

Abstract
Background
In beef cattle, maternal care is critical for calf survival and growth. Our objective was to evaluate the major sources of additive genetic variation in maternal behavior and suckling performance in two genetically close beef breeds.
Methods
Maternal performance was assessed based on maternal behavior (MB), milk yield (MY) and udder swelling score (US) of 1236 Blonde d’Aquitaine cows and 1048 Limousin cows. MB was scored just after calving to describe the intensity of the dam’s protective behavior towards her calf. Most of the cows were genotyped using the low-density chip EuroG10K BeadChip, and imputed to the high-density 770K panel within breed. Genetic parameters for each trait were estimated for each breed under a multi-trait best linear unbiased prediction animal model. Genomic analysis was performed for each breed using the high-density genotypes and a Bayesian variable selection method.
Results
Heritabilities were low for MB (0.11–0.13), intermediate for MY (0.33–0.45) and high for US (0.47–0.64). Genetic correlations between the traits ranged from 0.31 to 0.58 and 0.72 to 0.99 for the Blonde d’Aquitaine and Limousin breeds, respectively. Two quantitative trait loci (QTL) were detected for MB in Blonde d’Aquitaine with NPY1R and ADRA2A as candidate causative genes. Thirty to 56 QTL were detected for MY and US in both breeds and 12 candidate genes were identified as having a role in the genetic variation of suckling performance. Since very few pleiotropic QTL were detected, there was little biological explanation for the moderate (0.57) to very high (0.99) genetic correlations estimated between MY and US in the Blonde d’Aquitaine and Limousin cows, respectively. In Blonde d’Aquitaine, the correlation was largely due to the pleiotropic QTL detected in the region upstream of the CG gene, while in Limousin, this region was only identified for US, thus attesting the difference in genetic architecture between the breeds.
Conclusions
Our findings question the assumption that two populations that have close genetic links share many QTL. Nevertheless, we identified four candidate genes that may explain a substantial amount of the genetic variation in suckling performance of these two breeds.
Electronic supplementary material
The online version of this article (doi:10.1186/s12711-016-0223-z) contains supplementary material, which is available to authorized users.
Background
Developments in molecular biology and statistical methodologies have provided new tools to unravel the genetics of complex traits in farm animals. Maternal traits in cattle are of particular economic importance to breeders, but because performance records for these traits are limited in the beef sector, there is a lack of efficient selection tools to improve such traits. Traits related to the udder and milk production influence calf growth and weaning weight, which have a direct effect on the income of livestock farmers [1]. Creation of the maternal bond generates diverse behaviors in cows such as sniffing, licking and protecting the neonate to ensure the growth and survival of their calves [2]. It has been reported that nulliparous dams immediately before or during fetal delivery develop an intense interest for the neonate, due to the action of several hormones [3], some of which (such as oxytocin) are also linked to milk release after teat stimulation by suckling [4]. Therefore, traits that affect maternal care and milk production may be genetically linked. In addition, the maternal behavior of the dam impacts calf survival [5] which is one of the main traits that affects the economic performance of suckler production systems [6]. The aim of this study was to increase knowledge about the major sources of additive genetic variance for some important maternal traits in beef cows that are linked to their behavior and milk production. Rare phenotypes for primiparous beef cows were available for this study: udder swelling before calving, maternal behavior after calving, and milk production. These traits were recorded in two distinct but genetically close breeds [7]: Blonde d’Aquitaine and Limousin. These two breeds are relatively close since the Blonde d’Aquitaine breed was granted an official new breed status in 1962 by associating three branches of this breed from the south-west of France: Garonnaise, Pyrénéenne and Blonde du Quercy (the latter being very similar to Limousin) [8].
The primary objective of our study was to validate the existence of strong genetic correlations between the three maternal traits mentioned above in these two breeds, based on the classic infinitesimal polygenic model. The second aim was to identify the main chromosomal regions that play a role in each of the three maternal traits within the breeds, and to identify putative candidate genes that might have a pleiotropic effect on these traits. The third aim was to compare the results between the breeds in order to determine whether the same quantitative trait loci (QTL) for maternal traits segregate in these two genetically close cattle populations.
Methods
Approval from the Animal Care and Use Committee was not required for this study because the data were collected routinely as part of the breeding program and the determination of phenotypes did not violate the integrity of the animals.
Animals and phenotypes
Data were collected from the French progeny-testing programs for artificial insemination (AI) using bulls from the Blonde d’Aquitaine and Limousin breeds. Three traits that are linked to maternal care were recorded on 2284 purebred females that were born between 2002 and 2011 and were the daughters of 70 Blonde d’Aquitaine sires and 57 Limousin sires. The number of records, raw means and raw variances for each trait are in Table 1.
Table 1.
Number of records, mean and variance of traits for Blonde d’Aquitaine and Limousin primiparous cows
| Blonde d’Aquitaine | Limousin | |||||
|---|---|---|---|---|---|---|
| Trait | US | MB | MY | US | MB | MY |
| Number of records | 1236 | 1212 | 1127 | 1048 | 1048 | 1036 |
| Mean | 3.01 | 4.45 | 5.54 | 3.64 | 4.14 | 6.61 |
| Variance | 0.75 | 1.42 | 1.91 | 0.82 | 0.63 | 3.13 |
MB Maternal Behavior, US Udder Score, MY Milk Yield
The udder score (US) that assesses udder swelling was scored 1 week before calving according to a five-point scale ranging from 1 for the least swollen udder to 5 for the most swollen udder (see Additional file 1). Maternal behavior (MB) was scored during the first hour after parturition and it aimed at describing the intensity of the dam’s protective behavior towards her calf. A score of 1 was assigned to a dam that did not pay attention to her calf, while a score of 5 was assigned to a dam that actively stimulated the newborn calf to suck, particularly by licking it immediately after calving. Milk yield (MY) was assessed using the calf weigh-suckle-weigh technique to estimate milk yield by calculating the difference in calf weights before and after suckling (see Additional file 1).
Genotypes
In total, 2269 females (1259 Limousin and 1010 Blonde d’Aquitaine cows) were genotyped. Most of the females were genotyped using the customized low-density chip EuroG10K BeadChip®. Only 249 Blonde d’Aquitaine cows were genotyped with the Bovine SNP50 BeadChip® [medium-density (MD) chip]. Most of the sires (69 of 78 Blonde d’Aquitaine and 39 of 57 Limousin sires) were genotyped using the Bovine HD BeadChip® corresponding to a high-density (HD) chip with 777K single nucleotide polymorphisms (SNPs). The remaining sires were genotyped with the MD chip. All female genotypes were imputed to HD by applying a two-step procedure, from low-density to MD, then from MD to HD, using the BEAGLE 3.3.0 software [9]. The procedures used for genotype editing and imputation are described in Hozé et al. [10]. Error rates of allelic imputation from low-density to MD were estimated at 1.3 % for Blonde d’Aquitaine and 1.6 % for Limousin [11] and those from MD to HD were estimated at 1.8 and 1.1 %, respectively [10]. After editing, 706,791 SNPs were used for the analysis. SNPs were mapped to the UMD 3.1 bovine genome sequence assembled by the Center of Bioinformatics and Computational Biology at the University of Maryland.
