

Abstract
Eosinophilic esophagitis (EoE) is a recently recognized inflammatory disease of the esophagus with clinical symptoms derived from esophageal dysfunction. The etiology of EoE is now being elucidated, and food hypersensitivity is emerging as the central cornerstone of disease pathogenesis. Herein, we present a thorough picture of the current clinical, pathologic, and molecular understanding of the disease with a focus on disease mechanisms.
Keywords: eosinophilic esophagitis, atopy, barrier dysfunction, epithelial inflammation, fibrosis
INTRODUCTION
The esophagus is normally devoid of eosinophils, but as far back as 1962, the occurrence of esophageal eosinophilia was noted. Eosinophilic esophagitis (EoE) was first described in 1977 by Dobbins et al. (1) as a variant of eosinophilic gastroenteritis, and in 1993, EoE became recognized as a separate disease state (2).
EoE remains relatively uncommon, with annual incidence rates varying between 0.1 and 1.2 cases per 10,000 persons in several studies, representing the second-most common cause of chronic esophagitis. EoE has been reported worldwide, with prevalence rates that continue to increase and reach 1:1,000 in several studies (3). This increase in prevalence is in agreement with a general trend of increasing prevalence of allergic diseases over the last half-century (4). Notably, though, the increase also appears to be a consequence of improved recognition of this disorder, at least since the mid-1980s, in part related to increasing frequency of endoscopic procedures in the pediatric population, rather than a fundamental increase in a new disease entity (5).
This disorder occurs in both the pediatric and adult populations, typically in male patients with evidence of atopy. The primary symptoms of the disorder vary with the age of the patient and progress with age in the following order: difficulty with eating, failure to thrive, vomiting, chest and/or abdominal pain, dysphagia, and food impaction (6, 7) (Figure 1).
Figure 1.
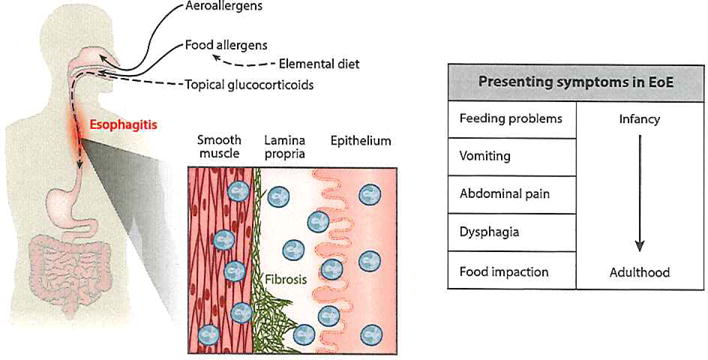
The clinical manifestations of eosinophilic esophagitis (EoE). Both aeroallergens and food allergens have been implicated in EoE. Elemental diet and topical glucocorticoids reverse both symptoms and microscopic features of the disease. The presenting symptoms are age dependent.
CLINICAL ASPECTS OF EoE
Endoscopy
Endoscopically, EoE in adults is characterized by esophageal linear furrows with loss of vascularity, mucosal rings (trachealization), a small-caliber lumen, strictures, mucosal exudates, and, less commonly, polyps and ulcerations (8). The pediatric population can present with similar endoscopic features, though they may be more subtle, and up to one-third of pediatric patients with EoE have a normal endoscopy (4).
Diagnosis
The diagnosis of EoE includes assessment of food and aeroallergen sensitization and exclusion of other eosinophilic diseases of the esophagus, including gastroesophageal reflux disease (GERD) (see the sidebar entitled Differential Diagnoses for Esophageal Eosinophilia). Skin testing may assist in identification of food allergy and result in improved dietary therapy. The primary histologic feature used in diagnosing EoE is an esophageal biopsy with at least 15 eosinophils per high-power field; this is referred to as the peak eosinophil count. Other histopathologic changes consistent with EoE include basal layer hyperplasia, papillary lengthening, and lamina propria fibrosis (8). In addition, the high level of esophageal eosinophilia should be resistant to a proton-pump inhibitor (PPI) trial of 8 weeks (4). Although PPI-responsive esophageal eosinophilia has been thought to be an acid-induced disease, there is emerging evidence that this disease entity may be a subtype more closely aligned with EoE (9). Histologic response to dietary antigen elimination and/or to topical corticosteroids may suggest EoE but is not considered a diagnostic criterion.
Recently, the EoE diagnostic panel (EDP), a set of 94 genes differentially expressed in the esophagus of EoE patients, was introduced (10). The EDP identifies EoE with ~96% sensitivity and ~98% specificity in adult and pediatric patients and distinguishes patients with EoE in remission from non-EoE controls, as well as identifies patients who have been exposed to swallowed glucocorticoids. The EDP demonstrates predicative capacity for patients with subclinical histology (1–14 eosinophils per high-power field), suggesting that these patients should be tightly monitored as an EoE high-risk population and indicating the prospective utility of the EDP as a prognostic approach in personalized medicine. The EDP has the potential to overcome the limitations of histologic analysis, as it provides potentially deeper insight into tissue processes that are not visible microscopically or that may be microscopically patchy, highlighting the transformative value of using molecular parameters rather than histology for the diagnosis of inflammatory diseases. Furthermore, the EDP has the capacity to reveal EoE pathogenesis that could vary from patient to patient, forming the basis for practicing personal medicine. Notably, the EDP has a high performance after only one distal esophageal biopsy—compared with conventional histology, which currently requires at least four biopsies according to consensus recommendations (4, 10).
GERD has symptoms that closely overlap with those of EoE, but EoE is an antigen-driven disorder whose symptoms and pathology are responsive to either dietary control or steroid therapies. Additionally, these disorders are distinct in their gene expression profiles, heritability, genetic underpinnings, and linkage with atopy. However, EoE and GERD are not mutually exclusive and may coexist in the same patient (Table 1).
Table 1.
Features of EoE and GERD
| Associated features | EoEa | GERDa |
|---|---|---|
| Clinical | ||
| Atopy | Yes | No |
| Food sensitivity | Yes | No |
| Gender preference | Male | No |
| Food impaction | Common | Uncommon |
| Procedural findings | ||
| pH probe | Neutral | Acidic |
| Endoscopic furrowing | Yes | No |
| Endoscopic rings | Sometimes | No |
| Decreased luminal distention | Yes | Unknown |
| Radiographic small caliber | Sometimes | No |
| Histopathology | ||
| Proximal disease | Yes | No |
| Distal disease | Yes | Yes |
| Epithelial hyperplasia | Severe | Moderate |
| Eosinophils/HPF | ≥15 | <15 |
| EoE diagnostic panel positiveb | Yes | No |
| Treatment effectiveness | ||
| H-2 blockers | No | Yes |
| Proton-pump inhibitors | Partial | Yes |
| Glucocorticoids | Yes | No |
| Food elimination | Yes | No |
| Elemental diet | Yes | No |
EoE and GERD can co-occur in the same patient.
First approximation.
Abbreviations: EoE, eosinophilic esophagitis; GERD, gastroesophageal reflux disease; HPF, high-power field; H-2, histamine receptor 2.
Therapy
Disease remission typically occurs with treatment, which may include dietary exclusion, topical corticosteroids, or both. Topical steroids represent the principal drug-based therapies in current use for EoE. Oral steroids are effective for acute, severe, or difficult-to-control EoE (4). Allergen avoidance by dietary measures is successfully used for the treatment of EoE (11). Elemental formulas represent the most effective therapy in terms of both histologic and symptomatic response, with a response rate of 97%. The empiric elimination diet may also be recommended for patient care. In this diet, foods commonly associated with immunoglobulin E (IgE)-mediated food allergy (typically the top six foods: milk, egg, wheat, soy, fish/shellfish, and nuts) are eliminated regardless of allergen test results. Multiple empiric elimination diet studies have reported similar response rates of about 75% for both histology and symptoms. The high variability and low predictive value of skin prick tests (SPTs) and serum IgE measurements, as demonstrated in these studies, suggest that the clinical utility for standardized assessment of food-specific reactivity in patients with EoE remains to be determined. Antigen-directed diets appear to be as effective as the empiric elimination diet. There is controversy over whether an antigen-directed diet leads to a less restrictive diet; however, as the six-food elimination diet is difficult to achieve and maintain because it is highly restrictive, antigen testing may retain its value to specific patients as both a means to design patient-specific elimination diets and as a guide for the food reintroduction process (11).
In difficult-to-treat and oral steroid–dependent cases, antimetabolite therapy (e.g., azathioprine and 6-mercaptopurine) has also been shown to be effective (12). Another secondary therapy for EoE is esophageal dilation, a mechanical procedure with proven efficacy in improving symptoms related to strictures and luminal narrowing in the majority of patients with EoE (13). Notably, however, endoscopic dilation has no effect on the underlying inflammatory process (14), so patients often must undergo subsequent procedures.
Prognosis
Currently, therapy for EoE is chronic, with relapse of disease occurring rapidly after the discontinuation of either dietary or drug-based therapies (15). Though the natural history of EoE has not been fully studied, children with EoE are more likely to have a parent with a history of esophageal strictures and/or a formal diagnosis of EoE, and biopsies of these parents have often revealed EoE. Furthermore, pediatric patients diagnosed with EoE by retrospective biopsy analysis are at increased risk of developing persistent disease characterized by dysphagia, food impaction, a need for esophageal dilation, and food allergy (5, 16). Thus, if left untreated, EoE is likely to progress to esophageal scarring and dysfunction. The development of Barrett esophagus in EoE has not been found, although this has not been vigorously studied. EoE increases the risk of developing other forms of eosinophilic gastrointestinal disorders such as eosinophilic gastritis. Thus, routine endoscopic surveillance of the entire gastrointestinal tract, guided by symptoms, is recommended.
EPIDEMIOLOGY
Age, Gender, and Family History
Research is beginning to shed light on the natural history of EoE, its strong association with specific ethnicities and the male sex, and the genetic and environmental factors involved (Figure 2). As has been noted across multiple epidemiologic studies, males comprise approximately three-quarters of all EoE cases. Interestingly, a coding variant in the cytokine receptor–like factor 2 (CRLF2) gene, which encodes the receptor for thymic stromal lymphopoietin (TSLP) and is located on pseudoautosomal region 1 of the X and Y chromosomes, shows a sex-specific association with EoE risk in men only. Though the mechanism is not understood, these data are intriguing because they link the male predilection for EoE with a male risk variant on sex chromosomes (8). Pediatric EoE diagnosis peaks within the first 3 years of life and presents with feeding disorder (17), most likely resulting from antigen hypersensitivity as solid foods are introduced. Most adults are not diagnosed until the third decade of life (4), presenting most commonly with dysphagia, food impaction, heartburn, or chest pain. A number of previous reports demonstrated a high preponderance of EoE in individuals with Caucasian ancestry (up to 90% of cases), though the appreciation for the racial demographics of EoE may be broadening. In a recent retrospective survey, the numbers of Caucasian and African American patients with EoE were identical. Additionally, the male gender bias found in early studies of mainly the Caucasian population persisted in the African American population. Another survey in the adult population also found similar prevalence of EoE in the Caucasian and African American populations (3). EoE often occurs in multiple family members in a non-Mendelian pattern, indicating that the heritable component of EoE is likely complex in nature. In fact, Alexander et al. (18) recently reported data on a large familial cohort. This study analyzed 914 pediatric probands (within 2,192 first-degree family members) and reported relative risk ratios for EoE in family members, which ranged from 10 to 64 depending on the relationship, with higher values for brothers (64-fold), fathers (43-fold), and men (51-fold) than sisters, mothers, and women, respectively (18); this is compared with a relative risk of 2 in siblings of patients with asthma (19). Overall, EoE is observed in 1.8–2.4% of patients’ relatives, depending on their relationship and sex (18).
Figure 2.
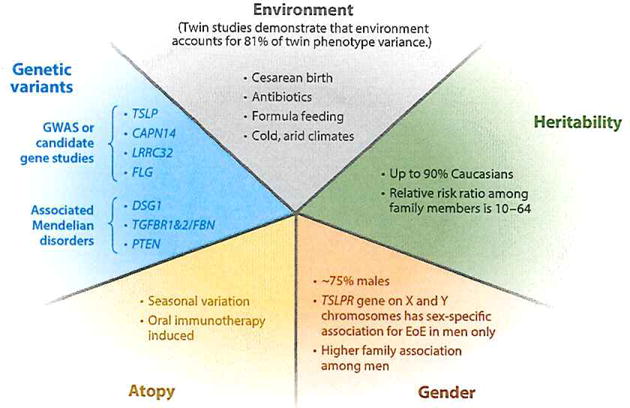
Risk factors for EoE. The main categories of risk are environmental exposure and genetic predisposition. Genetic predisposition can be described through multiple subcategories including genetic variants, atopy, gender, and heritability. Abbreviations: EoE, eosinophilic esophagitis; GWAS, genome-wide association study.
Environment
Environmental factors also appear to be important in establishing risk for EoE. Several early life exposures, including cesarean birth, antibiotics, and formula feeding, have been linked to increased risk of EoE (20). Also, in a recent study of a twins cohort, analysis revealed that phenotype variance among twins was accounted for more by environmental factors (81.0%) than by additive genetic heritability (14.5%) (18). It was demonstrated that environmental factors, such as food and penicillin allergies and discordant birth weight among twins, increase EoE risk and that fall birth season and breast-feeding may reduce risk. In addition, Spergel et al. (21) found that in the United States, EoE is diagnosed more often in urban areas and in the Northeast. Supporting this, Hurrrel et al. (22) found an increasing prevalence of EoE in cold and arid climates in the United States, and seasonal variations in disease suggest environmental influences. Almansa et al. (23) studied 78 patients with EoE retrospectively and found that mere was a significant increase in diagnosis during the spring and summer months. Also, Helicobacter pylori infection appears to be inversely related to an EoE diagnosis (24). In line with these data, colonization by immune-shaping commensal microbiota, in the gut and also in the esophagus, could be a key determinant of environmental risk (25, 26).
