

Abstract
Tuberculosis remains a global public health problem, and its immunopathogenesis is still poorly understood. In this study, 25 single nucleotide polymorphisms (SNPs) in the WNT pathway were evaluated in relation to tuberculosis risk in a Chinese Han discovery set, and 6 candidate susceptible SNPs were further validated in a Chinese Tibetan cohort. Luciferase reporter assay, RT-qPCR and Western blot were used to assess the functionality of the important WNT polymorphisms. Five polymorphisms were associated with tuberculosis susceptibility after Bonferroni correction: SFRP1 rs4736958, CTNNB1 rs9859392, rs9870255 and rs3864004 showed decreased tuberculosis risk; SFRP1 rs7832767 was related to an increased risk (OR = 1.81, 95% CI = 1.30–2.52, p = 0.010). Patients with TT genotype of rs4736958 and rs7832767 correlated with higher CRP concentrations (p = 0.003, <0.001, respectively). Functional assays revealed that mutant alleles of rs9859392 (G), rs9870255 (C) and rs3864004 (A) were associated with significantly decreased transcriptional activity, lower CTNNB1 mRNA expression and p-β-catenin level, which were consistent with their effects of decreasing TB risk. Our results provide evidences that WNT pathway polymorphisms influence tuberculosis susceptibility and host immune response to Mycobacterium tuberculosis, suggesting that these variations may serve as novel markers for identifying the risk of developing tuberculosis.
Tuberculosis (TB) remains a leading infectious cause of morbidity and mortality worldwide, with an estimated 9.6 million cases and 1.5 million deaths occurring in 20141. More than one-third of the global population is infected with Mycobacterium tuberculosis (MTB), however, only 5–10% of infections will develop into clinical TB2, suggesting that genetic factors play an important role in the disease outcome after MTB infection. Previous studies have suggested that host genetic variations may contribute to the heterogeneity in disease susceptibility and clinical responses through modulating the immune reaction against MTB3,4. Over the years, association studies have evaluated many candidate loci that may be associated with susceptibility to TB, including the genes encoding human leukocyte antigen5, cytokines and chemokines6, toll-like receptor and others7. However, most of the studies only investigated a few polymorphisms and were unable to draw definitive conclusions. Thus, more systematic and comprehensive studies are needed to fully clarify the genetic components affecting TB progression. Recently, pathway-based association studies based on the biological functions of related signaling pathway networks were found to be a powerful approach to identify causal genes underlying complex diseases8.
The WNT signaling pathway is known as a key transduction cascade governing ontogeny and homeostatic processes9, as well as modulating host immune responses against microbial pathogens10. It is activated when Wnt proteins bind to Frizzled receptors and co-receptors through a series of cascade reactions, which causes β-catenin co-activating transcription factors to regulate the downstream gene transcription. Antagonists, including the secreted frizzled-related protein (SFRP) family, Wnt inhibitory protein one (WIF1) and Dickkopf (DKK), inhibit WNT pathway signaling by binding to Wnt or Wnt receptors and negatively regulating target gene expression9. Recent evidence has indicated that the WNT pathway has an etiological role in TB disease11,12,13,14,15. Wu et al. found that activation of Wnt/β-catenin signaling induced mycobacteria-infected cell apoptosis and promoted pro-inflammatory TNF-α and IL-6 production11. A study by Li Y et al. revealed that Wnt/β-catenin acted as a negative feedback loop to suppress inflammation stimulated by Mycobacterium bovis Bacillus Calmette-Guerin (BCG), thus alleviating TLR-induced pro-inflammatory responses in alveolar epithelial cells12. Neumann et al. reported that Wnt/Frizzled signaling was vital in bridging innate and adaptive immunity in MTB infection13,14. Schaale K. et al.15 further found that Wnt6 expressed in granulomatous lesions participated in macrophage differentiation and proliferation by driving macrophage polarization toward an M2-like phenotype. The specific molecular mechanism of the WNT pathway in the progression of MTB has garnered interest recently but remains poorly understood. What we do know is that genetic variations within the WNT pathway have been associated with the development of various diseases, such as cancers16 and infectious diseases17, suggesting that polymorphisms within the WNT pathway may similarly affect TB susceptibility.
China is experiencing a serious epidemic of TB and has the highest annual number of MDR-TB cases worldwide, ranking second among the 22 high-burden countries18. In West China, the annual incidence of pulmonary tuberculosis is significantly higher than that in the rest of China, reaching 695 cases per 100,000 inhabitants19. Screening a high-risk population among exposed/infected individuals is of great importance in the TB control program established to reduce the burden of disease. We previously investigated a small amount of SNPs in CTNNB1 gene and SFRP1 gene in a small sample of Western Chinese, and we identified that two SNPs (CTNNB1 rs4135385 and SFRP1 rs7832767) were associated with TB risk, indicating a potential role of these variants in susceptibility to TB20. Hence, it is necessary and rational to screen more TB-associated gene loci based on the WNT signaling pathway. To provide a full understanding of the relationship between polymorphisms within the WNT signaling pathway and TB risk, we conducted a 2-stage case-control study (832 cases and 1084 controls in Chinese Han discovery set; 480 cases and 450 controls in Chinese Tibetan validation set) and functional assays for susceptible polymorphisms at both mRNA and protein level.
Results
General characteristics of the study population
Demographic characteristics of cases and controls are presented in Table 1. In general, both groups mainly consisted of middle-aged males, and statistically significant differences were observed for BMI and smoking (p < 0.001, 0.023, respectively), and not found in BCG status (p = 0.089). TB patients involved 367 pulmonary tuberculosis (PTB, 44.11%), 120 extra-pulmonary TB (EPTB, 14.42%) and 345 PTB combined with EPTB (PTB&EPTB, 41.47%).
Table 1. Demographic characteristics of study participants in Chinese Han population.
| Characteristics | TBa (n = 832) | HCb (n = 1084) | P |
|---|---|---|---|
| Age, mean ± SD (years) | 41.10 ± 19.04 | 40.54 ± 10.69 | 0.574 |
| Male/female | 496/336 | 629/455 | 0.484 |
| BMI (kg/m2) | 20.99 ± 2.55 | 23.03 ± 2.06 | <0.001 |
| BCG scar n (%) | 0.089 | ||
| Yes | 376 (45.19) | 453 (41.79) | |
| No | 361 (43.39) | 524 (48.34) | |
| Uncertain | 95 (11.42) | 107 (9.87) | |
| Smoking n (%) | 0.023 | ||
| Smoking | 262 (31.49) | 283 (26.10) | |
| Ever Smoking | 135 (16.23) | 207 (19.09) | |
| Nonsmoking | 435 (52.28) | 594 (54.81) | |
| TB subtype n (%) | |||
| PTBc | 367 (44.11) | ||
| EPTBd | 120 (14.42) | ||
| PTB & EPTB | 345 (41.47) | ||
aTuberculosis patients.
bHealthy controls.
cPulmonary tuberculosis (PTB).
dExtra pulmonary tuberculosis (EPTB).