Statistical model used to estimate genetic parameters
The analyses were performed using a multi-trait linear model, considering the animal genetic effect for each trait i:
where is the vector of observations, is the vector of fixed effects, is the vector of random effects of cow, is the vector of random residual effects, and , and are the corresponding incidence matrices for trait i. It is assumed that , and . Noting and , the genetic and residual variance–covariance matrices for the multi-trait model were defined as follows:
and
where is the additive genetic variance for trait , is the genetic covariance between traits and , is the residual variance for trait , is the residual covariance between traits and , is the relationship matrix, and is the identity matrix.
The relationship matrices included 6657 Blonde d’Aquitaine and 8836 Limousin animals with pedigrees that could be traced back five generations. The discrete nature of the US and MB scores was not taken into account because linear models can perform evenly or better than threshold models when assessing genetic values for which several ordinal categories are considered or the amount of information per level of fixed effect is small [12, 13]. Moreover, estimation of genetic correlations is not affected by the statistical treatment (linear or threshold model) of the categorical trait [14].
The fixed effects considered for the three traits were the birth region in France (six and five levels for the Blonde d’Aquitaine and Limousin breeds, respectively), birth year-season of the heifers (four seasons), gender of the calf and effect of the mating bull (two bulls per breed). The age at calving (in days) was also fitted as a co-variable for all traits. With respect to the US and MB traits, the calving period (three levels) within the year was added to the previous model. For MY, the suckling batch (four and six levels for the Blonde d’Aquitaine and Limousin breeds, respectively) within 1 year and the calving difficulty score (two levels: easy calving versus hard pull or caesarean section) were added to the model.
Analyses were performed using WOMBAT software [15].
Statistical approach for the detection of QTL
Because the number of SNP effects that had to be estimated was larger than the number of records, detection of QTL was based on a Bayesian variable selection approach [16]. We considered a BayesC strategy [17] where a fraction of the SNPs, , was assumed to have a non-zero effect at each iteration. SNP effects were estimated using a mixture of a proportion of SNPs with a normal effect distribution and a proportion of SNPs with mass point distribution at 0. The general linear mixture model was defined as follows:
where is the phenotype adjusted for environmental effects of animal , the mean for the trait considered, the number of SNPs, the genotype at locus for animal (with for the homozygote with allele 1 at locus , for the opposite heterozygote and for the heterozygote), the effect for SNP , the indicator variable ( if SNP was selected at a given iteration, , otherwise), and the random residual effect. We used GS3 software [18] to perform these analyses. A total of 100,000 iterations were performed, with a burn-in of 20,000 iterations. The retained value was equal to 0.025 %, corresponding to 177 SNPs selected at each iteration among all the SNPs on the HD chip, in order to have a smaller number of SNP effects to estimate at each iteration than the number of phenotypic records. This value of was sufficient to completely capture the additive variance derived under the pedigree BLUP animal model. We verified that adding a polygenic component to the sum of the SNP effects in the model did not help to capture any spurious associations and did not modify the results of the pure genomic model. Therefore, a pure genomic model was retained to present the results.
Definition of the QTL regions
The degree of association between each SNP and the phenotypes was assessed using the Bayes factor (BF) [19]. This involves and as in the following equation [20], being the probability for the SNP of having a non-zero effect:
The Bayes factor offers a rigorous and clear framework to compare competing models. It is the recommended statistical criterion when using the Bayesian method to detect QTL [21, 22].
A transformation of BF (logBF), which is computed as twice the natural logarithm of the BF, was considered in order to obtain a clearer visual appraisal of all QTL regions at the scale of the chromosome (see Fig. 1). Since this logarithmic scale produced values within the same usual range as the deviance and likelihood ratio test values, it facilitated the determination of thresholds to define QTL as proposed by Kass and Raftery [20]. Indeed, these authors suggested the following categories to classify the strength of the evidence provided by logBF: evidence in favor of the existence of the effect is positive for values within the interval (3, 6], strong for values within the interval (6, 10] and very strong for values higher than 10. In this study, we assumed that a chromosomal region qualified as a QTL when it contained at least one SNP with a peak logBF above a threshold of 8 (BF ≈ 55). We considered that the evidence for this QTL was strong when the peak logBF was above a threshold of 12 (BF ≈ 400). Finally, we considered a chromosomal region as a “putative QTL” when it contained at least one SNP with a logBF value within the interval (6, 8[.
Fig. 1.
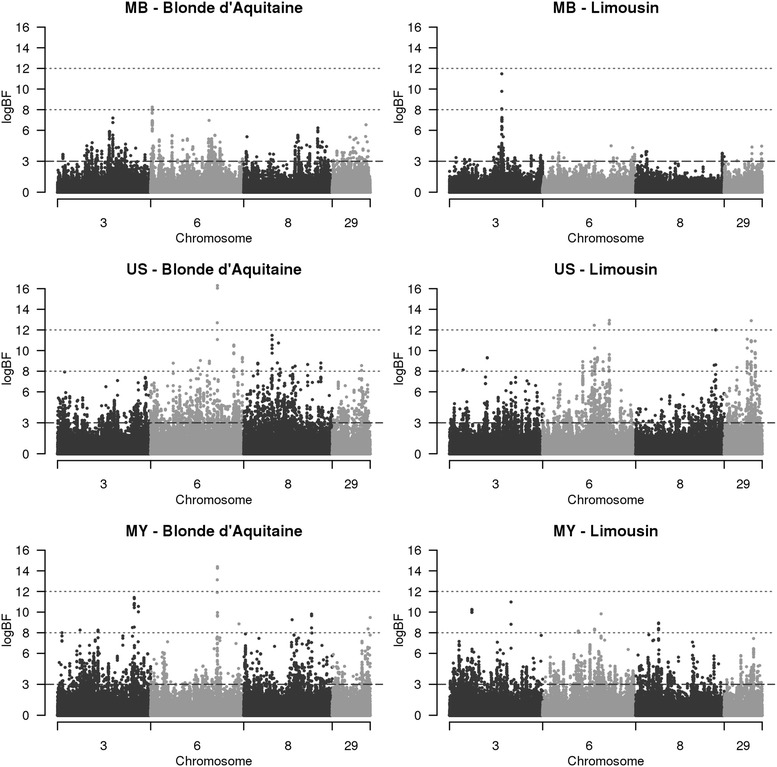
Chromosome plots of logBF for maternal behavior, udder swelling score and milk yield for the two breeds. The logBF values for all SNPs were plotted for chromosomes 3, 6, 8, and 29 for the detection of QTL for maternal behavior (MB), udder swelling score (US) and milk yield (MY)
Accuracy of genomic prediction in the reference population was estimated by calculating the correlation coefficient r between the phenotypes and the genomic predicted breeding values that were derived from the effects of the 706,791 SNPs. This estimated accuracy is a proxy for the true accuracy of genomic prediction which is the correlation between the genomic predicted breeding values and the true breeding values (instead of the phenotypes). The contribution of each QTL in the total genomic prediction was assessed by considering the ratio of the correlation between the phenotypes and the partial genomic values derived from the sum of the effects of SNPs within the QTL region over the total accuracy r. This ratio aims at quantifying the impact of the QTL on the estimated accuracy of genomic prediction for each trait.