Atopy
The majority (50–80%) of patients with EoE have atopy and other allergic diseases, such as atopic dermatitis, asthma, oral allergy syndrome, and allergic rhinitis, or have specific IgE to food antigens and aeroallergens (8). These clinical findings have been supported through multiple murine studies demonstrating that skin, lung, and intranasal exposure to various antigens can induce EoE-like symptoms (27–29). In addition to the aforementioned link to allergic rhinitis and aeroallergen-specific IgE, patients with EoE commonly report seasonal variations in symptoms and diagnosis (23, 30). It also seems that aeroallergens may contribute to food hypersensitivity due to cross-reactivity or cross sensitization. In a recent study, IgE antibodies against food-specific allergen components were rare in patients with EoE, but cross-reactive responses to aeroallergen IgE were common (31, 32). The allergen families predominantly responsible for this cross-reactivity were profilins, pathogenesis-related-10 proteins, and lipid transfer proteins. These proteins remain intact until being degraded in the stomach (31, 33), a process that may limit hypersensitivity responses to the esophagus. This atopic link is further supported by the striking effectiveness of elemental formulas in disease therapy. Although food elimination or elemental diet therapy can reduce or eradicate disease symptoms, relapse almost universally occurs after reintroduction of food allergens or discontinuation of treatment, again suggesting that chronic food antigen hypersensitivity is a fundamental feature of EoE. Additionally, a subset of patients with EoE (15%) have a history of food anaphylaxis (15,17, 34, 35). There are also multiple reports of patients who develop EoE while undergoing oral food immunotherapy (36). However, in a subset of patients with EoE, no allergen sensitization or history of allergic diseases can be identified, suggesting again that multiple factors (including environment and genetics) affect disease predisposition and development (8).
Other Associated Disorders
In addition to the frequent co-occurrence of EoE with atopic disorders, there has been increasing recognition of EoE in association with several Mendelian and non-Mendelian diseases (37). A few of these diseases have other associated atopic features, and as a whole, these comorbid conditions highlight important candidate genes or pathways related to EoE.
The most studied of these disease associations is that of EoE with inherited connective tissue disorders (CTD) that involve hypermobility syndromes [e.g., Loeys-Dietz syndrome (LDS), Marfan syndrome type II, and Ehlers-Danlos syndrome]. The co-occurrence of EoE with these disorders is now called EoE-CTD. EoE increases the risk for CTD approximately eightfold. EoE and CTD share excessive production of transforming growth factor β (TGF-β) and TGF-β signaling. In fact, LDS is caused by gain-of-function mutations in the TGF-β receptors, and Marfan syndrome type II is caused by mutations in connective tissue proteins that bind to TGF-β, such as fibrillin 1 (type I). Additionally, patients with LDS have elevated IgE levels, eosinophil counts, and T helper type 2 (Th2) cell cytokines, and their naive CD4+ T cells skew to Th2 upon TGF-β stimulation. Ehlers-Danlos syndrome is caused by mutations in collagens, and although the syndrome has not been directly associated with excess TGF-β1 levels, direct interactions between TGF-β1 and collagen have been reported (37).
A Mendelian disease that frequently co-occurs with EoE is severe dermatitis, multiple allergies, and metabolic wasting (SAM) syndrome. SAM syndrome is caused by homozygous mutations in desmoglein 1 (DSG1). DSG1 is a major constituent of desmosomes, which connect the cell surface to the keratin cytoskeleton to help maintain epidermal integrity and barrier function. SAM syndrome is a rare disorder reported in only two consanguineous families and one nonconsanguineous family (38, 39). This association is interesting and substantiates further investigation of this gene, as it has been shown that DSGl is decreased in EoE and is associated with an impaired barrier phenotype (40).
Associations of EoE with a few other diseases have also been noted. Other atopic Mendelian disorders include autosomal dominant hyper-IgE syndrome, caused by loss-of-function mutations in signal transducer and activator of transcription 3 (STAT3), and a syndrome characterized by increased levels of mast cell tryptase in the blood and associated with CTD. Notably, patients with PTEN hamartoma tumor syndrome have a >200-fold increase in risk for EoE and other eosinophil-associated gastrointestinal disorders, but the mechanism of this association is unknown (37). Finally, multiple recent high-powered studies have linked celiac disease with EoE (41, 42).
METHODS TO UNCOVER MOLECULAR PATHWAYS IN EoE
Multiple cutting-edge methods have been used recently to discover molecular pathways involved in the pathophysiology of EoE (43–46). These methods include gene expression profiling of patient tissue and screening for disease risk genetic variants by genome-wide association studies (GWAS), candidate gene studies, and studies of the epigenome via microRNA (miRNA) arrays, DNA methylation profiling, and chromatin immunoprecipitation sequencing technologies. With the exception of candidate gene studies, these methods are unbiased approaches, and they are beginning to reveal many of the critical molecular pathways underlying EoE pathogenesis.
GWAS performed on both discovery and replication cohorts have identified important new candidate genes, demonstrated association with known candidates, and replicated findings from previous studies. Gene expression profiling of esophageal biopsy specimens demonstrated a striking transcript signature that distinguishes patients with EoE from healthy control subjects and from patients with chronic esophagitis (44). This signature includes 574 genes collectively termed the EoE transcriptome. Remarkably, 98% of this transcriptome normalizes with topical steroid therapy. As previously mentioned, a set of 94 signature EoE transcriptome genes (the EDP) has promise as a diagnostic tool capable of differentiating patients with EoE from those with other forms of esophagitis and patients with active EoE from those with EoE in remission (10). More recently, a more sensitive method of whole-transcriptome sequencing (RNA-seq) of biopsy transcripts identified over 1,000 new EoE-dysregulated genes not noted in the previous microarray data, providing further opportunities for insight into EoE’s molecular underpinnings.
Gene-environment interactions, central to environmentally driven allergic inflammatory diseases such as EoE, are now being explored via epigenetics. Emerging epigenetic data are beginning to provide clues as to how environmental factors may be affecting genetic dysregulation in EoE, which ultimately affects disease pathophysiology (47). Remarkably, the molecular pathways involved in EoE are relatively similar between males and females, sporadic and familial cases, pediatric and adult patients, and allergic and nonallergic patients, despite varying clinical manifestations (48, 49). The candidate genes (Table 2) and associated molecular pathways discovered with these approaches are discussed with regard to EoE pathophysiology in further detail below.
Table 2.
Genes associated with EoE
| Gene | Approach | Modification | Associated Mendelian disease | Plausible biological mechanism |
|---|---|---|---|---|
| CCL26 | Candidate gene | SNP in 3′UTR | Enhancement of activity or expression of eotaxin-3 | |
| TGFB1 | Candidate gene | SNP in promoter | Increase in expression and steroid resistance | |
| FLG | Candidate gene | Nonsense and missense mutations | Impairment of barrier function | |
| TSLP | GWAS | SNP in promoter and introns | Increase in expression of potent Th2-polarizing cytokine; association with basophil responses | |
| CRLF2 | Candidate gene | Missense SNP | Male-specific association; TSLP receptor increase; TSLP signaling | |
| DSG1 | Mapping/sequencing | Missense SNP | SAM | Impairment of barrier function; increase in production of TSLP and periostin |
| TGFBR1/TGFBR2/FBN | Phenotype association | Missense SNP | LDS and MF | Increase in TGF-β levels and/or signaling and Th2 cell skewing |
| CAPN14 | GWAS | SNP in promoter, introns and intergenic | Regulation of expression of calpain-14, a calcium-activated intracellular regulatory protease | |
| LRRC52 | GWAS | SNP in introns and intergenic | Altered TGF-β signaling, as LRRC32 (also known as GARP) is a membrane-bound TGF-β binding protein | |
| PTEN | Phenotype association | Missense SNP | PHTS | Gain of function of phosphatase activity; regulation of eosinophil responses |
Abbreviations: CAPN14, calpain-14; CCL26, chemokine C-C motif ligand 26; CRLF2, cytokine receptor-like factor 2; DSG1, desmoglein-1; FBN, fibrillin; FLG, filaggrin; GARP, glycoprotein-A repetitions predominant protein; GWAS, genome-wide association study; LDS, Loeys-Dietz syndrome; LRRC32, leucine rich repeat containing 32; MF, Marfan syndrome; PHTS, PTEN hamartoma tumor syndrome; PTEN, phosphatase and tensin homolog; SAM: severe dermatitis, multiple allergies and metabolic wasting syndrome; SNP, single-nucleotide polymorphism; TGF-β, transforming growth factor β; Th2, T helper type 2; TSLP, thymic stromal lymphopoietin.
IMMUNOPATHOGENESIS OF EoE
Hypersensitivity
EoE is a disease that is associated with food antigen–driven hypersensitivity, as evidenced by its response to dietary therapy. Hypersensitivity reactions can occur via multiple immune mechanisms including IgE (immediate type) and/or T cell–mediated (delayed type). Most patients with EoE develop sensitivity to foods, according to food-specific serum IgE or SPTs (17, 50, 51). Fold-induced anaphylaxis occurs in only approximately 15% of patients with EoE. Thus the role of IgE-mediated hypersensitivity remains unclear. In contrast, delayed-type, T cell-mediated reactions are increasingly understood to participate in EoE, which appears to have an immunologic response similar to delayed-type hypersensitivity characterized by Th2 responses involving interleukin 4 (IL-4), IL-5, and IL-13 and the associated eosinophilic infiltrate (52). It is currently not fully understood how allergic sensitization occurs in EoE, but mechanisms involving loss of immunologic tolerance are being elucidated. Furthermore, murine models of EoE suggest that sensitization can potentially occur through multiple modes, including epicutaneous (29, 53) and pulmonary routes (27). Below, we explore these concepts of loss of tolerance and allergic sensitization as we consider the immunologic data in EoE (Figure 3).
Figure 3.
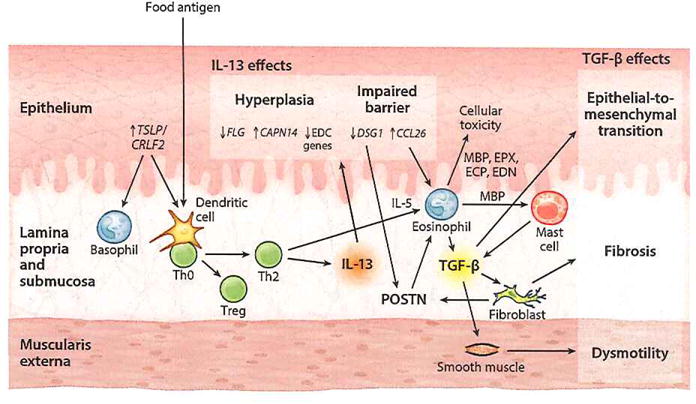
Pathophysiology of EoE. TSLP is released from the epithelium and activates basophils and food antigen–presenting dendritic cells to induce Th2 polarization of naive CD4+ T cells. These Th2 cells then secrete JL-13 that increases CCL26, CAPN14, and periostin (POSTN) expression and decreases DSG1, FLG, and DC gene expression in the epithelium. Decreased DSG1 level impairs barrier function, thereby forming a propagation loop by allowing further penetration of food antigen, and also leads to increased POSTN levels. The increased CCL26 and POSTN promote eosinophil recruitment from the bloodstream. The accumulating activated eosinophils cause epithelial cell cytotoxicity. TGF-β released by both eosinophils and mast cells increases POSTN expression and stimulates fibrotic response and smooth muscle dysmotility. Abbreviations: CAPN14, calpain 14; CCL26, chemokine (C-C motif) ligand 26; CRLF2, cytokine receptor-like factor 2; DSG1, desmoglein 1; ECP, eosinophil cationic protein; EDC, epidermal differentiation complex; EDN, eosinophil-derived neurotoxin; EoE, eosinophilic esophagitis; EPX, eosinophil peroxidase; FLG, filaggrin; IL, interleukin; MBP, major basic protein; TGF-β, transforming growth factor β; Th0, naive T helper; Th2, T helper type 2; Treg, regulatory T cell; TSLP, thymic stromal lymphopoietin.
Innate Immune Cells in EoE
Innate immune cells are important mediators of EoE pathology. These cells include eosinophils (cells that are required for diagnosis), mast cells (sentinel Th2 cells lining the esophageal mucosa), and less studied, though not less important, cells including dendritic cells and basophils. These are individually discussed below.
Eosinophils
The esophagus is normally devoid of eosinophils, and the presence of esophageal eosinophilia is a defining pathologic feature of EoE. Despite esophageal eosinophilia not being pathognomonic of EoE, murine models demonstrate the relative importance of the eosinophil in the disease. Mice genetically engineered to lack eosinophils have a decrease in allergen-induced basal hyperplasia and lamina propria thickness and also do not develop strictures (54, 55), yet these mice still have esophageal motility dysfunction, suggesting that the entire disease process is not fully dependent on eosinophils (54). Furthermore, mice with selective deletion of eosinophils via anti–SIGLEC F antibody treatment have decreased esophageal eosinophilia, angiogenesis, basal zone hyperplasia, and fibronectin deposition (56) in an antigen-induced model of EoE.
A number of eosinophil products have been shown to have direct effects on immune signaling and tissue dysfunction (3). The eosinophil granule proteins major basic protein (MBP), eosinophil peroxidase, and eosinophil cationic protein (ECP) are cytotoxic to epithelium at concentrations similar to those found in biological fluids from patients with eosinophilia. In vitro, the granule components of eosinophils are toxic to many tissues, including the intestinal epithelium. ECP can render cell membranes porous, and MBP increases smooth muscle reactivity via vagal muscarinic M2 receptors and can trigger mast cell and basophil degranulation. Evidence of eosinophil activation and release of granule components such as MBP has been directly observed in patients with EoE; therefore, eosinophils could exacerbate epithelial damage in EoE.
Eosinophils also have the capacity to initiate antigen-specific immune responses by acting as antigen-presenting cells (APCs). Consistent with this role, eosinophils express relevant co-stimulatory molecules (CD40, CD28, CD86, B7), secrete cytokines capable of inducing T cell proliferation and maturation (TL-2, IL-4, IL-6, IL-10, IL-12), and can be induced to express major histocompatibility complex class II molecules (57). Interestingly, experimental adoptive transfer of antigen-pulsed eosinophils induces antigen-specific T cell responses in vivo. In addition, tissue eosinophils have distinct cytokine expression patterns under inflammatory versus noninflammatory conditions. For example, esophageal eosinophils from patients with EoE express high levels of Th2 cytokines and TGF-β (3). Activated eosinophils generate a wide range of cytokines including IL-1, IL-3, IL-4, IL-5, and IL-13; GM-CSF; TGF-α and TGF-β; tumor necrosis factor α; RANTES (regulated on activation, normal T cells expressed and secreted); macrophage inflammatory protein lα; and eotaxin (3). This diverse cytokine production indicates that eosinophils have the potential to sustain or augment multiple aspects of the immune response, inflammatory reaction, and tissue repair processes.