Polymorphisms association with tuberculosis in Chinese Han population
Single SNP association
Of the 23 SNPs successfully genotyped, six were found to be significantly associated with TB (Tables 2 and 3). In the CTNNB1 gene, rs9859392, rs9870255 and rs3864004 were significantly different between patients and controls. Subjects with mutant alleles (G, C and A) and dominant model of the three SNPs were associated with decreased TB susceptibility, and the OR ranged from 0.69 to 0.78 (p < 0.05). Of the four SNPs in SFRP1, two were found to be significant. A reduced risk of TB was related with the mutant C allele, the additive and dominant pattern of rs4736958 with an estimated OR of 0.78, 0.78 and 0.73 (p = 0.033, 0.040 and 0.017, after Bonferroni correction). The minor T allele, additive and recessive model of rs7832767 were more frequent in the patients than in the controls (1.30-, 1.29- and 1.81-fold, respectively). A potential beneficial effect was also identified for the additive and dominant model of rs752107 in WNT3A [ORadd = 0.85 (0.73–0.99), padd = 0.040; ORdom = 0.82 (0.68–0.98), pdom = 0.033], however, the potential association disappeared after Bonferroni correction in rs752107. No significant differences among the frequencies of the other 18 SNPs were obtained between the patients and the controls.
Table 2. Comparison of frequency distributions of WNT pathway polymorphisms in Chinese Han population.
| Gene | SNP | Group | Allele n (%) |
OR (95% CI) | P* | P** | Genotype n (%) |
P* | P** | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 11 | 12 | 22 | ||||||||
| CTNNB1 | rs9859392 | TB | 316 (19.41) | 1312 (80.59) | 0.75 (0.64–0.88) | 4.07 ×10−4 | 0.009 | 38 (4.67) | 240 (29.48) | 536 (65.85) | 6.57 × 10−4 | 0.013 |
| C > G | HC | 515 (24.25) | 1609 (75.75) | 60 (5.65) | 395 (37.19) | 607 (57.16) | ||||||
| rs9870255 | TB | 323 (19.84) | 1305 (80.16) | 0.76 (0.65–0.89) | 7.66 × 10−4 | 0.018 | 34 (4.18) | 255 (31.32) | 525 (64.50) | 0.003 | 0.092 | |
| G > C | HC | 522 (24.46) | 1612 (75.54) | 64 (6.00) | 394 (36.92) | 609 (57.08) | ||||||
| rs3864004 | TB | 322 (19.80) | 1304 (80.20) | 0.77 (0.66–0.91) | 0.001 | 0.033 | 36 (4.43) | 250 (30.75) | 527 (64.82) | 0.007 | 0.149 | |
| G > A | HC | 509 (24.19) | 1595 (75.81) | 67 (6.37) | 375 (35.65) | 610 (57.98) | ||||||
| WIF1 | rs34203757 | TB | 280 (17.11) | 1356 (82.89) | 0.92 (0.78–1.09) | 0.327 | 28 (3.47) | 224 (27.72) | 556 (68.81) | 0.592 | ||
| C > G | HC | 385 (18.14) | 1737 (81.86) | 44 (4.19) | 297 (28.31) | 708 (67.49) | ||||||
| rs34505206 | TB | 270 (16.52) | 1364 (83.48) | 0.87 (0.74–1.03) | 0.116 | 29 (3.55) | 212 (25.95) | 576 (70.50) | 0.311 | |||
| C > G | HC | 390 (18.50) | 1718 (81.50) | 46 (4.36) | 298 (28.27) | 710 (67.37) | ||||||
| rs56900803 | TB | 200 (12.27) | 1430 (87.73) | 1.00 (0.82–1.22) | 0.987 | 15 (1.84) | 170 (20.86) | 630 (77.30) | 0.359 | |||
| G > T | HC | 260 (12.25) | 1862 (87.75) | 12 (1.13) | 236 (22.24) | 813 (76.63) | ||||||
| DKK1 | rs11001553 | TB | 119 (7.48) | 1471 (92.52) | 1.04 (0.81–1.34) | 0.737 | 7 (0.88) | 105 (13.21) | 683 (85.91) | 0.925 | ||
| C > T | HC | 156 (7.2) | 2012 (92.80) | 8 (0.73) | 140 (12.92) | 936 (86.35) | ||||||
| rs1896367 | TB | 494 (30.31) | 1136 (69.69) | 0.98 (0.85–1.13) | 0.792 | 72 (8.83) | 350 (42.94) | 393 (48.23) | 0.272 | |||
| G > A | HC | 662 (30.71) | 1494 (69.29) | 115 (10.67) | 432 (40.07) | 531 (49.26) | ||||||
| rs1896368 | TB | 623 (38.46) | 997 (61.54) | 0.96 (0.84–1.09) | 0.499 | 114 (14.07) | 395 (48.77) | 301 (37.16) | 0.131 | |||
| G > A | HC | 825 (39.55) | 1261 (60.45) | 178 (17.06) | 469 (44.97) | 396 (37.97) | ||||||
| SFRP1 | rs3242 | TB | 101 (6.17) | 1535 (93.83) | 0.96 (0.74–1.25) | 0.776 | 13 (1.59) | 75 (9.17) | 730 (89.24) | 0.788 | ||
| C > T | HC | 138 (6.4) | 2018 (93.60) | 7 (0.65) | 124 (11.50) | 947 (87.85) | ||||||
| rs4736958 | TB | 471 (28.79) | 1165 (71.21) | 0.78 (0.69–0.92) | 0.001 | 0.033 | 79 (9.66) | 313 (38.26) | 426 (52.08) | 0.003 | 0.092 | |
| T > C | HC | 727 (33.66) | 1433 (66.34) | 125 (11.57) | 477 (44.17) | 478 (44.26) | ||||||
| rs72643819 | TB | 648 (39.80) | 980 (60.20) | 1.12 (0.98–1.28) | 0.088 | 154 (18.92) | 340 (41.77) | 320 (39.31) | 0.063 | |||
| G > T | HC | 798 (37.08) | 1354 (62.92) | 160 (14.87) | 478 (44.42) | 438 (40.71) | ||||||
| rs7832767 | TB | 483 (29.45) | 1157 (70.55) | 1.30 (1.11–1.48) | 6.93 × 10−4 | 0.016 | 89 (10.85) | 305 (37.20) | 426 (51.95) | 8.81 × 10−4 | 0.021 | |
| C > T | HC | 529 (24.54) | 1627 (75.46) | 68 (6.31) | 393 (36.46) | 617 (57.24) | ||||||
| WNT1 | rs4760662 | TB | 808 (49.88) | 812 (50.12) | 1.01 (0.88–1.15) | 0.924 | 210 (25.93) | 388 (47.90) | 212 (26.17) | 0.778 | ||
| C > T | HC | 1064 (49.72) | 1076 (50.28) | 267 (24.95) | 530 (49.53) | 273 (25.52) | ||||||
| rs4760663 | TB | 778 (47.67) | 854 (52.33) | 1.01 (0.89–1.14) | 0.922 | 204 (24.85) | 380 (46.28) | 237 (28.87) | 0.297 | |||
| C > T | HC | 1036 (47.83) | 1130 (52.17) | 248 (22.90) | 540 (49.86) | 295 (27.24) | ||||||
| WNT3A | rs13373831 | TB | 339 (20.60) | 1307 (79.40) | 0.94 (0.80–1.10) | 0.409 | 43 (5.22) | 253 (30.74) | 527 (64.04) | 0.423 | ||
| A > G | HC | 464 (21.70) | 1674 (78.30) | 42 (3.93) | 380 (35.55) | 647 (60.52) | ||||||