Because SNPs with the highest logBF are not necessarily those that are closest to the causal mutation, SNPs that had a logBF higher than 3 and were located close to the peak SNP were also included in the QTL region when these SNPs were within a sliding window of 0.5 Mb on each side of the peak SNP. The sliding window approach was applied when at least one SNP with a logBF higher than 3 was found in the current window. The start and end bounds of the QTL regions were defined by the locations of the last SNP that had a logBF higher than 3 and was within the QTL region.
Results and discussion
Genetic parameters
Genetic parameters and their standard errors (SE) are in Table 2 for the three traits and the two breeds. Heritabilities (on the diagonal, Table 2) covered a broad range of values from 0.11 to 0.64. Regardless of the breed, MB had a lower heritability (0.11 to 0.13) than MY (0.33 to 0.45), and MY had a lower heritability than US (0.47 to 0.64). While the heritability of MB was very similar between the Blonde d’Aquitaine and Limousin breeds, the heritability of MY and US was higher in the Limousin (0.45 and 0.64, respectively) than in the Blonde d’Aquitaine breed (0.33 and 0.47, respectively). Regarding the low heritability of MB, two very different estimates are found in the literature, i.e. Phocas et al. [23] estimated a heritability of 0.36 (SE = 0.06) in an earlier study based on the progeny-testing records for the French Limousin breed and Vallée et al. [24] estimated an extremely low heritability of 0.02 (SE = 0.01) for the same trait recorded on-farm in a population of French Charolais cows. Udder morphology has not been extensively studied in beef cows because it is not an easy trait to score, unlike in dairy cows that visit the milking parlor. Our results based on station data led to high heritabilities for US in both breeds. Previous studies reported lower heritabilities based on field records of beef cows: 0.12 (SE = 0.02), 0.20 (SE = 0.04), 0.23 (SE = 0.05) and 0.43 (SE = 0.03), respectively for udder development in Asturiana de los Valles [25], udder volume in Charolais [24], udder score in Hereford [26] and udder width in the Rendena dual-purpose breed [27]. Heritability estimates for MY were moderate and considerably higher than those for maternal weaning weight: 0.13 in the Blonde d’Aquitaine and 0.12 in the Limousin breed, based on farm records [28]. Although maternal effects on calf growth are generally expected to be due to the dam’s milk production [29], a maternal effect on weaning weight is a more complex trait than milk production and accounts for milk quality, maternal bonding and calf behavior. In addition, assessing phenotypes on a station rather than on a farm can markedly limit any environmental effects and therefore provide higher heritability estimates than those based on field data. Using a similar measurement of milk production as that used in our study, MacNeil and Mott [26] reported a heritability of 0.25 (SE = 0.06) for MY in Hereford cattle.
Table 2.
Genetic parameters for the three traits in Blonde d’Aquitaine and Limousin cattle
| Blonde d’Aquitaine | Limousin | |||||
|---|---|---|---|---|---|---|
| Trait | US | MB | MY | US | MB | MY |
| US | 0.470 (0.099) | 0.312 (0.240) | 0.572 (0.151) | 0.640 (0.121) | 0.790 (0.228) | 0.994 (0.072) |
| MB | 0.042 | 0.135 (0.067) | 0.584 (0.244) | 0.194 | 0.108 (0.068) | 0.720 (0.260) |
| MY | 0.281 | 0.163 | 0.331 (0.092) | 0.305 | 0.161 | 0.455 (0.113) |
Heritabilities are underlined on the diagonal, genetic correlations are above the diagonal with standard errors in brackets, phenotypic correlations are in italics below the diagonal (standard errors ranging from 0.03 to 0.04)
MB maternal behavior, US udder score, MY milk yield
The phenotypic correlations (Table 2) were positive between the three traits in both breeds. The strongest phenotypic correlations were around 0.30 between MY and US in both breeds. Genetic correlations (Table 2) were generally stronger. Genetic correlations between the three traits ranged from 0.31 to 0.58 in the Blonde d’Aquitaine breed and from 0.72 to 0.99 in the Limousin breed. Due to the small number of records, SE of the genetic correlations were relatively large, ranging from 0.07 to 0.26.
The environmental correlations were all less than 0.10 in absolute value, except between US and MY in the Limousin breed with a value of −0.52. Persson Waller et al. [30] reported that beef cows with pendulous udders were at risk of subclinical mastitis. Marked swelling of the udder may then induce mastitis and consequently reduce milk production. This phenomenon may explain the strong negative environmental correlation between US and MY.
The three traits were clearly more strongly genetically correlated in the Limousin than in the Blonde d’Aquitaine breed. In particular, US and MY phenotypes are expected to be controlled by the same pools of genes in the Limousin cows, which may not be the case for the Blonde d’Aquitaine cow for which only a moderate genetic association (0.57, SE = 0.15) was estimated. MacNeil and Mott [26] estimated a moderately positive genetic correlation (0.36, SE = 0.16) between udder score and milk production in Hereford cattle, while Berry et al. [31] also derived positive but moderate correlations (ranging from 0.32 to 0.48) between MY and some udder traits (fore-udder attachment, udder support and rear-udder height) in Holstein primiparous cows.
Detection of QTL within breed
Regardless of the breed, very few QTL were detected for MB, which is probably due to its low heritability compared to the moderate to high heritabilities estimated for US and MY. Thirty to 56 QTL were detected for US and MY with a threshold logBF level equal to 8.
A strong correlation (0.98) between the estimates of (underlying the BF) and the absolute value of the estimated effects for each SNP was computed for all traits. Therefore, a QTL that is supported by strong evidence based on the BF value is expected to be a QTL with an important effect on performance.
QTL detected for the Blonde d’Aquitaine breed
In the Blonde d’Aquitaine population, 56, 53 and two QTL were detected for US, MY and MB, respectively. The main QTL region for MB was on chromosome 6 (Table 3; Fig. 1) and it explained 18.8 % of the estimated total accuracy of genomic prediction for MB (r = 0.72). The NPY1R and NPY5R genes (Table 6) are located close to the peak SNP of this QTL and represent good candidate genes. Longo et al. [32] showed that these two genes were co-expressed and linked to stress and anxiety in mice. In addition, Muroi and Ishii [33] demonstrated the relationship between NPY1R and maternal behavior by assessing mice that crouch over their pups after a period of separation. The second QTL for MB was located on chromosome 26 (Table 3) and explained 21.5 % of the accuracy of genomic prediction. The pseudogene ADRA2A (Table 6) present in the corresponding human, murine and rat genomic regions that align with the QTL region detected here, encodes an adrenergic receptor which is related to several behavior traits [34], and thus, it is a likely candidate gene for MB.
Table 3.
