

Abstract
Despite improved methods of cancer detection and disease management over the last few decades, cancer remains a major public health problem in many societies. Conventional therapies, such as chemotherapy, radiation, and surgery, are not usually sufficient to prevent disease recurrence. Therefore, efforts have been focused on developing novel therapies to manage metastatic disease and to prolong disease-free and overall survival, by modulating the immune system to alleviate immunosuppression, and to enhance antitumor immunity. This review discusses protumor mechanisms in patients that circumvent host immunosurveillance, and addresses current immunotherapy modalities designed to target these mechanisms. Given the complexity of cancer immunosuppressive mechanisms, we propose that identification of novel disease biomarkers will drive the development of more targeted immunotherapy. Finally, administration of different classes of immunotherapy in combination regimens, will be the ultimate route to impact low survival rates in advanced cancer patients.
Keywords: cancer, immunotherapy, immunosurveillance, immunosuppression, dendritic cells, T-cells
Video abstract
Introduction
Cancer is a global health problem that affects all socioeconomic groups and people of all ages. Each year, there are approximately 7.6 million cancer deaths worldwide.1,2 The precise causes of cancer have not been defined. It is known that gene mutations induced by host internal or by environmental factors can lead to the growth of cancer cells. Additionally, cancer cells have unique biological properties, including replicative immortality, metastatic capability, and escaping immunosurveillance.3–5 Investigators have also proposed the idea of cancer stem cells as one of the possible causes of cancer.6–9
Many early cancers do not exhibit defined symptoms, and are only detected in the later stages of disease. By this time, there is usually significant spread to other organs and to lymph nodes. If detected early, conventional therapies, such as chemotherapy, radiation, and surgery are generally successful, resulting in excellent 5-year survival rates. However, cancers such as digestive, ovarian, and pancreatic cancer, which lack easily recognizable symptoms for early stage disease and have inefficient screening procedures, are usually discovered at the stage of advanced metastatic disease. At this time, conventional therapy is not sufficiently effective for management, and typically there is a poor response to therapy, a high rate of disease recurrence, and a poor 5-year survival rate.10 To advance the care of cancer patients, over the past few decades, efforts have focused on developing successful immunotherapeutic agents.
Immunotherapeutic agents used to treat cancer began as early as 1891, when streptococcal organisms were injected into a patient with inoperable cancer, resulting in reduced size of the tumor.11–14 Today, much more is known about immune escape mechanisms in cancer, and immunotherapy is more targeted and increasingly more successful. Strategies for cancer immunotherapy vary with the affected organ, and include the use of antitumor cytokine administration, monoclonal antibodies to inhibit tumor-cell growth by inducing apoptosis or other mechanisms, targeting of tumor-associated antigens, blocking of immune-checkpoint molecules, dendritic cell (DC) vaccines to boost antitumor immunity, and adoptive transfer of genetically engineered T-cells.
Recently, the era of modern immunotherapy has been marked by significant triumphs, including breakthroughs with the antigen-presenting cell vaccine sipuleucel-T for castration-resistant prostate cancer,15 antibody-blocking checkpoint inhibitor cytotoxic T lymphocyte-associated antigen (CTLA)-4 (Yervoy® [ipilimumab]) for metastatic melanoma,16,17 and genetically engineered T-cells for lymphomas and leukemias. These immune approaches significantly enhance the management of cancer therapy, resulting in improved survival, and pave the way for expanded use of these or similar immunotherapy regimens for other cancers.
This review provides an up-to-date account of immunosurveillance in cancer, promising immune-based clinical trials against various cancers, US Food and Drug Administration (FDA)-approved immunotherapies, and future directions that may facilitate the development of novel immunotherapeutic agents, which we hope will be sufficiently cost-effective and available to all patients who require these treatments.
Cancer immunosurveillance
One in four deaths in the US is due to cancer. It is projected that 1,665,540 new cancer cases of cancer and 585,720 cancer deaths will occur in 2014.10 Even so, there has been a decline in cancer deaths over the last two decades, and we anticipate that as scientists unravel more clues to the causes of cancer, combined with the development of novel immunotherapy for management of advanced and recurrent disease, there will be further improvement in survival rates.
The precise causes of cancer are unknown. In 1909, Paul Ehrlich predicted that the immune system repressed the growth of carcinomas that would otherwise occur at high frequency.4,18 This initiated the debate in the field of cancer immunosurveillance. With further understanding in tumor immunology, transplantation, and immunogenetics, a broader concept of “cancer immunoediting” emerged. Cancer immunoediting encompasses a complex network of immunosuppressive factors within the host, tumor-evasion mechanisms, and host-elicited antitumor mechanisms.
