

Abstract
The small G proteins Cdc42, Rac1, and Rac2 regulate the rearrangements of actin and membrane necessary for Fcγ receptor-mediated phagocytosis by macrophages. Activated, GTP-bound Cdc42, Rac1, and Rac2 bind to the p21-binding domain (PBD) of PAK1, and this interaction provided a basis for microscopic methods to localize activation of these G proteins inside cells. Fluorescence resonance energy transfer-based stoichiometry of fluorescent chimeras of actin, PBD, Cdc42, Rac1, and Rac2 was used to quantify G protein activation relative to actin movements during phagocytosis of IgG-opsonized erythrocytes. The activation dynamics of endogenous G proteins, localized using yellow fluorescent protein-labeled PBD, was restricted to phagocytic cups, with a prominent spike of activation over an actin-poor region at the base of the cup. Refinements of fluorescence resonance energy transfer stoichiometry allowed calculation of the fractions of activated GTPases in forming phagosomes. Cdc42 activation was restricted to the leading margin of the cell, whereas Rac1 was active throughout the phagocytic cup. During phagosome closure, activation of Rac1 and Rac2 increased uniformly and transiently in the actin-poor region of phagosomal membrane. These distinct roles for Cdc42, Rac1, and Rac2 in the component activities of phagocytosis indicate mechanisms by which their differential regulation coordinates rearrangements of actin and membranes.
INTRODUCTION
The Rho family GTPases Cdc42, Rac1, and RhoA regulate cell shape, cell motility, and phagocytosis. In their GTP-bound, active state, they interact with effectors that alter the actin cytoskeleton, contractility, and vesicle fusion (Ridley, 2001; Takai et al., 2001). Each of these GTPases drives distinct morphological rearrangements. The regulation of these component activities arises from activation and deactivation of Rho GTPases by regulatory interactions with three classes of molecules: guanine nucleotide exchange factors (GEFs), which activate G proteins; GTPase accelerating proteins (GAPs), which deactivate G proteins; and GDP disassociation inhibitors (GDIs), which inhibit the activation of Rho GTPases by sequestering them from membranes. These regulatory interactions are predicted to result in differential spatial and temporal activation of Rho family proteins, which in turn regulate cellular movements (Etienne-Manneville and Hall, 2002). Accordingly, complex movements of cytoskeleton and membrane should contain distinct underlying patterns of activation and deactivation for each of the participating GTPases.
Fcγ receptor (FcγR)-mediated phagocytosis is a spatially and temporally regulated process that requires the functions of Rac1 and Cdc42 (Cox et al., 1997; Caron and Hall, 1998; Massol et al., 1998). Particles coated with IgG activate FcγRs on the surfaces of macrophages that subsequently induce actin polymerization, myosin activity, and membrane fusion, leading to the extension of membrane over the particle and a constriction process that closes the phagosome (Greenberg, 1999; Swanson et al., 1999). Phagocytosis is essentially comprised of three morphological phases: 1) particle binding, 2) pseudopod extension, and 3) phagosome closure. FcγR-mediated phagocytosis also includes Rac-regulated assembly of the NADPH oxidase complex for generating superoxide in the phagosomal lumen (Bokoch and Diebold, 2002; Myers and Swanson, 2002). It has been speculated that Rac1 and Cdc42 regulate distinct processes during phagocytosis, with Cdc42 regulating pseudopod extension and Rac1 regulating phagosome closure (Castellano et al., 2001). Recent work also has indicated different roles for Rac1 and Rac2 in phagocytosis, chemotaxis, and superoxide production (Gu et al., 2003). However, current approaches lack the resolution needed to correlate the timing and localization of Rho GTPase signaling with the phases of phagocytosis.
The mechanisms of activation and deactivation of Cdc42, Rac1, and Rac2 during FcγR-mediated phagocytosis are largely unknown. First, ligation of the FcγR activates a phosphorylation cascade involving Src family and Syk tyrosine kinases (Cox and Greenberg, 2001). The GEF Vav can be phosphorylated by Syk and activates Rac during phagocytosis (Patel et al., 2002). Second, Syk can phosphorylate the p85 subunit of the type I phosphatidylinositol 3-kinase (PI3K), leading to local production of phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3] at the site of phagocytosis (Marshall et al., 2001; Srinivasan et al., 2003).
Once activated, Cdc42, Rac1, and Rac2 regulate phagocytic functions by their interactions with effector molecules. For example, Cdc42 interacts with Wiscott-Aldrich Syndrome Protein, whose deficiency in Wiscott-Aldrich Syndrome patients results in defective phagocytosis (Lorenzi et al., 2000). Rac interacts with PAK1, which localizes to phagocytic cups and macropinosomes (Dharmawardhane et al., 1999; Diakonova et al., 2002) and may regulate macropinocytosis (Dharmawardhane et al., 2000; Srinivasan et al., 2003). Rac interaction with p67phox subunit regulates the catalytic activity of the NADPH oxidase complex (Bokoch and Diebold, 2002).
Although the list of molecules that interact with the Rho GTPases provides insight into the mechanisms of Rho GTPase activation and downstream signaling, the signal transduction organizing phagocytosis will only be understood when the timing and organization of these interactions are defined. In this study, the p21-binding domain (PBD) from PAK1, which can detect GTP-bound Cdc42, Rac1, and Rac2 in biochemical and morphological assays (Kraynov et al., 2000; Gardiner et al., 2002), was used to localize endogenous, activated Cdc42, Rac1, or Rac2 during phagocytosis. Ratiometric microscopy of yellow fluorescent protein (YFP)-PBD and cyan fluorescent protein (CFP) identified two phases of G protein activation that correlated with distinct phases of phagocytosis. The first phase colocalized with actin in the extending pseudopod, and the second phase was marked by a prominent, transient spike of activity behind the actin-rich region as the phagocytic cup closed. We also used a novel fluorescence resonance energy transfer (FRET)-based imaging method to localize and measure the fractions of activated GTPases in living cells. The contributions of Cdc42, Rac1, and Rac2 to the phases of PBD localization were estimated by measuring the fraction of YPF-labeled GTPase bound to CFP-PBD and the fraction of activated GTPase in various regions of the forming phagosome. Activation of Cdc42 occurred early and preferentially at the tips of extending pseudopodia, activation of Rac1 occurred in phagocytic cups and during closure, and Rac2 was primarily active during phagosome closure.
MATERIALS AND METHODS
Constructs and Protein Purification
All constructs named YFP were either citrine (Griesbeck et al., 2001) or monomeric citrine (Zacharias et al., 2002). YFP-actin, YFP-Akt, and YFP-PBD were made with citrine. All YFP-GTPases were made with monomeric citrine, and CFP-PBD was made with monomeric CFP. pCitrine-N1 was created as described previously (Hoppe et al., 2002). To reduce the possibility of fluorescent protein-mediated dimerization, monomeric fluorescent proteins (Zacharias et al., 2002) were generated with the QuikChange method (Stratagene, La Jolla, CA) to yield the A206K mutation GCC→AAA, producing pmCit-N1 and pmCFP-N1. Plasmids encoding human Rac1, Rac1L61, and PBD were generous gifts from Klaus Hahn (Scripps Research Institute, La Jolla, CA). cDNA for human Rac1N17, Rac2, Rac2V12, Rac2N17, Cdc42, Cdc42V12, and Cdc42N17 were obtained from the Guthrie Institute (Sayre, PA). Polymerase chain reaction was used to amplify the PBD region of PAK1 or PAK1(H83,86L) (from Gary Bokoch, Scripps Research Institute) coding for amino acid residues 70–118 with the addition of BglII and HindIII restriction sites. This product was then cloned into pmCFP-N1 to create CFP-PBD with linking amino acids SGLRS. Three sets of primers were designed to amplify Rac1, Rac2, and Cdc42, with BglII and HindIII sites for insertion into pmCit-N1 to create YFP-Rac1 (wt, L61, N17), YFP-Rac2 (wt, V12,N17), YFP-Cdc42 (wt, V12, N17) with linker SGLRS. The mutation Y40H was introduced into YFP-Rac1(L61) by the QuikChange method (Stratagene) to yield YFP-Rac1(L61,H40). GFP-AktPH was a gift from Tobias Meyer (Stanford, Palo Alto, CA). YFP-AktPH was generated by replacement of the GFP with citrine. YFP-actin was generated by replacing YFP in pEYFP-actin (BD Biosciences Clontech, Palo Alto, CA) with citrine.