QTL regions (in Mb) with strong evidence for the peak SNP and corresponding candidate genes in the Blonde d’Aquitaine breed
| Trait | Chra | Start–end positions (in Mb) | Peak position (Mb) | Peak logBF | Contribution to rb (%) | Candidate gene |
|---|---|---|---|---|---|---|
| MB | 6 | 2.362–2.974 | 2.486743 | 8.3 | 18.8 | NPY1R, NPY5R |
| MB | 26 | 31.569–32.67 | 32.099795 | 8.2 | 21.5 | ADRA2A |
| US | 6 | 88.485–88.959 | 88.922396 | 16.3 | 42.5 | GC |
| MY | 5 | 28.577–29.137 | 29.072132 | 13.4 | 15.2 | SLC11A2 |
| MY | 6 | 88.485–89.223 | 88.919352 | 14.4 | 20.6 | GC |
| MY | 10 | 69.747–72.705 | 70.306697 | 12.8 | 20.8 | – |
| MY | 13 | 82.728–84.013 | 83.805618 | 12.1 | 14.7 | DOK5 |
| MY | 20 | 3.861–7.327 | 5.504819 | 13.2 | 19.3 | – |
| MY | 27 | 42.375–43.266 | 42.896895 | 13.4 | 15.9 | UBE2E2 |
MB maternal behavior, US udder score, MY milk yield
aChromosome
br is the accuracy of the genomic prediction for the reference population
Table 6.
Symbol, name and position of candidate genes
| Gene symbol | Gene name | Chra | Position (Mb) |
|---|---|---|---|
| OTOL1 | Otolin 1 | 1 | 106.700432–106.731202 |
| CASQ2 | Calsequestrin 2 | 3 | 27.658862–27.729416 |
| SLC11A2 | Solute Carrier Family 11 Member 2 | 5 | 28.887967–28.914607 |
| NPY5R | Neuropeptide Y Receptor Type 5 | 6 | 2.385668–2.396509 |
| NPY1R | Neuropeptide Y Receptor Type 1 | 6 | 2.415744–2.423445 |
| CORIN | Corin Serine Peptidase | 6 | 67.925293–68.176596 |
| GC | group-specific component | 6 | 88.695940–88.739180 |
| RGP1 | Retrograde Golgi Transport Homolog | 8 | 60.328033–60.333812 |
| PALM2 | Paralemmin 2 | 8 | 101.018440–101.182488 |
| DOK5 | Docking Protein 5 | 13 | 82.696138–82.803966 |
| PABPC1 | Poly(A) Binding Protein, Cytoplasmic 1 | 14 | 65.816006–65.833756 |
| RPL27A | Ribosomal Protein L27a | 15 | 44.469327–44.472127 |
| SCO1 | Cytochrome C Oxidase Assembly Protein | 19 | 30.276156–30.296404 |
| ADRA2A | Adrenoceptor Alpha 2A | 26 | 31.870225–31.871586 |
| UBE2E2 | Ubiquitin-Conjugating Enzyme E2E 2 | 27 | 42.558023–42.569249 |
aChromosome
Only one QTL with strong evidence was detected for US and was located on chromosome 6 (Table 3). Its peak logBF value (16.3) was the highest among all the QTL detected during this analysis. This QTL explained 42.5 % of the estimated total accuracy of genomic prediction for US (r = 0.81). This QTL region was also detected with strong evidence for MY in the Blonde d’Aquitaine population (Fig. 1) and explained 20.6 % of the estimated total accuracy of genomic prediction for MY (r = 0.85). The peak SNPs for these two QTL were separated by a distance of 0.003 Mb on the HD chip, which indicates that they are probably associated with the same causal mutation that has a pleiotropic effect on US and MY. Goddard et al. [35] also detected this region for milk yield in the Holstein breed based on bull sequence data. The region that contains the peak SNP is intergenic but close (<0.2 Mb) to the GC gene (Table 6), which encodes the vitamin D binding protein (Fig. 2). This gene, or its regulatory sequence, can be considered as a good candidate because vitamin D is involved in blood calcium homeostasis, which is required for stable milk production. As shown by Horst et al. [36], a deregulation of calcium homeostasis reduces milk production and can lead to milk fever.
Fig. 2.
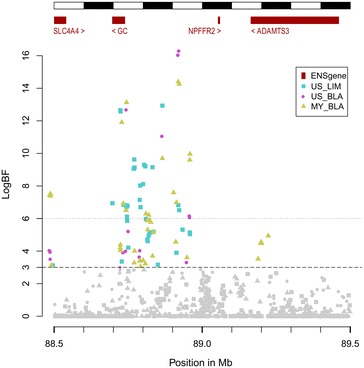
Plot of logBF for SNPs in a 1-Mb QTL region on chromosome 6 for udder score and milk yield. The logBF values for all SNPs were plotted between 88.5 and 89.5 Mb on chromosome 6 for the detection of QTL for milk yield (MY_BLA) and udder score (US_BLA) in Blonde d’Aquitaine and Limousin (US_LIM) cows. The locations of known genes (ENSgene) within the region are indicated at the top of the figure
In addition to this pleiotropic QTL on chromosome 6, five other QTL with strong evidence were detected for MY in the Blonde d’Aquitaine breed and explained 15.2 to 20.8 % of the accuracy of genomic prediction. However, combined together, the six QTL regions only explained 31.0 % of the estimated total accuracy of genomic prediction for MY.
On chromosome 5, three sequence alignments of the SLC11A2 gene (Table 6) were found in the vicinity of the peak SNP (29.072 Mb) of a QTL with strong evidence for MY (Table 3). The SLC11A2 gene is a likely candidate for this QTL because it encodes a divalent metal transporter protein that is involved in regulating essential nutrients such as iron in the milk [37].
The DOK5 gene (Table 6) which was the only gene identified in the QTL region that was detected with strong evidence for MY on chromosome 13. DOK5 encodes a member of the DOK family of membrane proteins, which are adapter proteins involved in signal transduction, and DOK5 has a potential role in insulin and IGF-1 action [38]. This gene can be considered as a candidate gene because IGF-1 stimulates protein synthesis in bovine mammary epithelial cells [39].
On chromosome 27, a QTL with strong evidence for MY (Table 3) corresponded to a single gene that aligned with mouse, rat and sheep sequences: UBE2E2 (Table 6). This gene is a good candidate for MY because it encodes a protein that plays an important role in the synthesis and secretion of insulin and may influence fat and protein content in milk [40, 41].
Two large regions with no particular identified genes were detected with strong evidence for MY on chromosomes 10 and 20 (Table 3). The QTL on chromosome 20 was also detected for maternal weaning weight based on Blonde d’Aquitaine field records (unpublished results).
QTL detected for the Limousin breed
For the Limousin breed, 54, 30 and one QTL were detected for US, MB and MY, respectively. The only QTL detected for MB in the Limousin breed was on chromosome 3 (Table 4) and explained 24.6 % of the estimated total accuracy of genomic prediction for MB (r = 0.75). No particular candidate gene was identified in the region that might play a role in MB (Fig. 1).
Table 4.