Cancer immunoediting consists of three dynamic phases. Briefly, normal cells are transformed by oncogenic stimuli, express distinct tumor specific molecules, and generate proinflammatory danger signals, which initiate phase I: the elimination or immunosurveillance phase.3,4 According to Burnet, the thymus-dependent cells of the body constantly survey host tissues for nascently transformed cells.19 Components of the innate and adaptive immune system detect and eradicate transformed cells before they are clinically apparent. In healthy individuals, the immunosurveillance network consists of immune parameters, such as CD4+, CD8+, and γδ T-cells, natural killer (NK) cells, macrophages, IFNγ, perforin, and TRAIL (TNF-related apoptosis inducing ligand), which function effectively and prevent the growth of tumors, and hence in theory they should never become clinically apparent.4,20–23
However, if these transformed cells are not completely eliminated by a combination of existing immunity, they enter phase 2 – the equilibrium phase – in which the immune system controls net tumor-cell outgrowth, with immune responses primarily mediated by CD8+ T-cells and IL-12p70 produced by DCs. If at this stage there is lack of sufficiently strong host antitumor immunity, or if tumor cells become less immunogenic via tumor-antigen loss, or there is major histocompatibility complex (MHC)-molecule downregulation,1,24 these cells can avoid T-cell attack and enter phase 3, the escape phase. In the escape phase, tumors begin to grow progressively in an unrestrained manner, ie, become malignant, establish an immunosuppressive tumor microenvironment, and eventually become clinically apparent tumors.4,21,22 Understanding immunosuppressive networks in the tumor will guide the development of novel and effective cancer immunotherapy.
Immune components of the cancer microenvironment
Immunosuppressive events occurring in the tumor microenvironment are critical to the clinical onset of cancer and to survival. Firstly, as shown by recent investigations, not all cellular events in the tumor are detrimental to tumor outcome. In this respect, the cell type, abundance, and location of lymphocytes in tumor beds have been identified as useful factors in predicting disease outcome.25–28 In colorectal cancer, for example, a diagnosis of stage I cancer with patients having few tumor-infiltrating lymphocytes (TILs) had a similar outcome to patients with stage IV and metastatic disease.26 Therefore, a high frequency of CD3+ or of CD8+ T-cells at the tumor site is beneficial to outcome. Similar findings were reported in ovarian and other cancers, where the T-cell infiltration in tumor beds is favorable to disease outcome.29,30
On the contrary, elevated numbers of CD4+ T-regulatory cells (Tregs) expressing the transcription molecule FoxP3 in cancer immune infiltrates and in peripheral blood generally have detrimental consequences on cancer outcome.31–36 It should be noted, however, that there are exceptions to this protumor effect of Tregs, as in the case of colon cancer, in which they may have a positive correlation with survival.37 Cytokines and chemokines produced by cancer cells and by infiltrating immune cells may confer a protumor cytokine polarization in the tumor beds (Figure 1).
Figure 1.

Immunosuppressive components of the tumor microenvironment.
Notes: At tumor sites, immune cells express soluble molecules that cause the recruitment or differentiation of Tregs, M2 macrophages, and immature DCs to the tumor microenvironment, conferring an immunosuppressive polarization. Administration of immunotherapy to patients reprograms the tumor promoting environment to a Th1 antitumor polarization, favoring tumor regression.
Abbreviations: CCL, chemokine C–C motif ligand; CXCL, chemokine C–X–C motif ligand; IDO, indoleamine 2,3-dioxygenase; Th, T helper; Tregs, T-regulatory cells; MDSC, myeloid-derived suppressor cell; M1 macrophages, antitumor macrophages; M2 macrophages, protumor macrophages; DC, dendritic cell; pDC, plasmacytoid DC; CTL, cytolytic T-cell.
Characteristically, the tumor microenvironment produces high levels of cytokines IL-10 and TGFβ, which are associated with worsening of cancer.38–40 Many cell types also contribute to this immunosuppression. For example Tregs are attracted into the tumor beds by the chemokine CCL22.41–43 Additionally, chemokine receptor CCR4 is highly expressed on some Tregs. Tumors may also promote the release of CCL17 and CCL21, which also causes these Tregs to be recruited to tumor sites. Similarly, Tregs expressing VEGF-A (CXCR4) are recruited into tumors expressing the cognate ligand CXCL12.44,45 In diseases where Tregs are detrimental to tumor outcome, these cells may be enriched by the conversion of immunocompetent T-cells into Tregs, or by preferential expansion of resident Tregs over other cell types, as a consequence of the existing protumor cytokine environment. Other immunosuppressive contributing cells in tumors include plasmacytoid DCs, suppressive macrophages, immature myeloid-derived DCs, and myeloid-derived suppressor cells (MDSCs).46–52
The maturation, function, and ability of DCs to migrate may be suppressed by MDSCs, M2 (alternatively activated, protumor) macrophages, Tregs, and other factors in the tumor microenvironment.53,54 Therefore, agents that can reprogram M2 macrophages to M1 (classically activated, antitumor) macrophages may be useful in alleviating immunosuppression in the tumor.55 M1 macrophages produce antitumor cytokines and are associated with prolonged survival, whereas M2 macrophages secrete protumor IL-10 and TGFβ, and are associated with poor outcome in cancer. The chemotherapeutic agent gemcitabine was shown to selectively reduce myeloid-suppressor cells (Gr1+CD11b+) and enhance antitumor activity of CD8+ and NK cells.56
Immature myeloid-derived DCs, which express low levels of costimulatory molecules (CD40, CD80 [B7-1] and CD86 [B7-2]), and high levels of B7-H1 inhibit T-cell proliferation and induce FoxP3 Tregs.57 Also, other investigations showed that patients with a low expression of B7-H4 on tumor-associated macrophages had a higher 5-year survival than those with high expression of B7-H4 on these cells.58,59 Furthermore, myeloid DCs and macrophages can induce Th17 cells, which secrete IL-17, a cytokine associated with protumor or antitumor immune responses depending on the nature of the cancer.60–62 Plasmacytoid DCs express the enzyme indoleamine 2,3-dioxygenase, which is associated with poor outcomes in cancer.63–67
In addition to immunosuppression by host immune mechanisms, there are direct immunosuppressive elements imposed by tumor cells in an effort to induce immune evasion. Therefore, tumor cells are capable of downregulating MHC class I, thereby making cross-presentation of tumor-derived antigens (eg, from dead tumor cells) less efficient.24,68,69
Another immunosuppressive mechanism in the tumor and in the periphery is imposed at immune-checkpoint junctions. Immune checkpoints are necessary to prevent the overstimulation of the immune system; however, coinhibitory molecules that mediate this function can have detrimental consequences in the cancer immunosuppressive environment, inducing protumor immune responses.70 There are several coinhibitory molecules at immune checkpoints in humans, but notable examples that have recently come full circle as cancer immunotherapy targets are two members of the immunoglobulin gene superfamily, CTLA-4 and programmed death (PD)-1 (CD279) (Figure 2).