| rs708113 | TB | 590 (36.88) | 1010 (63.12) | 1.04 (0.91–1.19) | 0.607 | 118 (14.75) | 354 (44.25) | 328 (41.00) | 0.288 | |||
| T > A | HC | 760 (36.05) | 1348 (63.95) | 132 (12.52) | 496 (47.06) | 426 (40.42) | ||||||
| rs74672629 | TB | 164 (9.98) | 1480 (90.02) | 0.92 (0.74–1.14) | 0.440 | 7 (0.85) | 150 (18.25) | 665 (80.90) | 0.530 | |||
| C > G | HC | 226 (10.75) | 1876 (89.25) | 7 (0.67) | 212 (20.17) | 832 (79.16) | ||||||
| rs752107 | TB | 398 (24.54) | 1224 (75.46) | 0.86 (0.74–1.00) | 0.047 | > 0.999 | 45 (5.55) | 308 (37.98) | 458 (56.47) | 0.101 | ||
| C > T | HC | 590 (27.44) | 1560 (72.56) | 69 (6.42) | 452 (42.05) | 554 (51.53) | ||||||
| WNT5A | rs2076831 | TB | 597 (36.67) | 1031 (63.33) | 1.04 (0.91–1.19) | 0.605 | 112 (13.76) | 373 (45.82) | 329 (40.42) | 0.727 | ||
| C > G | HC | 768 (35.85) | 1374 (64.15) | 148 (13.82) | 472 (44.07) | 451 (42.11) | ||||||
| rs3732750 | TB | 189 (11.60) | 1441 (88.40) | 1.23 (1.00–1.51) | 0.055 | 9 (1.10) | 171 (20.98) | 635 (77.91) | 0.143 | |||
| G > A | HC | 208 (9.67) | 1944 (90.33) | 7 (0.65) | 194 (18.03) | 875 (81.32) | ||||||
| rs504849 | TB | 603 (36.90) | 1031 (63.10) | 1.00 (0.87–1.14) | 0.971 | 144 (17.00) | 375 (44.27) | 328 (38.73) | 0.999 | |||
| A > G | HC | 788 (36.96) | 1344 (63.04) | 149 (13.98) | 490 (45.97) | 427 (40.06) | ||||||
| rs566926 | TB | 612 (38.25) | 988 (61.75) | 1.01 (0.89–1.16) | 0.849 | 114 (14.25) | 384 (48.00) | 302 (37.75) | 0.268 | |||
| A > C | HC | 812 (37.94) | 1328 (62.06) | 169 (15.79) | 474 (44.30) | 427 (39.91) | ||||||
SNPs were bolded and underlined if they showed a significance of p < 0.05; “1” designates the mutant allele and “2” designates the wild allele; 11 = mutant homozygote, 12 = heterozygote, 22 = wild homozygote; OR = odds ratio, followed by 95% CI in parentheses. P*: p value after adjusting for age, gender and BMI; P**: p value after Bonferroni correction.
Table 3. Association of WNT pathway polymorphisms with TB risk in Chinese Han population.
| Gene | SNP | Additive Model |
Dominant Model |
Recessive Model |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OR | P* | P** | OR | P* | P** | OR | P* | P** | ||
| CTNNB1 | rs9859392C > G | 0.76 (0.65–0.88) | 4.00 × 10−4 | 0.010 | 0.69 (0.57–0.84) | 1.35 × 10−4 | 0.003 | 0.82 (0.54–1.24) | 0.344 | |
| rs9870255G > C | 0.77 (0.65–0.90) | 8.00 × 10−4 | 0.019 | 0.73 (0.61–0.88) | 0.001 | 0.026 | 0.68 (0.45–1.05) | 0.079 | ||
| rs3864004G > A | 0.78 (0.67–0.91) | 0.002 | 0.039 | 0.75 (0.62–0.90) | 0.003 | 0.063 | 0.68 (0.45–1.03) | 0.071 | ||
| WIF1 | rs34203757C > G | 0.92 (0.78–1.09) | 0.340 | 0.92 (0.76–1.13) | 0.434 | 0.81 (0.50–1.31) | 0.391 | |||
| rs34505206C > G | 0.88 (0.74–1.04) | 0.130 | 0.86 (0.71–1.05) | 0.146 | 0.81 (0.50–1.30) | 0.374 | ||||
| rs56900803G > T | 1.00 (0.82–1.22) | 0.990 | 0.96 (0.77–1.20) | 0.731 | 1.64 (0.76–3.52) | 0.205 | ||||
| DKK1 | rs11001553C > T | 1.04 (0.82–1.33) | 0.740 | 1.04 (0.80–1.35) | 0.787 | 1.20 (0.43–3.31) | 0.732 | |||
| rs1896367G > A | 0.98 (0.86–1.13) | 0.790 | 1.04 (0.87–1.25) | 0.655 | 0.81 (0.60–1.11) | 0.186 | ||||
| rs1896368G > A | 0.96 (0.84–1.09) | 0.500 | 1.04 (0.86–1.25) | 0.722 | 0.80 (0.62–1.03) | 0.079 | ||||
| SFRP1 | rs3242C > T | 0.97 (0.75–1.24) | 0.790 | 0.87 (0.65–1.16) | 0.347 | 2.47 (0.98–6.22) | 0.055 | |||
| rs4736958T > C | 0.78 (0.70–0.92) | 0.002 | 0.040 | 0.73 (0.61–0.88) | 7.41 × 10−4 | 0.017 | 0.82 (0.61–1.10) | 0.183 | ||
| rs72643819G > T | 1.11 (0.98–1.26) | 0.100 | 1.06 (0.88–1.28) | 0.544 | 1.34 (1.05–1.70) | 0.019 | 0.447 | |||
| rs7832767C > T | 1.29 (1.10–1.46) | 0.001 | 0.023 | 1.26 (1.03–1.49) | 0.022 | 0.505 | 1.81 (1.30–2.52) | 4.29 × 10−4 | 0.010 | |
| WNT1 | rs4760662C > T | 1.01 (0.89–1.14) | 0.920 | 0.97 (0.78–1.19) | 0.747 | 1.05 (0.85–1.30) | 0.631 | |||
| rs4760663C > T | 1.01 (0.89–1.14) | 0.920 | 0.92 (0.75–1.13) | 0.433 | 1.11 (0.90–1.38) | 0.323 | ||||
| WNT3A | rs13373831A > G | 0.94 (0.80–1.10) | 0.410 | 0.86 (0.71–1.04) | 0.119 | 1.35 (0.87–2.08) | 0.179 | |||
| rs708113T > A | 1.04 (0.91–1.18) | 0.610 | 0.98 (0.81–1.18) | 0.801 | 1.21 (0.93–1.58) | 0.165 | ||||
| rs74672629C > G | 0.92 (0.74–1.14) | 0.430 | 0.90 (0.71–1.13) | 0.352 | 1.28 (0.45–3.67) | 0.644 | ||||
| rs752107C>T | 0.85 (0.73–0.99) | 0.040 | 0.928 | 0.82 (0.68–0.98) | 0.033 | 0.766 | 0.86 (0.58–1.26) | 0.433 | ||
| WNT5A | rs2076831C > G | 1.03 (0.91–1.18) | 0.610 | 1.07 (0.89–1.29) | 0.461 | 1.00 (0.76–1.30) | 0.93 | |||
| rs3732750G > A | 1.23 (1.00–1.52) | 0.053 | 1.23 (0.98–1.55) | 0.068 | 1.71 (0.63–4.60) | 0.292 | ||||
| rs504849A > G | 1.00 (0.87–1.14) | 0.970 | 1.00 (0.83–1.20) | 0.968 | 1.00 (0.77–1.30) | 0.988 | ||||
| rs566926A > C | 1.01 (0.89–1.16) | 0.850 | 1.10 (0.91–1.32) | 0.344 | 0.89 (0.68–1.15) | 0.357 | ||||
We further examined whether the six candidate SNPs were preferentially associated with specific tubercular subtypes, as described by Fernando et al.21. The results are summarized in Table S6. Rs4736958 seemed to have a stronger magnitude of decreased risk with PTB as compared to all forms of TB, and rs9859392, rs9870255 and rs3864004 showed slightly lower OR values with PTB&EPTB as compared to all forms of TB. Rs7832767 was most powerfully associated with a higher risk of PTB&EPTB (OR = 2.19, 95% CI = 1.47–3.26, p = 0.001, after Bonferroni correction) in the recessive model.