QTL regions with strong evidence for the peak SNP and corresponding candidate genes in the Limousin breed
| Trait | Chra | Start–end position (in Mb) | Peak position (Mb) | Peak logBF | Contribution to rb (%) | Candidate gene |
|---|---|---|---|---|---|---|
| MB | 3 | 66.755–68.481 | 67.907266 | 11.5 | 24.6 | – |
| US | 2 | 128.497–129.297 | 128.921997 | 12.2 | 16.5 | – |
| US | 6 | 67.175–69.578 | 68.004090 | 12.5 | 28.8 | CORIN |
| US | 6 | 87.542–88.959 | 88.865430 | 12.9 | 20.6 | GC |
| US | 8 | 100.655–102.859 | 101.161417 | 12 | 19.5 | PALM2 |
| US | 19 | 30.26–30.315 | 30.259765 | 13.9 | 19.4 | SCO1 |
| US | 29 | 33.826–35.441 | 34.797963 | 12.9 | 23.9 | – |
| MY | 5 | 104.195–105.769 | 104.616203 | 12.8 | 23.2 | – |
| MY | 15 | 43.616–45.452 | 44.477700 | 14.4 | 20.1 | RPL27A |
MB maternal behavior, US udder score, MY milk yield
aChromosome
br is the accuracy of the genomic prediction for the reference population
Six QTL were detected with strong evidence for US in the Limousin breed and explained 16.5 to 28.8 % of the accuracy of genomic prediction. Combined together, these six QTL explained 37.4 % of the estimated total accuracy of genomic prediction for US (r = 0.87). The QTL on chromosome 6 had already been reported for MY and US in the Blonde d’Aquitaine breed (Fig. 2). As discussed above, it was the only QTL detected with strong evidence in both breeds. It explained 20.6 % of the accuracy of genomic prediction for US in the Limousin breed. A second QTL was detected on chromosome 6 with strong evidence (Table 4). The peak SNP was within the CORIN gene (Table 6) which plays various roles in the regulation of blood pressure. The CORIN gene is up-regulated in the decidua of the pregnant uterus, which suggests a potential role for CORIN during pregnancy [42].
Another QTL with strong evidence was detected on chromosome 19 (Table 4). This QTL contained the SCO1 gene (Table 6), which encodes a protein involved in the mitochondrial respiratory chain that is essential for adenosine triphosphate (ATP) synthesis [43]. Because of the possible link between this metabolic pathway and udder development, SCO1 is a putative candidate gene. Concerning the QTL with strong evidence on chromosome 8 (Table 4), the PALM2 gene (Table 6), which is involved in the process of cell formation [44], was located near the peak SNP within the QTL region. Finally, two QTL with strong evidence for US but without any particular candidate genes were detected on chromosomes 2 and 29 (Table 4).
Two QTL were detected with strong evidence for MY on chromosomes 5 and 15 in the Limousin breed (Table 4). We did not identify candidate genes in these two QTL regions, although they explained 23.2 and 20.1 % of the accuracy of genomic prediction. Sukegawa et al. [45] reported a significant association between marbling in Japanese Black cattle and the RPL27A gene (Table 6), located next to the peak of the QTL of chromosome 15. Because marbling is related to the metabolic pathway of fat, a possible link may exist between this gene and MY.
QTL shared by genetically-correlated traits within each breed
In this study, the three traits that were considered to be linked to maternal care and suckling performance were moderately to strongly correlated within breeds. Therefore, we assumed that a set of common QTL with a role in at least two of the three traits under study would be found. However, this was not the case between MB and either of the other two traits. The absence of common QTL might be related to the very small number of QTL that were detected for MB in the Limousin and Blonde d’Aquitaine breeds.
Four of the QTL regions that were identified for the Blonde d’Aquitaine breed were common to those detected for US and MY (Table 5). Combined together, these four QTL explained 44.4 % of the estimated total accuracy of genomic prediction for US (r = 0.81) and 22.9 % of the estimated total accuracy of genomic prediction for MY (r = 0.85). As previously noted, the only QTL that was detected with strong evidence for US on chromosome 6 was one of these four common QTL. The contributions of this QTL to the accuracies of genomic predictions for US and MY (42.5 % for US and 20.6 % for MY) were nearly the same as the overall contributions of the four QTL combined together (44.4 % for US and 22.9 % for MY). Thus, the positive genetic correlation that was found between US and MY (Table 2) seems to be mainly explained by this single QTL region on chromosome 6, probably linked to GC gene variants in the Blonde d’Aquitaine breed. Analysis of the three common regions that were detected with a lower logBF peak led to the identification of another putative candidate gene that affects milk production (Fig. 1). On chromosome 8, the RGP1 gene (Table 6) was identified in the vicinity of the peak SNP (at 0.02 Mb) within the QTL for MY (Table 5). This gene is involved in the conversion of guanosine diphosphate (GDP) into guanosine triphosphate (GTP) and is linked to milk production [46].
Table 5.
Common QTL regions between traits and/or breeds and corresponding candidate genes
| Trait 1–trait 2 | Chra | Common region (Mb) | Peak position trait 1 | Peak position trait 2 | Candidate gene |
|---|---|---|---|---|---|
| US_BLA–MY_BLA | 4 | 44.177–44.917 | 44.260073 | 44.198598 | |
| US_BLA–MY_BLA | 6 | 88.485–88.959 | 88.922396 | 88.919352 | GC |
| US_BLA–MY_BLA | 8 | 60.348–60.353 | 61.044151 | 60.352572 | RGP1 |
| US_BLA–MY_BLA | 28 | 43.511–44.63 | 43.242413 | 44.036312 | |
| US_LIM–MY_LIM | 1 | 106.449–106.515 | 106.73685 | 106.008757 | OTOL1 |
| US_LIM–MY_LIM | 6 | 68.405–68.664 | 68.00409 | 68.488326 | CORIN |
| US_BLA–US_LIM | 6 | 52.894–52.978 | 52.814243 | 52.897667 | |
| US_BLA–US_LIM | 6 | 63.663–65.139 | 65.495791 | 63.440877 | |
| US_BLA–US_LIM | 6 | 88.485–88.959 | 88.922396 | 88.86543 | GC |
| US_BLA–US_LIM | 14 | 65.751–65.862 | 65.806497 | 65.802602 | PABPC1 |
| US_BLA–US_LIM | 29 | 37.426–37.505 | 37.459691 | 40.655558 | |
| MY_BLA–MY_LIM | 3 | 27.53–28.035 | 27.842537 | 27.657684 | CASQ2 |
MB maternal behavior, US udder score, MY milk yield
aChromosome
Only two common QTL were detected for US and MY in the Limousin breed. Combined together, these two QTL regions explained 31.6 % of the estimated total accuracy of genomic prediction for US (r = 0.87) and 22.6 % of the estimated total accuracy of genomic prediction for MY (r = 0.84). The QTL region detected on chromosome 1 (Table 5) had a peak SNP that was the same for the two traits. The peak SNP was located in the OTOL1 gene (Table 6) that has variants that affect body mass index in human Japanese or Korean populations [47]. This gene appears to play a role in the energy storage process, and, thus, may impact milk production.