The DNAs for YFP-Rac1, YFP-Rac1(L61), and CFP-PBD were cloned into pET-28a (Novagen, Madison, WI) prokaryotic expression vector by polymerase chain reaction between the NheI and HindIII sites to add a six-His tag at the N terminus. All constructs were verified by sequencing.
Prokaryotic expression plasmids were transferred to Codon Plus BL21(RIL) Escherichia coli (Stratagene) for protein purification. The cells were grown with shaking (150 rpm) at 33°C in LB, to an OD600 of 0.7, and induced with isopropyl β-d-thiogalactoside for 5 h. After induction, the culture was chilled on ice 15 min, pelleted by centrifugation (5000 × g, 15 min), and resuspended in G protein-specific lysis buffer (Self and Hall, 1995b) with 1 mg/ml lysozyme for 15 min. Lysates were passed through a French press, treated with DNase and RNase for 10 min at 4°C, cleared by centrifugation (15,000 × g, 30 min), and then the proteins were purified on Ni2+-NTA agarose according to the manufacturer's protocol (QIAGEN, Valencia, CA). SDS-PAGE stained with Coomassie Blue showed the proteins to be >98% pure.
In Vitro FRET Titration
A FluoroMax-2 spectrofluorometer, fit with a 96-well plate reader and controlled by a computer running Data Max software (Instruments SA, Edison, NJ), was used to measure FRET between His-YFP-Rac1 and His-CFP-PBD. Equal volumes of 0.5 μM His-CFP-PBD in phosphate-buffered saline with 1 mg/ml bovine serum albumin were placed into the wells of a fluorescence plate. His-YFP-Rac1 or His-YFP-Rac1(L61) were serially diluted through the wells containing His-CFP-PBD. The plate was then read using 435-nm excitation and 475-nm emission light. Background was measured from wells containing PBD/bovine serum albumin and was subtracted from all wells. FRET efficiency was calculated as the decrease in donor fluorescence E = 1 - FDA/FD. Data were fit with Prism Software to a single site mass action binding equation that accounted for ligand depletion.
Cell Culture and Transfection
RAW264.7 cells from American Type Culture Collection (Manassas, VA) were grown in DMEM supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) (heat-inactivated at 56°C for 45 min) and 100 U/ml penicillin/streptomycin mixture (Sigma-Aldrich, St. Louis, MO) at 37°C with 5% CO2. Macrophages were plated on coverglasses 4 h before transfection. Transfection was carried out 24 h before the experiment with 1 μg of total plasmid DNA and 2 μl of FuGene6 (Roche Diagnostics, Indianapolis, IN). During microscopic observation, the cells were maintained at 37°C on a heated stage in Ringer's buffer (155 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, 10 mM HEPES, 10 mM glucose). Sheep erythrocytes opsonized with rabbit anti-sheep erythrocyte IgG were prepared as described previously (Swanson, 2002).
FRET Imaging
The FRET microscope consisted of an inverted fluorescence microscope (Nikon TE-300; Nikon, Tokyo, Japan), equipped with a temperature-controlled stage, a 175-W xenon arc lamp, shutters for trans- and epifluorescence illumination, filter wheels for both excitation and emission filters, dichroic mirrors that allowed detection of multiple fluorophores, a 60× numerical aperture 1.4 Planapo objective, and a cooled digital charge-coupled device camera (Cool Snap; Photometrics, Tucson, AZ), all of which were controlled by MetaMorph Image processing software (version 4.6.2; Universal Imaging, Malvern, PA). Excitation and emission filters were selected using two filter wheels (Sutter Instrument, Novato, CA) and a double pass dichroic mirror bandpass combination JP4 filter set, Chroma (Chroma Technology, Brattleboro, VT). The microscope and imaging system were calibrated for FRET stoichiometry as described previously (Hoppe et al., 2002). The component images consisted of epifluorescence filter combinations for CFP (ID: excitation 435 nm; emmission 490 nm), YFP (IA: excitation 505 nm; emmission 540 nm), and FRET (IF: excitation 435 nm; emmission 540 nm), as well as phase-contrast images. Coefficients for image processing were determined from RAW264.7 cells expressing YFP only (α), CFP only (β), and a linked CFP-YFP chimera whose FRET efficiency was independently measured by fluorescence lifetime spectroscopy (γ and ξ). Shading-correction images (to correct differences in illumination across the image plane) were collected from solutions of purified CFP and YFP sandwiched between two coverslips.
During experiments, cells were maintained in a heated observation chamber at 37°C. Macrophages expressing fluorescent probes were identified, and then phagocytosis was initiated by adding ∼105 IgG-opsonized erythrocytes to the chamber. Image collection began as erythrocytes landed on cells. Image series' consisting of phase-contrast, IA, ID, and IF for FRET microscopy, or phase-contrast, IA, and ID for Ratio imaging, were collected every 30 s. These primary images were shading corrected and used to obtain the processed images EA, ED, and R, as defined for FRET stoichiometry (Hoppe et al., 2002). For each pixel of the image, R represented the molar ratio of YFP to CFP, EA was the fraction of YFP chimera in complex with CFP, times the characteristic FRET efficiency of the YFP/CFP complex (or EA = ([YFP–CFP complex]/[YFP]) × EC), and ED was the fraction of CFP in complex with YFP, times the characteristic FRET efficiency of the YFP/CFP complex (or ED = ([YFP–CFP complex]/[CFP]) × EC). Conditions were chosen such that photobleaching was minimized. In general, the photobleaching as measured by loss of fluorescence intensity for either YFP or CFP constructs was <10% for the duration of data collection; differential photobleaching was generally <5%.
Ratiometric Imaging
A ratiometric imaging approach based on the imaging principles of FRET stoichiometry (Hoppe et al., 2002) was used to measure the molar ratios of two fluorescent chimeras expressed in macrophages. In its simplest form, chimeric YFP, which localized according to the protein it was linked to, was coexpressed with CFP, which served as a general marker of soluble cytosol. The Ratio image reported the concentrations of YFP-chimera relative to CFP, thereby correcting for variations in optical pathlength related to cell shape. Ratiometric imaging has been used to image the localization of molecules within living cells (Swanson, 2002; Henry et al., 2004). FRET stoichiometry provides a method for quantifying the molar ratio of an acceptor, (YFP)-labeled molecule to a donor, (CFP)-labeled molecule from three images, the acceptor image IA, donor image ID, and the “FRET” image IF as in Hoppe et al. (2002):
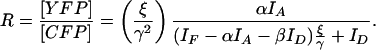 |
In the case where there is no FRET, the IF-αIA-βID term is zero, which allows use of an expression that requires only two images:
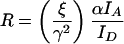 |
where α, γ, and ξ are defined as for FRET stoichiometry (Hoppe et al., 2002).