Multiple chemotactic factors for eosinophils are important in EoE. The most studied is eotaxin-3, also known as chemokine (C-C motif) ligand 26 (CCL26). The CCL26 gene is the most upregulated gene in the esophagus of patients with EoE (44), its expression increasing 53-fold in esophageal biopsy specimens from patients with EoE compared with normal esophageal biopsy specimens and correlating with disease activity (44). Additionally, CCL26 expression is upregulated by 279-fold after IL-13 stimulation of primary esophageal epithelial cells grown in culture ex vivo (58), and mice lacking the eotaxin receptor, CCR3, were protected from developing experimental EoE. The esophageal epithelium is the main source of eotaxin-3 production (44), which is notably induced by IL-13 stimulation. Eotaxin-3 belongs to the eotaxin family (eotaxtn-1 to eotaxin-3) of CC chemokines and is the only family member that is upregulated in EoE. Eotaxin-3 binds CCR3 and activates G protein signaling to drive eosinophil chemotaxis and activation. In the earliest work demonstrating genetic factors that play a role in EoE, Blanchard et al. (44) identified the first EoE risk variant in a likely candidate, eotaxin-3 (CCL26). The single-nucleotide polymorphism (SNP) (rs2302009) was shown to be highly associated with disease (p < 0.001) in a case-control cohort. Researchers replicated this finding by genotyping rs2302009 in 117 EoE cases and 225 unrelated healthy controls, also showing a significant association with disease (p = 6.9 × 10−3, OR= 1.63) (44). Additionally, the observed association between rs2302009 and EoE was independent of atopic status, indicating a direct link with EoE susceptibility.
Much of what is known about the involvement of epigenetics in EoE has been attained from biochemical studies of the eotaxin-3 promoter. Lim et al. (59) demonstrated that the CCL26 promoter is regulated by DNA methylation. Methylation occurs on cytosine nucleotides located within CpG (cytosine-guanine) dinucleotide motifs and, like other epigenetic marks, is dynamically regulated (60). Two CpG sites in the CCL26 promoter were identified as hypomethylated in patients with EoE (59). Methylation of these sites affected both STAT6 and CBP binding to the promoter region and was remarkably stable and heritable in cell passage, remaining detectable after multiple passages. Chromatin immunoprecipitation assays by Lim et al. (61) suggest that IL-13 induces the formation of a multiprotein complex on the CCL26 promoter that includes CBP and STAT6 and leads to increases in acetylated histone 3 and opening of the CCL26 promoter for additional transcriptional machinery (61). PPIs have been suggested to dampen the levels of histone methylation and of STAT6 bound to the CCL26 promoter, resulting in decreased eotaxin-3 expression (62). These findings could explain the emerging observation of PPI-responsive EoE, in which PPI therapy yields partial resolution of symptoms (63). Collectively, these findings suggest that a coordinated interaction involving DNA demethylation followed by histone acetylation occurs at the CCL26 promoter in response to IL-13 and leads to increased eotaxin-3 expression.
Other eosinophil-directed cytokines that are likely important in EoE pathogenesis include IL-5 and prostaglandin D2 (PGD2). Eosinophils selectively express the receptor for IL-5, a cytokine that regulates eosinophil expansion and eosinophil survival and primes eosinophils to respond to appropriate activating signals. Aspects of IL-5 signaling in EoE will be discussed in the cytokine section. Additionally, PGD2, a prostanoid largely produced by mast cells, is sufficient to attract eosinophils to the esophagus (64, 65). Lastly, an orally active small-molecule inhibitor of the eosinophil and Th2 cell chemoattractant receptor, PGD2 receptor (also known as CRTH2), modestly reduces esophageal eosinophilia (65).
Mast cells
Mast cells normally reside in the mucosa and submucosa of the esophagus. Mast cell activation classically occurs when a multivalent allergen cross-links IgE molecules bound to the high-affinity IgE receptor (FcεRI). The significant increases in mast cell degranulation and mastocytosis within the epithelium, lamina propria, and smooth muscle layer (66,67) observed in EoE can be ameliorated with steroid therapy (66), further implicating these cells in the local inflammatory milieu within the esophagus. Thus, it is likely that an immediate hypersensitivity response occurs locally in the esophagus in EoE, similar to what is observed in oral allergy syndrome.
Mast cells generate cytokines, proteases, and bioactive compounds that can activate eosinophils and lead to the fibrosis that is often apparent across the range of pediatric and adult patients with EoE (68–70). Murine models of EoE using mast cell–deficient mice have demonstrated reduced muscle cell hyperplasia and hypertrophy consistent with a role for mast cells in remodeling, but there was no effect on eosinophil recruitment (71).
Additionally, mast cell–specific genes, specifically carboxypeptidase 3A, high-affinity IgE receptor, and tryptase α, are abundantly represented in the EoE transcriptome, and two unique mast cell–related transcriptomes have been generated on the basis of counts of intact and degranulated mast cells. These transcriptomes demonstrate an unusual mast cell-protease profile with evidence of increased expression of carboxypeptidase A3 (25-fold increase) and tryptase, with very little or no expression of either chymase or tryptase γ1 (66).
Similar to its role for eosinophils, IL-5 may participate in increased induction of mast cells in the tissue, as CD2-IL5 transgenic mice had increased levels of esophageal mast cells (54). Furthermore, anti-IL-5 therapy in clinical trials found reduced esophageal mast cells in treated patients (72). Use of immunohistochemistry to analyze biopsies of patients with EoE enrolled in an anti-IL-5 trial (73) showed that activated MBP+ eosinophils and other unidentified cells that are adjacent to the tryptase+ mast cells in the esophagus produced IL-9. IL-9, a pleiotropic cytokine, can promote the activation and maturation of mast cells (74). Notably, the expression of the IL9 gene transcript is elevated in the esophagus of patients with EoE (75). Other cells that have been shown to be important sources of IL-9 include T helper type 9 (Th9) cells (76), which also produce other Th2 cytokines (IL-4, BL-5, and IL-13), and innate lymphoid cells (77). The anti-IL-5 clinical trial also showed that the severity of EoE symptoms correlated with mast cell number, whereas the reduction of eosinophil numbers alone did not correlate with symptom severity (72). Because multiple studies have demonstrated a role of mast cells in regulating smooth muscle contractility and vagal nerve activity (67, 78, 79), it is reasonable to speculate that the activated mast cells in the esophagus are the primary drivers of the pathophysiology of EoE in a subgroup of patients. In summary, these studies revealed that esophageal eosinophils may promote esophageal mastocytosis via IL-9 and that this interaction between mast cells and eosinophils may regulate the severity of EoE symptoms.
Dendritic cells
The understanding of dendritic cells in EoE is limited, but there is evidence that both dendritic cells and nonprofessional APCs, such as epithelial cells (80) and possibly eosinophils, play a role (81–83). The primary professional APC in the esophagus seems to be the Langerhans cell, a type of dendritic cell found in all squamous epithelia (32), particularly the epidermis. Langerhans cells of the esophagus express FcεRI (84), which increases in active EoE (85). FcεRI expression on dendritic cells in human disease and mouse models of asthma and atopic dermatitis is correlated with increased Th2 effector response (86–88). It is possible that IgE signaling on APCs facilitates antigen uptake and enhances development of allergen-specific T cells (89). In addition to the importance of dendritic cells in sensitization, selective depletion of dendritic cells during allergen challenge in both asthma and allergic rhinitis murine models has demonstrated the importance of dendritic cells in the effector phase of these diseases (90, 91). A similar mechanism likely occurs in EoE.
Allergens can induce a Th2-mediated response, either alone (with a self-adjuvant effect) or in combination with other environmental adjuvants (viral or bacterial infections or air pollution) (92), and this effect likely occurs via communication by resident stromal cells and dendritic cells (93, 94). The stimulation of dendritic cells via environmental adjuvants may be one direct mechanism by which environment is contributing to EoE.
Basophils
Basophils are known to be important in allergic inflammation, but there is a paucity of data on basophil function in EoE. Siracusa et al. (95) showed that TSLP could lead to basophil hematopoiesis and that basophils that express the TSLP receptor are present in the esophagus of patients with EoE. Shortly following this report, Noti and colleagues (29) found that sensitization to egg or peanut protein could occur during skin inflammation or injury (tape stripping) in a TSLP-dependent, basophil-dependent, and IgE-independent manner. This study also showed increased basophil levels in EoE biopsies. TSLP is an interesting link to this basophil axis, as it is known to be an alarmin molecule released by stressed epithelium and is considered a master regulator of Th2 immune responses. TSLP has previously been linked to EoE, and further details of TSLP’s role in EoE are discussed in the epithelium section below.
Adaptive Immune Cells in EoE
In addition to the innate immune system, the adaptive arm is also integral to the Th2 immune response in EoE. B cells are important mediators via IgE and IgG4 secretion. Multiple subtypes of T cells, including Th2, CD8+, and invariant natural killer T (iNKT) cells, are relevant to pathophysiology. Finally, intercellular communicators, cytokines, help orchestrate the concerted response.
B cells and IgE
Lymphocytes represent another component of the inflammatory infiltrate that is consistently elevated in patients with active EoE relative to normal controls and patients with inactive EoE (44, 82, 96, 97). A process central to the pathogenesis of many atopic disorders, including anaphylaxis, allergic bronchospasm, and urticaria, is generation of antigen-specific IgE via Th2 cell–mediated class switching of B cells. As noted above, this IgE can then bind FcεRI on mast cells and basophils to carry out its function. Allergen-specific serum IgE and SPT are commonly abnormal, providing evidence of immediate hypersensitivity in EoE, but the role of B cells in EoE is not clear. In addition to being increased in number, B cells undergo class-switch recombination and generation of IgE locally within the esophagi of both atopic and nonatopic patients with EoE (98–100). However, though B cells appear to be generating IgE in patients with EoE, in murine models of EoE utilizing B cell–deficient, IgE-deficient mice and an IgE-independent aeroantigen (Aspergillus), the B cell and its repertoire appear to be dispensable in initiating the primary characteristics of EoE. This suggests that IgE may play a role in maintenance rather than initiation of the disorder in humans (29, 81). In clinical studies using a monoclonal antibody directed against IgE in EoE, the use of the drug demonstrated clinical but not histologic or endoscopic improvement (101), suggesting that some acute symptoms in EoE may be associated with IgE but that the chronic inflammation may not be as dependent (29, 81). Interestingly, a recent study of anti-IgE in EoE showed a lack of response in terms of either symptoms or biopsy eosinophil count (102). This study showed that esophageal tissues from patients with EoE had a 45-fold increase in IgG4 compared with controls and that this IgG4 could be detected extracellularly in biopsy specimens. Additionally, the study showed that patients with EoE had increased food-specific serum IgG4 in response to the foods that are most associated with EoE (milk, egg, wheat, and nuts).
T cells
Studies of mice lacking various components of the adaptive immune system have established a critical role for T cells in EoE (53, 81, 103). Similar to the case in other atopic diseases, tissue inflammation in EoE is characterized by a Th2-type inflammatory response (58, 75, 82, 98). This immune response includes an increase in both CD4+ and CD8+ T cells and an increase in the CD8+ T celI/CD4+ T cell ratio in the esophagus (96, 97). Regulatory T cells (Tregs), characterized by expression of FOXP3, are vital to maintenance of tolerance. Studies in the pediatric and adult EoE populations are in disagreement with regard to Treg numbers in EoE. Increased numbers of Tregs were seen in pediatric cases, whereas there was a relative lack of Tregs in the adult cases (104). This may point to a fundamental difference in disease processes in pediatric versus adult cases of EoE. Regardless, further studies are needed to decipher the general roles of both Tregs and T cells in EoE.
Recently, the importance of iNKT cells in EoE pathophysiology has begun to be elucidated. A study of early-onset EoE has suggested that insufficient immune imprinting by environmental microorganisms results in an esophageal increase of CXCL16, an iNKT cell chemokine (105). iNKT cells respond to lipid and glycolipid antigens presented by CD1d molecules (106, 107). It has been previously shown that mucosal iNKT cell tolerance to later environmental exposures is formed at an early age (26) and that if this tolerance does not develop, these cells can mediate allergic sensitization and tissue inflammation (108). iNKT cells from peripheral blood of pediatric patients with EoE have been shown to be activated by milk sphingolipids (109). Additionally, CD1d-deficient mice were protected from EoE disease induction (110).
Cytokines
The Th2 cytokines (IL-4, IL-5, and IL-13) are elevated in patients with EoE (75, 82), as well as in murine models of EoE (111). IL-4 induces naive Th cells into Th2 cells and activates B cell class switching to produce IgE, thus initiating a Th2-mediated immune response. It appears that TSLP-elicited basophils, Th2 cells, and iNKT cells are important sources of IL-4 in EoE (82, 112–114). IL-5 is one of the most well-studied Th2 cytokines in EoE and seems to be central to the disease. IL-5 is produced by Th2 cells, mast cells, and eosinophils. IL-5 regulates eosinophil expansion and eosinophil survival and primes eosinophils to respond to appropriate activating signals (115). EL-5 overexpression in mice induces EoE, and IL-5 neutralization completely blocks allergen- or IL-13-induced EoE in mice (116). However, anti-IL-5 therapy in humans has notyet been shown to be effective at ameliorating clinical aspects of the disease, although esophageal eosinophilia improves (117).
Among the Th2 cytokines, EL-13 plays a unique role in EoE. Highlighting its importance, a recent study found the genetic locus of STAT6, a downstream transcription factor of the IL-13 signaling pathway (118), to have a major genetic association in EoE. IL-13 is overproduced in the esophagus of patients with EoE. In addition, in experimental systems—such as IL-13 lung transgenic systems—IL-13 induces EoE and tissue remodeling with features both dependent and independent of eosinophils (119). Ex vivo microarray analysis showed that treatment of biopsy-derived primary esophageal epithelial cells with IL-13, which is upregulated at the mRNA level in patients with EoE, can largely recapitulate the EoE transcriptome (58). Th2 cells and activated eosinophils are important sources of EL-13 in EoE (82, 120, 121). IL-13 also recruits eosinophils by increasing an eosinophil chemokine, eotaxin-3, and by promoting fibroblasts to produce periostin (POSTN), which increases eosinophil adhesion to fibronectin (122). IL-13 induces tissue remodeling by promoting collagen deposition, angiogenesis, and epithelial hyperplasia (119). The esophageal remodeling in this model occurs independently of eosinophilia and is inhibited by the type 2 IL-13 receptor (119). IL-13 is also important in barrier function. It has been shown to down-regulate DSG1, filaggrin (FLG), and involucrin, genes important in epithelial differentiation and barrier function (40, 123). Mice treated with anti-IL-13 antibody demonstrated improvement in esophageal inflammation (124); furthermore, a Phase II, double-blind, randomized control trial has shown the safety and efficacy of an anti-IL-13 antibody (QAX576, Novartis, Switzerland), which produced improvement in both esophageal eosinophilia and disease-related transcripts, as well as a trend for improved symptoms (125). These studies demonstrate that IL-13 is a central mediator and link between the immunologic and histologic changes that are germane to EoE, largely through its effects on the esophageal epithelium.