Linkage analysis and haplotype construction
Linkage disequilibrium (LD) was estimated by calculating the pairwise r2 coefficient22. Figure 1 displays the LD plot of SNPs on the same chromosome. With a pairwise r2 > 0.8, three polymorphisms of CTNNB1 (rs9859392/rs9870255/rs3864004) and three of WNT5A (rs566926/rs504849/rs2076831) on chromosome 3, as well as two SNPs in WNT1 (rs4760663/rs4760662) and two in WIF1 (rs34203757/rs34505206) on chromosome 12, were in strong linkage disequilibrium with one another, respectively. We constructed the haplotype by using the SNPs which were in strong LD state from Fig. 1. As seen in Table 4, the [GCA] haplotype frequency of CTNNB1 was significantly lower in the patients than in the controls for rs9859392/rs9870255/rs3864004 (OR = 0.65, 95% CI = 0.60–0.83, p = 0.016, after Bonferroni correction). The distribution of haplotype frequencies of WNT5A, WNT1 and WIF1 did not differ significantly between the patients and the controls.
Figure 1. Linkage disequilibrium (LD) plots of SNPs.

(A) LD results of SNPs in the CTNNB1 and WNT5A genes on chromosome 3. (B) LD results of SNPs in the WNT1 and WIF1 genes on chromosome 12. Notes: Pairwise r2 values for all pairs of SNPs are presented as percentages in diamonds, and shading from white to black indicates the intensity of r2 from 0 to 1. Strong LD is represented by a high percentage (>80) and a darker square.
Table 4. Haplotype association of SNPs in WNT pathway and tuberculosis.
| Gene | Haplotypea | Frequency |
OR (95% CI) | P | P** | ||
|---|---|---|---|---|---|---|---|
| All | TB | HC | |||||
| CTNNB1 | CGG | 0.764 | 0.785 | 0.748 | 1.00 | ||
| GCA | 0.212 | 0.175 | 0.240 | 0.69 (0.60–0.83) | 5.85 × 10−4 | 0.016 | |
| WNT5A | AAC | 0.617 | 0.614 | 0.619 | 1.00 | ||
| CGG | 0.359 | 0.366 | 0.354 | 1.03 (0.90–1.17) | 0.700 | ||
| CAC | 0.014 | 0.019 | 0.010 | 2.04 (1.15–3.62) | 0.054 | ||
| WNT1 | CC | 0.502 | 0.496 | 0.508 | 1.00 | ||
| TT | 0.475 | 0.480 | 0.47 | 1.01 (0.89–1.15) | 0.840 | ||
| CT | 0.023 | 0.024 | 0.022 | 1.13 (0.73–1.76) | 0.590 | ||
| WIF1 | CC | 0.819 | 0.830 | 0.811 | 1.00 | ||
| GG | 0.175 | 0.126 | 0.185 | 0.88 (0.74–1.03) | 0.120 | ||
P**: p value after Bonferroni correction.
aHaplotype frequency <0.01 in both cases and controls has been dropped; significant associations were denoted in bold. Wild haplotype was set as the reference, and the OR of other haplotypes were calculated.
Gene-gene interaction evaluation
The one-dimensional interactions (including multiplicative and additive interactions) between the six significant SNPs were tested, but they did not yield additional positive information (p all > 0.05). High-dimensional interactions among all SNPs were also performed, but no significant interactions were found from the one to four-way models (p all > 0.05). Together, gene-gene interactions failed to reveal potential synergistic or antagonistic effects among the polymorphisms in different genes. Details are shown in Tables S7–S9.
Correlation between genotypes and clinical phenotypes
We investigated whether the six candidate susceptibility SNPs affected patients’ clinical manifestations. Common indices of disease severity for TB consisted of symptom intensity (fever, hemoptysis), cavities in the chest radiograph, smear acid-fast and culture of MTB, hemoglobin concentration, lymphocyte count, erythrocyte sedimentation rate, and C-reactive protein (CRP) level, which was regarded as a regular inflammatory marker for host defense and innate immunity23. Due to the low frequencies of some minor genotypes, six SNPs were stratified based on the dominant or recessive model. Subgroup analysis attested that rs4736958 and rs7832767 were closely associated with plasma levels of CRP (p = 0.003, <0.001), and patients carrying the TT genotype corresponded to higher levels of CRP (Fig. 2). Beyond that, no nominally significant relation between other clinical indices and genotypes were obtained (p all > 0.05, data not shown, available on request).
Figure 2. Serum concentration of CRP in relation to polymorphisms of SFRP1 in TB patients.
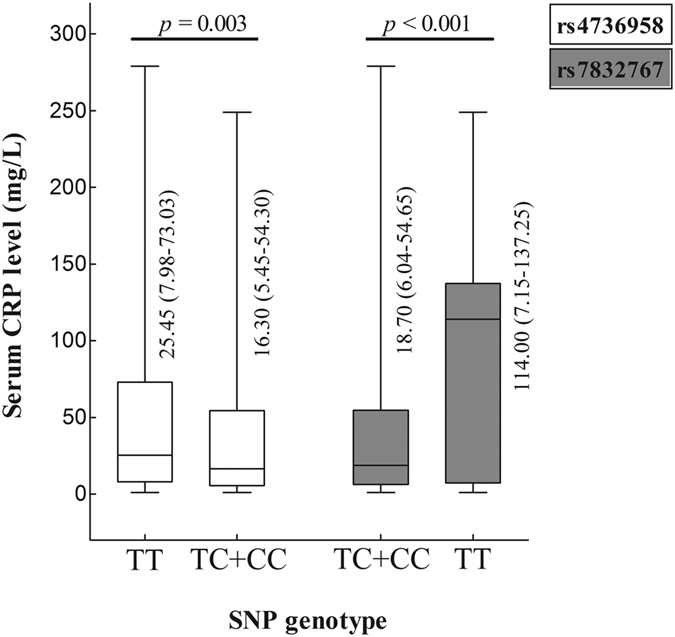
Notes: rs4736958 (T > C) and rs7832767 (C > T) genotype was stratified based on the significant dominant and recessive model, respectively; rs4736958: TC + CC vs TT, rs7832767: TT vs TC + CC.