Particle Tracking
To quantify signaling events from multiple phagocytic events, a particle-tracking image analysis algorithm was developed in MetaMorph software (Universal Imaging). This algorithm measured the phagosome-associated signals by tracking the center of the opsonized erythrocyte in the phase-contrast image by the cross-correlation centroid-tracking algorithm TRACOBJ in MetaMorph (Universal Imaging). The algorithm used the position outputs of TRACOBJ to position 5-μm-diameter circular measurement regions in the computed images, and the phase-contrast images at each frame in the time series. A threshold was applied over the cell, and measurements were collected from a logical AND of the binary threshold and the gray-scale images, such that noncellular regions (zeros) were not included in the computed averages. For ratiometric imaging, the tracking algorithm determined the center of the erythrocyte and then positioned the measurement region in the phase-contrast and Ratio images (RP). The measurement region consisted of a 5-μm-diameter circle that included the entire erythrocyte and a small amount of surrounding cytoplasm. A second region was drawn around the entire cell and then Ratio values for the cell were determined (RC). This same process also was applied to the FRET stoichiometry data for the Ratio, EA, and ED. The output of these tracking algorithms included the particle- and cell-associated phase intensity, Ratio, EA, and ED. Phagocytic events were aligned by the phase-contrast grayscale values, which decreased as the particle changed from phase-bright to phase-dark during phagosome closure (Diakonova et al., 2002).
Recruitment Index RP/RC
Cells transfected with two separate plasmids express widely varying concentrations of CFP and YFP probes. A localization index was used to compare YFP chimera recruitment to phagosomes in different cells. Assuming that CFP distributes evenly throughout cytoplasm, the concentration of CFP should be the same throughout the cell. Dividing the Ratio on the phagosome RP by the Ratio for the entire cell RC should report the relative concentration of YFP-chimera on the phagosome, such that
 |
Determining the Fraction of Active G Protein
To compare signaling events among cells with varying expression levels of labeled molecules, a relationship had to be defined the between EA, ED, Ratio, and the fraction of active G protein. The relative concentrations of CFP-PBD and YFP-GTPase affect the fraction of active GTPases bound by CFP-PBD. To compare signals from cells expressing various ratios of CFP and YFP chimeras, we used empirical data from constitutively active GTPases to define a relationship between EA, ED, R, and the fraction of active GTPase.
Constitutively active GTPase chimeras were used to determine the effects of relative expression levels on the maximum values of EA or ED. Assuming that for constitutively active GTPases the fraction of active G protein is 1, the relationship between molar ratio [e.g., YFP-Rac1(V12)/CFP-PBD] and EA or ED can be obtained by measuring Ratio, EA, and ED in cells expressing different levels of YFP and CFP chimeras. In RAW264.7 macrophages, expression of YFP-Cdc42(V12), YFP-Rac1(Q61L), or YFP-Rac2(V12) in combination with CFP-PBD gave EA and ED values that were sigmoidal functions of the Ratio (Figure 5). At higher ratios, ED approached maximal values that indicated the characteristic FRET efficiency (EC) for that CFP-YFP complex (Hoppe et al., 2002). These plots allowed rescaling of the FRET images from cells expressing wild-type YFP-GTPases. That is, fractions of donor or acceptor in complex (fD and fA, respectively) could then be obtained by dividing the ED or EA images by EC (Hoppe et al., 2002). Data from the video sequences were recalculated to obtain the fraction of YFP-labeled G protein in the GTP-bound form, by using an empirical relationship between Ratio, and EA or ED defined by fitting the data in Figure 5 to single-site binding equations:
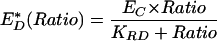 |
1 |
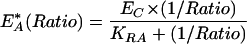 |
2 |
where KRD or KRA are fitting constants (not to be confused with dissociation constants) defined as the Ratio or 1/Ratio at which half of the donor or acceptors are in complex, respectively. To obtain the fraction of active GTPase (G*) for each component image of phagocytosis, the measured EA was divided by EA* at the mean cellular Ratio as determined by fitting the data in Figure 5 with equation 2:
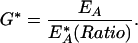 |
3 |
It should be emphasized that these calculations did not involve further processing of the ED and EA images. They simply changed the calibration scales applied to the already processed images.
Figure 5.

Estimation of EC for FRET pairs. In vivo titration of YFP-Cdc42(V12), YFP-Rac1(L61), and YFP-Rac2(V12) with CFP-PBD. Populations of cells were imaged by FRET stoichiometry and then average cellular EA (A, C, and E) or ED (B, D, and F) of entire cells were plotted against Log(1/Ratio) or Log(Ratio), respectively. At high 1/Ratio, CFP-PBD was present in excess and the plateau of EA indicated the characteristic FRET efficiency (EC). Conversely, at high Ratio, YFP-GTPase was present in excess and the maximal ED indicated EC. Data were fit to single site equilibrium binding equations and the parameters determined as indicated (see MATERIALS AND METHODS). Cells expressed CFP-PBD plus YFP-Cdc42(V12) (A and B), YFP-Rac(L61) (C and D), or YFP-Rac2(V12) (E and F).
RESULTS
PBD Dynamics during Phagocytosis
Time-lapse ratiometric imaging of phagocytosis by RAW264.7 macrophages expressing YFP-PBD and CFP allowed measurement of the distributions of endogenous GTP-bound G proteins that bind PBD. YFP-PBD was distributed uniformly throughout the cytoplasm, but it was concentrated in some ruffles and lamellipodia (our unpublished data). During phagocytosis, YFP-PBD redistributed to the forming phagosome and demonstrated a prominent burst of localization during phagosome closure (Figure 1A). Closure was recognizable in phase-contrast images by the particle's transition from phase-bright to phase-dark (Diakonova et al., 2002).
Figure 1.

Organization of PBD binding sites relative to actin localization during FcγR-mediated phagocytosis. (A) Time series showing phase-contrast and Ratio images of a macrophage internalizing an IgG-opsonized erythrocyte. The color bar indicates the molar ratio (YFP/CFP). YFP-PBD was recruited to the forming phagosome (1.5–4.5 min) to a much greater extent during closure (5.0–7.5 min) and was cleared from the closed phagosome (8.5 min). (B) Particle-tracking analysis of YFP-actin (open circles) and YFP-PBD (closed circles) indicated that actin was recruited to the phagosome after particle binding (0–1 min) and during extension (1–5 min), and YFP-PBD was recruited throughout phagocytosis, with a pronounced increase in recruitment during closure (5.0–8 min). Data are mean ± SEM for 10 phagocytic events; no more than three events were taken from any one cell, and at least five different cells made up the 10 traces. (C) Simultaneous imaging of YFP-PBD and CFP-actin indicated that actin and the majority of PBD binding sites formed a discrete interface during constriction of the opsonized erythrocyte (constriction is inferred from the deformation of the erythrocyte; see arrow). CFP-actin was recruited to the forming phagosome, moved as a concentrated band over the particle during the extension phase (1.5–6.0 min), and condensed at the point of closure (6–7.5 min). YFP-PBD accumulated significantly on the base of the phagosome as constriction of the particle began (4.5–5.0 min), closely followed the moving band of actin during closure (5.5–7.5 min) and then rapidly dissipated (7.5–8.5 min). Bars, 3 μm. Also see Movie 1.
A particle-tracking method was developed that could follow movement of the opsonized erythrocyte through images of a time series and guide measurements on the phagosome in the Ratio image (see MATERIALS AND METHODS). By tracking the phase-intensity of the particle and identifying the frames in which particles became phase-dark, movies of phagocytic events could be aligned temporally for quantitative analysis. To compare cells with varying expression levels of YFP and CFP probes, the YFP/CFP Ratio measured on the phagosome (RP) was divided by the YFP/CFP Ratio for the entire cell (RC). For all of the molecules studied, RC varied little over time, and CFP indicated the distributions of cytoplasmic volume. Therefore, the ratio of the two ratios, RP/RC, indicated the fractional concentrations of YFP-labeled molecules near the phagosome (see MATERIALS AND METHODS). Cells expressing nonchimeric YFP and CFP demonstrated uniform ratios throughout phagocytosis, with RP/RC values of 1, indicating that cell movements did not create ratiometric artifacts (Henry et al., 2004; our unpublished data).