IL-15 appears to mediate CD4+ T and iNKT cells (114) and drive the synthesis of Th2 cytokines IL-5 and IL-13, all four of which are believed to play significant roles in EoE (55, 58, 114, 123). IL-15 has also been demonstrated to be elevated in patients with EoE, as well as in the Aspergillus-driven experimental model of EoE.
ESOPHAGEAL TISSUE IN EoE
Apart from the inflammatory response, the resident tissue of the esophagus itself plays a fundamental role in the pathophysiology of disease. Genetic variants associated with both barrier function and immune signaling have been linked to the epithelium. Finally, both the lamina propria and smooth muscle are key tissue types that produce symptoms of EoE via fibrosis and dysmotility, respectively. The contributions of these tissue types are explored in more detail below.
Epithelium
The epithelium is a protective barrier and key interface for immune signaling. In addition to genetic variants in barrier function and immune signaling being linked to the epithelium, it is also a source of disease biomarkers, including transcript and miRNA such as CCL26 and miR-375.
Barrier
The esophagus is composed of stratified squamous epithelium. In contrast to the skin, the esophagus lacks a cornified layer but does have a mucus layer that provides additional protection. This esophageal epithelium is composed of a proliferating basal layer of one to three cells in depth and a differentiating suprabasal layer migrating toward the esophageal lumen (126). The epithelium forms a protective barrier against environmental antigen exposure that, when compromised, can lead to antigen hypersensitivity and exacerbated immune responses (127). The structure of the epithelium is disrupted in EoE, and a similar disruption occurs in an in vitro esophageal epithelium model when stimulated with IL-13. These structural changes include basal cell hyperplasia, dilated intracellular spaces, and impaired barrier function (128–130). These changes may be linked to derangements in processes such as epithelial differentiation and epithelial barrier formation, including cellular junction formation.
The epidermal differentiation complex (EDC) on human chromosome lq21 is a cluster of genes that regulates terminal differentiation (131). The EDC locus contains the highest density of dysregulated genes in the EoE transcriptome (123). FLG, involucrin and several small proline-rich repeat family members (2C, 2D, and 3) are expressed in esophageal epithelial cells and are downregulated in response to IL-13 ex vivo (123), suggesting a homeostatic role for the EDC in the esophageal epithelium. FLG dysfunction has been associated with defects in epidermal barrier function in patients with atopic dermatitis (132,133), a disease that frequently co-occurs with EoE. It is also important to note that 2% of the EoE transcriptome is not fully reversible after disease remission (58). Interestingly, these transcripts include genes whose protein products are involved in regulating homeostatic and pathogenic responses in the epithelium, such as cadherin-like 26 (CDH26), uroplakin 1B, POSTN, and DSG1 (58).
The epithelial barrier gene FLG represents yet another genetic locus linked with numerous allergic diseases, including EoE. FLG has been shown to play an important role in barrier function of the skin (134, 135). FLG is negatively regulated by IL-13 and is decreased in the esophageal mucosa of patients with EoE, and two coding variants (R501X and 2282del4) in FLG associate with EoE risk irrespective of atopic status (123).
Sherrill et al. (40) reported that expression of DSG1 is decreased in active EoE (22-fold) compared with healthy control subjects and that DSG1 deficiency in an esophageal epithelial in vitro model leads to impaired barrier function and acantholysis and intraepidermal clefting. This impaired barrier function was also seen in EoE patient biopsies compared with controls. DSGl is a transmembrane desmosomal cadherin component and facilitates the calcium-dependent homotypic interactions between adjacent cells that impart both structure and mechanical strength to the epithelium. Notably, DSGl autoantibodies are found in pemphigus foliaceus and pemphigus vulgaris, squamous epithelial diseases that demonstrate decreased cellular adhesion resulting in epidermal blistering, with some patients displaying eosinophilic infiltration (136). Additionally, two recent reports have shown that families with recessive mutations in DSG1 have a severe allergy phenotype, including one patient with EoE (38, 39). Collectively, these findings substantiate the significance of alterations in DSGl in a spectrum of human diseases, including EoE. Therefore, it appears that tissue-specific decreases in DSGl may be pathogenic in EoE. Other junctional proteins, including E-cadherin and claudin-1, are also reduced in EoE (137). Thus, in active EoE, the mucosal integrity is altered due to defects in desmosomal and tight junction adhesion proteins. These findings place the esophageal epithelium as a central location for the basic defect in EoE and as one essentially involved in the Th2 cytokine signaling cascade, as IL-13 promotes germane EoE-related responses directly in esophageal epithelial cells.
Recently, the largest and most comprehensive GWAS in patients with EoE identified calpain 14 (CAPN14) as the gene most highly associated with EoE (41). This finding was soon replicated in a separate GWAS (118). CAPN14 is a member of the calpain family—a group of intracellular, calcium-activated proteases. CAPN14 was shown to be specifically expressed in the esophagus, dynamically regulated as a function of disease activity and genetic haplotype and after exposure of esophageal epithelial cells to IL-13, and located in an epigenetic hot spot modified by IL-13 (41). Recently, it has also been shown that CAPN14 has a significantly disruptive effect on the esophageal epithelial barrier, and its expression and activity appear to lead to cleavage of DSG1 (138), providing a unifying mechanism that connects genetics with an essential disease susceptibility mechanism.
MicroRNAs
A select set of miRNAs—short, noncoding RNAs that fine-tune the expression of target genes by repressing translation through binding complementary sequences in the 3′ untranslated region of target mRNAs (139)—has been shown to be dynamically altered in the esophageal mucosa of patients with EoE. The miRNA signature associated with EoE includes 21 upregulated and 11 downregulated miRNAs, and this dysregulation is largely reversed with steroid therapy (140,141). Some of these miRNAs have been shown to correlate with esophageal eosinophil levels in patients with EoE (140) and regulation of IL-13-induced transcriptional responses (142). These findings identify miRNAs as potential biomarkers for EoE diagnosis and steroid responsiveness.
Epithelial inflammatory response
Besides its barrier function, the esophageal epithelium can also induce inflammation. As noted above, the epithelium is a potent reservoir of cytokines and lipid mediators (143). IL-13 stimulates esophageal epithelial cells to produce eotaxin-3 (44), which signals eosinophils to traffic to the esophagus. Esophageal epithelial cells also express Toll-like receptors (144, 145) and produce proinflammatory cytokines in response to both pathogen-associated and danger-associated molecular patterns (82, 144). EoE-derived epithelial cells can produce RANTES (CCL5), a chemotactic factor for T cells, eosinophils, and basophils (109), and CXCL16 (105), which may play a role in the migration and activation of iNKT cells in EoE. In addition, others have found that esophageal epithelial cells may function as nonprofessional APCs (80). Resident innate immune cells are also present within the esophageal epithelium at baseline (97, 105), and their roles in EoE are being actively investigated (105).
Multiple GWAS (41,45,118) and candidate gene studies (46) have implicated genetic variants of TSLP in genetic susceptibility for EoE. These variants encode an epithelial protein expressed in response to cytokines (146), noxious substances (147), and mechanical stress (148). TSLP has been termed a master regulator of Th2 responses (149) and has been shown to target dendritic cells and promote their Th2-polarizing ability (150). It also exerts effects on nearly every cell type involved in Th2 inflammation, including eosinophils (151) and mast cells (152). TSLP is increased in the esophageal tissue of patients with EoE (45, 46). Patients carrying the risk allele for the variant most associated with EoE have elevated TSLP expression (45). TSLP has recently been shown to be key in the development and maintenance of EoE in a murine model and is associated with an increased number of basophils in the esophageal tissue (29). In addition, TSLP risk genotypes correlate with increased levels of basophils (29). Effects in the TSLP signaling pathway as a result of variants either increasing TSLP gene expression or altering receptor function could potentially amplify innate inflammatory responses to food antigens—the exact processes involved in allergen sensitization that underscore the EoE phenotype.
Lamina Propria in EoE
The lamina propria is important for the inflammatory response as it is a site of considerable inflammatory infiltration. It is also an important site for the major symptoms of EoE because of the fibrotic response leading to fibrostenosis and dysphagia. This fibrotic response includes key mediators and processes such as TGF-β, POSTN, and epithelial-to-mesenchymal transition (EMT), which are further reviewed below.
Fibrotic remodeling
On the basis of the paradigm of other eosinophilic inflammatory conditions, the natural history of EoE has been described as a progression from an inflammatory to a fibrostenotic disease (153). Fibrotic remodeling, including metaplasia of the mucosal glands, smooth muscle hypertrophy, angiogenesis, and subepithelial collagen deposition or fibrosis, occurs in pediatric (69, 70) and adult (154, 155) patients with EoE. The myofibroblast, which shares properties of smooth muscle cells and fibroblasts, is the key effector cell in fibrosis. The myofibroblast contracts the extracellular matrix and secretes extracellular matrix components, including type 1 collagen (156).
Esophageal remodeling is associated with stricture formation and, as a result, dysphagia and food bolus impaction (153, 157). In fact, EoE is currently the leading cause of emergency endoscopy for esophageal food impaction in adults (158). Both histopathologic fibrosis (154) and gross fibrostenosis increase with age (155). Symptoms of fibrosis range from signs of progressive solid food dysphagia to food impaction.
Esophageal inflammation leads to deposition of subepithelial fibrous tissue and tissue remodeling correlated with eosinophil degranulation (69). In fact, eosinophil-released MBP has been found to increase the expression of FGF9 in biopsies of patients with EoE (159). Additionally, eosinophils secrete high levels of CCL18, the expression of which is highly increased in EoE (154). Both of these cytokines, FGF9 and CCL18, are important in fibroblast activation and extracellular matrix deposition.
TGF-β1 is a key cytokine for epithelial growth, fibrosis, and tissue remodeling and is reportedly generated not only by mast cells but also by eosinophils and epithelial cells of patients with EoE. Its expression is elevated in esophageal biopsy samples of patients with active EoE relative to control patients (55, 70, 117, 155). Furthermore, the number of TGF-β1-positive cells is increased in biopsy samples from patients with EoE relative to either control patients or patients with GERD (70). TGF-β stimulates myofibroblast differentiation (67) and expression of various genes responsible for the fibroblast phenotype, including α smooth muscle actin, collagen, and POSTN (122). Moreover, TGF-β has recently been shown to stimulate esophageal smooth muscle contractility and potentially contribute to esophageal dysmotility in patients with EoE (67). Also, in a small cohort of patients with steroid-treated EoE, a genetic variant in the promoter of TGFB1 was associated with steroid unresponsiveness and correlated with increased TGF-β-positive cells in the esophagus (43). Remarkably, there is a high rate of EoE with CTD (160), such as Marfan syndrome, Ehlers-Danlos syndrome, and LDS, which has been associated with variants in TGF-β receptors 1 and 2 (161). In addition toTGF-β, IL-4, IL-5, and IL-13 have been shown to mediate the remodeling processes (55, 162, 163).
POSTN, which functions as a cell adhesion molecule that regulates extracellular matrix deposition (164, 165), is dramatically upregulated (approximately 52-fold) in patients with EoE; both TGF-β and IL-13 are able to induce POSTN expression in primary esophageal fibroblasts and epithelial cells. Studies of POSTN-deficient mice have demonstrated that POSTN facilitates eosinophil recruitment into the esophagus (122), increasing eosinophil adhesion to fibronectin.
Epithelial-to-mesenchymal transition
EMT is a process in which epithelia lose epithelial markers (e.g., E-cadherin and keratins 8 and 14) and acquire markers of mesenchymal cells (e.g., α smooth muscle actin, vimentin, fibronectin, and N-cadherin), which share features of both fibroblasts and smooth muscle cells (myofibroblasts) and participate in the synthesis, deposition, and degradation of extracellular matrix along with the contraction of wound tissue (166–168). TGF-β, MMP-9, IL-13, and MBP released by eosinophils or damaged epithelium may induce EMT and contribute to fibrosis (167,169). The extent of EMT in patients with EoE correlates with eosinophils, TGF-β, and fibrosis (169). The dedifferentiation of epithelial cells in EoE appears to be effected by another TGF-β superfamily member, bone morphogenetic protein, and one of its inhibitors, follistatin (170). Follistatin is increased in EoE, increases with IL-13, and correlates with basal cell hyperplasia and loss of differentiation markers (171).
Fibrosis response to therapy
It appears that both swallowed topical corticosteroids (43, 172) and elimination diets (173) can prevent and even reverse the esophageal remodeling process in children; however, there is conflicting evidence in adults (154). This difference may be due to either the populations studied or the differences in topical steroid formulations. Thus, untreated eosinophilic inflammation leads to esophageal remodeling with stricture formation, and the use of early, consistent anti-inflammatory therapy may protect against this outcome.
Losartan, an angiotensin II receptor blocker and FDA-approved antihypertensive medication, may reduce TGF-β responsiveness via an effect either on signaling or on transcription of TGF-β and its receptor (174); thus, losartan is a potential treatment for EoE. A Phase II trial to evaluate losartan therapy in patients with EoE is currently under way (175).
Smooth Muscle
EoE is associated with esophageal dysmotility and dysphagia, which may be related to motor dysfunction of the esophagus rather than to physical narrowing. Esophageal ultrasound shows dysfunctional muscularis mucosa in EoE (176). Tissue remodeling also involves both morphologic and functional changes in smooth muscle components. In fact, esophageal muscle cells respond to various profibrogenic stimuli. Eosinophils and mast cells infiltrate the submucosal and myenteric neuronal plexus and the smooth muscle, respectively, in EoE (67, 177). Additionally, both eosinophils and mast cells and their secreted mediators affect smooth muscle function and innervation (176,178–181). Particularly, TGF-β has been shown to increase smooth muscle contractibility over time (67) but decrease its force of contraction (178). Thus, TGF-β may be a key mediator of esophageal dysmotility in EoE.