Figure 2.
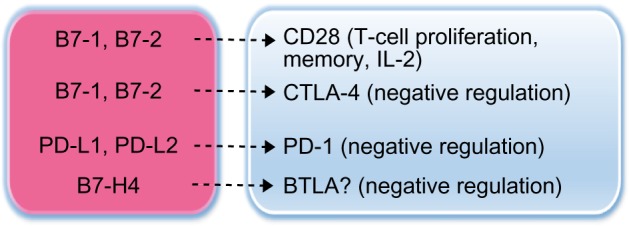
Immunosuppression by B7-family molecules in cancer.
Notes: Tumors may escape from immune surveillance by ligation of immune-checkpoint inhibitory molecules. Binding of B7-1/B7-2 to CD28 results in T-cell proliferation, IL-2 secretion, and T-memory-cell development. Conversely, binding of B7-1/B7-2 to CTLA-4 in cancer results in immune coinhibition and immunosuppression. Binding of PD-1 to PD-L1/PD-L2 results in immunosuppression in peripheral tissues. Ligation of B7-H4 on macrophages to its cognate receptor, possibly BTLA (not fully characterized) on T-cells, inhibits T-cell proliferation and IL-2 secretion. Agents targeting these immune-checkpoints in cancer are currently being optimized for maximum therapeutic benefit.
Abbreviations: CTLA, cytotoxic T-lymphocyte-associated antigen; PD, programmed death; BTLA, B- and T-lymphocyte attenuator.
Antigen-specific T-cell activation requires at least two signals. Firstly, an antigen is presented to a T-cell receptor (TCR) by an MHC molecule on antigen-presenting cells (APCs). Secondly, B7-1 and B7-2 on APCs bind to CD28 on T-cells and enhance T-cell stimulation. Coinhibitory molecules, such as CTLA-4, on T-cells are useful to prevent inappropriate stimulation of T-cells and possibly autoimmune disease; however, upregulation of CTLA-4 can promote the preferential binding of this molecule (CTLA-4) to B7-1 or B7-2, inducing suppression of T-cell activity. Similarly, binding of PD-1, primarily expressed on tumor-infiltrating T-cells, to PD ligand (PD-L)-1, (B7-H1, CD274) or to PD-L2, represents another coinhibitory and immunosuppressive T-cell mechanism in cancer.70–72
In the clinic, a recent strategy in use to overcome immunosuppression is by antibody blockade of immune-checkpoint inhibitory molecules, such as CTLA-4 and PD-1,16,17,73,74 and several companies are currently designing molecules to optimize the recently FDA-approved targeted-therapy mechanism of immune-checkpoint antibody blocking.
Scope of cancer immunotherapy
Immunotherapy aims to diminish existing immunosuppressive mechanisms in cancer by blocking these mechanisms and/or to potentiate specific memory T-cell or antibody responses by vaccine administration or cellular immunotherapy, with the ultimate goal of disease management or cure, and improved survival. Unfortunately, there are few identified biomarkers in cancer that can be universally targeted in the treatment of a specific disease. Therefore, current immunotherapy approaches primarily target the immunosuppressive mechanisms discussed in the foregoing section. Immunotherapeutic strategies range from the use of tumor cell-based vaccines, virus-based immunotherapy, antibody immunotherapy, and cellular immunotherapy. We now cover these areas and discuss related preclinical studies and clinical trials.
Tumor cell-based vaccines
Whole tumor cells have been employed as a useful tool against various forms of cancer. For example, the most clinically advanced trial for pancreatic cancer is the use of algenpantucel-L immunotherapy. This is an irradiated live combination of two human allogeneic pancreatic cancer cell lines modified to express the murine enzyme α-1,3 galactosyltransferase, which is needed for the synthesis of α-galactosyl epitopes on surface proteins and glycolipids of such cell lines.75–77 The mouse gene causes the cells to be recognized as foreign to patients’ immune system, and thus the immune system attacks these cancer cells and destroys many of them. This is classified as HyperAcute™ immunotherapy (designed by NewLink Genetics, Ames, IA, USA), and similar products are currently being developed with specificity for other types of cancer (non-small-cell lung cancer [NSCLC], advanced melanoma, metastatic castrate-resistant prostate cancer, and renal cancer). HyperAcute immunotherapies do not require tissue from individual patients, but use intact whole cells rather than cell fragments.