The recruitment of YFP-PBD was assessed from multiple phagocytic events by particle-tracking-based quantification. Localization of YFP-PBD to the phagosome was biphasic, with limited recruitment during the binding (0–1 min) and extension phases (0–5 min), followed by a transient localization to the phagosome during the closure phase (5–8 min) (Figure 1B).
In macrophages coexpressing YFP-actin and CFP, YFP-actin redistributed to the phagosome (0–3 min), and then was mostly cleared by 7 min (Figure 1B). Similar to previous imaging studies (Araki et al., 2003; Henry et al., 2004), YFP-actin localized as a concentrated, narrow band that moved over the opsonized erythrocyte at the leading edge of the extending pseudopod, attaining localized molar ratios greater than 10 times that of the cell body (our unpublished data). Quantitation of multiple phagocytic events in cells expressing YFP-actin indicated that actin was cleared from phagosomes before or during the transient increase in PBD (Figure 1B).
To localize PBD relative to actin, macrophages expressing YFP-PBD plus CFP-actin were imaged during phagocytosis. CFP-actin in the forming phagosome moved over the particle in a narrow band that was spatially distinct from, and preceded, the prominent labeling with YFP-PBD (Figure 1C and Movie 1). Much more of the cellular CFP-actin was recruited to the forming phagosome than YFP-PBD (compare scales of Figure 1B), but a color overlay of the CFP and YFP images showed that YFP-PBD localized to the base of the phagosomal membrane, following the CFP-actin band as it passed over the particle (Figure 1C and Movie 1). The boundary between the YFP-PBD and CFP-actin defined a region of constriction, as seen by the deformation of the opsonized erythrocyte, which moved over the particle as it was drawn into the cell (Figure 1C, 5–7 min). This boundary therefore coincided with the circumferential contractile activities of phagosome closure (Swanson et al., 1999; Araki et al., 2003).
Localization of Rac1, Rac2, Cdc42, and PI3K
To determine whether the dynamics of actin and PBD in forming phagosomes could be explained by regulated redistribution of G proteins, the distributions of YFP-Cdc42, YFP-Rac1, and YFP-Rac2 were measured by ratiometric imaging. YFP-Cdc42 localized to the tips of extending pseudopodia in the phagocytic cup and to the entire phagosome during closure (Figure 2A). Tracking analysis indicated that the net concentration of YFP-Cdc42 on the phagosomal membrane remained constant and elevated (Figure 2B). A similar pattern of increased but constant association was measured for YFP-Rac1 (Figure 2C). Importantly, YFP-Rac1 was often partially localized on the plasma membrane, as indicated by increased ratios near the cell periphery. Because the phagocytic cup is membrane rich (i.e., two membranes folded around a very thin layer of cytoplasm), membrane-associated YFP molecules could have seemed to be recruited to the site of phagocytosis simply as a result of the increase in membrane density. YFP-Rac1 demonstrated a small decline in localization to the phagosome during the extension phase of phagocytosis (Figure 2D), probably indicating a transition from the tightly folded plasma membrane of the pseudopod to the uniform membrane surrounding the particle. YFP-Rac2 also was present on the pseudopod; however, unlike YFP-Cdc42 or YFP-Rac1, its association was restricted to the base of the forming phagosome (Figure 2E). In general, YFP-Cdc42, YFP-Rac1, and YFP-Rac2 were slightly enriched on the phagosome (Figure 2B, D, and F) and displayed minor differences in their spatial arrangements. These small changes during phagocytosis indicated that their localization was not sufficient to coordinate the moving band of YFP-actin or the localization of YFP-PBD.
Figure 2.

Ratiometric imaging and tracking analysis of YFP-Cdc42, YFP-Rac1, YFP-Rac2, and YFP-AtkPH domain relative to CFP during phagocytosis. (A, C, E, and G) Phase-contrast, YFP, and Ratio image time series of RAW macrophages phagocytosing IgG-coated erythrocytes. Color bars indicate the ranges of the Ratio values. (B, D, F, and H) Plots of RP/RC indicating the dynamics of YFP-chimera localization to phagosomes, averaged for 10 phagocytic events each. Error bars are the SE of the mean. (A) YFP-Cdc42 was present at the site of binding (0.5 min), localized to the tips of the advancing pseudopod (1.5–4.5 min) and then remained on the phagosome during and following closure (5.5–8.5 min). (B) Tracking analysis indicated the enhancement of YFP-Cdc42 on multiple phagosomes, but it did not indicate a significant change in localization. (C) YFP-Rac1 was present on plasma membranes before phagocytosis as seen by the Ratio image (0.5 min). The ratio increased as membrane extended around the particle (1.5–4.5 min) and then diminished somewhat during internalization (5.5–8.5 min). (D) Cumulative tracking data indicated that the association of YFP-Rac1 with the phagosome was variable, decreased until closure (>8.0 min) and then remained slightly elevated. (E) YFP-Rac2 localized to the base of the phagosome during extension and closure. (F) Tracking analysis showed YFP-Rac2 slightly increased on the phagosome. (G) The YFP-AktPH domain localized rapidly to the site of particle contact (0.5–1.5 min), continually increased (1.5–4.5 min), and then was cleared from the plasma membrane after closure (5.5–8.5 min). (H) YFP-AktPH was localized to phagosomes throughout formation and closure and then was cleared slowly. Bar, 3 μm.
We next asked whether the dynamics of 3′ phosphatidylinositols (PIs) could explain the patterns of actin and PBD. 3′ PIs have been implicated in guiding actin polymerization and in localizing activities to the forming phagosome (Marshall et al., 2001), and PI3K has been proposed to function in a positive feedback loop with Rac1 (Srinivasan et al., 2003). Ratiometric imaging was performed in cells expressing YFP-AktPH, which binds to phosphatidylinositol 3,4-diphosphate (PI(3,4)P2)and PI(3,4,5)P3 and is recruited to phagosomes (Marshall et al., 2001). YFP-AktPH was recruited to phagosomes rapidly after binding of the opsonized erythrocyte to the surface of the macrophage. It remained on the phagosomal membrane throughout phagocytosis (Figure 2G). The tracking analysis indicated that YFP-AktPH was rapidly recruited to the phagosome; was present throughout the binding, extension, and closure phases; and then was cleared slowly (Figure 2H). This uniform pattern of YFP-AktPH labeling indicated that distributions of PI(3,4,5)P3 and PI(3,4)P2 were insufficient to explain the dynamics of YFP-actin and YFP-PBD.
FRET-based Detection of Rho GTPase Activation
To determine the contributions of activated Cdc42, Rac1, and Rac2 to the localization dynamics of YFP-PBD during phagocytosis, we localized active G proteins by using FRET stoichiometry. To measure the affinity of CFP-PBD/YFP-Rac1 interactions, purified His-CFP-PBD was titrated with purified GDP-bound His-YFP-Rac1 or a GTP-bound His-YFP-Rac1(L61). In vitro FRET measurements indicated that His-YFP-Rac1(L61) bound to His-CFP-PBD with a KD value of ∼200 nM (Figure 3A). This was ∼10-fold lower affinity than that reported for PBD binding to Rac1(L61) (Thompson et al., 1997), but higher affinity than reported for chemically labeled PBD and GFP-Rac1(L61) (Kraynov et al., 2000). Therefore, the constructs were adequate for low-affinity detection of GTPase activation.
Figure 3.