Nevertheless, these two QTL alone, which each accounted only for a limited contribution to the accuracy of genomic predictions, cannot explain the very strong genetic correlation between MY and US. To investigate further this strong genetic correlation, we estimated the contributions of all the SNPs present in the two QTL or 20 putative QTL regions that were common to US and MY, and they accounted for 55.5 and 59.9 % of the accuracy of genomic predictions for US and MY, respectively. These contributions were a little larger than those estimated for the Blonde d’Aquitaine breed (52.8 and 46.1 % for US and MY, respectively), but could not explain the large discrepancy between the genetic correlations in the Limousin and the Blonde d’Aquitaine populations. As a consequence, we hypothesize that a very large number of small polygenic effects explain the very strong correlation observed between US and MY in the Limousin breed. In addition, our results indicate that the sources of additive genetic variance of US and MY clearly differ in the Limousin and Blonde d’Aquitaine breeds.
QTL shared by the two breeds for the same trait
Blonde d’Aquitaine and Limousin are genetically close breeds because of their common geographical origin in south-western France. By studying the genetic history of 47 cattle breeds, Gautier et al. [7] showed that Limousin was the closest breed to Blonde d’Aquitaine based on the FST approach using 50K SNP genotyping data. Servin et al. [48] confirmed these results using 700K SNP genotyping data. Therefore, we assumed that a large set of common QTL for the same trait would be found across the two breeds. Indeed, during the second part of the twentieth century, selective breeding was essentially focused on improving growth and conformation traits rather than maternal traits, thus little divergence was expected to have affected the sources of additive genetic variance of maternal traits between the two breeds. However, our results on the within-breed detection of QTL strongly questioned this initial assumption.
First, we found no common QTL for MB across the two breeds. Second, our results provided strong evidence (logBF > 8) for only one QTL for MY, which was shared by the two breeds. This QTL was located on chromosome 3 (Table 5) and explained only 16.5 and 18.5 % of the accuracy of genomic predictions for the Blonde d’Aquitaine and Limousin breeds, respectively. The peak SNP for MY in the Limousin dataset was located 0.001 Mb upstream of the CASQ2 gene (Table 6). This gene encodes a calcium binding protein that stores calcium for muscle function [49]. In dairy cows, Horst et al. [36] demonstrated the relationship between calcium storage and MY, which makes CASQ2 a possible candidate gene. However, this common QTL was not the major source of genetic variation in MY for the two breeds. If all common (1) and putative QTL (17) for MY are considered, only 43.8 and 49.5 % of the accuracy of genomic predictions was explained for Blonde d’Aquitaine and Limousin, respectively.
Among the 55 QTL detected for US with strong evidence within breeds, five were shared by the two breeds. Combined together, these five QTL explained 44.0 and 34.0 % of the accuracy of genomic predictions for US in the Blonde d’Aquitaine and Limousin breeds, respectively; if the 33 putative common QTL were added, contributions of about 54 % to the genomic prediction accuracies were estimated for each breed.
The QTL for US on chromosome 6 that was supported by strong evidence in both breeds contributed substantially to the accuracy of genomic prediction, although the proportion was smaller for the Limousin (20.6 %) than for the Blonde d’Aquitaine (42.5 %) breed. Detecting this QTL in both breeds further supported the GC gene as a major candidate gene for US in beef cattle. No candidate gene related to US was identified for three of the four other QTL shared by the two breeds. Only one candidate gene was proposed for the common QTL located on chromosome 14 (Table 5). The two peak SNPs were very close to each other (0.004 Mb) and were located about 0.01 Mb from the PABPC1 gene (Table 6), which regulates multiple aspects of mRNA translation and stability [50].
Conclusions
Apart from maternal behavior, which had a low heritability in both breeds (0.11 to 0.13), heritabilities for the two suckling traits, MY and US, ranged from moderate values (0.33 to 0.47) in the Blonde d’Aquitaine to high values (0.45 to 0.64) in the Limousin breed. Genetic correlations between these three traits were moderately positive (0.31 to 0.58) in the Blonde d’Aquitaine cows, but strongly positive (0.72 to 0.99) in the Limousin cows. These results allow for the first time to evaluate the major sources of additive genetic variation of maternal traits in both breeds. They indicate that the polymorphisms of interest for selective breeding may differ substantially between these two breeds although the genetic distance between them is short compared to bovine populations as a whole. Regarding MB, strong evidence supported two QTL in the Blonde d’Aquitaine and one QTL in the Limousin breed and we identified two major candidate genes i.e. NPY1R and ADRA2A. Regarding MY, 30 and 53 QTL were detected for the Limousin and Blonde d’Aquitaine breeds, respectively; of these, only two (Limousin) and six (Blonde d’Aquitaine) were supported by strong evidence. Several good candidate genes were identified for four of the main QTL for MY in the Blonde d’Aquitaine breed: in particular, the upstream region close to the CG gene, but also SLC11A2, DOK5 and UBE2E2. However, no convincing candidate genes were found for the two main QTL for MY in the Limousin breed. In addition, only one QTL region for MY was shared by the two breeds and we did not identify any likely candidate gene to explain this common effect.
For US, 54 and 56 QTL were detected in the Limousin and Blonde d’Aquitaine breeds, respectively. The same QTL with the strongest evidence was found for the Blonde d’Aquitaine and Limousin breeds and it was located next to the CG gene, which was also identified for MY in the Blonde d’Aquitaine breed. Four other common QTL were involved in udder swelling in both breeds. However, apart from the CG gene, only the PABPC1 gene was identified as a putative candidate gene underlying the common QTL. These common QTL between the Limousin and Blonde d’Aquitaine breeds that are involved in the genetic variation of US were the only clear evidence that agreed with their genetic proximity.
Very few pleiotropic QTL were detected for both US and MY. In Blonde d’Aquitaine, the genetic correlation between the two traits was largely due to the QTL located near the CG gene and that had a strong effect on both US and MY. Genotyping data for the Limousin breed did not allow us to clarify the very high genetic correlation between MY and US that was estimated based on performance and pedigree information. As a consequence, we assume that numerous pleiotropic regions with minor effects must explain the polygenic correlation, without any statistical possibility of detecting the corresponding QTL because of the limited size of our dataset.
Nevertheless, we identified 15 candidate genes that could explain the major sources of additive genetic variation in maternal traits in continental beef breeds. Among these, four genes may have a role in the significant variations in suckling performance of both Limousin and Blonde d’Aquitaine cows. Future studies based on the analysis of sequence data for the highlighted regions may allow us to determine the causal mutations.
Authors’ contributions
EV, RS and AM prepared the data. The data were analyzed by AM and FP. AM and FP wrote the paper. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank all the MIDATEST and Idele technicians for performance records collected at the test stations in Casteljaloux and Moussours. Financial support for this work was provided by the MATERGEN project funded by the Conseil Régional Midi-Pyrénées Aquitaine.
Competing interests
The authors declare that they have no competing interests.
Additional file
10.1186/s12711-016-0223-z Description of the recorded traits. Description: Description of the way the three traits were recorded on a station.
