

Abstract
Introduction
A wide variety of DNA lesions such as ultraviolet light-induced photoproducts and chemically induced bulky adducts and crosslinks (intrastrand and interstrand) interfere with replication and lead to mutations and cell death. In the human body, these damages may cause cancer, inborn diseases, and aging. So far, mutation-related actions of DNA polymerases during replication have been intensively studied. However, DNA lesions also block RNA synthesis, making the detection of their effects on transcription equally important for chemical safety assessment. Previously, we established an in vivo method for detecting DNA damage induced by ultraviolet light and/or chemicals via inhibition of RNA polymerase by visualizing transcription.
Results
Here, we present an in vitro method for detecting the effects of chemically induced DNA lesions using in vitro transcription with T7 RNA polymerase and real-time reverse transcription polymerase chain reaction (PCR) based on inhibition of in vitro RNA synthesis. Conventional PCR and real-time reverse transcription PCR without in vitro transcription can detect DNA lesions such as complicated cisplatin DNA adducts but not UV-induced lesions. We found that only this combination of in vitro transcription and real-time reverse transcription PCR can detect both cisplatin- and UV-induced DNA lesions that interfere with transcription.
Conclusions
We anticipate that this method will be useful for estimating the potential transcriptional toxicity of chemicals in terminally differentiated cells engaged in active transcription and translation but not in replication.
Keywords: DNA lesion, Inhibition of RNA synthesis, Genetic toxicity assay
Introduction
Genomic DNA is continuously damaged by various exogenous and endogenous agents [1, 2]. The induced DNA lesions interfere with replication, leading to mutations and cell death, and have been associated with cancer, inborn diseases, and aging. In addition, DNA lesions interfere with transcription, inhibiting elongation by RNA polymerase and leading to reduced transcription and/or mutations of the transcript [3, 4]. Therefore, DNA lesions can induce cytotoxicity by inhibiting replication and/or transcription.
Under laboratory conditions, cell lines are maintained in an environment favorable to growth, DNA repair, prevention of apoptosis, and other aspects of cellular metabolism [3, 5–7]. As cancer and stem cells divide rapidly and constantly, proper experimental conditions for such cell lines are focused on replication. However, most cells within the human body are not exposed to such experimental conditions and are terminally differentiated, and post-mitotic cells engage in active transcription and translation but not in replication. Hence, transcription is assumed to be as important as replication for estimating the genetic toxicity of DNA lesions in human organs.
Inhibition of transcription can be effected by RNA polymerase stalling due to DNA lesions or RNA polymerase II inhibitors such as alpha-amanitin from the death cap [8, 9] and actinomycin D from Streptomyces bacteria, which intercalate into DNA and activate an inducer of apoptosis in many cell lines [10, 11].
All known organisms have repair systems to remove DNA lesions and maintain genomic integrity. When RNA polymerase II encounters DNA damage that blocks transcription during the elongation phase, transcription-coupled repair (TCR) immediately counteracts the interference. Previous studies have demonstrated TCR responses to bulky and helix-distorting lesions such as ultraviolet light (UV)-induced photolesions, e.g., cyclobutane pyrimidine dimers (CPD) [12, 13] and 6–4 photoproducts (6–4 pp) [14], cisplatin intrastrand crosslinks [15], and benzopyrene adducts [16].
Assessment of the biological risk and toxicity of newly synthesized chemicals is hampered by the large number of substances and the complexities of living cells. Current methods for detecting toxic substances are based on the DNA damage response in replication and cell proliferation. For example, the micronucleus assay that detects small nuclei is a well-established method for monitoring genetic toxicity of test substances in the environment [17], and the comet assay (single-cell gel electrophoresis) is a simple and sensitive method for measuring chemically induced DNA damage [18, 19]. Despite the availability of numerous assays to detect genotoxic chemicals, there was no simple method to estimate their effects on transcription until recently.
In our previous work, we established a method for detecting the effects of chemically induced DNA damage on transcription using the nucleotide analog 5-bromouridine and a anti-5-bromouridine antibody and/or the nucleotide analog 5-ethynyluridine [20] and a click chemistry reaction [21, 22] without radioisotopes [23]. Our method is based on a model for transcription-coupled nucleotide excision repair (NER) triggered by blocked transcription at DNA lesions [24, 25]. The method employs common human cells without genetic modifications and terminally differentiated PC12 cells that actively undergo transcription but not replication, and it can detect a wide range of DNA lesions within 8 h of exposure to UV and/or chemicals, e.g., camptothecin, etoposide, 4-nitroquinoline 1-oxide (4NQO), mitomycin, and cisplatin.
Here we present an in vitro assay for detecting the effects of chemically induced DNA lesions using in vitro transcription and real-time reverse transcription polymerase chain reaction (PCR). The method is based on the inhibition of in vitro RNA synthesis for chemically treated DNA templates and allows evaluation of the potential effects of chemicals on transcription in terminally differentiated human cells.
Materials and methods
Enzymes and chemicals
T7 RNA polymerase (T7 RNAP) and reverse transcriptase were purchased from TOYOBO (Osaka, Japan). RNase inhibitor was from Wako (Osaka, Japan), restriction enzymes were from New England Biolabs (Ipswich, MA, USA), Fast SYBR Green Master Mix was from Life Technologies (Carlsbad, CA, USA), and cisplatin was from Sigma-Aldrich (St. Louis, MO, USA).
