for basiliximab is the difference in patient populations, multiple sclerosis vs. renal transplant patients. Disease–drug interaction could be an important factor for therapeutic proteins that modulate cytokines due to the association between elevated pro‐inflammatory cytokine levels at baseline (i.e. before therapeutic protein treatment) and CYP activity 19, 20. Lack of published literature on baseline IL‐2 levels in renal transplant patients does not allow for comparison between the two populations. Furthermore, the magnitude of increase in IL‐2 levels following basiliximab therapy in transplant patients is not known.
Because the potential impact of therapeutic proteins on CYP enzymes is believed to be mediated mainly through modulation of cytokine levels (either elevation of cytokines post treatment or normalization of cytokine levels altered by disease state), assessment of the TP‐DI in vitro, using current available pre‐clinical approaches, is of limited predictive value 21. This is because these in vitro systems cannot reliably capture the involved complex in vivo pharmacology. To our knowledge, the most well‐documented clinical effects on CYPs to date are those therapeutic proteins modulating IL‐6. A TP‐DI study using a cocktail approach showed that treatment with sirukumab, an mAb targeting IL‐6, in patients with rheumatoid arthritis had modest effects on the activity of CYP3A (30–35% reduction), CYP2C19 (37–45% reduction) and CYP2C9 (18–19% reduction) 22. Similarly, tocilizumab, an mAb targeting the IL‐6R, was shown to decrease the in vivo activity of CYP2C19 and CYP3A by 28% and 57%, respectively 23, 24. For both of these drugs, the effects on CYP activity were considered likely to be relevant to drugs with a narrow therapeutic index. Inhibition of the cytokine nuclear factor kappa‐B ligand (RANKL) by denosumab, an mAb that binds to and neutralizes RANKL activity, did not affect the exposure of midazolam, a CYP3A substrate, in post‐menopausal women with osteoporosis 25.
In conclusion, multiple dosing of the proposed clinical regimen of DAC HYP 150 mg SC every 4 weeks in patients with multiple sclerosis had no effect on CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A. No dosage adjustments are needed for drugs that are substrates of these CYP enzymes when given concomitantly with DAC HYP. The results from this study also provide important clinical information on the potential impact of cytokine modulators on CYP enzyme activities, contributing to the clinical database to guide future recommendation on TP‐DI assessment.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: JQT was a full‐time employee of and stockholder in Biogen when this study was conducted and during the development of the manuscript. PW and JE are employees of and stockholders in Biogen. AAO is an employee of and a stockholder in AbbVie. The authors declare no other relationships or activities that could appear to have influenced the submitted work.
The study was funded by Biogen and AbbVie Biotherapeutics Inc. Biogen and AbbVie Biotherapeutics Inc. provided funding for medical writing support in the development of this paper; Rebecca Jarvis from Excel Scientific Solutions, Southport, Connecticut, USA, provided editorial support, and Kristen DeYoung from Excel Scientific Solutions, Southport, Connecticut, USA, copyedited and styled the manuscript per journal requirements. Biogen reviewed and provided feedback on the paper to the authors. The authors had full editorial control of the paper, and provided their final approval of all content. JQT and AAO designed the study. PW and JE monitored the study. JQT and AAO analysed the data and interpreted the results. JQT wrote the manuscript. AAO, PW and JE reviewed, provided edits and approved the manuscript.
Tran, J. Q. , Othman, A. A. , Wolstencroft, P. , and Elkins, J. (2016) Therapeutic protein–drug interaction assessment for daclizumab high‐yield process in patients with multiple sclerosis using a cocktail approach. Br J Clin Pharmacol, 82: 160–167. doi: 10.1111/bcp.12936.
References
- 1. Huang S‐M, Zhao H, Lee J‐I, Reynolds K, Zhang L, Temple R, et al. Therapeutic protein–drug interactions and implications for drug development. Clin Pharmacol Ther 2010; 87: 497–503. [DOI] [PubMed] [Google Scholar]
- 2. Lee J‐I, Zhang L, Men AY, Kenna LA, Huang S‐M. CYP‐mediated therapeutic protein–drug interactions: clinical findings, proposed mechanisms and regulatory implications. Clin Pharmacokinet 2010; 49: 295–310. [DOI] [PubMed] [Google Scholar]
- 3. US Department of Health and Human Services, US Food and Drug Administration, Center for Drug Evaluation and Research . Guidance for industry. Drug interaction studies – study design, data analysis, implications for dosing and labeling recommendations. Available at http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf (last accessed 7 October 2015).