For pancreatic cancer, Phase II clinical trials with this technology were very encouraging, where data on 69 patients showed that overall survival at 3 years was 39%. This prompted two Phase III trials for pancreatic cancer using algenpantucel-L immunotherapy (HyperAcute pancreas). IMPRESS (IMmunotherapy for Pancreatic REsectable cancer Survival Study) involves up to 722 patients with surgically resected pancreatic cancer (ClinicalTrials.gov identifier: NCT01072981). PILLAR (Pancreatic Immunotherapy with algenpantucel-L for Locally Advanced non-Resectable; given with or without chemotherapy and radiation) (ClinicalTrials.gov identifier: NCT01836432), is currently enrolling patients with locally advanced pancreatic cancer. Therapeutic advances in pancreatic cancer are desperately needed, as with existing therapy this disease has a 5-year survival rate of below 10%.
Another tumor-cell therapy regimen consisting of autologous tumor cells conjugated to dinitrophenyl, called M-Vax, was used for the treatment of malignant melanoma (ClinicalTrials.gov identifier: NCT00257465).
OncoVAX, a tumor-cell product, is in Phase III clinical trials for colon cancer patients. The OncoVAX regimen consists of intradermally injecting two doses of autologous irradiated (200,000 rads) tumor cells, mixed with fresh-frozen mycobacteria of the Tice strain of bacillus Calmette–Guérin, followed by two injections of irradiated tumor cells alone.78 Bacillus Calmette–Guérin is a live but weakly pathogenic bacterium, and induces a strong immune response, and hence it is incorporated in the first two vaccines to boost the immune response.
BiovaxID (ClinicalTrials.gov identifier: NCT00091676) is in the regulatory approval process in Canada and Europe for late-stage indolent follicular lymphoma.
Other tumor-cell products that may be used as immunotherapeutic agents are the heat-shock protein (HSP) family molecules. HSPs act as molecular chaperones. They can be induced or released during cellular stress and necrosis. They bind potential antigens on cell death, and deliver them to APCs through several mechanisms.79 Tumor cells can secrete HSP70-containing exosomes, which recruit MDSCs, thereby contributing to immunosuppression. On the contrary, HSP70 peptide complexes are also secreted from necrotic tumor cells and can trigger anticancer CTL after entering APCs and cross-presenting to CD4 T-cells in afferent lymph nodes, leading to inhibition of tumor growth. HSPs can also bind tumor-associated antigens and deliver them to APCs through MHC I and MHC II molecules, inducing the activation of antitumor CD4+ and CD8+ T-cells. The antitumor effects of HSP have been investigated in several preclinical studies, and due to the positive results it has advanced to clinical trials.79–81 To mention a few examples, clinical trials with HSP96 are currently in progress for the immunotherapy of diseases, such as glioblastoma (ClinicalTrials.gov identifiers: NCT01814813 and NCT02122822).
Virus-based immunotherapy
Some virus infections can lead to cancer, but on the contrary, virus technology is often of significant benefit in cancer immunotherapy. Engineering of viruses to express novel cancer-specific proteins and other molecules is currently being employed in several clinical trials. ProstVac-VF, a prostate-specific antigen (PSA)-targeted therapeutic vaccine, is a combination of recombinant vaccinia and fowlpox virus vaccine that delivers PSA and three costimulatory signals – B7-1, ICAM-1, and LFA-1 – known as Tricom. This strategy is designed to enhance antigen uptake by DCs and subsequent antigen presentation to T-cells. A Phase I clinical trial using ipilimumab and PSA-Tricom showed clinical benefit and development of specific immune responses in patients.82,83 A detailed summary of the scope of viral vector immunotherapy strategies is outlined elsewhere.84
Oncolytic viruses infect and kill cancer cells and associated endothelial cells, preferentially to normal cells.85–89 The death of cancer cells occurs by immunogenic apoptosis, autophagic cell death, necrosis, and pyroptosis, and cell proteins are processed and presented by DCs stimulating antitumor and antiviral immune responses. Current technology allows the manipulation of the viral genome to improve safety, and the insertion of transgenes to augment antitumor activity.85,88 Ongoing clinical trials with modified adenovirus, herpesvirus, reovirus, measles, and other viruses will provide critical information regarding the safety and efficacy of oncolytic virus immunotherapy. In general, toxicity with virus clinical trials has been minor.