DNA treatment
Plasmid (pBluescript II SK (−) containing the T7 promoter; Stratagene, La Jolla, CA, USA) DNA templates were purified using the QIAGEN Midi Kit (QIAGEN, Hilden, Germany). For UV irradiation of DNA templates, UV-light (254 nm, 450 J/m2) was used. For cisplatin treatment, DNA templates were incubated with cisplatin (drug/nucleotide ratio = 0.005) at 37 °C overnight.
Polymerase chain reaction (PCR)
For PCR, UV-irradiated or cisplatin-treated DNA samples were used with primers 2140–2159 (5′-tatcagcaataaaccagcca-3′) and 2440–2421 (5′-gcggccaacttacttctgac-3′) and EmeraldAmp PCR Master Mix (TaKaRa, Otsu, Shiga, Japan) according to the manufacturer’s instructions. PCR products were analyzed by 1 % agarose gel electrophoresis.
In vitro transcription
For in vitro transcription, 50-μL reactions containing 100 ng DNA template, 4 mM NTP mixture (ATP, CTP, GTP, and UTP), and 5 units thermo T7 RNAP in buffer (40 mM Tris–HCl, pH 8.0, 50 mM NaCl, 8 mM MgCl2, 5 mM dithiothreitol, 20 units RNase inhibitor) were incubated at 37 °C for 1 h. RNA transcripts were purified using an RNeasy Mini Kit (QIAGEN) with RNase-Free DNase (QIAGEN) according to the manufacturer’s instructions. Transcription products were analyzed by 1 % agarose gel electrophoresis.
Reverse transcription (RT)-PCR
cDNAs were generated from purified RNA samples using primer 2440–2421 (5′-gcggccaacttacttctgac-3′) and ReverTra Ace reverse transcriptase (TOYOBO) according to the manufacturer’s instructions.
Real-time quantitative PCR (qPCR)
Real-time quantitative PCR (qPCR) was performed on a StepOne System (Life Technologies) using Fast SYBR Green Master Mix (Life Technologies) with primers 2140–2159 (5′-tatcagcaataaaccagcca-3′) and 2440–2421 (5′-gcggccaacttacttctgac-3′) to ensure the appearance of a single product peak (301 bp) from mock mixtures in the melting curve analysis. Each reaction was run in triplicate, and the data were plotted as ∆Rn versus cycle number.
Results and discussion
Previously, we demonstrated that our in vivo method for visualizing transcription in mammalian cells can detect UV- and/or chemically (e.g., camptothecin, etoposide, 4NQO, cisplatin) induced damage in genomic DNA by inhibiting RNA polymerase during transcription elongation [20, 22, 23]. Here, we modified this in vivo method to establish a new in vitro method (Fig. 1) using T7 RNAP and DNA templates containing a T7 promoter. RNA polymerase synthesizes new transcripts from a DNA template, which can be detected by RT-PCR (Fig. 1a). However, RNA polymerase stops at chemically induced DNA lesions, and no products are detected by RT-PCR (Fig. 1b). First, we tested the detection of UV-induced DNA lesions (e.g., CPD and 6–4 pp that trigger NER) using conventional PCR. Under these experimental conditions, approximately 12 DNA lesions were expected in plasmid DNA [26]. Although 6–4 pp is more frequently induced than CPD, both lesions are thought to inhibit mammalian RNA polymerase II transcription almost completely [14, 27]. However, conventional PCR (Fig. 2a) could obscure the difference between UV-irradiated and non-irradiated DNA (Fig. 2b), as Taq DNA polymerase might synthesize DNA from the undamaged region (301 bp) in the UV-irradiated DNA template and/or beyond UV lesions such as CPD or 6–4 pp in the PCR cycles [28]. In contrast, T7 RNAP covers the DNA sequence between the T7 promoter and the PCR-detecting region and shows inefficient RNA primer extension from arrested RNAs in a single reaction [29]. This approach seems to be more suitable for detecting DNA lesions than conventional PCR as it reveals the effects of DNA lesions on transcription.
Fig. 1.
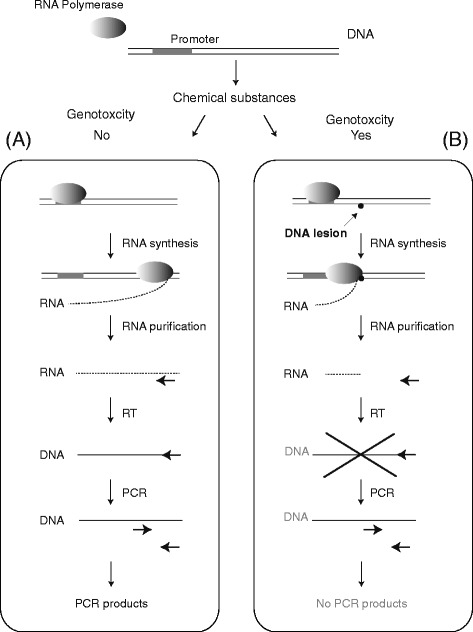
Experimental design. a In the absence of DNA damage, RNA polymerase generates RNA transcripts from DNA templates (normal transcription). After purifying RNA, RT-PCR is performed, and the PCR products are analyzed. b If chemical substances damage DNA, the resulting lesions inhibit RNA synthesis, as RNA polymerase cannot synthesize transcripts from damaged templates, and PCR products will not be detected
Fig. 2.