Contributor Information
Alexis Michenet, Email: alexis.michenet@jouy.inra.fr.
Romain Saintilan, Email: romain.saintilan@jouy.inra.fr.
Eric Venot, Email: eric.venot@jouy.inra.fr.
Florence Phocas, Email: florence.phocas@jouy.inra.fr.
References
- 1.Goonewardene LA, Wang Z, Price MA, Yang RC, Berg RC, Makarechian M. Effect of udder type and calving assistance on weaning traits of beef and dairy × beef calves. Livest Prod Sci. 2003;81:47–56. doi: 10.1016/S0301-6226(02)00194-X. [DOI] [Google Scholar]
- 2.von Keyserlingk MAG, Weary DM. Maternal behavior in cattle. Horm Behav. 2007;52:106–113. doi: 10.1016/j.yhbeh.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 3.Williams GL, Gazal OS, Leshin LS, Stanko RL, Anderson LL. Physiological regulation of maternal behavior in heifers: roles of genital stimulation, intracerebral oxytocin release, and ovarian steroids. Biol Reprod. 2001;65:295–300. doi: 10.1095/biolreprod65.1.295. [DOI] [PubMed] [Google Scholar]
- 4.Akers RM, Lefcourt AM. Effect of presence of calf on milking-induced release of prolactin and oxytocin during early lactation of dairy cows. J Dairy Sci. 1984;67:115–122. doi: 10.3168/jds.S0022-0302(84)81274-6. [DOI] [PubMed] [Google Scholar]
- 5.Barrier AC, Ruelle E, Haskell MJ, Dwyer CM. Effect of a difficult calving on the vigour of the calf, the onset of maternal behaviour, and some behavioural indicators of pain in the dam. Prev Vet Med. 2012;103:248–256. doi: 10.1016/j.prevetmed.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Phocas F, Bloch C, Chapelle P, Bécherel F, Renand G, Ménissier F. Developing a breeding objective for a French purebred beef cattle selection programme. Livest Prod Sci. 1998;57:49–65. doi: 10.1016/S0301-6226(98)00157-2. [DOI] [Google Scholar]
- 7.Gautier M, Laloë D, Moazami-Goudarzi K. Insights into the genetic history of French cattle from dense SNP data on 47 worldwide breeds. PLoS One. 2010;5:e13038. doi: 10.1371/journal.pone.0013038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.OS Blonde d’Aquitaine. http://www.blonde-aquitaine.fr/site-upra–fr-/la-blonde-d-aquitaine/historique/historique-430.aspx (2015). Assessed 1 Sept 2015.
- 9.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hozé C, Fouilloux MN, Venot E, Guillaume F, Dassonneville R, Fritz S, et al. High-density marker imputation accuracy in sixteen French cattle breeds. Genet Sel Evol. 2013;45:33. doi: 10.1186/1297-9686-45-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saintilan R, Capitan A, Benoit M, Barbier S, Fritz S. Développement d’un outil génomique de présélection sur aptitudes bouchères et facilité de naissance en races Charolaise, Limousine et Blonde d’Aquitaine. Renc Rech Rumin. 2014;21:241–4. http://www.journees3r.fr/IMG/pdf/Texte_3_Genetique_R-Saintilan.pdf.
- 12.Meijering A, Gianola D. Linear versus nonlinear methods of sire evaluation for categorical traits: a simulation study. Genet Sel Evol. 1985;17:115–132. doi: 10.1186/1297-9686-17-1-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phocas F, Laloë D. Evaluation models and genetic parameters for calving difficulty in beef cattle. J Anim Sci. 2003;81:933–938. doi: 10.2527/2003.814933x. [DOI] [PubMed] [Google Scholar]
- 14.Kadarmideen HN, Thompson R, Coffey MP, Kossaibati MA. Genetic parameters and evaluations from single and multiple trait analysis of dairy cow fertility and milk production. Livest Prod Sci. 2003;81:183–195. doi: 10.1016/S0301-6226(02)00274-9. [DOI] [Google Scholar]
- 15.Meyer K. ‘‘WOMBAT’’—digging deep for quantitative genetic analyses by restricted maximum likelihood. In: Proceedings of the 8th world congress on genetics applied to livestock production, 13–18 August 2008, Belo Horizonte (2006).
- 16.Habier D, Fernando RL, Kizilkaya K, Garrick DJ. Extension of the Bayesian alphabet for genomic selection. BMC Bioinformatics. 2011;12:186. doi: 10.1186/1471-2105-12-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kizilkaya K, Fernando RL, Garrick DJ. Genomic prediction of simulated multibreed and purebred performance using observed fifty thousand single nucleotide polymorphism genotypes. J Anim Sci. 2010;88:544–551. doi: 10.2527/jas.2009-2064. [DOI] [PubMed] [Google Scholar]
- 18.Legarra A, Ricard A, Filangi O. GS3, User manual. http://snp.toulouse.inra.fr/~alegarra/manualgs3_last.pdf (2013). Accessed 1 Dec 2014.