Vaccines against pathogens that cause cancer
Prophylactic vaccination therapy is effective against some viruses that cause cancer. In 1981, the FDA approved the hepatitis B virus vaccine, now given to infants. This vaccine reduces hepatitis B virus infections and incidences of hepatocellular carcinoma.90
Human papillomavirus (HPV) causes almost all cases of cervical cancer, as well as some anal, vulval, vaginal, penile, and oropharyngeal cancers.91,92 Courses of the FDA-approved quadrivalent vaccine Gardasil (Merck Pharmaceuticals, 2006) against HPV types 6, 11, 16, and 18, and the bivalent vaccine Cervarix (GlaxoSmithKline, 2009) against HPV types 6 and 11 are efficient in preventing cervical cancer and genital warts in individuals at risk. Gardasil is approved in many countries for the prevention of cervical cancer. In these vaccines, immune memory is primarily mediated by B cells.91–93 The shortcoming with these current prophylactic HPV vaccines, however, is that they are not effective in individuals who already have HPV infections. Therefore, vaccines such as VGX-3100 are being tested in women with cervical dysplasia. This vaccine induces antibodies to HPV serotypes, and specific CD8+ T-cells with granzyme B, perforin, and cytolytic potential.94,95
Cytokine immunotherapy
The use of cytokine treatment to repolarize the immune system was one of the early forms of immunotherapy. IFNα was one of the first cytokines approved for use in leukemia96,97 and melanoma98,99 patients. IL-2, FDA-approved in 1998, is effective in metastatic melanoma and in renal cell carcinoma. IL-2 in combination with granulocyte macrophage colony-stimulating factor (GM-CSF) or with chemotherapy may have added benefits.5,100,101 Cytokine GM-CSF stimulates the differentiation of stem cells to granulocytes and monocytes, and this treatment has been used in combination with primed lymphocytes in hematopoietic diseases, such as acute myeloid leukemia.102 One of the drawbacks in cytokine immunotherapy is toxic side effects. For example, TNFα used in the therapy of melanoma patients may induce a septic shock-like condition if not administered at the right doses.103 Recent preclinical studies showed that cytokine IL-12 therapy combined with blocking anti-CTLA-4 therapy causes a decrease in FoxP3+ Tregs and an increase in effector CD4+ T-cells.104
Antibody immunotherapy to reduce immunosuppression
There are several mechanisms whereby antibody immunotherapy can be used to overcome immunosuppression in cancer or to potentiate antitumor immunity. Some examples follow.
Anticytokine antibodies
IL-6 promotes early colitis-associated cancer, and thus anti-IL-6 antibody immunotherapy may be useful in the treatment of such diseases as colorectal cancer.105
Depleting CD47 on tumor cells
CD47 on tumor cells binds to its receptor – signal regulatory protein-α – on phagocytic cells, and prevents phagocytic cells from ingesting tumor cells. This process is mediated by the selective expression of calreticulin on tumor cells. Antibody blocking of CD47 in mouse models of different cancers results in dramatic improvements or failure to induce disease.106–109 Engulfment of tumor cells by macrophages after anti-CD47 antibody blockade results in improved antigen-specific CD8+ T-effector cell immunity.110
Abrogation of Tregs
Daclizumab, an anti-CD25 antibody was effective in inducing a prolonged decrease in Tregs in breast cancer patients.111,112 Denileukin diftitox, an IL-2 diphtheria-toxin fusion protein, was used to treat cutaneous T-cell lymphoma and melanoma113 and for clinical trials in several other cancers, including one with ovarian cancer by the authors (ClinicalTrials.gov identifier: NCT00703105). Cyclophosphamide is frequently used in clinical trials as a mechanism of targeting Tregs.114–118
Enhancement of immune responses
CD40 expressed in high levels on the surface of mature DCs binds to CD40 ligand (CD40L) on T-cells, and increases costimulation and effective T-cell immune responses. Ligation of CD40 to CD40L mediates IL-12 production in DCs. Anti-CD40 monoclonal antibodies and IL-2 or IL-15, an NK- and T-cell activator, was effective in the treatment of a murine model of colon cancer.119,120 CD137 is expressed on activated T-cells, DCs, and NK cells. It increases T-cell proliferation and IL-2 secretion, and these properties make it a good candidate for antitumor immunotherapy. Clinical trials with anti-CD137 (BMS-663513) have been conducted in melanoma, ovarian cancer, and NSCLC.121
Immune-checkpoint antibody-blocking immunotherapy
CTLA-4 and PD-1 prevent immune mediated damage to normal tissues in healthy individuals. In cancer, interaction of these molecules with their ligands B7-1 and B7-2 (for CTLA-4) and PD-L1 or PD-L2 (for PD-1) have been identified as significant immunosuppressive pathways.71,122–125 A blocking anti-CTLA-4 antibody, ipilimumab, is FDA-approved (2011) for treating metastatic malignant melanoma.16,17,72 Ipilimumab is a monoclonal antibody designed to block CTLA-4, thereby preventing the development of tolerance, and augmenting immune responses. Clinical trials are ongoing with this treatment for prostate cancer, NSCLC, renal cancer, and pancreatic cancer, as well as hematologic malignancies.73,126–129 In preclinical studies of a murine tumor model, CTLA-4 blockade in synergy with a GM-CSF expressing tumor-derived vaccine enhanced T-cell activation and memory and elicited antitumor T-cell responses in the early stages of tumor growth.130