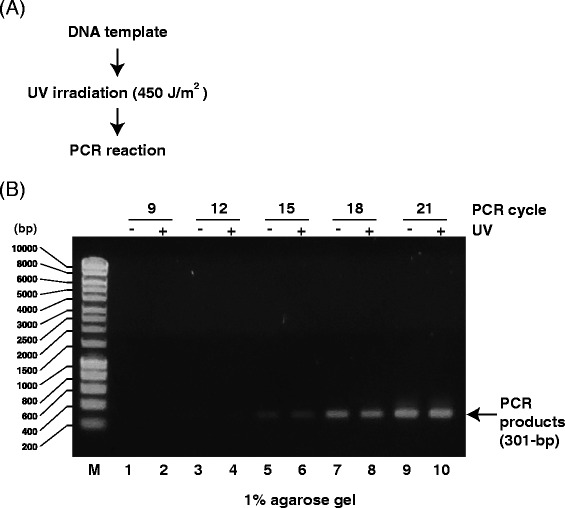
Analysis of PCR products from UV-irradiated DNA templates. a Scheme for detecting DNA damage using PCR products from UV-irradiated (450 J/m2) DNA templates. b Agarose gel (1 %) demonstrating PCR products (301 bp) from UV-irradiated DNA templates. M: size marker. Odd lanes: PCR products from mock DNA templates. Even lanes: PCR products from UV-irradiated DNA templates. DNA was amplified for 9 (lanes 1 and 2), 12 (lanes 3 and 4), 15 (lanes 5 and 6), 18 (lanes 7 and 8), or 21 (lanes 9 and 10) PCR cycles
When we tested this method to detect transcription inhibition by DNA lesions in vitro (Fig. 3a), we identified newly synthesized RNA transcripts (Fig. 3b) and cDNA products from those transcripts using RT-PCR (Fig. 3c), but we could not detect any PCR products without using reverse transcriptase (Fig. 3c, lanes 11 and 12). As shown in Fig. 3c, the amounts of PCR products (301 bp) from non-irradiated DNA (UV -) were higher than those from UV-irradiated DNA (UV +) until 15 PCR cycles (lanes 5 and 6), indicating that UV-induced DNA lesions blocked RNA synthesis by T7 RNAP. However, after 18 PCR cycles, no difference between RT-PCR products from non-irradiated and UV-irradiated templates could be detected (Fig. 3c, lanes 7–10). These results indicate that RT-PCR using agarose gel electrophoresis for detection (Fig. 3a) might not be suitable for determining the effects of UV-induced DNA damage on transcription because it is necessary to optimize the number of PCR cycles.
Fig. 3.
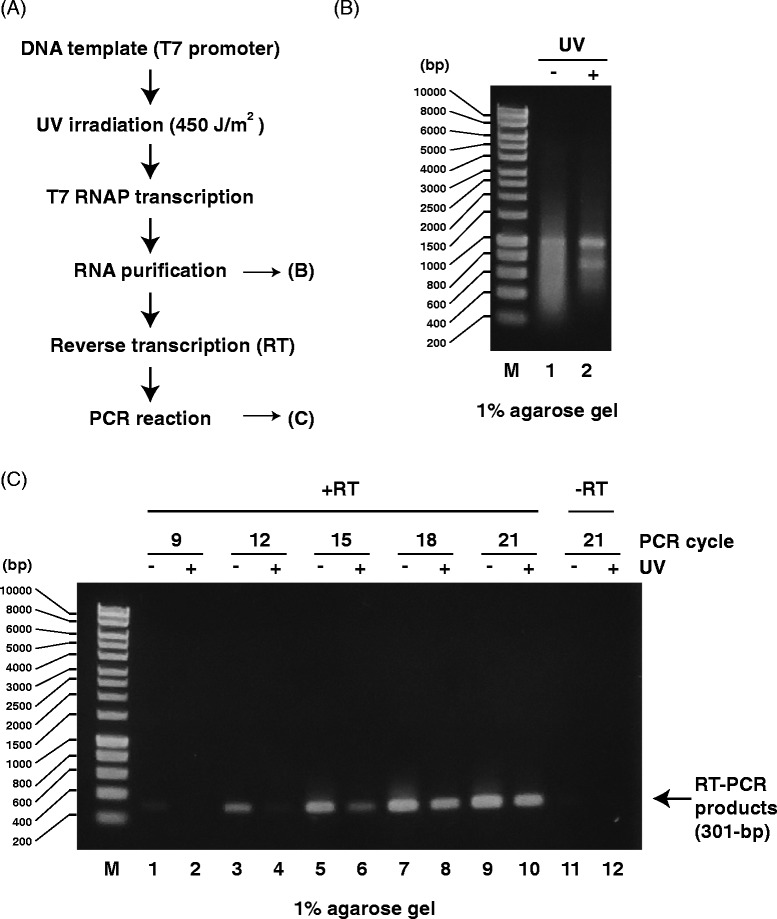
Analysis of PCR products generated by RT-PCR from RNA transcripts of UV-irradiated DNA templates. a Scheme for detecting transcription inhibition using RNA transcripts from UV-irradiated (450 J/m2) DNA templates. b Agarose gel (1 %) demonstrating RNA transcripts from UV-irradiated DNA templates. M: size marker. Lane 1: RNA transcripts from mock DNA templates. Lane 2: RNA transcripts from UV-irradiated DNA templates. c Agarose gel (1 %) demonstrating RT-PCR products (301 bp) from UV-irradiated DNA templates. M: size marker. Odd lanes: RT-PCR products from mock DNA templates. Even lanes: RT-PCR products from UV-irradiated DNA templates. DNA was amplified for 9 (lanes 1 and 2), 12 (lanes 3 and 4), 15 (lanes 5 and 6), 18 (lanes 7 and 8), or 21 (lanes 9 and 10) PCR cycles. Lanes 11 and 12: no RT reaction