FRET measures YFP-Rac1 binding to CFP-PBD in vitro and in vivo. (A) Titration of His-CFP-PBD with His-YFP-Rac1 and His-YFP-Rac1(L61). Curve-fitting and correcting for ligand depletion yielded KD ∼ 200 nM for His-YFP-Rac(L61) (mean ± SD). (B) Cells expressing YFP-Rac1(L61) and CFP-PBD were imaged by FRET stoichiometry. Left, primary data: ID(CFP), IA(YFP) and IF images. Right, computed FRET images: ED, EA, and Ratio. Color bars indicate the magnitude of each. Expression of YFP-Rac1(61L) induced significant ruffling and lamellipod formation (primary images). YFP-Rac1(61L) localized primarily to the plasma membrane (IA image), whereas CFP-PBD was mostly soluble, with less of it recruited to the plasma membrane. Note that the cell had an average Ratio of less than one, indicating a molar excess of CFP-PBD. EA produced a uniform image with high EA values, indicating that most of the YFP-Rac(L61) was bound to CFP-PBD. ED varied, similar to the Ratio image, indicating that the ED signal was composed of the free (soluble) and bound (membrane-associated) CFP-PBD. (C) FRET signals from various combinations of YFP and CFP chimeras inside macrophages. FRET is indicated as (EA+ ED)/2, averaged over the entire cell. Significant FRET signals were detected from combinations of CFP-PBD plus the constitutively active mutants YFP-Cdc42(V12), YFP-Rac1(L61) and YFP-Rac2(V12). Little or no FRET was detected from cells expressing constitutively active mutants plus either CFP or CFP-PBD(LL). Small but significant FRET signals were detected from cells expressing CFP-PBD plus the dominant negative mutants YFP-Cdc42(N17), YFP-Rac1(N17), and YFP-Rac2(N17). Likewise, YFP-Rac1(L61,H40) gave reduced but detectable FRET. Errors bars are the propagated SD. n values are indicated on the right side of the figure.
FRET stoichiometry is an analytical imaging method that can quantify FRET between separately expressed fluorescent proteins inside cells (Hoppe et al., 2002). From three fluorescence images of a cell, FRET stoichiometry determines three essential elements of a protein–protein interaction: 1) Ratio, the molar ratio of acceptor to donor (e.g., [YFP-Rac1]/[CFP-PBD]); 2) ED, which is proportional to the fraction of donor (CFP) in complex with acceptor (YFP); and 3) EA, which is proportional to the fraction of acceptor (YFP) in complex with donor (CFP) (e.g., the fraction of YFP-Rac1 bound to CFP-PBD).
RAW 264.7 macrophages expressing YFP-Rac1(L61) and CFP-PBD showed dramatically increased ruffling and lamellipodia formation (Figure 3B). YFP-Rac1(L61) was localized to the plasma membrane, whereas CFP-PBD was largely soluble. FRET stoichiometry indicated that the fraction of YFP-Rac1(L61) in complex was uniform throughout the plasma membrane (Figure 3B, EA image). The fraction of CFP-PBD in complex displayed variations that matched the Ratio image, indicating differences in membrane-versus cytosol-rich regions (Figure 3B, compare Ratio with ED). Expression of YFP-Cdc42(V12) or YFP-Rac2(V12) also caused morphological rearrangements in cells (our unpublished data). However, YFP-Cdc42(V12) and YFP-Rac2(V12) localized differently than YFP-Rac1(L61) and indicated FRET on internal membranes, vesicles and to a lesser extent the plasma membrane (our unpublished data).
To measure the effects of mutant Rho-family proteins on the ability of FRET to detect the interaction of Rho GTPases with CFP-PBD, various combinations of YFP and CFP chimeras were expressed in macrophages and their FRET signals were analyzed. FRET was measured as (EA + ED)/2, which allows for comparison of FRET signals from cells with different ratios of protein expression. Constitutively active mutants of Cdc42, Rac1 and Rac2 (YFP-Cdc42(V12), YFP-Rac1(L61), and YFP-Rac2(V12), respectively) exhibited strong FRET signals when coexpressed with CFP-PBD, but very little FRET when coexpressed with CFP or with CFP-PBD mutated in amino acids essential for PBD interaction with G proteins (CFP-PBD(LL)) (Bokoch, 2003) (Figure 3C). Unexpectedly, dominant negative GTPases (YFP-Cdc42(N17), YFP-Rac1(N17), and YFP-Rac2(N17), exhibited low but detectable FRET when coexpressed with CFP-PBD, but no FRET when expressed with CFP, indicating that, in the context of the living cell, N17 mutants of Cdc42 or Rac permit interaction with PBD. Importantly, the N17 mutants were largely localized to membranes, suggesting that they were not bound to GDI (our unpublished data). Also, the Y40H mutation was introduced into YFP-Rac1(L61), which interferes with Rac binding to PAK (Joneson et al., 1996). This mutant displayed low but detectable FRET with CFP-PBD. Together, the measurements indicated that FRET stoichiometry detected CFP-PBD interactions with GTP-bound forms of YFP-Cdc42, YFP-Rac1, and YFP-Rac2.
Activation of G Proteins during Phagocytosis
To localize the activation of Rho GTPases during phagocytosis, macrophages expressing CFP-PBD plus either YFP-Cdc42, YFP-Rac1, or YFP-Rac2 were imaged during phagocytosis of IgG-coated erythrocytes. YFP-Cdc42 and CFP-PBD reported high EA at the site of particle attachment, immediately after contact with the macrophage (Figure 4A, 0.5 min, and Movie 2). YFP-Cdc42 was active only at the advancing edge of the pseudopod (Figure 4A and Movie 2). Application of the particle-tracking analysis to the FRET stoichiometry data allowed quantitative comparisons of the patterns of activation. The cumulative data from 10 phagocytic events indicated that YFP-Cdc42 gave high EA as soon as the particle bound. This signal persisted throughout the extension phase, but it decreased before the closure phase (Figure 4B).
Figure 4.

FRET stoichiometric imaging of YFP-Cdc42, YFP-Rac1, and YFP-Rac2 activation during phagocytosis of E-IgG. (A) Phase-contrast and EA images for cells expressing YFP-Cdc42 and CFP-PBD. YFP-Cdc42 produced an EA signal as soon as the erythrocyte contacted the macrophage. The high EA was restricted to the advancing tip of the pseudopod as it moved over the particle (1.5–5.5 min) and diminished during the closure phase (5.5–8.5 min). (B) Tracking analysis indicated the rapid association of YFP-Cdc42 with CFP-PBD and persistent FRET throughout pseudopod extension. (C and D) FRET microscopy and stoichiometry of macrophages expressing YFP-Rac1 and CFP-PBD. YFP-Rac1 interacted with CFP-PBD shortly after particle binding and throughout the pseudopod during extension (1.5–5.5 min). The quantity of YFP-Rac1 in complex with CFP-PBD increased transiently on the base of the pseudopod during the closure phase (5.5–7.5 min) and was deactivated on the closed phagosome (8.5 min). (D) This response was consistent when averaged over multiple phagocytic events. (E and F) FRET microscopy of cells expressing YFP-Rac2 and CFP-PBD indicated that Rac2 activation was delayed until closure. (G) Control cell expressing YFP-Cdc42 and CFP showed a uniform value of EA = 0 throughout phagocytosis. (H) Averaged traces from control cells expressing CFP plus YFP-Cdc42 (red), YFP-Rac1 (blue), or YFP-Rac2 (green) never indicated FRET. Bar, 3 μm. Also see Movies 2–4.
As the pseudopod filled the particle-tracking region of interest, the area over which the signal was averaged increased. Therefore, for a signal such as EA for Cdc42, which was localized at the advancing tips, the tracking algorithm reported a declining average signal, even though the intensity of the localized signal remained constant. This effect can be seen by comparing the images in the time series Figure 4A with the tracking analysis trace in Figure 4B.
Macrophages expressing YFP-Rac1 and CFP-PBD displayed increased EA shortly after particle contact, throughout the extending pseudopod and over the base of the phagosome during closure (Figure 4C and Movie 3). Compiled tracking data indicated that these dynamics were consistent from one event to the next (Figure 4D). The fraction of YFP-Rac1 bound to CFP-PBD was greatest over the base of the cup during phagosome closure, coincident with the large peak in YFP-PBD recruitment during phagosome closure (compare Figures 1B and 4D).