CONCLUSION
In a relatively short period of time, diligent investigation of both patient cohorts and animal models and the utilization of cutting-edge technology (e.g., GWAS, expression arrays, and epigenetic studies) to investigate the pathophysiology of EoE have led to tremendous strides in piecing together the features of this enigmatic disease (Figure 4). Disease pathophysiology is associated with allergic hypersensitivity to food, heritability via genetic risk variants [e.g., 5q22 (TSLP) and 2p23 (CAPN14)], and early-life environmental risk factors. On the molecular level, activation of innate epithelial inflammatory pathways (TSLP and eotaxin-3), impaired barrier function (mediated by loss of DSG1 via IL-13 and CAPN14 mechanisms), and allergic inflammation likely mediated by eosinophils and mast cells, which produce increased TGF-β, lead to fibrosis and dysmotility that together contribute to the major symptom of dysphagia. Collectively integrating these data, we propose a two-hit mechanism for the etiology of EoE that involves allergic sensitization and esophagus-specific pathways including CAPN14.
Figure 4.
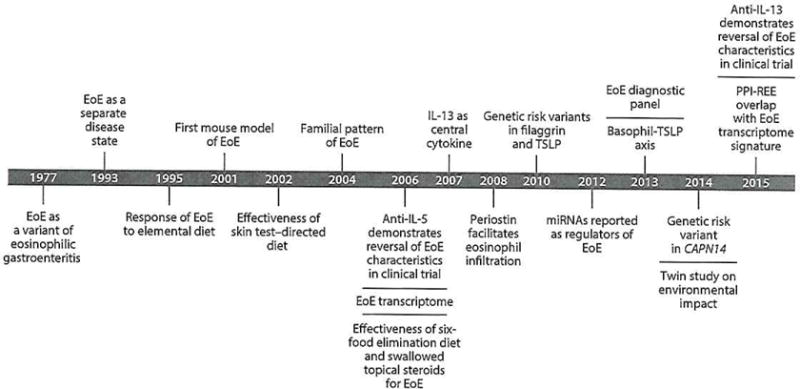
Timeline of EoE research. EoE has been studied over the last 20 years, with the majority of findings occurring within the last decade. These findings have advanced clinical and mechanistic insights into disease epidemiology, pathogenesis, and treatment. Abbreviations: CAPN14, calpain 14; EoE, eosinophilic esophagitis; IL, interleukin; TGF-β, transforming growth factor β; TSLP, thymic stromal lymphopoietin; miRNAs, microRNAs; PPI-REE, proton-pump inhibitor-responsive esophageal eosinophilia.
Further dissection of the molecular mechanisms of EoE represents a promising area for translational research aimed at discovering novel therapies, noninvasive diagnostics, and biomarkers for therapy response. A better understanding of EoE-specific pathways (e.g., calpain proteases) and how early-life exposures (e.g., altered microbiome) increase susceptibility to EoE is predicted to impact future treatment. Notably, oral desensitization trials for food anaphylaxis have demonstrated that a major side effect of therapy includes the possible development of EoE. This finding demonstrates an entangled relationship between IgE-mediated food allergy and the chronic, Th2 cell–associated inflammation of EoE (36). A better understanding of the factors involved in the balance between anaphylaxis and EoE may lead to tolerance-inducing protocols for treatment and to an eventual cure for EoE. Though the findings from the early-stage clinical trials for IL-5 (mepolizumab and reslizumab) and IL-13 (QAX576) mentioned in this review did not meet primary end points, they did demonstrate tissue responses, provide needed insight into the pathology of disease, and prompt further studies of these and related biological agents. Additional agents in development for treatment of EoE include inhibitors of TSLP (e.g., AMG 157), CCR3, eotaxin (e.g., bertilimumab), and IL-4Rα (the common receptor for IL-4 and IL-13; e.g., dupilumab). Other agents include eosinophil-depleting antibodies, such as those against IL-5Rα (e.g., benralizurnab) or sialic acid–binding Ig-like lectin 8 (182). A molecular diagnostic test for EoE that is now clinically available could improve diagnosis and clinical monitoring, patient-specific therapy, and predictive and personalized medicine approaches to EoE. Additionally, therapeutic agents such as drugs and microorganisms are easy to deliver to the esophagus. Therefore, it may one day be possible to prevent EoE in individuals with high risk for this disease. We hope that this review will aid basic scientists and clinical investigators alike in their future endeavors to continue to advance the field, thereby promoting research that has the potential to change the natural course of this disease and, in turn, countless patients’ lives.
DIFFERENTIAL DIAGNOSES FOR ESOPHAGEAL EOSINOPHILIA.
There are several differential diagnoses for esophageal eosinophilia, as eosinophil activation and accumulation is a triggered immune response to a variety of stimuli. Thus, diagnosis of eosinophilic esophagitis (EoE) includes exclusion of other eosinophilic diseases of the esophagus (e.g., GERD) and of other more general immune system–inducing conditions (e.g., infection):
-
■
EoE
-
■
Proton pump inhibitor–responsive esophageal eosinophilia (PPI-REE)
-
■
Celiac disease
-
■
Crohn’s disease
-
■
Infection
-
■
Hypereosinophilic syndrome
-
■
Achalasia
-
■
Drug hypersensitivity
-
■
Vasculitis
-
■
Pemphigoid vegetans
-
■
Inherited connective tissue disease
-
■
Graft-versus-host disease
-
■
Gastroesophageal reflux disease
Acknowledgments
We thank Shawna Hottinger for her editorial assistance. This work was supported by NIH R37 AI045898, R01 DK067255, U19 AI070235, U19 AI066738 (CoFAR supported by NIAID and NIDDK), R01 DK076893, R01 AI057803, T32 HL7752-19, P30 DK078392 (Gene and Protein Expression Core), U54 All 17804 [CEGIR is part of the Rare Disease Clinical Research Network (RDCRN), an initiative of the Office of Rare Disease Research (ORDR), NCATS, and is funded through collaboration among NCATS, NIAID, and NIDDK], the Campaign Urging Research for Eosinophilic Disease (CURED), the Buckeye Foundation, and Sunshine Charitable Foundation and its supporters, Denise A. Bunning and David G. Bunning.
Footnotes
DISCLOSURE STATEMENT
M.E.R. serves on the Boards of the International Eosinophil Society and the American Partnership for Eosinophilic Disorders (APFED); consults for Immune Pharmaceuticals, Receptos, Celsus Therapeutics, Genentech, Roche, and Novartis; has submitted patents owned by Cincinnati Children’s Hospital Medical Center; has a royalty interest in reslizumab (Teva Pharmaceuticals); and owns stock or stock options in Immune Pharmaceuticals, Receptos, Celsus Therapeutics, and NKT Therapeutics.
Contributor Information
Benjamin P. Davis, Email: ben-davis@uiowa.edu.
Marc E. Rothenberg, Email: rothenberg@cchmc.org.
LITERATURE CITED
- 1.Dobbins JW, Sheahan DG, Behar J. Eosinophilic gastroenteritis with esophageal involvement. Gastroenterology. 1977;72(6):1312–16. [PubMed] [Google Scholar]
- 2.Attwood SE, Smyrk TC, DeMeester TR, Jones JB. Esophageal eosinophilia with dysphagia. A distinct clinicopathologic syndrome. Dig Dis Sri. 1993;38(1):109–16. doi: 10.1007/BF01296781. [DOI] [PubMed] [Google Scholar]
- 3.Abonia JP, Rothenberg ME. Eosinophilic esophagitis: rapidly advancing insights. Annu Rev Med. 2012;63(1):421–34. doi: 10.1146/annurev-med-041610-134138. [DOI] [PubMed] [Google Scholar]
- 4.Liacouras CA, Furuta GT, Hirano I, Atkins D, Attwood SE, et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. J Allergy Clin Immunol. 2011;128(1):3–20.e6. doi: 10.1016/j.jaci.2011.02.040. [DOI] [PubMed] [Google Scholar]
- 5.DeBrosse CW, Collins MH, Buckmeier Butz BK, Allen CL, King EC, et al. Identification, epidemiology, and chronicity of pediatric esophageal eosinophilia, 1982–1999. J Allergy Clin Immunol. 2010;126(1):112–19. doi: 10.1016/j.jaci.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh SV, Antonioli DA, Goldman H, Fox VL, Bousvaros A, et al. Allergic esophagitis in children: a clinicopathological entity. Am J Surg Pathol. 1999;23(4):390–96. doi: 10.1097/00000478-199904000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Liacouras CA, Spergel JM, Ruchelli E, Verma R, Mascarenhas M, et al. Eosinophilic esophagitis: a 10-year experience in 381 children. Clin Gastroenterol Hepatol. 2005;3(12):1198–206. doi: 10.1016/s1542-3565(05)00885-2. [DOI] [PubMed] [Google Scholar]
- 8.Sherrill JD, Rothenberg ME. Genetic dissection of eosinophilic esophagitis provides insight into disease pathogenesis and treatment strategies. J Allergy Clin Immunol. 2011;128(1):23–32. doi: 10.1016/j.jaci.2011.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wen T, Dellon ES, Moawad FJ, Furuta GT, Aceves SS, Rothenberg ME. Transcriptome analysis of proton pump inhibitor-responsive esophageal eosinophilia reveals proton pump inhibitor-reversible allergic inflammation. J Allergy Clin Immunol. 2015;135(1):187–97.e4. doi: 10.1016/j.jaci.2014.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen T, Stucke EM, Grotjan TM, Kemme KA, Abonia JP, et al. Molecular diagnosis of eosinophilic esophagitis by gene expression profiling. Gastroenterology. 2013;145(6):1289–99. doi: 10.1053/j.gastro.2013.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis BP, Rothenberg ME. Emerging concepts of dietary therapy for pediatric and adult eosinophilic esophagitis. Expert Rev Clin Immunol. 2013;9(4):285–87. doi: 10.1586/eci.13.15. [DOI] [PubMed] [Google Scholar]
- 12.Netzer P, Gschossmann JM, Straumann A, Sendensky A, Weimann R, Schoepfer AM. Corticosteroid-dependent eosinophilic oesophagitis: Azathioprine and 6-mercaptopurine can induce and maintain long-term remission. Eur J Gastroenterol Hepatol. 2007;19(10):865–69. doi: 10.1097/MEG.0b013e32825a6ab4. [DOI] [PubMed] [Google Scholar]
- 13.Ally MR, Dias J, Veerappan GR, Maydonovitch CL, Wong RK, Moawad FJ. Safety of dilation in adults with eosinophilic esophagitis. Dis Esophagus. 2013;26(3):241–45. doi: 10.1111/j.1442-2050.2012.01363.x. [DOI] [PubMed] [Google Scholar]
- 14.Schoepfer AM, Gonsalves N, Bussmann C, Conus S, Simon HU, et al. Esophageal dilation in eosinophilic esophagitis: effectiveness, safety, and impact on the underlying inflammation. Am J Gastroenterol. 2010;105(5):1062–70. doi: 10.1038/ajg.2009.657. [DOI] [PubMed] [Google Scholar]
- 15.Spergel JM, Brown-Whitehorn TF, Beausoleil JL, Franciosi J, Shuker M, et al. 14 years of eosinophilic esophagitis: clinical features and prognosis. J Pediatr Gastroenterol Nutr. 2009;48(1):30–36. doi: 10.1097/MPG.0b013e3181788282. [DOI] [PubMed] [Google Scholar]
- 16.DeBrosse CW, Franciosi JP, King EC, Butz BK, Greenberg AB, et al. Long-term outcomes in pediatric-onset esophageal eosinophilia. J Allergy Clin Immunol. 2011;128(1):132–38. doi: 10.1016/j.jaci.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Assa’ad AH, Putnam PE, Collins MH, Akers RM, Jameson SC, et al. Pediatric patients with eosinophilic esophagitis: an 8-year follow-up. J Allergy Clin Immunol. 2007;119(3):731–38. doi: 10.1016/j.jaci.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 18.Alexander ES, Martin LJ, Collins MH, Kottyan LC, Sucharew H, et al. Twin and family studies reveal strong environmental and weaker genetic cues explaining heritability of eosinophilic esophagitis. J Allergy Clin Immunol. 2014;134(5):1084–92.e1. doi: 10.1016/j.jaci.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blanchard C, Wang N, Rothenberg M. Eosinophilic esophagitis: pathogenesis, genetics, and therapy. J Allergy Clin Immunol. 2006;118(5):1054–59. doi: 10.1016/j.jaci.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 20.Jensen ET, Kappelman MD, Kim HP, Ringel-Kulka T, Dellon ES. Early life exposures as risk factors for pediatric eosinophilic esophagitis. J Pediatr Gastroenterol Nutr. 2013;57(1):67–71. doi: 10.1097/MPG.0b013e318290d15a. [DOI] [PubMed] [Google Scholar]
- 21.Spergel JM, Book WM, Mays E, Song L, Shah SS, et al. Variation in prevalence, diagnostic criteria, and initial management options for eosinophilic gastrointestinal diseases in the United States. J Pediatr Gastroenterol Nutr. 2011;52(3):300–6. doi: 10.1097/MPG.0b013e3181eb5a9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hurrell JM, Genta RM, Dellon ES. Prevalence of esophageal eosinophilia varies by climate zone in the United States. Am J Gastroenterol. 2012;107(5):698–706. doi: 10.1038/ajg.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Almansa C, Krishna M, Buchner AM, Ghabril MS, Talley N, et al. Seasonal distribution in newly diagnosed cases of eosinophilic esophagitis in adults. Am J Gastroenterol. 2009;104(4):828–33. doi: 10.1038/ajg.2008.169. [DOI] [PubMed] [Google Scholar]
- 24.Dellon ES, Peery AF, Shaheen NJ, Morgan DR, Hurrell JM, et al. Inverse association of esophageal eosinophilia with Helicobacter pylori based on analysis of a US pathology database. Gastroenterology. 2011;141(5):1586–92. doi: 10.1053/j.gastro.2011.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fillon SA, Harris JK, Wagner BD, Kelly CJ, Stevens MJ, et al. Novel device to sample the esophageal microbiome—the esophageal string test. PLoS ONE. 2012;7(9):e42938. doi: 10.1371/journal.pone.0042938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olszak T, An D, Zeissig S, Vera MP, Richter J, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336(6080):489–93. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rayapudi M, Mavi P, Zhu X, Pandey AK, Abonia JP, et al. Indoor insect allergens are potent inducers of experimental eosinophilic esophagitis in mice. J Leukoc Biol. 2010;88(2):337–46. doi: 10.1189/jlb.0110025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pope SM, Fulkerson PC, Blanchard C, Akei HS, Nikolaidis NM, et al. Identification of a cooperative mechanism involving interleukin-13 and eotaxin-2 in experimental allergic lung inflammation. J Biol Chem. 2005;280(14):13952–61. doi: 10.1074/jbc.M406037200. [DOI] [PubMed] [Google Scholar]