Therefore, we tested the utility of qPCR for detecting RNA transcription inhibition by UV-induced DNA lesions (Fig. 4a). qPCR is both powerful and sensitive and is used for a broad range of applications. Combined with reverse transcription, it can quantify RNA in cells or tissues. To adapt the new qPCR method, we first assessed primer sets using melting curve analysis (Fig. 4b) and confirmed that these primers generated one PCR product under our experimental conditions. This primer design is a crucial step because inefficient or non-specific primer annealing will negatively affect the quality and reliability of the assay. The amplification plot of the qPCR analysis showed a delay in the accumulation of qPCR products to later cycles, indicating fewer RNA transcripts from UV-irradiated templates (Fig. 4c). Normalized to non-irradiated templates, the amount of RNA transcripts from UV-irradiated templates was markedly decreased by 0.052-fold (Fig. 4d). As expected, qPCR improved detection of the effects of DNA damage on RNA transcription. Without reverse transcriptase, no specific qPCR products were detected (Fig. 4c and d), confirming the origin of these products from T7 RNAP transcription of UV-irradiated DNA templates. Considering the melting curves shown in Fig. 4c and d, however, we cannot rule out the possibility that PCR products derived from DNA remaining after RNA purification. As qPCR proved suitable for detecting damaged DNA templates, we tried to directly apply this method to UV-irradiated templates (Fig. 5). However, while the amplification plot showed slight differences, the method was not sensitive enough to significantly detect damage of UV-irradiated DNA templates (Fig. 5c and d).
Fig. 4.
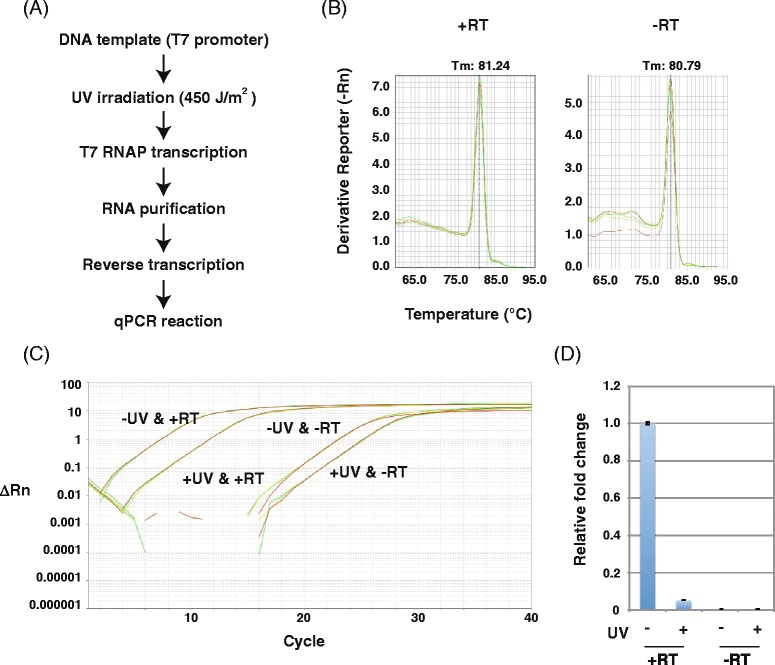
RT-qPCR analysis of transcripts from UV-irradiated DNA templates. a Scheme for detecting transcription inhibition from UV-irradiated (450 J/m2) DNA templates. b Melting curve of RT-qPCR products from RNA transcripts of UV-irradiated DNA templates. Each reaction was run in triplicate. c Amplification plot of RT-qPCR analysis of RNA transcripts of UV-irradiated DNA templates. Each reaction was run in triplicate. d Relative fold change of transcripts from mock (set as 1.0) and UV-irradiated DNA templates. Data show the mean of three samples ± standard deviation (SD)
Fig. 5.