Phase I, II, or III clinical trials are in progress with ipilimumab, or with another anti-CTLA-4 antibody blocker, tremelimumab, in combination with cellular vaccines, chemotherapy, radiation, cytokine treatment, or adoptive T-cell-transfer therapy, and are reviewed in Gelao et al.73 Both agents recognize CTLA-4 and prevent ligation of CTLA-4 with B7 molecules. Even though these treatments show significant promise, ipilimumab therapy is not without side effects, many of which are gastrointestinal.72,131,132
In September 2014, Keytruda (pembrolizumab), to be used for the treatment of unresectable or metastatic melanoma, was the first anti-PD-1 receptor therapy to be FDA-approved. An anti-PD-1 monoclonal antibody, nivolumab, has been investigated in Phase I and II trials in 296 patients with pretreated NSCLC, prostate cancer, renal cell carcinoma, colorectal cancer, and melanoma.133,134 Like anti-CTLA-4 antibody-blocking therapy, anti-PD-1 and anti-PD-L1 antibodies are used in single or combination therapy, resulting in some encouraging findings, but with fewer adverse gastrointestinal, skin, liver, and endocrine system side effects than ipilimumab. Combination therapy with these checkpoint inhibitors and other therapy shows more promise than either therapy alone, without additional side effects.73,74,130,133,134
Glioblastoma is a deadly cancer in which patients are expected to survive 1 year after diagnosis.135 This disease is characterized by a highly suppressive Treg environment.136,137 In a recent mouse-model study of this disease, IL-12 fusion protein (IL-12 Fc) was delivered directly into cerebral tumors via osmotic minipumps over a 28-day period.104 This was combined with systemic administration of anti-CTLA-4 antibody treatment. This combined treatment led to full remission in most mice and greatest improved histological response, unlike the outcome for either monotherapy. Additionally, IL-12 Fc treatment led to a dramatic increase in CD4 T-cells, whereas both treatments resulted in increases in CD8 T-cells. Fluorescence-activated cell-sorting analysis showed that between days 21 and 35 after injection, there was an increase in total CD4+ T-cells, and a strong increase in the frequency of these cells that produced IFNγ, but a profound reduction in FoxP3+ cells. There was a long-lasting improved effect even after 91 days of therapy. Interestingly, IL-12 Fc increased the expression of perforin in CD8+ T-cells and in NK cells, but not in CD4+ T-cells, indicating that CTLs are primarily responsible for tumor control.104 Other studies indicate that selective opsonization of Tregs with anti-CTLA-4 antibodies can induce potent antibody-dependent cell-mediated cytotoxicity within the tumor site.138
Cellular immunotherapy in cancer
Since the FDA approval of the DC immunotherapy sipuleucel-T for castration-resistant prostate cancer, investigators have been prompted to initiate clinical trials and preclinical studies with novel ex vivo manipulations of DCs, and to determine appropriate combination therapies that would enhance and optimize DC-vaccine regimens. Additionally, the field of T-cell immunotherapy for cancer has been rapidly expanding.139 Hematopoietic stem cell transplant (HSCT) from a healthy donor is a potent form of cellular immunotherapy to treat hematologic malignancies, and is sometimes the last option in children who have relapsed malignancy.140 In some trials, TILs are isolated and used for adoptive cell transfer.141 Recent advances in the field of T-cell immunotherapy have been marked by genetic manipulations of infused T-cells, either by TCR or chimeric antigen receptor (CAR) gene transfer.142 The following sections will discuss advances in DC-based and T-cell immunotherapy.
Dendritic cell-based vaccines
DCs are the most potent APCs in the immune system. They were first reported in the skin in 1948,143 and later characterized in spleens of mice in 1973.144 DCs capture, process, and present antigens, and induce the generation of specific effector and memory T-cells. DCs express costimulatory molecules that regulate immune activation and anergy, T-cell differentiation, and cytokine polarization. DCs prime both T-cell and humoral immune responses.145–147
DCs recognize tumor-associated antigens, and have been used in several clinical trials over the past two decades exploiting their inherent properties.148–150 The most successful DC immunotherapy to date is the FDA-approved sipuleucel-T (Provenge; Dendreon) for castration-resistant prostate cancer.15 Prostatic acid phosphatase is restricted to prostate tissue, and is thus a specific target in metastatic prostate cancer.151 In this personalized vaccine, sipuleucel-T is prepared with autologous APCs treated with recombinant fusion protein PA2024. PA2024 is a fusion protein consisting of full-length human prostatic acid phosphatase and full-length human GM-CSF.152,153 A complete course of sipuleucel-T therapy, resulting in improved survival in these prostate cancer patients, consists of three doses of the vaccine at about 2-week intervals.15,153
Recently, a novel approach was used to treat malignant glioma in a cohort of 22 patients.154 Mature DC classified as alpha-DC-1 were prepared in cytokines GM-CSF, IL-4, and a potent cytokine-maturation cocktail, consisting of IL-1β, TNFα, IFNγ, IFNα, and poly-I-C, as previously described.155,156 Two hours before harvesting DCs, cells were loaded with Pan-DR epitope peptide (PADRE) optimized for Th-cell response, and with glomerular-associated peptides. Alpha-DC-1 were administered intranodally in the right or left inguinal and axillary lymph nodes for alternate injections at 2-week intervals (one to four vaccines), and polyinosinic–polycytidylic acid stabilized by lysine and carboxymethyl cellulose (poly-ICLC; 20 μg/kg) was given intramuscularly twice weekly to all patients for 8 weeks. Poly-ICLC enhances the efficacy of glioma-associated antigen-targeting vaccinations.157,158 Vaccine regimens were subject to later repeat courses in patients who did not have adverse responses. IL-12p70 secreted by patient DCs correlated positively with time to progression, ie, high DC IL-12 secretion was associated with improved outcome. Nine patients achieved progression-free status lasting at least 12 months, while 58% of patients showed improved Th1 (eg, IFNγ) responses in peripheral blood mononuclear cells to at least one of the vaccination-targeted glioma-associated antigens.154