- 29.Noti M, Wojno EDT, Kim BS, Siracusa MC, Giacomin PR, et al. Thymic stromal lymphopoietin-elicited basophil responses promote eosinophilic esophagitis. Nat Med. 2013;19(8):1005–13. doi: 10.1038/nm.3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prasad GA, Alexander JA, Schleck CD, Zinsmeister AR, Smyrk TC, et al. Epidemiology of eosinophilic esophagitis over three decades in Olmsted County, Minnesota. Clin Gastroenterol Hepatol. 2009;7(10):1055–61. doi: 10.1016/j.cgh.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Rhijn BD, van Ree R, Versteeg SA, Vlieg-Boerstra BJ, Sprikkelman AB, et al. Birch pollen sensitization with cross-reactivity to food allergens predominates in adults with eosinophilic esophagitis. Allergy. 2013;68(11):1475–81. doi: 10.1111/all.12257. [DOI] [PubMed] [Google Scholar]
- 32.de Fraissinette A, Schmitt D, Thivolet J. Langerhans cells of human mucosa. J Dermatol. 1989;16(4):255–62. doi: 10.1111/j.1346-8138.1989.tb01261.x. [DOI] [PubMed] [Google Scholar]
- 33.Lopez-Torrejon G, Crespo JF, Sanchez-Monge R, Sanchez-Jimenez M, Alvarez J, et al. Allergenic reactivity of the melon profilin Cuc m 2 and its identification as major allergen. Clin Exp Allergy. 2005;35(8):1065–72. doi: 10.1111/j.1365-2222.2005.02303.x. [DOI] [PubMed] [Google Scholar]
- 34.Noel RJ, Putnam PE, Rothenberg ME. Eosinophilic esophagitis. N Engl J Med. 2004;351(9):940–41. doi: 10.1056/NEJM200408263510924. [DOI] [PubMed] [Google Scholar]
- 35.Chehade M, Aceves SS. Food allergy and eosinophilic esophagitis. Curr Opin Allergy Clin Immunol. 2010;10(3):231–37. doi: 10.1097/ACI.0b013e328338cbab. [DOI] [PubMed] [Google Scholar]
- 36.Lucendo AJ, Arias A, Tenias JM. Relation between eosinophilic esophagitis and oral immunotherapy for food allergy: a systematic review with mexs-amlysis. Ann Alleigy Asthma Immunol. 2014;113(6):624–29. doi: 10.1016/j.anai.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 37.Rothenberg ME. Molecular, genetic, and cellular bases for treating eosinophilic esophagitis. Gastroenterology. 2015;148(6):I143–57. doi: 10.1053/j.gastro.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet. 2013;45(10):12, 44–48. doi: 10.1038/ng.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Has C, Jakob T, He Y, Kiritsi D, Hausser I, Bruckner-Tuderman L. Loss of desmoglein 1 associated with palmoplantar keratoderma, dermatitis and multiple allergies. Br J Dermatol. 2014;172(1):257–61. doi: 10.1111/bjd.13247. [DOI] [PubMed] [Google Scholar]
- 40.Sherrill JD, KC K, Wu D, Djukic Z, Caldwell JM, et al. Desmoglein-1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis. Mucosal Immunol. 2014;7(3):718–29. doi: 10.1038/mi.2013.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kottyan LC, Davis BP, Sherrill JD, Liu K, Rochman M, et al. Genome-wide association analysis of eosinophilic esophagitis provides insight into the tissue specificity of this allergic disease. Nat Genet. 2014;46(8):895–900. doi: 10.1038/ng.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Firszt R, Peterson K. Celiac disease and immune disorders in patients with eosinophilic esophagitis. J Allergy Clin Immunol. 2015;135(2):AB130. [Google Scholar]
- 43.Aceves SS, Newbury RO, Chen D, Mueller J, Dohil R, et al. Resolution of remodeling in eosinophilic esophagitis correlates with epithelial response to topical corticosteroids. Allergy. 2010;65(1):109–16. doi: 10.1111/j.1398-9995.2009.02142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blanchard C, Wang N, Stringer KF, Mishra A, Fulkerson PC, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest. 2006;116(2):536–47. doi: 10.1172/JCI26679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rothenberg ME, Spergel JM, Sherrill JD, Annaiah K, Martin LJ, et al. Common variants at 5q22 associate with pediatric eosinophilic esophagitis. Nat Genet. 2010;42(4):289–91. doi: 10.1038/ng.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sherrill JD, Gao P-S, Stucke EM, Blanchard C, Collins MH, et al. Variants of thymic stromal lymphopoietin and its receptor associate with eosinophilic esophagitis. J Allergy Clin Immunol. 2010;126(1):160–63. doi: 10.1016/j.jaci.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330(6004):612–16. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collins MH, Blanchard C, Abonia JP, Kirby C, Akers R, et al. Clinical, pathologic, and molecular characterization of familial eosinophilic esophagitis compared with sporadic cases. Clin Gastroenterol Hepatol. 2008;6(6):621–29. doi: 10.1016/j.cgh.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Straumann A, Aceves SS, Blanchard C, Collins MH, Furuta GT, et al. Pediatric and adult eosinophilic esophagitis: similarities and differences. Allergy. 2012;67(4):477–90. doi: 10.1111/j.1398-9995.2012.02787.x. [DOI] [PubMed] [Google Scholar]
- 50.Spergel JM, Andrews T, Brown-Whitehorn TF, Beausoleil JL, Liacouras CA. Treatment of eosinophilic esophagitis with specific food elimination diet directed by a combination of skin prick and patch tests. Ann Allergy Asthma Immunol. 2005;95(4):336. doi: 10.1016/S1081-1206(10)61151-9. [DOI] [PubMed] [Google Scholar]
- 51.Spergel JM. Eosinophilic esophagitis in adults and children: evidence for a food allergy component in many patients. Curr Opin Allergy Clin Immunol. 2007;7(3):274–78. doi: 10.1097/ACI.0b013e32813aee4a. [DOI] [PubMed] [Google Scholar]
- 52.Uzzaman A, Cho SH. Chapter 28: classification of hypersensitivity reactions. Allergy Asthma Proc. 2012;33(Suppl. 1):96–99. doi: 10.2500/aap.2012.33.3561. [DOI] [PubMed] [Google Scholar]
- 53.Akei HS, Brandt EB, Mishra A, Strait RT, Finkelman FD, et al. Epicutaneous aeroallergen exposure induces systemic TH2 immunity that predisposes to allergic nasal responses. J Allergy Clin Immunol. 2006;118(1):62–69. doi: 10.1016/j.jaci.2006.04.046. [DOI] [PubMed] [Google Scholar]
- 54.Mavi P, Rajavelu P, Rayapudi M, Paul RJ, Mishra A. Esophageal functional impairments in experimental eosinophilic esophagitis. Am J Physiol Gastrointest Liver Physiol. 2012;302(11):G1347–55. doi: 10.1152/ajpgi.00013.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mishra A, Wang M, Pemmaraju VR, Collins MH, Fulkerson PC, et al. Esophageal remodeling develops as a consequence of tissue specific EL-5-induced eosinophilia. Gastroenterology. 2008;134(1):204–14. doi: 10.1053/j.gastro.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rubinstein E, Cho JY, Rosenthal P, Chao J, Miller M, et al. Siglec-F inhibition reduces esophageal eosinophilia and angiogenesis in a mouse model of eosinophilic esophagitis. J Pediatr Gastroenterol Nutr. 2011;53(4):409–16. doi: 10.1097/MPG.0b013e3182182ff8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davis BP, Rothenberg ME. Antigen presentation by eosinophils in eosinophilic esophagitis? J Pediatr Gastroenterol Nutr. 2013;56(3):242. doi: 10.1097/MPG.0b013e31827ab8d3. [DOI] [PubMed] [Google Scholar]
- 58.Blanchard C, Mingler MK, Vicario M, Abonia JP, Wu YY, et al. IL–13 involvement in eosinophilic esophagitis: transcriptome analysis and reversibility with glucocorticoids. J Allergy Clin Immunol. 2007;120(6):1292–300. doi: 10.1016/j.jaci.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 59.Lim E, Rothenberg ME. Demethylation of the human eotaxin-3 gene promoter leads to the elevated expression of eotaxin-3. J Immunol. 2014;192(1):466–74. doi: 10.4049/jimmunol.1302454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 61.Lim EJ, Lu TX, Blanchard C, Rothenberg ME. Epigenetic regulation of the IL-13-induced human eotaxin-3 gene by CREB-binding protein-mediated histone 3 acetylation. J Biol Chem. 2011;286(15):13193–204. doi: 10.1074/jbc.M110.210724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang X, Cheng E, Huo X, Yu C, Zhang Q, et al. Omeprazole blocks STAT6 binding to the eotaxin-3 promoter in eosinophilic esophagitis cells. PLoS ONE. 2012;7(11):e50037. doi: 10.1371/journal.pone.0050037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dohil R, Newbury RO, Aceves S. Transient PPI responsive esophageal eosinophilia may be a clinical sub-phenotype of pediatric eosinophilic esophagitis. Dig Dis Sci. 2012;57(5):1413–19. doi: 10.1007/s10620-011-1991-5. [DOI] [PubMed] [Google Scholar]
- 64.Zhang S, Wu X, Yu S. Prostaglandin D2 receptor D-type prostanoid receptor 2 mediates eosinophil trafficking into the esophagus. Dis Esophagus. 2014;27(6):601–6. doi: 10.1111/dote.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Straumann A, Hoesli S, Bussmann CH, Stuck M, Perkins M, et al. Anti-eosinophil activity and clinical efficacy of the CRTH2 antagonist OC000459 in eosinophilic esophagitis. Allergy. 2013;68(3):375–85. doi: 10.1111/all.12096. [DOI] [PubMed] [Google Scholar]
- 66.Abonia JP, Blanchard C, Butz BB, Rainey HF, Collins MH, et al. Involvement of mast cells in eosinophilic esophagitis. J Allergy Clin Immunol. 2010;126(1):140–49. doi: 10.1016/j.jaci.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aceves SS, Chen D, Newbury RO, Dohil R, Bastian JF, Broide DH. Mast cells infiltrate the esophageal smooth muscle in patients with eosinophilic esophagitis, express TGF-β1, and increase esophageal smooth muscle contraction. J Allergy Clin Immunol. 2010;126(6):1198–204.e4. doi: 10.1016/j.jaci.2010.08.050. [DOI] [PubMed] [Google Scholar]
- 68.Straumann A, Spichtin H-P, Grize L, Bucher KA, Beglinger C, Simon H-U. Natural history of primary eosinophilic esophagitis: a follow-up of 30 adult patients for up to 11.5 years. Gastroenterology. 2003;125(6):1660–69. doi: 10.1053/j.gastro.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 69.Chehade M, Sampson HA, Morotti RA, Magid MS. Esophageal subepithelial fibrosis in children with eosinophilic esophagitis. J Pediatr Gastroenterol Nutr. 2007;45(3):319–28. doi: 10.1097/MPG.0b013e31806ab384. [DOI] [PubMed] [Google Scholar]
- 70.Aceves SS, Newbury RO, Dohil R, Bastian JF, Broide DH. Esophageal remodeling in pediatric eosinophilic esophagitis. J Allergy Clin Immunol. 2007;119(1):206–12. doi: 10.1016/j.jaci.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 71.Niranjan R, Mavi P, Rayapudi M, Dynda S, Mishra A. Pathogenic role of mast cells in experimental eosinophilic esophagitis. Am J Physiol Gastrointest Liver Physiol. 2013;304(12):1087–94. doi: 10.1152/ajpgi.00070.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Otani IM, Anilkumar AA, Newbury RO, Bhagat M, Beppu LY, et al. Anti-IL-5 therapy reduces mast cell and IL-9 cell numbers in pediatric patients with eosinophilic esophagitis. J Allergy Clin Immunol. 2013;131(6):1576–82. doi: 10.1016/j.jaci.2013.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Assa’ad AH, Gupta SK, Collins MH, Thomson M, Heath AT, et al. An antibody against IL-5 reduces numbers of esophageal intraepithelial eosinophils in children with eosinophilic esophagitis. Gastroenterology. 2011;141(5):1593–604. doi: 10.1053/j.gastro.2011.07.044. [DOI] [PubMed] [Google Scholar]
- 74.Goswami R, Kaplan MH. A brief history of IL-9. J Immunol. 2011;186(6):3283–88. doi: 10.4049/jimmunol.1003049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blanchard C, Stucke EM, Rodriguez-Jimenez B, Burwinkel K, Collins MH, et al. A striking local esophageal cytokine expression profile in eosinophilic esophagitis. J Allergy Clin Immunol. 2011;127(1):208–17.e7. doi: 10.1016/j.jaci.2010.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 77.Wilhelm C, Hirota K, Stiegutz B, Van Snick J, Tolaini M, et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol. 2011;12(11):1071–77. doi: 10.1038/ni.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bara I, Ozier A, Tunon de Lara JM, Marthan R, Berger P. Pathophysiology of bronchial smooth muscle remodelling in asthma. Eur Respir J. 2010;36(5):1174–84. doi: 10.1183/09031936.00019810. [DOI] [PubMed] [Google Scholar]
- 79.Van Nassauw L, Adriaensen D, Timmermans J-P. The bidirectional communication between neurons and mast cells within the gastrointestinal tract. Anton Neurosci. 2007;133(1):91–103. doi: 10.1016/j.autneu.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 80.Mulder DJ, Pooni A, Mak N, Hurlbut DJ, Basta S, Justmich CJ. Antigen presentation and MHC class II expression by human esophageal epithelial cells: role in eosinophilic esophagitis. Am J Pathol. 2011;178(2):744–53. doi: 10.1016/j.ajpath.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mishra A, Schlotman J, Wang M, Rothenberg ME. Critical role for adaptive T cell immunity in experimental eosinophilic esophagitis in mice. J Leukoc Biol. 2007;81(4):916–24. doi: 10.1189/jlb.1106653. [DOI] [PubMed] [Google Scholar]
- 82.Straumann A, Bauer M, Fischer B, Blaser K, Simon H-U. Idiopathic eosinophilic esophagitis is associated with a TH2-type allergic inflammatory response. J Allergy Clin Immunol. 2001;108(6):954–61. doi: 10.1067/mai.2001.119917. [DOI] [PubMed] [Google Scholar]