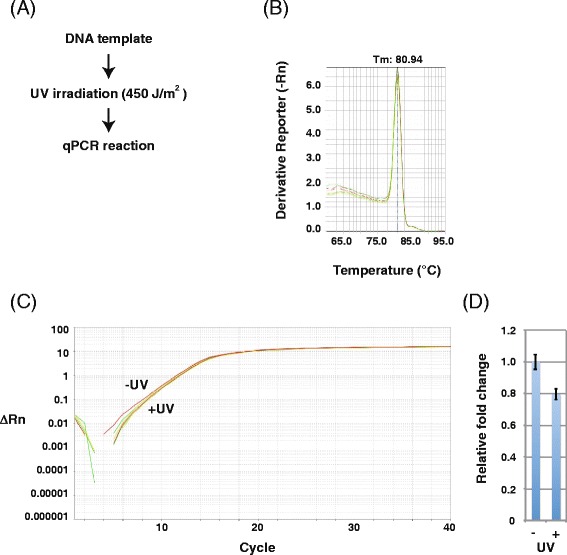
qPCR analysis of UV-irradiated DNA templates. a Scheme for detecting DNA damage using qPCR products from UV-irradiated (450 J/m2) DNA templates. b Melting curve of qPCR products from UV-irradiated DNA templates. Each reaction was run in triplicate. c Amplification plot of qPCR analysis of UV-irradiated DNA templates. Each reaction was run in triplicate. d Relative fold change of PCR products from mock (set as 1.0) and UV-irradiated DNA templates. Data show the mean of three experiments ± standard deviation (SD)
Next, we used cisplatin to directly induce DNA adducts [30, 31] as intrastrand or interstrand crosslinks and monoadducts, which interfere with replication and transcription. Although these adducts are mainly eliminated by NER [32], they are thought to mediate the cytotoxic activity of cisplatin in tumor cells. Using cisplatin-treated DNA samples, we investigated the generation of PCR products from damaged templates. Unlike with UV-damaged templates, conventional PCR (Fig. 6a) revealed differences between cisplatin-treated and untreated DNA (Fig. 6b). These results suggest that cisplatin DNA adducts efficiently block DNA synthesis by Taq DNA polymerase and/or prevent primer annealing. Consistent with this observation, we obtained similar results using qPCR to detect cisplatin-induced DNA adducts, revealing a clear difference between damaged and non-damaged DNA (Fig. 7).
Fig. 6.
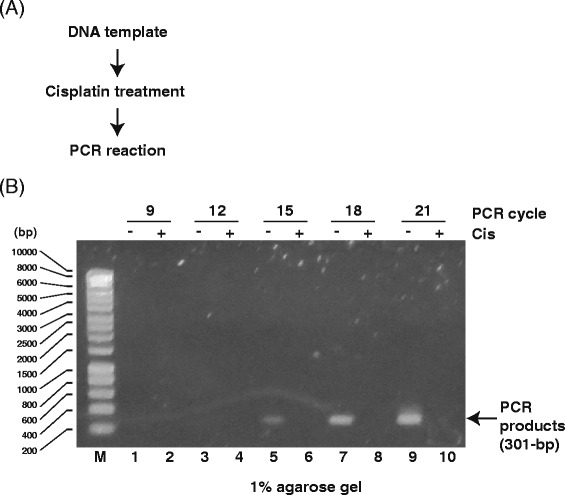
Analysis of PCR products generated from cisplatin-treated DNA templates. a Scheme for detecting DNA damage using PCR products from cisplatin-treated DNA templates (drug/nucleotide ratio = 0.005). b Agarose gel (1 %) demonstrating PCR products (301 bp) from cisplatin-treated DNA templates. M: size marker. Odd lanes: PCR products from mock DNA templates. Even lanes: PCR products from UV-irradiated DNA templates. DNA was amplified for 9 (lanes 1 and 2), 12 (lanes 3 and 4), 15 (lanes 5 and 6), 18 (lanes 7 and 8), or 21 (lanes 9 and 10) PCR cycles
Fig. 7.
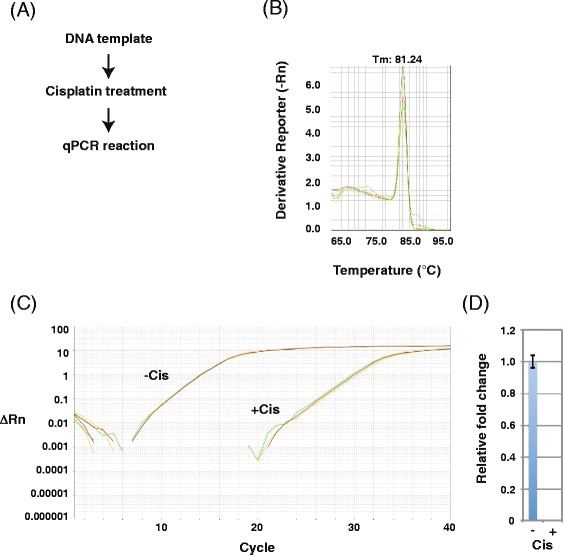
qPCR analysis of cisplatin-treated DNA templates. a Scheme for detecting DNA damage using PCR products from cisplatin-treated DNA templates (drug/nucleotide ratio = 0.005). b Melting curve of qPCR products from cisplatin-treated DNA templates. Each reaction was run in triplicate. c Amplification plot of qPCR analysis of cisplatin-treated DNA templates. Each reaction was run in triplicate. d Relative fold change of PCR products from mock (set as 1.0) and cisplatin-treated DNA templates. Data show the mean of three experiments ± standard deviation (SD)
Previous biochemical studies demonstrated that T7 RNAP stalls at cisplatin DNA adducts [33, 34]. Accordingly, when we tested the effects of cisplatin on DNA templates via transcription and RT-PCR (Fig. 8a), we detected newly synthesized RNA transcripts from untreated DNA templates (Fig. 8b, lane 1) but not from cisplatin-treated templates (Fig. 8b, lane 2) and detected cDNA from untreated DNA templates (Fig. 8c, lanes 3, 5, 7, and 9) but not cisplatin-treated templates (Fig. 8c, lanes 4, 6, 8, and 10). No PCR products were detected in the absence of RT (Fig. 8c, lanes 11 and 12). These results indicate that cisplatin DNA adducts inhibited T7 RNAP transcription initiation and/or elongation. qPCR melting curve analysis (Fig. 9a) indicated that the primer set generated one PCR product after RNA transcription from cisplatin-treated DNA templates (Fig. 9b), suggesting that the PCR product is specific. The amplification plot (Fig. 9c) and the relative fold change (Fig. 9d) showed little T7 RNAP transcription of cisplatin-treated DNA templates, and insignificant amounts of PCR products were generated without RT, indicating that this method can detect the inhibition of RNA synthesis by cisplatin DNA damage.