Survival benefits of DC vaccines have been reported in several clinical trials,149,159 but like most immunotherapy there are still several hurdles to overcome in order to optimize this treatment for cancer and other diseases. Today, most investigators, including the authors who have ongoing ovarian cancer clinical trials with α-DC-1 (ClinicalTrials.gov identifier: NCT00703105), believe that a mature DC vaccine, such as α-DC-1, is superior to an immature DC for immunotherapy, but there are still several unanswered questions concerning methods of DC optimization for improved therapy. These questions relate to the cytokine culture environment of DCs, ex vivo manipulation and selection of antigens for loading, method of delivery of antigens, the route of vaccine administration, the frequency of administration, and dose of cells.160–162 The antigens used for loading DCs varies with the disease. Proteins, deoxyribonucleic acid (DNA), ribonucleic acid, or autologous tumor lysate can be used to load DCs.163–165 Some groups have used tumor-associated antigens or overexpressed tumor antigens, such as the cancer testis antigens NY-ESO-1, MAGE-family antigens, antigens derived from melanocyte-differentiation factors (eg, gp100 or MART), antigens encoded by oncogenes, MUC-1, HER-2/neu, and other antigens.1,165–170 DC incubation with the antigens or delivery of tumor-associated antigens with bacterial or viral vectors are strategies for loading DCs.171–173 As expected, a consideration of all of these parameters determines whether CD4+ and/or CD8+ T-cells are activated, and consequently the benefit of DC therapy to patients.
Another class of cancer antigens currently under investigation for immune targeting is neoantigens. These antigens are derived from mutated proteins present in tumors, but not found in normal individuals.174,175 Neoantigens may prove in future trials to be one of the most successful antigens for delivery to DCs for use in patient vaccines, since patients can mount a sustained antitumor immune response against these unique tumor antigens.
Hematopoietic stem cell transplants
Bone marrow transplants, peripheral blood stem cell transplants, or cord-blood transplants are grouped under the term HSCTs. Allogeneic HSCTs are the most widely used form of adoptive T-cell immunotherapy. This method was developed over 50 years ago to treat patients with anemias and immune deficiencies,140 and is now frequently used to treat hematologic malignancies and some solid cancers. HSCT has become standard immunotherapy for leukemia and lymphoma at several large centers, including that of the authors.
HSCT allows the delivery of myeloablative (high) doses of radiation or chemotherapy for increased killing of tumor cells, in comparison with conventional doses of these standard therapies. The patient’s bone marrow, which can no longer function, is rescued with intravenous infusion of HSCs. Peripheral blood SCTs account for 95% of adult autologous transplants, as well as 70% of adult allogeneic transplants.140,176 In children, bone marrow is commonly used for these allogeneic transplants, since the outcome is better.177 Several milestones have been accomplished in HSCT treatment since the start of this therapy in 1951. Notably, over the last 20 years, it has been found that cord blood harvested shortly after birth is very rich in HSCs. Today, there are many stem cell banks that provide stem cell products for allogeneic HSCT.178–181
The ultimate goal in HSCT cancer immunotherapy is to improve engraftment, decrease graft-versus-host disease, augment graft-versus-tumor effects, and increase survival. To this end, investigators are studying mechanisms to culture and expand HSCs, without further differentiation, by such techniques as using bone marrow stromal cell lines and various cytokine cocktails to optimize this expansion either on a small scale or commercially,182 or by regulation of molecular pathways in these cells. Since 2006, approximately 40% of allogeneic transplants have used nonmyeloablative conditioning (lower doses of chemotherapy and radiation) of patients. This strategy allows older patients and those with other diseases to benefit from HSCT. Improved techniques and knowledge concerning human leukocyte-antigen matching and posttransplant therapy, such as high-dose cyclophosphamide to deplete T-cells and suppress graft-versus-host disease, allows better identification of suitable donors, and consequently more patients to benefit from HSCT therapy.180 Furthermore, molecular characterization of the donor and recipient cells can give clues to better matching and to patient outcome after transplant. Additional measures that may improve the success of HSCT include the use of drugs to increase homing of CD34+ cells to the bone marrow. For example, CXCR4 and its ligand CXCL12 (SDF1) direct CD34+ stem cells to migrate to the bone marrow.183 CD34+ stem cells of bone marrow have the ability to engraft and give rise to stem cells of diverse hematopoietic origins.184 Plerixafor, a CXCR4 antagonist, given in combination with G-CSF, increases mobilization of stem cells.185 This combination therapy is FDA-approved for patients with non-Hodgkin’s lymphoma and multiple myeloma.