- 83.Tantibhaedhyangkul U, Tatevian N, Gilger MA, Major AM, Davis CM. Increased esophageal regulatory T cells and eosinophil characteristics in children with eosinophilic esophagitis and gastroesophageal reflux disease. Ann Clin Lab Sci. 2009;39(2):99–107. [PubMed] [Google Scholar]
- 84.Bannert C, Bidmon-Fliegenschnee B, Stary G, Hotzy F, Stift J, et al. Fc-epsilon-RI, the high affinity IgE-receptor, is robustly expressed in the upper gastrointestinal tract and modulated by mucosal inflammation. PLoS ONE. 2012;7(7):e4 2066. doi: 10.1371/journal.pone.0042066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yen EH, Hornick JL, Dehlink E, Dokter M, Baker A, et al. Comparative analysis of FcεRI expression patterns in patients with eosinophilic and reflux esophagitis. J Pediatr Gastroenterol Nutr. 2010;51(5):584–92. doi: 10.1097/MPG.0b013e3181de7685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hammad H, Plantinga M, Deswarte K, Pouliot P, Willart MAM, et al. Inflammatory dendritic cells—not basophils—are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J Exp Med. 2010;207(10):2097–111. doi: 10.1084/jem.20101563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sallmann E, Reininger B, Brandt S, Duschek N, Hoflehner E, et al. High-affinity IgE receptors on dendritic cells exacerbate Th2 -dependent inflammation. J Immunol. 2011;187(1):164–71. doi: 10.4049/jimmunol.1003392. [DOI] [PubMed] [Google Scholar]
- 88.Wollenberg A, Kraft S, Hanau D, Bieber T. Immunomorphological and ultrastructural characterization of Langerhans cells and a novel, inflammatory dendritic epidermal cell (IDEC) population in lesional skin of atopic eczema. J Invest Dermatol. 1996;106(3):446–53. doi: 10.1111/1523-1747.ep12343596. [DOI] [PubMed] [Google Scholar]
- 89.Bieber T. The pro- and anti-inflammatory properties of human antigen-presenting cells expressing the high affinity receptor for IgE (FcεRI) Immunobiology. 2007;212(6):499–503. doi: 10.1016/j.imbio.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 90.Beaty SR, Rose CE, Sung S-SJ. Diverse and potent chemokine production by lung CD11bhigh dendritic cells in homeostasis and in allergic lung inflammation. J Immunol. 2007;178(3):1882–95. doi: 10.4049/jimmunol.178.3.1882. [DOI] [PubMed] [Google Scholar]
- 91.Medoff BD, Seung E, Hong S, Thomas SY, Sandall BP, et al. CD11b+ myeloid cells are the key mediators of Th2 cell homing into the airway in allergic inflammation. J Immunol. 2009;182(1):623–35. doi: 10.4049/jimmunol.182.1.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kool M, Willart MAM, van Nimwegen M, Bergen I, Pouliot P, et al. An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity. 2011;34(4):527–40. doi: 10.1016/j.immuni.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 93.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15(4):410–16. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eiwegger T, Akdis CA. IL-33 links tissue cells, dendritic cells and Th2 cell development in a mouse model of asthma. Eur J Immunol. 2011;41(6):1535–38. doi: 10.1002/eji.201141668. [DOI] [PubMed] [Google Scholar]
- 95.Siracusa MC, Saenz SA, Hill DA, Kim BS, Headley MB, et al. TSLP promotes interIeukm-3-independent basophil haematopoiesis and type 2 inflammation. Nature. 2011;477(7363):229–33. doi: 10.1038/nature10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Teitelbaum JE, Fox VL, Twarog FJ, Nurko S, Antonioli D, et al. Eosinophilic esophagitis in children: immunopathological analysis and response to fluticasone propionate. Gastroenterology. 2002;122(5):1216–25. doi: 10.1053/gast.2002.32998. [DOI] [PubMed] [Google Scholar]
- 97.Lucendo AJ, Navarro M, Comas C, Pascual JM, Burgos E, et al. Immunophenotypic characterization and quantification of the epithelial inflammatory infiltrate in eosinophilic esophagitis through stereology: an analysis of the cellular mechanisms of the disease and the immunologic capacity of the esophagus. Am J Surg Pathol. 2007;31(4):598–606. doi: 10.1097/01.pas.0000213392.49698.8c. [DOI] [PubMed] [Google Scholar]
- 98.Vicario M, Blanchard C, Stringer KF, Collins MH, Mingler MK, et al. Local B cells and IgE production in the oesophageal mucosa in eosinophilic oesophagitis. Gut. 2010;59(1):12–20. doi: 10.1136/gut.2009.178020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Takhar P, Corrigan CJ, Smunhwaite L, O’Connor BJ, Durham SR, et al. Class switch recombination to IgE in the bronchial mucosa of atopic and nonatopic patients with asthma. J Allergy Clin Immunol. 2007;119(1):213–18. doi: 10.1016/j.jaci.2006.09.045. [DOI] [PubMed] [Google Scholar]
- 100.Takhar P, Smurthwaite L, Coker HA, Fear DJ, Banfield GK, et al. Allergen drives class switching to IgE in the nasal mucosa in allergic rhinitis. J Immunol. 2005;174(8):5024–32. doi: 10.4049/jimmunol.174.8.5024. [DOI] [PubMed] [Google Scholar]
- 101.Rocha R, Vitor AB, Trindade E, Lima R, Tavares M, et al. Omalizumab in the treatment of eosinophilic esophagitis and food allergy. Eur J Pediatr. 2011;170(11):1471–74. doi: 10.1007/s00431-011-1540-4. [DOI] [PubMed] [Google Scholar]
- 102.Clayton F, Fang JC, Gleich GJ, Lucendo AJ, Olalla JM, et al. Eosinophilic esophagitis in adults is associated with IgG4 and not mediated by IgE. Gastroenterology. 2014;147(3):602–9. doi: 10.1053/j.gastro.2014.05.036. [DOI] [PubMed] [Google Scholar]
- 103.Mishra A, Hogan SP, Brandt EB, Rothenberg ME. IL-5 promotes eosinophil trafficking to the esophagus. J Immunol. 2002;168(5):2464–69. doi: 10.4049/jimmunol.168.5.2464. [DOI] [PubMed] [Google Scholar]
- 104.Stuck MC, Straumann A, Simon HU. Relative lack of T regulatory cells in adult eosinophilic esophagitis—no normalization after corticosteroid therapy. Allergy. 2011;66(5):705–7. doi: 10.1111/j.1398-9995.2010.02525.x. [DOI] [PubMed] [Google Scholar]
- 105.Lexmond WS, Neves JF, Nurko S, Olszak T, Exley MA, et al. Involvement of the iNKT cell pathway is associated with early-onset eosinophilic esophagitis and response to allergen avoidance therapy. Am J Gastroenterol. 2014;109(5):646–57. doi: 10.1038/ajg.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Montalvillo E, Garrote JA, Bernardo D, Arranz E. Innate lymphoid cells and natural killer T cells in the gastrointestinal tract immune system. Rev Esp Enferm, Dig. 2014;106(5):334–45. [PubMed] [Google Scholar]
- 107.Berin MC, Shreffler WG. TH2 adjuvants: implications for food allergy. J Allergy Clin Immunol. 2008;121(6):1311–20. doi: 10.1016/j.jaci.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 108.Garn H, Renz H. Epidemiological and immunological evidence for the hygiene hypothesis. Immunobiology. 2007;212(6):441–52. doi: 10.1016/j.imbio.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 109.Jyonouchi S, Smith CL, Saretta F, Abraham V, Ruymann KR, et al. Invariant natural killer T cells in children with eosinophilic esophagitis. Clin Exp Allergy. 2013;44(1):58–68. doi: 10.1111/cea.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rajavelu P, Rayapudi M, Moffitt M, Mishra A. Significance of para-esophageal lymph nodes in food or aeroallergen-induced iNKT cell-mediated experimental eosinophilic esophagitis. Am J Physiol Gastrointest Liver Physiol. 2012;302(7):645–54. doi: 10.1152/ajpgi.00223.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Akei HS, Mishra A, Blanchard C, Rothenberg ME. Epicutaneous antigen exposure primes for experimental eosinophilic esophagitis in mice. Gastroenterology. 2005;129(3):985–94. doi: 10.1053/j.gastro.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 112.Blanchard C, Rothenberg ME. Basic pathogenesis of eosinophilic esophagitis. Gastrointest Endosc Clin N Am. 2008;18(1):133–43. doi: 10.1016/j.giec.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lucendo AJ, de Rezende L, Comas C, Caballero T, Bellón T. Treatment with topical steroids downregulates IL-5, eotaxin-1/CCL11, and eotaxin-3/CCL26 gene expression in eosinophilic esophagitis. Am J Gastroenterol. 2008;103(9):2184–93. doi: 10.1111/j.1572-0241.2008.01937.x. [DOI] [PubMed] [Google Scholar]
- 114.Zhu X, Wang M, Mavi P, Rayapudi M, Pandey AK, et al. Interleukin-15 expression is increased in human eosinophilic esophagitis and mediates pathogenesis in mice. Gastroenterology. 2010;139(1):182–87. doi: 10.1053/j.gastro.2010.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.O’Byrne PM, Inman MD, Parameswaran K. The trials and tribulations of IL-5, eosinophils, and allergic asthma. J Allergy Clin Immunol. 2001;108(4):503–8. doi: 10.1067/mai.2001.119149. [DOI] [PubMed] [Google Scholar]
- 116.Mishra A, Rothenberg ME. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology. 2003;125(5):1419–27. doi: 10.1016/j.gastro.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 117.Straumann A, Conus S, Grzonka P, Kita H, Kephart G, et al. Anti-interleukin-5 antibody treatment (mepolizumab) in active eosinophilic oesophagitis: a randomised, placebo-controlled, double-blind trial. Gut. 2010;59(1):21–30. doi: 10.1136/gut.2009.178558. [DOI] [PubMed] [Google Scholar]
- 118.Sleiman PMA, Wang M-L, Cianferoni A, Aceves S, Gonsalves N, et al. GWAS identifies four novel eosinophilic esophagitis loci. Nat Conrmun. 2014;5:5593. doi: 10.1038/ncomms6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zuo L, Fulkerson PC, Finkelman FD, Mingler M, Fischetti CA, et al. IL-13 induces esophageal remodeling and gene expression by an eosinophil-independent, IL-13Rα2-inhibited pathway. J Immunol. 2010;185(1):660–69. doi: 10.4049/jimmunol.1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yamazaki K, Murray JA, Arora AS, Alexander JA, Smyrk TC, et al. Allergen-specific in vitro cytokine production in adult patients with eosinophilic esophagitis. Dig Dis Sci. 2006;51(11):1934–41. doi: 10.1007/s10620-005-9048-2. [DOI] [PubMed] [Google Scholar]
- 121.Straumarm A, Kristl J, Conus S, Vassina E, Spichtin H-P, et al. Cytokine expression in healthy and inflamed mucosa: probing the role of eosinophils in the digestive tract. Inflamm Bowel Dis. 2005;11(8):720–26. doi: 10.1097/01.mib.0000172557.39767.53. [DOI] [PubMed] [Google Scholar]
- 122.Blanchard C, Mingler MK, McBride M, Putnam PE, Collins MH, et al. Periostin facilitates eosinophil tissue infiltration in allergic lung and esophageal responses. Mucosal Immunol. 2008;1(4):289–96. doi: 10.1038/mi.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Blanchard C, Stucke EM, Burwinkel K, Caldwell JM, Collins MH, et al. Coordinate interaction between IL-13 and epithelial differentiation cluster genes in eosinophilic esophagitis. J Immunol. 2010;184(7):4033–41. doi: 10.4049/jimmunol.0903069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Blanchard C, Mishra A, Saito-Akei H, Monk P, Anderson I, Rothenberg ME. Inhibition of human interleukin-13-induced respiratory and oesophageal inflammation by anti-human-interleukin-13 antibody (CAT-354) Clin Exp Allergy. 2005;35(8):1096–103. doi: 10.1111/j.1365-2222.2005.02299.x. [DOI] [PubMed] [Google Scholar]
- 125.Rothenberg ME, Wen T, Greenberg A, Alpan O, Enav B, et al. Intravenous anti-IL-13 mAb QAX576 for the treatment of eosinophilic esophagitis. J Allergy Clin Immunol. 2015;135(2):500–7. doi: 10.1016/j.jaci.2014.07.049. [DOI] [PubMed] [Google Scholar]
- 126.Odze RD. Pathology of eosinophilic esophagitis: what the clinician needs to know. Am J Gastroenterol. 2009;104(2):485–90. doi: 10.1038/ajg.2008.40. [DOI] [PubMed] [Google Scholar]
- 127.Kubo A, Nagao K, Amagai M. Epidermal barrier dysfunction and cutaneous sensitization in atopic diseases. J Clin Invest. 2012;122(2):440–47. doi: 10.1172/JCI57416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mueller S, Aigner T, Neureiter D, Stolte M. Eosinophil infiltration and degranulation in oesophageal mucosa from adult patients with eosinophilic oesophagitis: a retrospective and comparative study on pathological biopsy. J Clin Pathol. 2006;59(11):1175–80. doi: 10.1136/jcp.2005.031922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ravelli AM, Villanacci V, Ruzzenenti N, Grigolato P, Tobanelli P, et al. Dilated intercellular spaces: a major morphological feature of esophagitis. J Pediatr Gastroenterol Nutr. 2006;42(5):510–15. doi: 10.1097/01.mpg.0000215312.78664.b9. [DOI] [PubMed] [Google Scholar]
- 130.Lucendo AJ, Lucendo B. An update on the immunopathogenesis of eosinophilic esophagitis. Expert Rev Gastroenterol Hepatol. 2010;4(2):141–48. doi: 10.1586/egh.10.9. [DOI] [PubMed] [Google Scholar]
- 131.South AP, Cabral A, Ives JH, James CH, Mirza G, et al. Human epidermal differentiation complex in a single 2.5 Mbp long continuum of overlapping DNA cloned in bacteria integrating physical and transcript maps. J Invest Dermatol. 1999;112(6):910–18. doi: 10.1046/j.1523-1747.1999.00613.x. [DOI] [PubMed] [Google Scholar]