Fig. 8.
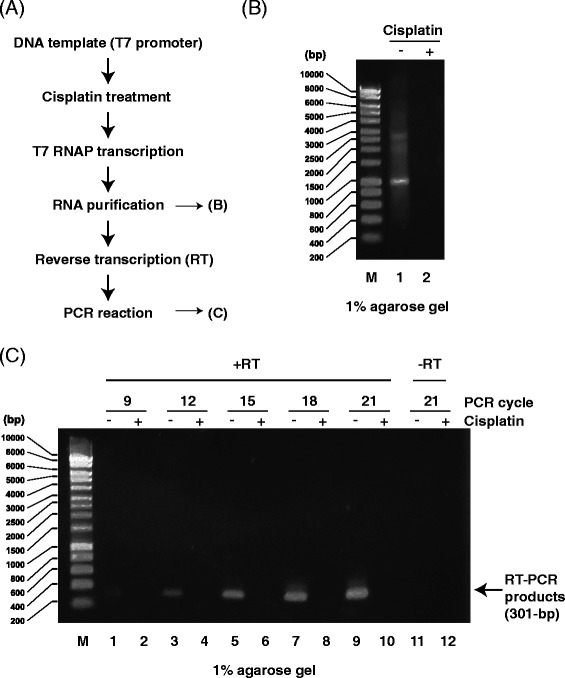
Analysis of PCR products generated by RT-PCR from RNA transcripts of cisplatin-treated DNA templates. a Scheme for detecting transcription inhibition using RNA transcripts from cisplatin-treated DNA templates (drug/nucleotide ratio = 0.005). b Agarose gel (1 %) demonstrating RNA transcripts from cisplatin-treated DNA templates. M: size marker. Lane 1: RNA transcripts from mock DNA templates. Lane 2: RNA transcripts from cisplatin-treated DNA templates. c Agarose gel (1 %) demonstrating RT-PCR products (301 bp) from cisplatin-treated DNA templates. M: size marker. Odd lanes: RT-PCR products from mock DNA templates. Even lanes: RT-PCR products from cisplatin-treated DNA templates. DNA was amplified for 9 (lanes 1 and 2), 12 (lanes 3 and 4), 15 (lanes 5 and 6), 18 (lanes 7 and 8), or 21 (lanes 9 and 10) PCR cycles. Lanes 11 and 12: no RT reaction
Fig. 9.
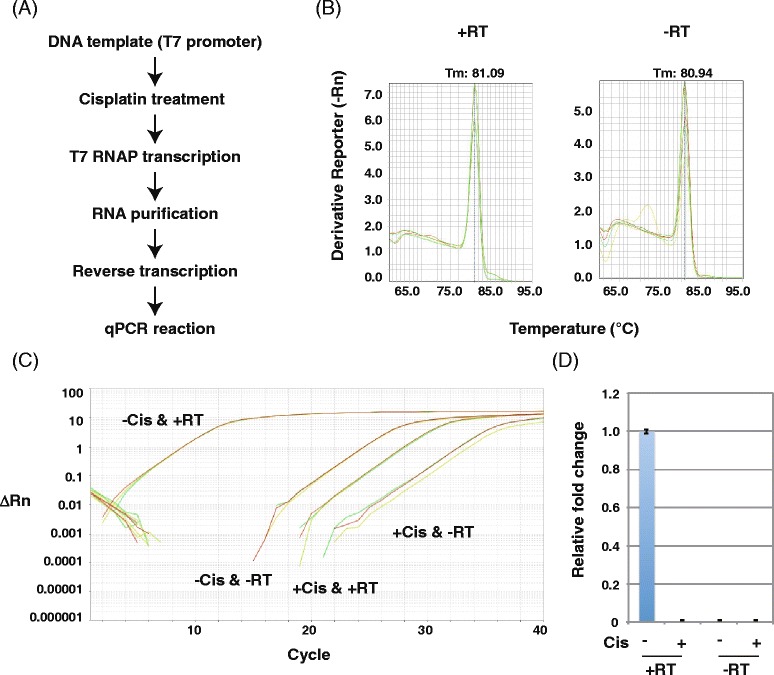
RT-qPCR analysis of transcripts from cisplatin-treated DNA templates. a Scheme for detecting transcription inhibition from cisplatin-treated DNA templates (drug/nucleotide ratio = 0.005). b Melting curve of RT-qPCR products from RNA transcripts of cisplatin-treated DNA templates. Each reaction was run in triplicate. c Amplification plot of RT-qPCR analysis of RNA transcripts of cisplatin-treated DNA templates. Each reaction was run in triplicate. d Relative fold change of transcripts from mock (set as 1.0) and cisplatin-treated DNA templates. Data show the mean of three samples ± standard deviation (SD)
Conclusions
In conclusion, conventional PCR and qPCR in the absence of T7 RNAP transcription can detect chemically induced DNA lesions such as cisplatin DNA adducts but not UV-induced lesions. Only the combination of T7 RNAP transcription and qPCR can detect both cisplatin- and UV-induced DNA lesions that interfere with transcription. Therefore, our results support the idea analysis of transcription products can be used to detect damage in DNA templates, consistent with the model of TCR [7, 25]. Our new method might reveal DNA lesions that cannot be detected by conventional replication-based methods and should facilitate research on DNA damage responses.