Phagocytosis by cells expressing YFP-Rac2 and CFP-PBD showed slightly increased EA in the vicinity of the particle and a pronounced transient increase in EA distributed over the base of the phagosome during closure (Figure 4E and Movie 4). The cumulative tracking analysis showed that, unlike YFP-Cdc42 or YFP-Rac1, YFP-Rac2 displayed only a minor activation during pseudopod extension. Its transient activation during closure coincided strikingly with the peaks of YFP-Rac1 activation and YFP-PBD localization (Figure 4, E and F).
Control cells expressing YFP-Cdc42 and CFP displayed EA values of zero throughout phagocytosis, indicating that FRET was dependent upon interaction between the GTPase and PBD (Figure 4G). Tracking analysis of phagocytic events for YFP-Cdc42, YFP-Rac1, and YFP-Rac2 coexpressed with CFP never indicated FRET (Figure 4H) nor did YFP-PBD coexpressed with CFP or with CFP-actin (our unpublished data). Thus, FRET signals were specific for the interactions between the YFP-GTPases and CFP-PBD.
Comparisons of Signal Timing and Amplitude
Recalculation of the imaging data quantified the contributions of each activated G protein to the various phases of PBD localization during phagocytosis. Data from cells expressing CFP-PBD and constitutively active YFP-Cdc42, YFP-Rac1, or YFP-Rac2 (Figure 5) allowed estimation of the characteristic FRET efficiency (EC) for each interaction. Determination of EC allowed recalculation of the EA data to obtain the fractions of PBD-bound G protein (see MATERIALS AND METHODS). For each time series of phagocytosis, the Ratio for the entire cell was used to correct the magnitude of EA for mass action effects, yielding the fraction of GTP-bound YFP-Cdc42, YFP-Rac1, and YFP-Rac2 (Figure 4, G* on color bars). The particle-tracking traces for each phagosome were corrected for mass action and averaged (Figure 6B). The corrected traces gave the same overall patterns of activation as the uncorrected traces (Figure 4B, D, and F), and the fractions of GTP-Cdc42, -Rac1, and -Rac2 reached similar magnitudes (Figure 6B). Therefore, the FRET stoichiometric analysis of component G protein signals indicated that the first phase of YFP-PBD localization to phagosomes (Figure 1) correlated with activation of Cdc42 and Rac1 at the leading edge of the phagocytic cup and that the prominent spike of PBD localization reflected transient, localized activation of Rac1 and Rac2 (Figure 6A and B).
Figure 6.

Summary of patterns of localization and activation on the forming phagosome. (A) Replotting of Figure 1B, showing the dynamics of YFP-actin (yellow) and YFP-PBD (black) on phagosomes relative to the phases of pseudopod extension and phagosome closure. (B) Data of Figure 4, B, D, and F, recalculated as fractions of active YFP-GTPase in the phagosome. Active Cdc42 (red) was maximal during particle binding and pseudopod extension, correlating with the strong recruitment of actin to the extending pseudopod. After a brief delay, Rac1 (blue) was active during pseudopod extension. The fraction of active Rac1 and Rac2 (green) increased transiently during closure, coincident with the increased PBD localization to the phagosome. (C) Cartoon summary of the spatial organization of the distributions of actin and activated GTPases during phagocytosis. YFP-actin (yellow) localized to the pseudopod tips during extension and was removed during closure. YFP-Cdc42 (red) was active at the advancing tip of the pseudopod. YFP-Rac1 (blue) demonstrated modest activation along the sides of the extending pseudopod, followed by prominent activation on the inner phagosomal membrane during contraction of the particle and phagosome closure. YFP-Rac2 (green) was active on the base of the phagosome during contraction and closure.
Figure 6C is a cartoon that summarizes the overall patterns of activation observed for YFP-Cdc42, YFP-Rac1, and YFP-Rac2. It is important to emphasize that the colors presented do not reflect the recruitment of the GTPases to the membrane as measured in Figure 2. Rather, they are intended to illustrate the temporal and spatial organization of the fractional activation and of each GTPase as observed from the FRET measurements in Figures 4 and 6B. From the traces in Figure 6B, we interpret the apparent order of GTPase activation to be Cdc42 activation followed by Rac1 activation followed by Rac2 activation. However, given the similarity in signal amplitude for Cdc42 and Rac1 activation we cannot quantitatively verify that Cdc42 activation completely precedes Rac1 activation.
DISCUSSION
This work applied new analytical imaging methods to identify distinct contributions from Cdc42, Rac1, and Rac2 to the component activities of phagocytosis in macrophages. The temporal and spatial resolution provided by these methods indicated that endogenous PBD-binding proteins were active in a precise pattern relative to actin during phagocytosis. In particular, endogenous GTP-bound GTPases were present during the actin-rich phase of phagocytic cup formation, and were present at greater concentrations over actin-poor regions during phagosome closure. FRET stoichiometry showed that although recruitment of GTPases to the phagosome was modest, the patterns of GTPase activation during phagocytosis were distinct for each type of protein.
The imaging methods localized protein interactions in various subregions of the forming phagosomes. The particle-tracking method of analysis allowed quantitative comparisons of separate phagocytic sequences. However, because that method averaged the signals from a 5-μm region encircling the entire phagosome, it lost much of the resolution available in the images. This loss of resolution was necessitated by the fact that different phagocytic events occurred with different orientations relative to the image plane; sometimes a particle was ingested as a side-view, other times as a top-view. For example, FRET stoichiometric imaging of Cdc42 activation, in side-views of phagocytosis (Movie 2), showed that it was activated primarily at the advancing edges of phagocytic cups. The quantitative analysis of the Cdc42 activation dynamics seemed to indicate instead that the protein was simply being deactivated during phagocytosis. However, the image sequences indicated that the measured decrease of activated Cdc42 simply reflected the gradual decrease in the fraction of that region occupied by the advancing edge. Thus, the dynamics discovered here are best represented by the images, especially the movies. The quantitative analyses from the tracking support those images by demonstrating the consistency of the signal timing in different phagocytic events.
FRET stoichiometry revealed the patterns of Cdc42, Rac1, and Rac2 activation underlying the recruitment of PBD to the phagosome. During phagocytosis, each GTPase displayed distinct patterns of activation that correlated with the phases of phagocytosis. GTP-Cdc42 and GTP-Rac1 localized to the band of actin in the extending pseudopod. GTP-Rac1 and GTP-Rac2 corresponded to contractile activities and closure of the phagosome. Together, these three GTPases displayed unique patterns of activation that could coordinate phagosome formation. The ability to quantify the interactions of independent donor- and acceptor-labeled molecules in cells has distinct advantages over methods that use linked, FRET-based sensors, because these sensors often lack essential localization domains. Importantly, the methods for measuring fractions of activated G protein in regions of the image (G*) allow unprecedented measurement of signal amplitudes. For example, the quantitative analyses revealed that Rac1 activation was consistently greater during the phagosome closure than during extension of the phagosomal cup.
A major limitation of this approach is that it requires the overexpression of fluorescently tagged molecules, which may perturb the endogenous signaling pathways. However, inhibitory effects due to overexpression can be discerned by measuring the effects of probe expression on the rates of phagocytosis. Any serious perturbation of the signaling should slow the process. Although expression of dominant negative Cdc42 and Rac1 chimeras inhibited phagocytosis, expression of wild-type GTPase or PBD chimeras did not measurably affect rates of phagocytosis (as measured by the timing of the particle's transition from phase-bright to phase-dark). This may be because the probes have sufficiently low affinities for endogenous proteins that they can report activities without measurably perturbing the endogenous interactions.