- 4. Bielekova B. Daclizumab therapy for multiple sclerosis. Neurotherapeutics 2013; 10: 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huss DJ, Mehta DS, Sharma A, You X, Riester KA, Sheridan JP, et al. In vivo maintenance of human regulatory T cells during CD25 blockade. J Immunol 2015; 194: 84–92. [DOI] [PubMed] [Google Scholar]
- 6. Elkahwaji J, Robin MA, Berson A, Tinel M, Lettéron P, Labbe G, et al. Decrease in hepatic cytochrome P450 after interleukin‐2 immunotherapy. Biochem Pharmacol 1999; 57: 951–4. [DOI] [PubMed] [Google Scholar]
- 7. Strehlau J, Pape L, Offner G, Nashan B, Ehrich JH. Interleukin‐2 receptor antibody‐induced alterations of ciclosporin dose requirements in paediatric transplant recipients. Lancet 2000; 356: 1327–8. [DOI] [PubMed] [Google Scholar]
- 8. Sifontis NM, Benedetti E, Vasquez EM. Clinically significant drug interaction between basiliximab and tacrolimus in renal transplant recipients. Transplant Proc 2002; 34: 1730–2. [DOI] [PubMed] [Google Scholar]
- 9. Ma JD, Nafziger AN, Villano SA, Gaedigk A, Bertino JS Jr. Maribavir pharmacokinetics and the effects of multiple‐dose maribavir on cytochrome P450 (CYP) 1 A2, CYP 2C9, CYP 2C19, CYP 2D6, CYP 3 A, N‐acetyltransferase‐2, and xanthine oxidase activities in healthy adults. Antimicrob Agents Chemother 2006; 50: 1130–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson BM, Song IH, Adkison KK, Borland J, Fang L, Lou Y, et al. Evaluation of the drug interaction potential of aplaviroc, a novel human immunodeficiency virus entry inhibitor, using a modified Cooperstown 5 + 1 cocktail. J Clin Pharmacol 2006; 46: 577–87. [DOI] [PubMed] [Google Scholar]
- 11. Goh BC, Reddy NJ, Dandamudi UB, Laubscher KH, Peckham T, Hodge JP, et al. An evaluation of the drug interaction potential of pazopanib, an oral vascular endothelial growth factor receptor tyrosine kinase inhibitor, using a modified Cooperstown 5 + 1 cocktail in patients with advanced solid tumors. Clin Pharmacol Ther 2010; 88: 652–9. [DOI] [PubMed] [Google Scholar]
- 12. Jenkins J, Williams D, Deng Y, Collins DA, Kitchen VS. Eltrombopag, an oral thrombopoietin receptor agonist, has no impact on the pharmacokinetic profile of probe drugs for cytochrome P450 isoenzymes CYP3A4, CYP1A2, CYP2C9 and CYP2C19 in healthy men: a cocktail analysis. Eur J Clin Pharmacol 2010; 66: 67–76. [DOI] [PubMed] [Google Scholar]
- 13. Flaherty KT, Lathia C, Frye RF, Schuchter L, Redlinger M, Rosen M, et al. Interaction of sorafenib and cytochrome P450 isoenzymes in patients with advanced melanoma: a phase I/II pharmacokinetic interaction study. Cancer Chemother Pharmacol 2011; 68: 1111–8. [DOI] [PubMed] [Google Scholar]
- 14. Morcos PN, Chang L, Kulkarni R, Giraudon M, Shulman N, Brennan BJ, et al. A randomised study of the effect of danoprevir/ritonavir or ritonavir on substrates of cytochrome P450 (CYP) 3 A and 2C9 in chronic hepatitis C patients using a drug cocktail. Eur J Clin Pharmacol 2013; 69: 1939–49. [DOI] [PubMed] [Google Scholar]
- 15. Tsunoda SM, Velez RL, von Moltke LL, Greenblatt DJ. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: effect of ketoconazole. Clin Pharmacol Ther 1999; 66: 461–71. [DOI] [PubMed] [Google Scholar]
- 16. Tran J, Othman A, Wu Y, Mikulskis A, Wolstencroft P, Elkins J. Pharmacokinetics of daclizumab high‐yield process (DAC HYP) with repeated administration of the clinical subcutaneous regimen in subjects with relapsing–remitting multiple sclerosis. Clin Pharmacol 2016; 8: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Turpault S, Brian W, Van Horn R, Santoni A, Poitiers F, Donazzolo Y, et al. Pharmacokinetic assessment of a five‐probe cocktail for CYPs 1A2, 2C9, 2C19, 2D6 and 3A. Br J Clin Pharmacol 2009; 68: 928–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kashuba AD, Nafziger AN, Kearns GL, Leeder JS, Gotschall R, Rocci ML Jr, et al. Effect of fluvoxamine therapy on the activities of CYP1A2, CYP2D6, and CYP3A as determined by phenotyping. Clin Pharmacol Ther 1998; 64: 257–68. [DOI] [PubMed] [Google Scholar]
- 19. Aitken AE, Richardson TA, Morgan ET. Regulation of drug‐metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol 2006; 46: 123–49. [DOI] [PubMed] [Google Scholar]
- 20. Morgan ET. Impact of infectious and inflammatory disease on cytochrome P450‐mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther 2009; 85: 434–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Evers R, Dallas S, Dickmann LJ, Fahmi OA, Kenny JR, Kraynov E, et al. Critical review of preclinical approaches to investigate cytochrome P450‐mediated therapeutic protein drug–drug interactions and recommendations for best practices: a White Paper. Drug Metab Dispos 2013; 41: 1598–609. [DOI] [PubMed] [Google Scholar]
- 22. Zhuang Y, de Vries DE, Xu Z, Marciniak SJ Jr, Chen D, Leon F, et al. Evaluation of disease‐mediated therapeutic protein–drug interactions between an anti‐interleukin‐6 monoclonal antibody (sirukumab) and cytochrome P450 activities in a phase 1 study in patients with rheumatoid arthritis using a cocktail approach. J Clin Pharmacol 2015; 55: 1386–94. [DOI] [PubMed] [Google Scholar]
- 23. Schmitt C, Kuhn B, Zhang X, Kivitz AJ, Grange S. Disease–drug–drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther 2011; 89: 735–40. [DOI] [PubMed] [Google Scholar]
- 24. Kim S, Östör AJ, Nisar MK. Interleukin‐6 and cytochrome‐P450, reason for concern? Rheumatol Int 2012; 32: 2601–4. [DOI] [PubMed] [Google Scholar]
- 25. Jang G, Kaufman A, Lee E, Hamilton L, Hutton S, Egbuna O, et al. A clinical therapeutic protein drug–drug interaction study: coadministration of denosumab and midazolam in postmenopausal women with osteoporosis. Pharmacol Res Perspect 2014; 2: e00033. [DOI] [PMC free article] [PubMed] [Google Scholar]