Acknowledgements
We would like to thank Editage (www.editage.jp) for English language editing. This work was supported by a Grant-in-Aid for Scientific Research (B) 25281018 from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
Abbreviations
- 4NQO
4-nitroquinoline 1-oxide
- 6-4 pp
6–4 photoproduct
- CPD
Cyclobutane pyrimidine dimer
- NER
Nucleotide excision repair
- PCR
Polymerase chain reaction
- qPCR
Real-time quantitative PCR
- RT
Reverse transcription
- T7 RNAP
T7 RNA polymerase
- TCR
Transcription-coupled repair
- UV
Ultraviolet light
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YS performed the experiments. SI designed the research. IK designed the research and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Yuina Sonohara, Email: yuina@bio.chem.es.osaka-u.ac.jp.
Shigenori Iwai, Email: iwai@chem.es.osaka-u.ac.jp.
Isao Kuraoka, Phone: +81-6-6850-6240, Email: kuraoka@chem.es.osaka-u.ac.jp.
References
- 1.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 2.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. Washington, DC: American Society for Microbiology Press; 2005. [Google Scholar]
- 3.Doetsch PW. Translesion synthesis by RNA polymerases: occurrence and biological implications for transcriptional mutagenesis. Mutat Res. 2002;510:131–40. doi: 10.1016/S0027-5107(02)00258-0. [DOI] [PubMed] [Google Scholar]
- 4.Kuraoka I. Effects of DNA lesions on transcription elongation by RNA polymerases. Genes Environ. 2008;30:63–70. doi: 10.3123/jemsge.30.63. [DOI] [Google Scholar]
- 5.Saxowsky TT, Doetsch PW. RNA polymerase encounters with DNA damage: transcription-coupled repair or transcriptional mutagenesis? Chem Rev. 2006;106:474–88. doi: 10.1021/cr040466q. [DOI] [PubMed] [Google Scholar]
- 6.Nouspikel T, Hanawalt PC. DNA repair in terminally differentiated cells. DNA Repair (Amst) 2002;1:59–75. doi: 10.1016/S1568-7864(01)00005-2. [DOI] [PubMed] [Google Scholar]
- 7.Kuraoka I. When and where polymerases encounter DNA lesions. Genes Environ. 2012;34:58–62. doi: 10.3123/jemsge.34.58. [DOI] [Google Scholar]
- 8.Chafin DR, Guo H, Price DH. Action of alpha-amanitin during pyrophosphorolysis and elongation by RNA polymerase II. J Biol Chem. 1995;270:19114–9. doi: 10.1074/jbc.270.32.19114. [DOI] [PubMed] [Google Scholar]
- 9.Bushnell DA, Cramer P, Kornberg RD. Structural basis of transcription: alpha-amanitin-RNA polymerase II cocrystal at 2.8 A resolution. Proc Natl Acad Sci U S A. 2002;99:1218–22. doi: 10.1073/pnas.251664698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sobell HM. Actinomycin and DNA transcription. Proc Natl Acad Sci U S A. 1985;82:5328–31. doi: 10.1073/pnas.82.16.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassé C, Giannoni F, Nguyen VT, Dubois MF, Bensaude O. The transcriptional Inhibitors, actinomycin D and alpha-amanitin, activate the HIV-1 promoter and favor phosphorylation of the RNA polymerase II C-terminal domain. J Biol Chem. 1999;274:16097–106. doi: 10.1074/jbc.274.23.16097. [DOI] [PubMed] [Google Scholar]
- 12.Selby CP, Drapkin R, Reinberg D, Sancar A. RNA polymerase II stalled at a thymine dimer: footprint and effect on excision repair. Nucleic Acids Res. 1997;25:787–93. doi: 10.1093/nar/25.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brueckner F, Hennecke U, Carell T, Cramer P. CPD damage recognition by transcribing RNA polymerase II. Science. 2007;315:859–62. doi: 10.1126/science.1135400. [DOI] [PubMed] [Google Scholar]
- 14.Mei Kwei JS, Kuraoka I, Horibata K, Ubukata M, Kobatake E, Iwai S, et al. Blockage of RNA polymerase II at a cyclobutane pyrimidine dimer and 6–4 photoproduct. Biochem Biophys Res Commun. 2004;320:1133–8. doi: 10.1016/j.bbrc.2004.06.066. [DOI] [PubMed] [Google Scholar]
- 15.Lainé JP, Egly JM. Initiation of DNA repair mediated by a stalled RNA polymerase IIO. EMBO J. 2006;25:387–97. doi: 10.1038/sj.emboj.7600933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schinecker TM, Perlow RA, Broyde S, Geacintov NE, Scicchitano DA. Human RNA polymerase II is partially blocked by DNA adducts derived from tumorigenic benzo[c]phenanthrene diol epoxides: relating biological consequences to conformational preferences. Nucleic Acids Res. 2003;31:6004–15. doi: 10.1093/nar/gkg771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirsch-Volders M, Decordier I, Elhajouji A, Plas G, Aardema MJ, Fenech M. In vitro genotoxicity testing using the micronucleus assay in cell lines, human lymphocytes and 3D human skin models. Mutagenesis. 2011;26:177–84. doi: 10.1093/mutage/geq068. [DOI] [PubMed] [Google Scholar]