Our interpretation of these data assumes that PBD can bind to GTP-bound GTPases in a manner comparable with their binding to endogenous effectors. However, many of the known effectors of Cdc42 and Rac also bind to other components that may regulate their localization. Importantly, the affinity of our CFP-PBD for GTP-bound GTPases (∼200 nM) was considerably less than that observed for GTPase binding to PAK (∼20 nM) (Thompson et al., 1998) or to p67phox (∼1 nM) (Diebold and Bokoch, 2001). This lower affinity indicates that, despite their overexpression, interactions of the probes with endogenous molecules were not seriously perturbed and may explain why expression of CFP-PBD did not significantly inhibit phagocytosis. It is likely that the dynamics of YFP-PBD in phagosomes missed some of the activated GTPases because of stronger interactions with nonfluorescent endogenous proteins.
Regulation of Cdc42, Rac1, and Rac2 Activation
The distinct patterns of Cdc42, Rac1, and Rac2 activation during phagocytosis displayed changes on the subminute time scale. This indicated that activation and deactivation of these GTPases was affected by coordinated activities of GEFs and GAPs. The rapid activation of these proteins requires the action of GEFs to catalyze the exchange of GDP for GTP. GTP-Cdc42 was detected at the site of phagocytosis as soon as the particle bound (Movie 2), indicating the existence of a GEF capable of acting on Cdc42 immediately after ligation of FcγR. GTP-Cdc42 remained at the tip of the extending pseudopod but was not present at the base, indicating either that Cdc42 was dynamically activated at the tip of the advancing pseudopod and deactivated at the phagosome base or that some mechanism confined GTP-Cdc42 to the tip of the advancing pseudopod.
The activation of Rac1 was biphasic, in that it was activated shortly after particle binding and in a second, stronger phase of activation coincident with the large peak of PBD recruitment and Rac2 activation. This pattern of Rac1 activation indicated that GEFs act both early and late in phagocytosis. The slight delay in generation of GTP-Rac1, along with the differences in localization on the phagosome, suggests that Rac1 was activated by a different mechanism than Cdc42. Indeed, recent work on phagocytosis signaling has indicated Vav1 as a GEF for Rac1 but not Cdc42 (Patel et al., 2002), consistent with independent mechanisms for Cdc42 and Rac1 activation. Given the timing difference in activation between Rac1 and Cdc42, it is possible that the activation of Rac1 requires generation of 3′ phosphoinositides and Cdc42 does not. In most systems, Rac1 activation is dependent on PI3K; thus, an explanation for the delay could be that 3′ phosphoinositides are required to first localize or activate the GEF for Rac1. Consistent with this notion is that the AktPH domain is recruited early to the forming phagosome, before activation of Rac1 and Rac2 (compare Figure 2H with 4D and 4H).
The second wave of Rac1 activation coincided with Rac2 activation, indicating that at this phase, the same GEF acts on both Rac1 and Rac2. Mechanisms for the differential regulation of Rac1 and Rac2 are unknown, although differences in their membrane localization may play a role (Michaelson et al., 2001).
Deactivation mechanisms seemed to be enhanced after phagosome closure. The intrinsic hydrolysis rate for Rac1 is t1/2 = 10 min (Self and Hall, 1995a), whereas the deactivation time for Rac1 and Rac2 during phagocytosis was on the order of t1/2 = 1 min (estimated from Figure 5B). This discrepancy suggests either that the GTPase activities of Rac1 and Rac2 are increased by the action of a GAP or that CFP-PBD is competed off as other binding partners become available. Myosin IXb is the only protein localized to the phagosome which is known to have Rho-GAP activity toward Rac (Post et al., 1998; Swanson et al., 1999; Diakonova et al., 2002).
Ratio imaging showed that YFP-Cdc42, -Rac1, and -Rac2 were largely cytosolic, whereas constitutively active forms were primarily localized to intracellular membranes. This indicates that GDIs affect the localization of these proteins. During phagocytosis, there was modest recruitment of YFP-Cdc42, YFP-Rac1, or YFP-Rac2 to the forming phagosomes (Figure 2). The enrichment of GTPases on phagosomes was not large (∼20–40%) relative to the cytosol. This indicates that in cells overexpressing a given GTPase, only a small fraction of the GTPases need to be localized to a given area and that signal amplification occurs downstream of Rac and Cdc42 activation.
Implications of Patterns of G Protein Activation
Imaging the activation patterns of Rho GTPases indicated the organization of their interactions with possible effectors. GTP-Cdc42 preceded actin recruitment and was localized to pseudopod tips, indicating that GTP-Cdc42 regulates actin polymerization during phagocytosis (Figure 4A). The Cdc42 effector WASP stimulates the polymerization of actin when bound to GTP-Cdc42 (Rohatgi et al., 2000) and could regulate actin polymerization in the forming phagosome (Lorenzi et al., 2000).
Rac1 activation followed shortly after Cdc42 activation on the forming phagosome. This early phase of Rac1 activation is consistent with GTP-Rac1 playing a role in actin polymerization. Rac1 activation was not restricted to the tip of the advancing pseudopod, indicating that it has activities in addition to regulating actin polymerization. In vitro data suggest that PAK1 may be a common effector for Cdc42 and Rac1 in the regulation of actin polymerization. PAK1 bound to Rac1 or Cdc42 can phosphorylate LIM kinase, which can phosphorylate cofilin/actin depolymerizing proteins and thereby promote pseudopod extension by inhibiting the depolymerization of actin filaments (Bokoch, 2003). Rac1 also could regulate actin polymerization without the aid of Cdc42, through activation of phosphatidylinositol-4-phosphate 5-kinase (Bishop and Hall, 2000; Botelho et al., 2000).
The correlation of Rac activation with particle deformation indicates that GTP-Rac regulates contractile mechanisms in the phagosome. Rac interactions with PAK1 can activate myosin light chain kinase to regulate myosin activity (Bokoch, 2003). However, there is little information about the molecular mechanisms governing this process. The strong localization of PBD to the actin-poor phagosomal membrane coincided with maximal Rac1 and Rac2 activation (Figures 1C and 4, C and E), indicating that active Rac1 and Rac2 are involved in regulating membrane-associated activities, such as activation of the NADPH oxidase complex (Bokoch and Diebold, 2002). If our speculation is correct that the transient activation of Rac1 and Rac2 coincides with the generation of reactive oxygen species, then it also would seem that those species are generated before full closure of the phagosome, when they may be released from the cell.
A model for how patterns of Cdc42, Rac1, and Rac2 activation correlate with the morphological rearrangements of phagocytosis consists of three phases (Figure 6C). In the first phase, receptor ligation increases Cdc42 activation, actin polymerization and PI3K activity. In the second phase, pseudopodia extend, with GTP-Cdc42 directing actin polymerization at the leading tip of the pseudopod, and with Rac1 inducing local contractile activities, facilitating pseudopod extension and possibly also insertion of membrane. In the third phase, Cdc42 is deactivated and Rac1 and Rac2 are activated transiently, directing contractile activities and NADPH oxidase functions. Because PI3K products are present on the phagosome throughout the process, they likely provide targets for 3′ PI-binding proteins to recruit signaling and effector molecules to the region of phagocytosis. Thus, the activation and deactivation patterns of Cdc42, Rac1, and Rac2 provide an essential framework for understanding the regulation of molecular activities during phagocytosis.
Acknowledgments
This work was supported by National Institute of Health grant AI35950 to J.A.S. A.D.H. was supported by National Institute of Health Cellular Biotechnology Training Program. We thank G. Bokoch, K. Hahn, and T. Meyer for DNAs.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E03-11-0847. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E03-11-0847.
Online version of this article contains supporting material. Online version is available at www.molbiolcell.org.