- 19.Schurink A, Janss LL, Heuven HC. Bayesian variable selection to identify QTL affecting a simulated quantitative trait. BMC Proc. 2012;6:S8. doi: 10.1186/1753-6561-6-S2-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kass RE, Raftery AE. Bayes factor. J Am Stat Assoc. 1995;90:773–795. doi: 10.1080/01621459.1995.10476572. [DOI] [Google Scholar]
- 21.Legarra A, Croiseau P, Sanchez MP, Teyssèdre S, Sallé G, Allais S, et al. A comparison of methods for whole-genome QTL mapping using dense markers in four livestock species. Genet Sel Evol. 2015;47:6. doi: 10.1186/s12711-015-0087-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wakefield J. Bayes factors for genome-wide association studies: comparison with P-values. Genet Epidemiol. 2009;33:79–86. doi: 10.1002/gepi.20359. [DOI] [PubMed] [Google Scholar]
- 23.Phocas F, Boivin X, Sapa J, Trillat G, Boissy A, Le Neindre P. Genetic correlations between temperament and breeding traits in Limousin heifers. Anim Sci. 2006;82:805–811. doi: 10.1017/ASC200696. [DOI] [Google Scholar]
- 24.Vallée A, Breider I, van Arendonk JA, Bovenhuis H. Genetic parameters for large-scale behavior traits and type traits in Charolais beef cows. J Anim Sci. 2015;93:4277–4284. doi: 10.2527/jas.2015-9292. [DOI] [PubMed] [Google Scholar]
- 25.Gutiérrez JP, Goyache F. Estimation of genetic parameters of type traits in Asturiana de los Valles beef cattle breed. J Anim Breed Genet. 2002;119:93–100. doi: 10.1046/j.1439-0388.2002.00324.x. [DOI] [PubMed] [Google Scholar]
- 26.MacNeil MD, Mott TB. Genetic analysis of gain from birth to weaning, milk production, and udder conformation in line 1 Hereford cattle. J Anim Sci. 2006;84:1639–1645. doi: 10.2527/jas.2005-697. [DOI] [PubMed] [Google Scholar]
- 27.Mazza S, Guzzo N, Sartori C, Berry DP, Mantovani R. Genetic parameters for linear type traits in the Rendena dual-purpose breed. J Anim Breed Genet. 2014;131:27–35. doi: 10.1111/jbg.12049. [DOI] [PubMed] [Google Scholar]
- 28.Phocas F, Laloë D. Genetic parameters for birth and weaning traits in French specialized beef cattle breeds. Livest Prod Sci. 2004;2004(89):121–128. doi: 10.1016/j.livprodsci.2004.02.007. [DOI] [Google Scholar]
- 29.Phocas F, Sapa J. Genetic parameters for growth, reproductive performance, calving ease and suckling performance in beef cattle heifers. Anim Sci. 2004;79:41–48. [Google Scholar]
- 30.Persson Waller K, Persson Y, Nyman AK, Stengärde L. Udder health in beef cows and its association with calf growth. Acta Vet Scand. 2014;56:9. doi: 10.1186/1751-0147-56-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berry DP, Buckley F, Dillon PP, Evans RD, Veerkamp RF. Genetic relationships among linear type traits, milk yield, body weight, fertility and somatic cell count in primiparous dairy cows. Irish J Agric Food Res. 2004;43:161–176. [Google Scholar]
- 32.Longo A, Mele P, Bertocchi I, Oberto A, Bachmann A, Bartolomucci A, et al. Conditional inactivation of neuropeptide Y Y1 receptors unravels the role of Y1 and Y5 receptors coexpressing neurons in anxiety. Biol Psychiatry. 2014;76:840–849. doi: 10.1016/j.biopsych.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Muroi Y, Ishii T. Neuropeptide Y is crucial for nutritional state-dependent regulation of maternal behavior. Psychoneuroendocrinology. 2015;51:392–402. doi: 10.1016/j.psyneuen.2014.09.022. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt KT, Weinshenker D. Adrenaline rush: the role of adrenergic receptors in stimulant-induced behaviors. Mol Pharmacol. 2014;85:640–650. doi: 10.1124/mol.113.090118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goddard ME, MacLeod IM, Kemper KE, Vander Jagt CJ, Savin K, Schrooten, et al. A research plan for the identification of QTL. In: Proceedings of the 10th world congress on genetics applied to livestock production, 17–22 August 2014, Vancouver (2014).
- 36.Horst RL, Goff JP, Reinhardt TA. Adapting to the transition between gestation and lactation: differences between rat, human and dairy cow. J Mammary Gland Biol Neoplasia. 2005;10:141–156. doi: 10.1007/s10911-005-5397-x. [DOI] [PubMed] [Google Scholar]
- 37.Montalbetti N, Dalghi MG, Albrecht C, Hediger MA. Nutrient transport in the mammary gland: calcium, trace minerals and water soluble vitamins. J Mammary Gland Biol Neoplasia. 2014;19:73–90. doi: 10.1007/s10911-014-9317-9. [DOI] [PubMed] [Google Scholar]
- 38.Cai D, Dhe-Paganon S, Melendez PA, Lee J, Shoelson SE. Two new substrates in insulin signaling, IRS5/DOK4 and IRS6/DOK5. J Biol Chem. 2003;278:25323–25330. doi: 10.1074/jbc.M212430200. [DOI] [PubMed] [Google Scholar]
- 39.Burgos SA, Cant JP. IGF-1 stimulates protein synthesis by enhanced signaling through mTORC1 in bovine mammary epithelial cells. Domest Anim Endocrinol. 2010;38:211–221. doi: 10.1016/j.domaniend.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 40.Kazakova EV, Wu Y, Zhou Z, Chen M, Wang T, Tong H, et al. Association between UBE2E2 variant rs7612463 and type 2 diabetes mellitus in a Chinese Han population. Acta Biochim Pol. 2015;62:241–245. doi: 10.18388/abp.2014_936. [DOI] [PubMed] [Google Scholar]
- 41.Winkelman LA, Overton TR. Long-acting insulins alter milk composition and metabolism of lactating dairy cows. J Dairy Sci. 2013;96:7565–7577. doi: 10.3168/jds.2012-6498. [DOI] [PubMed] [Google Scholar]
- 42.Kaitu’u-Lino TJ, Ye L, Tuohey L, Dimitriadis E, Bulmer J, Rogers P, et al. Corin, an enzyme with a putative role in spiral artery remodeling, is up-regulated in late secretory endometrium and first trimester decidua. Hum Reprod. 2013;28:1172–1180. doi: 10.1093/humrep/det028. [DOI] [PubMed] [Google Scholar]
- 43.Kadenbach B, Hüttemann M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion. 2015;24:64–76. doi: 10.1016/j.mito.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 44.Kutzleb C, Sanders G, Yamamoto R, Wang X, Lichte B, Petrasch-Parwez E, et al. Paralemmin, a prenyl-palmitoyl-anchored phosphoprotein abundant in neurons and implicated in plasma membrane dynamics and cell process formation. J Cell Biol. 1998;143:795–813. doi: 10.1083/jcb.143.3.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sukegawa S, Miyake T, Ibi T, Takahagi Y, Murakami H, Morimatsu F, et al. Multiple marker effects of single nucleotide polymorphisms in three genes, AKIRIN2, EDG1 and RPL27A, for marbling development in Japanese Black cattle. Anim Sci J. 2014;85:193–197. doi: 10.1111/asj.12108. [DOI] [PubMed] [Google Scholar]
- 46.Turner MD, Wilde CJ, Burgoyne RD. Exocytosis from permeabilized lactating mouse mammary epithelial cells. Stimulation by Ca2 + and phorbol ester, but inhibition of regulated exocytosis by guanosine 5′-[gamma-thio]triphosphate. Biochem J. 1992;286:13–15. doi: 10.1042/bj2860013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Croteau-Chonka DC, Marvelle AF, Lange EM, Lee NR, Adair LS, Lange LA, et al. Genome-wide association study of anthropometric traits and evidence of interactions with age and study year in Filipino women. Obesity (Silver Spring) 2011;19:1019–1027. doi: 10.1038/oby.2010.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Servin B, Boitard S, Chevalet C, Fariello MI, Phocas F, San Cristobal M. Accounting for population structure and haplotype diversity in whole genome scans for selection signatures. In: Proceedings of the 10th world congress on genetics applied to livestock production, 17–22 August 2014, Vancouver (2014).
- 49.Kalyanasundaram A, Viatchenko-Karpinski S, Belevych AE, Lacombe VA, Hwang HS, Knollmann BC, et al. Functional consequences of stably expressing a mutant calsequestrin (CASQ2D307H) in the CASQ2 null background. Am J Physiol Heart Circ Physiol. 2012;302:H253–H261. doi: 10.1152/ajpheart.00578.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gray NK, Hrabálková L, Scanlon JP, Smith RW. Poly(A)-binding proteins and mRNA localization: who rules the roost? Biochem Soc Trans. 2015;43:1277–1284. doi: 10.1042/BST20150171. [DOI] [PubMed] [Google Scholar]