- 18.Ostling O, Johanson KJ. Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem Biophys Res Commun. 1984;123:291–8. doi: 10.1016/0006-291X(84)90411-X. [DOI] [PubMed] [Google Scholar]
- 19.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–91. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 20.Morita Y, Iwai S, Kuraoka I. A method for detecting genetic toxicity using the RNA synthesis response to DNA damage. J Toxicol Sci. 2011;36:515–21. doi: 10.2131/jts.36.515. [DOI] [PubMed] [Google Scholar]
- 21.Jia N, Nakazawa Y, Guo C, Shimada M, Sethi M, Takahashi Y, et al. A rapid, comprehensive system for assaying DNA repair activity and cytotoxic effects of DNA-damaging reagents. Nat Protoc. 2015;10:12–24. doi: 10.1038/nprot.2014.194. [DOI] [PubMed] [Google Scholar]
- 22.Kametani Y, Iwai S, Kuraoka I. An RNA synthesis inhibition assay for detecting toxic substances using click chemistry. J Toxicol Sci. 2014;39:293–9. doi: 10.2131/jts.39.293. [DOI] [PubMed] [Google Scholar]
- 23.Hasegawa M, Iwai S, Kuraoka I. A non-isotopic assay uses bromouridine and RNA synthesis to detect DNA damage responses. Mutat Res. 2010;699:62–6. doi: 10.1016/j.mrgentox.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Svejstrup JQ. Mechanisms of transcription-coupled DNA repair. Nat Rev Mol Cell Biol. 2002;3:21–9. doi: 10.1038/nrm703. [DOI] [PubMed] [Google Scholar]
- 25.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–70. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 26.Wood RD. Repair of pyrimidine dimer ultraviolet light photoproducts by human cell extracts. Biochemistry. 1989;28:8287–92. doi: 10.1021/bi00447a005. [DOI] [PubMed] [Google Scholar]
- 27.van Hoffen A, Venema J, Meschini R, van Zeeland AA, Mullenders LH. Transcription-coupled repair removes both cyclobutane pyrimidine dimers and 6–4 photoproducts with equal efficiency and in a sequential way from transcribed DNA in xeroderma pigmentosum group C fibroblasts. EMBO J. 1995;14:360–7. doi: 10.1002/j.1460-2075.1995.tb07010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith CA, Baeten J, Taylor JS. The ability of a variety of polymerases to synthesize past site-specific cis-syn, trans-syn-II, (6–4), and Dewar photoproducts of thymidylyl-(3′-- > 5′)-thymidine. J Biol Chem. 1998;273:21933–40. doi: 10.1074/jbc.273.34.21933. [DOI] [PubMed] [Google Scholar]
- 29.Temiakov D, Anikin M, McAllister WT. Characterization of T7 RNA polymerase transcription complexes assembled on nucleic acid scaffolds. J Biol Chem. 2002;277:47035–43. doi: 10.1074/jbc.M208923200. [DOI] [PubMed] [Google Scholar]
- 30.Szymkowski DE, Yarema K, Essigmann JM, Lippard SJ, Wood RD. An intrastrand d(GpG) platinum crosslink in duplex M13 DNA is refractory to repair by human cell extracts. Proc Natl Acad Sci U S A. 1992;89:10772–6. doi: 10.1073/pnas.89.22.10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chijiwa S, Masutani C, Hanaoka F, Iwai S, Kuraoka I. Polymerization by DNA polymerase eta is blocked by cis-diamminedichloroplatinum(II) 1,3-d(GpTpG) cross-link: implications for cytotoxic effects in nucleotide excision repair-negative tumor cells. Carcinogenesis. 2010;31:388–93. doi: 10.1093/carcin/bgp316. [DOI] [PubMed] [Google Scholar]
- 32.Moggs JG, Yarema KJ, Essigmann JM, Wood RD. Analysis of incision sites produced by human cell extracts and purified proteins during nucleotide excision repair of a 1,3-intrastrand d(GpTpG)-cisplatin adduct. J Biol Chem. 1996;271:7177–86. doi: 10.1074/jbc.271.12.7177. [DOI] [PubMed] [Google Scholar]
- 33.Tornaletti S. Transcription arrest at DNA damage sites. Mutat Res. 2005;577:131–45. doi: 10.1016/j.mrfmmm.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 34.Tornaletti S, Patrick SM, Turchi JJ, Hanawalt PC. Behavior of T7 RNA polymerase and mammalian RNA polymerase II at site-specific cisplatin adducts in the template DNA. J Biol Chem. 2003;278:35791–7. doi: 10.1074/jbc.M305394200. [DOI] [PubMed] [Google Scholar]