- 132.Fallon PG, Sasaki T, Sandilands A, Campbell LE, Saunders SP, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet. 2009;41(5):602–8. doi: 10.1038/ng.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.O’Regan GM, Sandilands A, McLean WH, Irvine AD. Filaggrin in atopic dermatitis. J Allergy Clin Immunol. 2009;124(3 Suppl. 2):2–6. doi: 10.1016/j.jaci.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 134.McAleer MA, Irvine AD. The multifunctional role of filaggrin in allergic skin disease. J Allergy Clin Immunol. 2013;131(2):280–91. doi: 10.1016/j.jaci.2012.12.668. [DOI] [PubMed] [Google Scholar]
- 135.Simpson CL, Patel DM, Green KJ. Deconstructing the skin: cytoarchitectural determinants of epidermal morphogenesis. Nat Rev Mol Cell Biol. 2011;12(9):565–80. doi: 10.1038/nrm3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kottke MD, Delva E, Kowalczyk AP. The desmosome: cell science lessons from human diseases. J CellSci. 2006;119(Pt 5):797–806. doi: 10.1242/jcs.02888. [DOI] [PubMed] [Google Scholar]
- 137.Abdulnour-Nakhoul SM, Al-Tawil Y, Gyftopoulos AA, Brown KL, Hansen M, et al. Alterations in junctional proteins, inflammatory mediators and extracellular matrix molecules in eosinophilic esophagitis. Clin Immunol. 2013;148(2):265–78. doi: 10.1016/j.clim.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 138.Davis BP, Kottyan LC, Stucke EM, Sherrill JD, Rothenberg ME. Functional analysis of calpain-14 in eosinophilic esophagitis. J Allergy Clin Immunol. 2015;135(2):AB247. [Google Scholar]
- 139.Behm-Ansmant I, Rehwinkel J, Izaurralde E. MicroRNAs silence gene expression by repressing protein expression and/or by promoting mRNA decay. Cold Spring Harb Symp Quant Biol. 2006;71:523–30. doi: 10.1101/sqb.2006.71.013. [DOI] [PubMed] [Google Scholar]
- 140.Lu TX, Sherrill JD, Wen T, Plassard AJ, Besse JA, et al. MicroRNA signature in patients with eosinophilic esophagitis, reversibility with glucocorticoids, and assessment as disease biomarkers. J Allergy Clin Immunol. 2012;129(4):1064–69. doi: 10.1016/j.jaci.2012.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lu S, Mukkada VA, Mangray S, Cleveland K, Shillingford N, et al. MicroRNAprofiling in mucosal biopsies of eosinophilic esophagitis patients pre and post treatment with steroids and relationship with mRNA targets. PLoS ONE. 2012;7(7):e40676. doi: 10.1371/journal.pone.0040676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lu TX, Lim EJ, Wen T, Plassard AJ, Hogan SP, et al. MiR-375 is downregulated in epithelial cells after IL-I3 stimulation and regulates an IL-13-induced epithelial trans criptome. Mucosal Immunol. 2012;5(4):388–96. doi: 10.1038/mi.2012.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fillon S, Robinson ZD, Colgan SP, Furuta GT. Epithelial function in eosinophilic gastrointestinal diseases. Immunol Allergy Clin North Am. 2009;29(1):171–8. xii–iii. doi: 10.1016/j.iac.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 144.Lim DM, Narasimhan S, Michaylira CZ, Wang ML. TLR3-mediated NF- κB signaling in human esophageal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2009;297(6):1172–80. doi: 10.1152/ajpgi.00065.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mulder DJ, Lobo D, Mak N, Justdnich CJ. Expression of toll-like receptors 2 and 3 on esophageal epithelial cell lines and on eosinophils during esophagitis. Dig Dis Sci. 2012;57(3):630–42. doi: 10.1007/s10620-011-1907-4. [DOI] [PubMed] [Google Scholar]
- 146.Bogiatzi SI, Fernandez I, Bicher JC, Marloie-Provost MA, Volpe E, et al. Cutting edge: proinflammatory and Th2 cytokines synergize to induce thymic stromal lymphopoietin production by human skin keratinocytes. J Immunol. 2007;178(6):3373–77. doi: 10.4049/jimmunol.178.6.3373. [DOI] [PubMed] [Google Scholar]
- 147.Smelter DF, Sathish V, Thompson MA, Pahelick CM, Vassallo R, Prakash YS. Thymic stromal lymphopoietin in cigarette smoke-exposed human airway smooth muscle. J Immunol. 2010;185(5):3035–40. doi: 10.4049/jimmunol.1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Oyoshi MK, Larson RP, Ziegler SF, Geha RS. Mechanical injury polarizes skin dendritic cells to elicit a TH2 response by inducing cutaneous thymic stromal lymphopoietin expression. J Allergy Clin Immunol. 2010;126(5):976–84. doi: 10.1016/j.jaci.2010.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ziegler SF. The role of thymic stromal lymphopoietin (TSLP) in allergic disorders. Curr Opin Immunol. 2010;22(6):795–99. doi: 10.1016/j.coi.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ziegler SF, Artis D. Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol. 2010;11(4):289–93. doi: 10.1038/ni.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wong CK, Hu S, Cheung PF, Lam CW. Thymic stromal lymphopoietin induces chemotactic and prosurvival effects in eosinophils: implications in allergic inflammation. Am J Respir Cell Mol Biol. 2010;43(3):305–15. doi: 10.1165/rcmb.2009-0168OC. [DOI] [PubMed] [Google Scholar]
- 152.Allakhverdi Z, Comeau MR, Jessup HK, Yoon BR, Brewer A, et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potendy activates mast cells. J Exp Med. 2007;204(2):253–58. doi: 10.1084/jem.20062211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dellon ES, Kim HP, Sperry SLW, Rybnicek DA, Woosley JT, Shaheen NJ. A phenotypic analysis shows that eosinophilic esophagitis is a progressive fibrostenotic disease. Gastrointest Endosc. 2014;79(4):577–85.e4. doi: 10.1016/j.gie.2013.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lucendo AJ, Arias A, De Rezende LC, Yagüe-Compadre JL, Mota-Huertas T, et al. Subepithelial collagen deposition, profibrogenic cytokine gene expression, and changes after prolonged fluticasone propionate treatment in adult eosinophilic esophagitis: a prospective study. J Allergy Clin Immunol. 2011;128(5):1037–46. doi: 10.1016/j.jaci.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 155.Straumann A, Conus S, Degen L, Felder S, Kumrner M, et al. Budesonide is effective in adolescent and adult patients with active eosinophilic esophagitis. Gastroenterology. 2010;139(5):1526–37. 1537.e1. doi: 10.1053/j.gastro.2010.07.048. [DOI] [PubMed] [Google Scholar]
- 156.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3(5):349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 157.Schoepfer AM, Safroneeva E, Bussmann C, Kuchen T, Portmann S, et al. Delay in diagnosis of eosinophilic esophagitis increases risk for stricture formation in a time-dependent manner. Gastroenterology. 2013;145(6):1230–36. doi: 10.1053/j.gastro.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 158.Desai TK, Stecevic V, Chang CH, Goldstein NS, Badizadegan K, Furuta GT. Association of eosinophilic inflammation with esophageal food impaction in adults. Gastrointest Endosc. 2005;61(7):795–801. doi: 10.1016/s0016-5107(05)00313-5. [DOI] [PubMed] [Google Scholar]
- 159.Mulder DJ, Pacheco I, Hurlbut DJ, Mak N, Furuta GT, et al. FGF9-induced proliferative response to eosinophilic inflammation in oesophagitis. Gut. 2009;58(2):166–73. doi: 10.1136/gut.2008.157628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Abonia JP, Wen T, Stucke EM, Grotjan T, Griffith MS, et al. High prevalence of eosinophilic esophagitis in patients with inherited connective tissue disorders. J Allergy Clin Immunol. 2013;132(2):378–86. doi: 10.1016/j.jaci.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Frischmeyer-Guerrerio PA, Guerrerio AL, Oswald G, Chichester K, Myers L, et al. TGFβ receptor mutations impose a strong predisposition for human allergic disease. Sci Transl Med. 2013;5(195):195ra94. doi: 10.1126/scitranslmed.3006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hoeck J, Woisetschläger M. Activation of eotaxin-3/CCL126 gene expression in human dermal fibroblasts is mediated by STAT6. J Immunol. 2001;167(6):3216–22. doi: 10.4049/jimmunol.167.6.3216. [DOI] [PubMed] [Google Scholar]
- 163.Hoeck J, Woisetschlager M. STAT6 mediates eotaxin-1 expression in IL-4 or TNF-α-induced fibroblasts. J Immunol. 2001;166(7):4507–15. doi: 10.4049/jimmunol.166.7.4507. [DOI] [PubMed] [Google Scholar]
- 164.Conway SJ, Molkentin JD. Periostin as a heterofunctional regulator of cardiac development and disease. Curr Genom. 2008;9(8):548–55. doi: 10.2174/138920208786847917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Snider P, Hinton RB, Moreno-Rodriguez RA, Wang J, Rogers R, et al. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ Res. 2008;102(7):752–60. doi: 10.1161/CIRCRESAHA.107.159517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Muir AB, Lim DM, Benitez AJ, Modayur Chandramouleeswaran P, Lee AJ, et al. Esophageal epithelial and mesenchymal cross-talk leads to features of epithelial to mesenchymal transition in vitro. Exp Cell Res. 2013;319(6):850–59. doi: 10.1016/j.yexcr.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kagalwalla AF, Akhtar N, Woodruff SA, Rea BA, Masterson JC, et al. Eosinophilic esophagitis: Epithelial mesenchymal transition contributes to esophageal remodeling and reverses with treatment. J Allergy Clin Immunol. 2012;129(5):1387–96.e7. doi: 10.1016/j.jaci.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Roman S, Hirano I, Kwiatek MA, Gonsalves N, Chen J, et al. Manometric features of eosinophilic esophagitis in esophageal pressure topography. Neurogastroenterol Motil. 2010;23(3):208–14. doi: 10.1111/j.1365-2982.2010.01633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zeisberg M, Kalluri R. Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front Biosci. 2008;13:6991–98. doi: 10.2741/3204. [DOI] [PubMed] [Google Scholar]
- 170.Jiang M, Ku W-Y, Zhou Z, Dellon ES, Falk GW, et al. BMP-driven NRF2 activation in esophageal basal cell differentiation and eosinophilic esophagitis. J Clin Invest. 2015;125(4):1557–68. doi: 10.1172/JCI78850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fujiwara Y, Higuchi K, Takashima T, Hamaguchi M, Hayakawa T, et al. Roles of epidermal growth factor and Na+/H+ exchanger-1 in esophageal epithelial defense against acid-induced injury. Am J Physiol Gastrointest Liver Physiol. 2006;290(4):G665–73. doi: 10.1152/ajpgi.00238.2005. [DOI] [PubMed] [Google Scholar]
- 172.Straumann A, Conus S, Degen L, Frei C, Bussmann C, et al. Long-term budesonide maintenance treatment is partially effective for patients with eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2011;9(5):400–409. doi: 10.1016/j.cgh.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 173.Lieberman JA, Morotti RA, Konstantinou GN, Yershov O, Chehade M. Dietary therapy can reverse esophageal subepithelial fibrosis in patients with eosinophilic esophagitis: a historical cohort. Allergy. 2012;67(10):1299–307. doi: 10.1111/j.1398-9995.2012.02881.x. [DOI] [PubMed] [Google Scholar]
- 174.Lin F, Yang X. TGF-β signaling in aortic aneurysm: another round of controversy. J Genet Genom. 2010;37(9):583–91. doi: 10.1016/S1673-8527(09)60078-3. [DOI] [PubMed] [Google Scholar]
- 175.Cincinnati Children’s Research Foundation. Testing Effectiveness of Losartan in Patients With EoE With or Without a CTD. Cincinnati, OH: Cincinnati Children’s Research Foundation; 2013. http://clinicaltrials.gov/ct2/show/NCT01808196?term=Testing+effectiveness+of+losartan+in+patients+with+EoE+with+or+without+a+CTD&rank=1. [Google Scholar]
- 176.Fox VL, Nurko S, Teitelbaum JE, Badizadegan K, Furuta GT. High-resolution EUS in children with eosinophilic “allergic” esophagitis. Gastrointest Endosc. 2003;57(1):30–36. doi: 10.1067/mge.2003.33. [DOI] [PubMed] [Google Scholar]
- 177.Fontillón M, Lucendo AJ. Transmural eosinophilic infiltration and fibrosis in a patient with nontraumatic Boerhaave’s syndrome due to eosinophilic esophagitis. Am J Gastroenterol. 2012;107(11):1762. doi: 10.1038/ajg.2012.226. [DOI] [PubMed] [Google Scholar]
- 178.Rieder F, Nonevski I, Ma J, Ouyang Z, West G, et al. T-helper 2 cytokines, transforming growth factor β1, and eosinophil products induce fibrogenesis and alter muscle motility in patients with eosinophilic esophagitis. Gastroenterology. 2014;146(5):1266–77. e1–9. doi: 10.1053/j.gastro.2014.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Monnerat MM, Lemme EM. Eosinophilic esophagitis: manometric and pHmetric findings. Arq Gastroenterol. 2012;49(2):113–17. doi: 10.1590/s0004-28032012000200004. [DOI] [PubMed] [Google Scholar]
- 180.Lucendo AJ. Motor disturbances participate in the pathogenesis of eosinophilic oesophagitis, beyond the fibrous remodelling of the oesophagus. Aliment Pharmacol Ther. 2006;24(8):1264–67. doi: 10.1111/j.1365-2036.2006.03109.x. [DOI] [PubMed] [Google Scholar]
- 181.Martin-Munoz MF, Lucendo AJ, Navarro M, Letran A, Martin-Chavarri S, et al. Food allergies and eosinophilic esophagitis—two case studies. Digestion. 2006;74(1):49–54. doi: 10.1159/000096595. [DOI] [PubMed] [Google Scholar]
- 182.Fulkerson PC, Rothenberg ME. Targeting eosinophils in allergy, inflammation and beyond. Nat Rev Drug Discov. 2013;12(2):117–29. doi: 10.1038/nrd3838. [DOI] [PMC free article] [PubMed] [Google Scholar]