Candidate SNPs validation in Chinese Tibetan group
Next, we genotyped 6 SNPs (rs4736958, rs7832767, rs9859392, rs9870255, rs3864004 and rs752107) in an independent Chinese Tibetan cohort (480 TB cases and 450 healthy controls). After multiple testing adjustment, only CTNNB1 SNPs (rs9859392, rs9870255 and rs3864004) were significant referring to frequency distribution, the additive and dominant model (Table 5), which further demonstrated that carriers with mutant genotypes of these SNPs were less susceptible to TB disease.
Table 5. Association of six candidate SNPs with TB risk in Chinese Tibetan validation cohort.
| SNP | Group | Genotype |
Allele |
Additive Model |
Dominant Model |
Recessive Model |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (11/12/22) | P** | OR | P** | OR | P** | OR | P** | OR | P** | ||
| rs9859392 | TB | 19/131/327 | 0.014 | 0.69 (0.55–0.87) | 0.016 | 0.70 (0.56–0.88) | 0.018 | 0.64 (0.49–0.83) | 0.010 | 0.71 (0.39–1.32) | >0.999 |
| HC | 24/159/254 | ||||||||||
| rs9870255 | TB | 19/130/328 | 0.009 | 0.68 (0.54–0.86) | 0.011 | 0.70 (0.55–0.87) | 0.013 | 0.63 (0.49–0.83) | 0.008 | 0.68 (0.37–1.26) | >0.999 |
| HC | 25/158/254 | ||||||||||
| rs3864004 | TB | 19/130/328 | 0.014 | 0.69 (0.55–0.87) | 0.015 | 0.70 (0.56–0.88) | 0.018 | 0.64 (0.49–0.83) | 0.010 | 0.71 (0.39–1.32) | >0.999 |
| HC | 24/158/255 | ||||||||||
| rs4736958 | TB | 33/206/238 | >0.999 | 0.90 (0.74–1.10) | >0.999 | 0.90 (0.73–1.10) | >0.999 | 0.92 (0.71–1.19) | >0.999 | 0.74 (0.46–1.19) | >0.999 |
| HC | 40/188/209 | ||||||||||
| rs7832767 | TB | 100/242/135 | >0.999 | 0.93 (0.74–1.17) | >0.999 | 0.93 (0.73–1.18) | >0.999 | 0.90 (0.69–1.18) | >0.999 | 1.07 (0.49–2.34) | >0.999 |
| HC | 91/233/113 | ||||||||||
| rs752107 | TB | 28/131/318 | >0.999 | 1.18 (0.93–1.49) | >0.999 | 1.17 (0.93–1.47) | >0.999 | 1.06 (0.80–1.40) | >0.999 | 2.66 (1.27–5.55) | 0.080 |
| HC | 10/130/297 | ||||||||||
Numbers were bolded if they showed a significance of p < 0.05; 11 = mutant homozygous, 12 = heterozygote, 22 = wild homozygous; OR = odds ratio, followed by 95% CI in parentheses. P**: p value after Bonferroni correction.
Functional exploration of CTNNB1 polymorphisms
Investigation for the effect of polymorphisms on mRNA expression
We further explored the effect of six TB-associated SNPs on gene expression. The mRNA levels of candidate susceptibility genes (CTNNB1, SFRP1 and WNT3A) from a subset of 50 patients and 29 controls were compared, not only between the cases and the controls but also in different genotypes. The mRNA expressions of all three genes were significantly higher in the patients than in the controls [CTNNB1: 40.16 (20.13–107.17) vs 0.77 (0.52–2.53), p < 0.001; SFRP1: 10.22 (1.58–26.36) vs 2.23 (0.31–7.68), p = 0.008; WNT3A: 11.16 (2.24–58.23) vs 0.84 (0.39–2.37), p < 0.001], implying that their roles in the regulation of TB progression. The dominant pattern of three significant SNPs in CTNNB1 (rs3864004, rs9859392 and rs9870255) was associated with obviously decreased CTNNB1 mRNA levels both in all 79 subjects and in 50 TB patients (pall = 0.004, pTB = 0.034), however, no association was found in healthy controls (pHC = 0.080). Figure 3(A,B) show the example of the comparison of CTNNB1 mRNA expression based on the dominant genotype model of rs9859392 in these three groups. In addition, similarly significant effects were not observed in the three significant SNPs in SFRP1 and WNT3A (p all > 0.05, data not shown, available on request).
Figure 3. CTNNB1 polymorphisms affected CTNNB1 mRNA level and its transcriptional activity.

Notes: ALL: all subjects, TB: tuberculosis patients, HC: healthy controls. W H: wild homozygote, M H: mutant homozygote. (A,B) refer to the CTNNB1 mRNA levels in relation to different genotypes in all 79 subjects [GC + GG vs CC: 3.15 (0.64–33.68) vs 27.28 (4.37–103.38)], 50 TB patients [30.24 (6.93–62.69) vs 47.08 (21.59–131.98)] and 29 healthy controls [0.64 (0.53–2.85) vs 0.83 (0.50–2.31)], respectively. rs3864004, rs9859392 and rs9870255 were in strong linkage disequilibrium, and their genotypes had the same CTNNB1 mRNA expression level, therefore, we just listed rs9859392 as an example. Subjects were grouped for rs9859392 in CTNNB1 as genotype with (GG + GC) or without (CC) mutant G allele. (C) indicates the effect of three CTNNB1 polymorphisms on CTNNB1 promoter transcriptional activity in 293T cells. Each bar represents the mean of triplicate transfected plates plus standard deviation (rs3864004: 0.83 ± 0.04 vs 0.09 ± 0.01; rs9859392: 0.40 ± 0.05 vs 0.25 ± 0.02; rs9870255: 0.48 ± 0.01 vs 0.30 ± 0.08).
Influence of polymorphisms on transcriptional activity of CTNNB1 gene
To assess the effects of SNPs rs3864004, rs9859392 and rs9870255 on CTNNB1 gene transcription, we analyzed the transcriptional activity of its promoter and compared the activities of mutant and wild allele using luciferase report assay. As shown in Fig. 3(C), the pGL3-mutant homozygote reporters have significantly decreased luciferase activity as compared to pGL3-wild homozygote (9.22-folds for rs3864004, p < 0.001; 1.60-folds for rs9859392 and rs9870255, p = 0.007, 0.025, respectively). This finding indicates that these three SNPs are functional, and the substitution of the mutant allele for the wild allele can suppress CTNNB1 promoter transcriptional activity.
Effect of CTNNB1 polymorphisms on its protein expression
To verify whether the three SNPs could influence the endogenetic expression of CTNNB1 gene, we examined the protein products (β-catenin and p-β-catenin) in peripheral blood mononuclear cells (PBMC) of 16 TB patients and healthy controls. As shown in Fig. 4, compared with healthy controls, tuberculosis patients had significantly increased β-catenin and p-β-catenin protein expression (darker bands). In order to adjust the discordance of cell quantities among samples, relative gray intensity analysis of β-catenin and p-β-catenin protein bands was normalized by its β-actin expression and was further calculated using Image J software (NIH, MD, USA). Levels of p-β-catenin protein were significantly decreased in patients with mutant genotype (haplotype [GCA] for rs9859392/rs9870255/rs3864004) as compared with patients carrying wild genotype (haplotype [CGG]), indicating the similar trend with lower mRNA expression level and reduced TB risk.