References
- Araki, N., Hatae, T., Furukawa, A., and Swanson, J.A. (2003). Phosphoinositide-3-kinase-independent contractile activities associated with Fcγ-receptor-mediated phagocytosis and macropinocytosis in macrophages. J. Cell Sci. 116, 247-257. [DOI] [PubMed] [Google Scholar]
- Bishop, A.L., and Hall, A. (2000). Rho GTPases and their effector proteins. Biochem. J. 348, 241-255. [PMC free article] [PubMed] [Google Scholar]
- Bokoch, G.M. (2003). Biology of the p21-activated kinases. Annu. Rev. Biochem. 72, 743-781. [DOI] [PubMed] [Google Scholar]
- Bokoch, G.M., and Diebold, B.A. (2002). Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood 100, 2692-2696. [DOI] [PubMed] [Google Scholar]
- Botelho, R.J., Teruel, M., Dierckman, R., Anderson, R., Wells, A., York, J.D., Meyer,T., and Grinstein, S. (2000). Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J. Cell Biol. 151, 1353-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron, E., and Hall, A. (1998). Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science 282, 1717-1721. [DOI] [PubMed] [Google Scholar]
- Castellano, F., Chavrier, P., and Caron, E. (2001). Actin dynamics during phagocytosis. Semin. Immunol. 13, 347-355. [DOI] [PubMed] [Google Scholar]
- Cox, D., Chang, P., Zhang, Q., Reddy, P.G., Bokoch, G.M., and Greenberg, S. (1997). Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J. Exp. Med. 186, 1487-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, D., and Greenberg, S. (2001). Phagocytic signaling strategies: Fc(gamma) receptor-mediated phagocytosis as a model system. Semin. Immunol. 13, 339-345. [DOI] [PubMed] [Google Scholar]
- Dharmawardhane, S., Brownson, D., Lennartz, M., and Bokoch, G.M. (1999). Localization of p21-activated kinase 1 (PAK1) to pseudopodia, membrane ruffles, and phagocytic cups in activated human neutrophils. J. Leukoc. Biol. 66, 521-527. [DOI] [PubMed] [Google Scholar]
- Dharmawardhane, S., Schurmann, A., Sells, M.A., Chernoff, J., Schmid, S.L., and Bokoch, G.M. (2000). Regulation of macropinocytosis by p21-activated kinase-1. Mol. Biol. Cell 11, 3341-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diakonova, M., Bokoch, G., and Swanson, J.A. (2002). Dynamics of cytoskeletal proteins during Fcγ receptor-mediated phagocytosis in macrophages. Mol. Biol. Cell 13, 402-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold, B.A., and Bokoch, G.M. (2001). Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nat. Immunol. 2, 211-215. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and Hall, A. (2002). Rho GTPases in cell biology. Nature 420, 629-635. [DOI] [PubMed] [Google Scholar]
- Gardiner, E.M., Pestonjamasp, K.N., Bohl, B.P., Chamberlain, C., Hahn, K.M., and Bokoch, G.M. (2002). Spatial and temporal analysis of Rac activation during live neutrophil chemotaxis. Curr. Biol. 12, 2029-2034. [DOI] [PubMed] [Google Scholar]
- Greenberg, S. (1999). Modular components of phagocytosis. J. Leukoc. Biol. 66, 712-717. [DOI] [PubMed] [Google Scholar]
- Griesbeck, O., Baird, G.S., Campbell, R.E., Zacharias, D.A., and Tsien, R.Y. (2001). Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 276, 29188-29194. [DOI] [PubMed] [Google Scholar]
- Gu, Y., et al. (2003). Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science 302, 445-449. [DOI] [PubMed] [Google Scholar]
- Henry, R.M., Hoppe, A.D., Joshi, N., and Swanson, J.A. (2004). The uniformity of phagosome maturation in macrophages. J. Cell Biol. 164, 185-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe, A., Christensen, K., and Swanson, J.A. (2002). Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 83, 3652-3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joneson, T., McDonough, M., Bar-Sagi, D., and Van Aelst, L. (1996). RAC regulation of actin polymerization and proliferation by a pathway distinct from Jun kinase. Science 274, 1374-1376. [DOI] [PubMed] [Google Scholar]
- Kraynov, V.S., Chamberlain, C., Bokoch, G.M., Schwartz, M.A., Slabaugh, S., and Hahn, K.M. (2000). Localized Rac activation dynamics visualized in living cells. Science 290, 333-337. [DOI] [PubMed] [Google Scholar]
- Lorenzi, R., Brickell, P.M., Katz, D.R., Kinnon, C., and Thrasher, A.J. (2000). Wiskott-Aldrich syndrome protein is necessary for efficient IgG-mediated phagocytosis. Blood 95, 2943-2946. [PubMed] [Google Scholar]
- Marshall, J.G., Booth, J.W., Stambolic, V., Mak, T., Balla, T., Schreiber, A.D., Meyer, T., and Grinstein, S. (2001). Restricted accumulation of phosphatidylinositol 3-kinase products in a plasmalemmal subdomain during Fc gamma receptor-mediated phagocytosis. J. Cell Biol. 153, 1369-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massol, P., Montcourrier, P., Guillemot, J.C., and Chavrier, P. (1998). Fc receptor-mediated phagocytosis requires CDC42 and Rac1. EMBO J. 17, 6219-6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson, D., Silletti, J., Murphy, G., D'Eustachio, P., Rush, M., and Philips, M.R. (2001). Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 152, 111-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, J.T., and Swanson, J.A. (2002). Calcium spikes in activated macrophages during Fcγ receptor-mediated phagocytosis. J. Leukoc. Biol. 72, 677-684. [PubMed] [Google Scholar]
- Patel, J.C., Hall, A., and Caron, E. (2002). Vav regulates activation of Rac but not Cdc42 during FcγR-mediated phagocytosis. Mol. Biol. Cell 13, 1215-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post, P.L., Bokoch, G.M., and Mooseker, M.S. (1998). Human myosin-IXb is a mechanochemically active motor and a GAP for rho. J. Cell Sci. 111, 941-950. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J. (2001). Rho family proteins: coordinating cell responses. Trends Cell Biol. 11, 471-477. [DOI] [PubMed] [Google Scholar]
- Rohatgi, R., Ho, H.Y., and Kirschner, M.W. (2000). Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4, 5-bisphosphate. J. Cell Biol. 150, 1299-1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self, A.J., and Hall, A. (1995a). Measurement of intrinsic nucleotide exchange and GTP hydrolysis rates. Methods Enzymol. 256, 67-76. [DOI] [PubMed] [Google Scholar]
- Self, A.J., and Hall, A. (1995b). Purification of recombinant Rho/Rac/G25K from Escherichia coli. Methods Enzymol. 256, 3-10. [DOI] [PubMed] [Google Scholar]
- Srinivasan, S., Wang, F., Glavas, S., Ott, A., Hofmann, F., Aktories, K., Kalman, D., and Bourne, H.R. (2003). Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J. Cell Biol. 160, 375-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson, J.A. (2002). Ratiometric fluorescence microscopy. Method Microbiol. 31, 1-18. [Google Scholar]
- Swanson, J.A., Johnson, M.T., Beningo, K., Post, P., Mooseker, M., and Araki, N. (1999). A contractile activity that closes phagosomes in macrophages. J. Cell Sci. 112, 307-316. [DOI] [PubMed] [Google Scholar]
- Takai, Y., Sasaki, T., and Matozaki, T. (2001). Small GTP-binding proteins. Physiol. Rev. 81, 153-208. [DOI] [PubMed] [Google Scholar]
- Thompson, G., Chalk, P.A., and Lowe, P.N. (1997). Interaction of PAK with Rac: determination of a minimum binding domain on PAK. Biochem. Soc. Trans. 25, 509S. [DOI] [PubMed] [Google Scholar]
- Thompson, G., Owen, D., Chalk, P.A., and Lowe, P.N. (1998). Delineation of the Cdc42/Rac-binding domain of p21-activated kinase. Biochemistry 37, 7885-7891. [DOI] [PubMed] [Google Scholar]
- Zacharias, D.A., Violin, J.D., Newton, A.C., and Tsien, R.Y. (2002). Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296, 913-916. [DOI] [PubMed] [Google Scholar]