Figure 4. Effect of CTNNB1 polymorphisms on its endogenetic protein expression.

Notes: P1-P4: TB patients with mutant genotype of CTNNB1 polymorphisms (rs9859392/rs9870255/rs3864004), i.e. subjects with haplotype [GCA]. P5-P8: TB patients with wild genotype of CTNNB1 polymorphisms, i.e. subjects with haplotype [CGG]. Similarly, H1-H4 and H5-H6 refer to healthy controls with mutant genotype and wild genotype of CTNNB1 polymorphisms, respectively. (A,B) protein levels of β-catenin, phosphorylated β-catenin (p-β-catenin) and β-actin were determined by Western blot in the PBMC of 8 TB patients and 8 healthy controls, respectively. Each bar represents the mean grey value of each group plus standard deviation.
Discussion
The present study identified five SNPs within the WNT pathway (SFRP1 rs4736958 and rs7832767; CTNNB1 rs9859392, rs9870255 and rs3864004) that were associated with TB susceptibility. Further explorations preliminarily unveiled a relationship between two SFRP1 SNPs and serum CRP levels and a modulatory effect of three CTNNB1 polymorphisms on its mRNA and protein expression. To our knowledge, this is the first study to comprehensively interrogate the relationship between WNT pathway-based SNPs and TB risk, providing insight into the detailed mechanism for the pathogenesis of TB and a clue for clinical biomarker screening.
The CTNNB1 gene encodes the β-catenin protein, dually functioning in the coordination of cell adhesion and gene transcription. Compelling studies have indicated that aberrant β-catenin participates in an abnormal immune response to pathogens10 and disease course, including tuberculosis13. Recently, researchers have linked CTNNB1 mutations to the susceptibility and prognosis of various cancers24. However, the impact of the CTNNB1 variations on TB has not yet been corroborated. In four CTNNB1 SNPs, we identified mutant alleles of rs9859392 (G), rs9870255 (C) and rs3864004 (A) that might be protective factors for TB according to our discovery and validation cohorts. Haplotype analysis revealed a powerful reduced-risk haplotype, [GCA], composed of these minor alleles, thus supporting the reliability of single locus results and providing evidence of a synergistic beneficial effect of these SNPs on TB infection. Higher CTNNB1 expressions were observed in TB patients, pointing to a critical role of CTNNB1 in host immune response to MTB. Subsequent functional investigations found that mutant alleles at these SNPs significantly decreased CTNNB1 promoter transcriptional activity and correlated with lower CTNNB1 mRNA and phosphorylated β-catenin protein expression levels, identical to their effects of decreasing TB risk. Similar correlations were also reported by Sungmin Bae et al.25, who demonstrated that β-catenin haplotypes formed by these polymorphisms affected mRNA expression and were associated with asthma risk. Taken together, these observations supported the idea that these SNPs resulted in different recruits of transcription factors to CTNNB1 gene promoter region, which modulated CTNNB1 mRNA and protein expression and leaded to abnormal activation of the WNT pathway, participating in the pathogenesis of disease. Further functional experiments are warranted to confirm and elaborate these findings.
SFRP1 belongs to a well-established antagonist of the WNT pathway, and molecular abnormalities in its expression have been amply demonstrated to be involved in the development of carcinoma26. As a novel factor modulating inflammation27, SFRP1 may play an important role in the pathogenesis of TB. We explored five SFRP1 polymorphisms and found two (rs4736958 and rs7832767) that were significantly association with TB risk in Chinese Han population, but not in Tibetan group. This suggests that ethnic heterogeneity of these polymorphisms might exist among different populations, which is worthy of further investigation in other populations. Notably, we found that the risk genotype TT in these two SNPs was related to higher CRP levels, indicating that patients carrying the risk genotype TT may have a higher inflammatory state. When the two SNPs were analyzed for a possible association with gene expression, no significant differences were found, suggesting that these polymorphisms may not influence SFRP1 mRNA expression. Rs7832767 is located in the intron region, and rs4736958 lies in the 3′ untranslated region of SFRP1, neither of which directly code for the Sfrp1 protein, even though there seem to be some indirect links. As a tagSNP, rs7832767 may be in linkage disequilibrium with a functional mutation that controls the expression or function of SFRP1. According to the bioinformatics tool SNPinfo (NIH, SNPinfo Web Server. Available at: http://snpinfo.niehs.nih.gov/), rs4736958 is located in the seed binding site of miR-9 and SFRP1. The high-affinity miR-9-binding motif is predicted to be abrogated by an rs4736958 C-to-T transition, ultimately altering the regulation of miR-9 to inflammatory responses against invasive pathogens, as evidenced by Thulin et al.28. Although we do not currently know if these speculations conform to concrete mechanisms, ongoing studies will address these questions.
In this research, we found that rs752107 was associated with a reduced risk for TB in allele distribution, the additive and dominant model. This association did not remain after Bonferroni correction or in the validation cohort, therefore, we determined that rs752107 was not TB risk-related locus, even though other independent studies are needed. With regard to the rest of the SNPs and genes analyzed, no substantial associations with TB risk were obtained, suggesting that these polymorphisms may not predispose one to the disease. However, we cannot completely exclude their possible role as putative TB-risk genes on account of the imperceptible scrutiny of genotype frequency or mild effect of these polymorphisms. Despite these irrelevant results, we expect that our work will help to elucidate the genetic variations involved in the pathogenesis of TB.
Sample size in the present study was still limited, especially for some stratifications, therefore, some polymorphisms with mildly significant effects might not be detected. On the other hand, our study lacks some demographic information on the Chinese Tibetan cohort, and our results should be validated in larger population-based studies with more detailed information.
In conclusion, this study investigated the pathogenesis of TB from the perspective of pathway-based polymorphisms. Five SNPs in the WNT pathway (SFRP1 rs4736958 and rs7832767; CTNNB1 rs9859392, rs9870255 and rs3864004) were associated with TB susceptibility. Two SNPs in SFRP1 were correlated with serum CRP levels, and three CTNNB1 polymorphisms had a striking effect on CTNNB1 promoter transcriptional activity and modulated CTNNB1 mRNA and protein expression. Our findings suggest that genetic variations within the WNT pathway may play a significant role in TB risk and host inflammatory status.
Methods
Study population and samples
Discovery set: a total of 1916 participants (832 active TB cases and 1084 gender- and age-matched controls) were enrolled in this study. Cases were newly diagnosed TB patients consecutively recruited from the West China Hospital of Sichuan University between November 2011 and October 2015. Eligible cases were clinically diagnosed by experienced specialists using microbiological or pathological evidence of TB (smear/culture/TB-DNA positive) to confirm MTB infection. Patients who were HIV-positive, had other lung or infectious diseases or had an immunosuppressive condition were excluded. Controls were recruited from healthy blood donors with a negative interferon gamma release assay (TB-IGRA) result, no history of TB and a normal physical examination. All subjects in this study were unrelated Han Chinese in West China. Demographic and clinical data of cases and controls were obtained from interviews or extracted from medical records. Validation set was consist of 480 Tibetan cases and 450 gender- and age-matched Tibetan controls. This study was approved the Clinical Trials and Biomedical Ethics Committee of West China Hospital, Sichuan University [Reference No. 198 (2014)], and the methods were carried out in accordance with the approved guidelines. Informed consent was obtained from all individuals.
Candidate loci selection and genotyping
Key genes in the WNT signaling pathway were selected based on their biological functions as core components of this pathway, including CTNNB1, WIF1, DKK1, SFRP1, WNT1, WNT3A and WNT5A9,10,11,12,13,14,15. We selected candidate SNPs for this study through a thorough search of the International HapMap project, dbSNP database, 1000 Genomes database. SNPs were included if they were located in potential functional regions (exon, promoter and untranslated region) and could well represent those with minor allele frequency (MAF) >0.05 in a Chinese Han Beijing population. Finally, a total of 25 SNPs were selected for subsequent genotyping (Table S1).
Genomic DNA was extracted from 2- to 3-ml peripheral blood using a QIAamp® DNA blood Mini kit (Qiagen, Hilden, Germany). SNPs were genotyped using the MassARRAY platform (Sequenom, CA, USA) with primers and probes (Table S2) as described by Buetow et al.29, and SNP genotyping in validation cohort was performed by Shanghai Genesky Bio-Tech Genetic Core Lab (Shanghai, China) using multiplex ligation detection reaction as described by Thomas et al.30 (Primers and probes information presented in Tables S3 and S4). The laboratory staff was blinded to the case-control status of the samples. We included water (HPLC grade) as the negative control in each of the 96-well or 384-well plates. Fifty random samples were genotyped in duplicate with a concordance rate of 100%. SNPs with a clear genotype clustering and a call rate >95% were considered for further analysis.
Measurement of mRNA expression
We collected an additional 3 ml of peripheral blood from 85 participants and isolated peripheral blood mononuclear cells (PBMC) from 79 of them for mRNA measurement. Total RNA was isolated using TRIzol reagent (Invitrogen, CA, USA) and then converted to complementary DNA (cDNA) using Omniscript® Reverse Transcription Kit (Qiagen, CA, USA). The final 10-μl-volume reaction for RT-qPCR included 5 μl of SYBR® Premix Ex Taq™ II (Takara, Dalian, China), 0.8 μl of 10 μM forward and reverse primer (Table S5), 3.2 μl of water and 1 μl of cDNA template. RT-qPCR amplification was carried out in a LightCycler® 480 Real-Time PCR System (Roche Diagnostics, NJ, USA). The reaction setting consisted of an initial denaturation of 10 min at 95 °C and amplification for 40 cycles by denaturing at 95 °C for 20 s, annealing at 56–62 °C for 30 s and extension at 72 °C for 25 s. The samples were denatured at 95 °C for 30 s and then heated to 65 °C for 30 s at a rate of 0.2 °C/s. Fluorescence was measured to generate a denaturing curve of the amplified products. Raw data were analyzed using Gene Scanning v1.2 software. For each sample, the mRNA expression was normalized to the endogenous control GAPDH and calculated according to the 2−ΔΔCT method.
Luciferase report assay
Since SNP rs9859392, rs3864004, rs9870255 were suggested to be the most effective variant within or near the promoter of CTNNB1, we further tested their effects on the promoter activity of the gene using a Dual-Luciferase Reporter Assay System (Promega, WI, USA). Briefly, PCR fragments containing either wide or mutant allele of these SNPs were amplified using the following primers (the sequence in bold is a KpnI or XhoI restriction site): rs9870255: Sense: 5′-GGGGTACCCAGTGCTAGGGTGGTGAGTG-3′, Antisense: 5′-CCGCTCGAGAATCCCATCATGCAGGGTCC-3′; rs9859392: Sense: 5′-GGGGTACCATGGACAATCCCTTGAAGCAAC-3′, Antisense: 5′-CCGCTCGAGTCCTTGATAAAAATGAGGAAAGTGG-3′; rs3864004: Sense: 5′-CGGGGTACCGCTCTGGAGCTAATCCATTTCCA-3′, Antisense: 5′-CCGCTCGAGCTTATAAGTCGCGCAGAAGCC-3′. The cycling program consisted of 98 °C for 2 min, followed by 35 cycles of 98 °C for 10 s, 55 °C for 15 s and 72 °C for 60 s. PCR products were then cloned into the pGL3-Basic vector or pGL3-Promoter vector. All reporter constructs were verified by direct sequencing. For luciferase assays, 293T cells were plated in a 24-well plate and allowed to grow for 1 day prior to transfection (60–70% confluence). 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA) containing 10% heat-inactivated fetal bovine serum (Biowest, France). Transfection was performed using Lipofectamine 2000 Reagent (Invitrogen), with 0.2 μg of a given pGL-3 construct and 20 ng of renilla vector (pRLTK, as internal control) per well were co-transfected. After 48 h, cells were lysed in 16× Passive Lysis Buffer, and 30 μl of lysate was used to detect firefly luciferase (LAR II) and renilla luciferase (stop and glow buffer) activity. Fluorescence intensity was measured using BioTek Synergy 4 multi-mode microplate reader (BioTek Instruments Inc., USA). The relative luciferase activity was normalized to the cotransfected control Renilla luciferase. Each construct was transfected in triplicates and assayed in duplicate.
Western blot
PBMC from TB patients and healthy controls were freshly isolated using Human Lymphocyte Separation Tube Kit (Dakewe Biotech company limited, Shenzhen, China), and then proteins were extracted proteins were extracted in RIPA buffer and quantified by the BCA protein assay kit (Bio-Rad). The resulted proteins were subjected to 10% SDS-PAGE and transferred to PVDF membranes (Millipore). The membrane was blocked with 5% bovine serum albumin (BSA)-PBS and was incubated with rabbit anti-β-catenin and p-β-catenin (diluted 1:1000, Cell Signaling Technology, MA, USA) at 4 °C overnight, respectively. The blots were incubated with secondary antibody (diluted 1:2000; Zhongshan Biological) conjugated to horseradish peroxidase for 2 h. β-Actin was used as an internal control. Blots were visualized by enhanced chemiluminescence reagents (Amersham Biosciences, Shanghai, China) and analyzed with Image J Software (NIH, MD, USA).
Statistical analysis
Univariate analysis was used to analyze categorical variables with a Chi-square test and continuous variables with the Mann-Whitney U test. Hardy-Weinberg equilibrium (HWE) among the controls was evaluated using Fisher’s exact test. SNPs with monomorphism and significant HWE deviation were excluded from analysis. Associations between SNPs and TB risk were evaluated on the basis of allele frequency distributions and genetic models (additive, dominant and recessive model). Wild or major allele of each SNP was set as the reference, and odds ratio (OR) and 95% confidence interval (CI) were derived from unconditional logistic regression models, adjusting for age, gender and BMI. Linkage disequilibrium was assessed using Haploview v4.231. Haplotype frequencies and associations were calculated based on the expectation-maximization clustering algorithm using this software. The one-dimensional interactions between two associated SNPs were evaluated by logistic regression for multiplication interactions using the method suggested by Andersson et al.32. High-dimensional interactions among multiple SNPs were assessed using general multifactor dimensionality reduction (GMDR) software33.
Analyses were performed using SPSS v19.0, PLINK v1.07, Haploview v4.2 and GMDR. All tests were two-sided, and nominal statistical significance was established at an alpha level of 0.05 unless stated otherwise.
Additional Information
How to cite this article: Hu, X. et al. Pathway Analyses Identify Novel Variants in the WNT Signaling Pathway Associated with Tuberculosis in Chinese Population. Sci. Rep. 6, 28530; doi: 10.1038/srep28530 (2016).
Supplementary Material
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China [81472026, 81202375] and the Projects in the Science and Technology Department of Sichuan Province pillar program [2014SZ0208]. The authors also thank Dr. Chen for his guidance in statistics analysis, and Shanghai Genesky Bio-Tech Genetic Core Lab for assistance in genotyping techniques.
Footnotes
Author Contributions X.H., J.Z. and X.C. wrote the main manuscript text and participated in the experiment all the way. J.Z. and X.S. participated in modifying the manuscript and preparing Tables and figures; X.L. and B.C. participated in the analysis of data; X.L., B.Y. and Y.Z. engaged in the acquisition of data (laboratory or clinical); B.Y. and Y.Z. designed the study. All authors have reviewed the manuscript.
References
- WHO, Global tuberculosis report. http://www.who.int/tb/publications/global_report/en/, (Date of access: 25/12/2015) (2015).
- WHO, Guidelines on the Management of Latent Tuberculosis Infection. http://www.who.int/tb/publications/latent-tuberculosis-infection/en/, (Date of access: 20/10/2015) (2015). [PubMed]
- O’Garra A. et al. The immune response in tuberculosis. Annu Rev Immunol 31, 475–527 (2013). [DOI] [PubMed] [Google Scholar]
- Orme I. M., Robinson R. T. & Cooper A. M. The balance between protective and pathogenic immune responses in the TB-infected lung. Nat Immunol 16, 57–63 (2015). [DOI] [PubMed] [Google Scholar]
- Goldfeld A. E. et al. Association of an HLA-DQ allele with clinical tuberculosis. JAMA 279, 226–228 (1998). [DOI] [PubMed] [Google Scholar]
- Bellamy R. Inferferon-gamma and host suscepitibility to tuberculosis. Am J Respir Crit Care Med 167, 946–947 (2003). [DOI] [PubMed] [Google Scholar]
- Velez D. R. et al. NOS2A, TLR4, and IFNGR1 interactions influence pulmonary tuberculosis susceptibility in African-Americans. Hum Genet 126, 643–53 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabri A. et al. Association study of genes controlling IL-12-dependent IFN-γ immunity: STAT4 alleles increase risk of pulmonary tuberculosis in Morocco. J Infect Dis 210, 611–618 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama T. Wnt/beta-catenin signaling. Cytokine Growth Factor Rev 11, 273–282 (2000). [DOI] [PubMed] [Google Scholar]
- Manicassamy S. et al. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 329, 849–853 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X. et al. A caspase-dependent pathway is involved in Wnt/β-catenin signaling promoted apoptosis in Bacillus Calmette-Guerin infected RAW264.7 macrophages. Int J Mol Sci 15, 5045–5062 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. A Wnt/β-catenin negative feedback loop represses TLR-triggered inflammatory responses in alveolar epithelial cells. Mol Immuno l59, 128–135 (2014). [DOI] [PubMed] [Google Scholar]
- Neumann J. et al. Frizzled1 is a marker of inflammatory macrophages, and its ligand Wnt3A is involved in reprogramming Mycobacterium tuberculosis-infected macrophages. FASEB J 24, 4599–4612 (2010). [DOI] [PubMed] [Google Scholar]
- Blumenthal A. et al. The Wingless homolog Wnt5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood 108, 965–973 (2006). [DOI] [PubMed] [Google Scholar]
- Schaale K. et al. Wnt6 is expressed in granulomatous lesions of Mycobacterium tuberculosis-infected mice and is involved in macrophage differentiation and proliferation. J Immunol 191, 5182–5195 (2013). [DOI] [PubMed] [Google Scholar]
- Peters U. et al. Identification of Genetic Susceptibility Loci for Colorectal Tumors in a Genome-Wide Meta-analysis. Gastroenterology 144, 799–807 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H. et al. Beta-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am J Pathol 155, 1795–1801 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y. et al. National survey of drug-resistant tuberculosis in China. N Engl J Med 366, 2161–2170 (2012). [DOI] [PubMed] [Google Scholar]
- Wang Y. The compilation of the fifth national tuberculosis epidemiological sampling survey. (ed. Wang Y.) 23–5 (Military medical science press, Beijing, 2011).
- Hu X. et al. Association of genetic variants in Wnt signaling pathway with tuberculosis in Chinese Han population. PLoS One 9, e93841 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando S. L. A polymorphism in the P2X7 gene increases susceptibility to extrapulmonary tuberculosis. Am J Respir Crit Care Med 175, 360–366 (2007). [DOI] [PubMed] [Google Scholar]
- Ardlie K. G., Kruglyak L. & Seielstad M. Patterns of linkage disequilibrium in the human genome. Nat Rev Genet 3, 299–309 (2002). [DOI] [PubMed] [Google Scholar]
- Bottazzi B., Doni A., Garlanda C. & Mantovani A. An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol 28, 157–183 (2010). [DOI] [PubMed] [Google Scholar]
- Polakis P. The many ways of WNT in cancer. Curr Opin Genet Dev 17, 45–51 (2007). [DOI] [PubMed] [Google Scholar]
- Bae S. et al. Beta-catenin promoter polymorphism is associated with asthma risk in Korean subjects. Clin Biochem 45, 1187–1191 (2012). [DOI] [PubMed] [Google Scholar]
- Suzuki H. et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet 36, 417–422 (2004). [DOI] [PubMed] [Google Scholar]
- Barandon L. et al. Secreted frizzled-related protein-1 improves postinfarction scar formation through a modulation of inflammatory response. Arterioscler Thromb Vasc Biol 31, e80–e87 (2011). [DOI] [PubMed] [Google Scholar]
- Thulin P. et al. MicroRNA-9 regulates the expression of peroxisome proliferator-activated receptor δ in human monocytes during the inflammatory response. Int J Mol Med 31, 1003–1010 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetow K. H. et al. High-throughput development and characterization of a genomewide collection of gene-based single nucleotide polymorphism markers by chip-based matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Proc Natl Acad Sci USA 98, 581–584 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G. et al. Capillary and microelectrophoretic separations of ligase detection reaction products produced from low-abundant point mutations in genomic DNA. Electrophoresis 25, 1668–1677 (2004). [DOI] [PubMed] [Google Scholar]
- Kaur H. et al. Pharmacogenomics of neuropsychiatric disorders: analysis of genetic variability in 162 identified neuroreceptors using 1000 Genomes Project data. Pharmacogenomics 15, 1575–1587 (2014). [DOI] [PubMed] [Google Scholar]
- Andersson T., Alfredsson L., Kallberg H., Zdravkovic S. & Ahlbom A. Calculating measures of biological interaction. Eur J Epidemiol 20, 575–579 (2005). [DOI] [PubMed] [Google Scholar]
- Tunesi S. et al. Gene-asbestos interaction in malignant pleural mesothelioma susceptibility. Carcinogenesis 36, 1129–1135 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.