In a Phase II immunotherapy-combination trial, multiple myeloma patients were given DC-tumor infusions before and after autologous SCT.186 Twenty-four patients received serial vaccinations with DC-myeloma fusion cells following posttransplant hematopoietic cell recovery. A second group received pretransplant vaccine as well as posttransplant vaccine administration. Seventy-eight percent of patients achieved a best response of complete response + very good partial response. Immune monitoring of patients revealed that vaccination resulted in a marked expansion of myeloma-reactive CD4+ and CD8+ T-cells expressing IFNγ in response to autologous tumor lysate.186
Targeting of tumors with modified T-cells
TIL therapy has been used in preclinical models and in clinical trials with some success. However, the production of these cells is a complex process, generally limited to specialized centers. Isolated TILs (T-cells) are harvested, activated, and expanded in culture and then infused back into the patient, usually with IL-2 administration. In this form of adoptive cell transfer, these cells can traffic to the tumor and lead to prolonged tumor eradication.187,188
There are two main types of genetically engineered T-cells used for adoptive cell therapy for cancers: TCR gene-modified and CAR gene-modified T-cells. These cells have high avidity and high reactivity to tumor antigens.
T-cells may be genetically modified to enhance the expression of selected high-affinity TCR before infusion into patients (ClinicalTrials.gov identifier: NCT01586403). T-cells are isolated from patients and cultured and expanded in cytokine cocktails. Cells are genetically altered using vectors containing nucleic acids encoding molecules that enable T-cells to recognize and mount immune responses to cancer cells. These vectors, often replication-deficient retroviruses or lentiviruses, or DNA plasmid-based vectors, deliver the selected genes to T-cells; these genes are incorporated into the T-cell genome, and hence passed down to dividing cells. Genes inserted into T-cells can enhance tumor-cell recognition, tumor-cell killing, cause T-cells to migrate into tumors, increase T-cell proliferation, or overcome factors in the tumor-suppressive microenvironment.142,189–191
CARs combine antigen specificity with T-cell activation in a single fusion molecule. The structure consists of an antigen-binding domain, an extracellular domain spacer/hinge region, a transmembrane domain, and an intracellular signaling domain, leading to T-cell activation after antigen binding.142 For CAR T-cell therapy, a patient’s T-cells are collected and genetically altered to produce special receptors on their surface. These CAR proteins allow the T-cells to recognize a specific antigen on tumor cells. The altered cell is expanded in culture several-fold and then administered to the patient. These T-cells multiply in vivo and recognize and kill cancer cells bearing the target antigen independently of MHC I.192 This therapy has proved very effective, especially in children. There have been a limited number of successful cases so far, as this form of therapy is still developing. However, recently the CAR T-cell therapy CTL019 gained FDA breakthrough designation for the treatment of acute lymphoblastic leukemia.193 In this trial, the T-cells were released into the patient’s blood, where they proliferated and bound to the targeted CD19+ cells and destroyed them. CD19 is commonly expressed on B cells, and hence this is a critical tool against B-cell leukemia. There are several other ongoing clinical trials in this area for hematologic malignancies and solid cancer, but most genetically modified T-cell therapy is in the experimental stages.142,188,194,195
Conclusion
Since the original debate on cancer immunosurveillance about a century ago, there has been significant knowledge gained concerning events in the tumor microenvironment and immunosuppression in the host. Scientists have been able to use this information to design immunotherapy strategies to exploit the patient’s immune system to improve patient survival. Therefore, the use of anticytokine antibody therapy, immune-checkpoint inhibition, autologous DC immunotherapy, and T-cell immunotherapy are all designed to overcome immunosuppression, improve patient care, and ultimately prolong survival.
Today, immunotherapy has advanced from improvements in a few cases to FDA approval of several immunotherapeutic agents. However, even with achievements in DC immunotherapy, such as sipuleucel-T, immune-checkpoint blocking antibodies, such as ipilimumab and Keytruda (pembrolizumab) and the recent FDA breakthrough designation for CAR T-cell therapy CTL019, the greatest barrier to new immunotherapy developments is the lack of relevant disease biomarkers for each disease so that the immune system can be more effectively targeted.
Recently, investigators reported a detailed genomic analysis of adenocarcinoma, a cancer found in the tissues near the outer parts of the lungs. They identified 18 statistically significant gene mutations in a study of 230 adenocarcinomas,196 a cancer with very low 5-year survival. Similar genomic analysis has been done for ovarian, colon, and breast cancer and acute myeloid leukemia, findings that we hope will facilitate the identification of additional biomarkers for diagnosis and/or therapy.
Many immunotherapeutic agents are still in the experimental or clinical trial phase, but we are encouraged, because many of these trials have shown sufficient success to prompt Phase II and III clinical trials. Additionally, several large and mid-size biotechnology companies are providing experimental drugs for preclinical and clinical trial testing. Combined targeting of the immune system with different classes of immunotherapeutic agents, DC, and/or adoptive T-cell therapy, will deliver the optimum therapeutic force against cancer.
In the case of DC immunotherapy, it is likely that improved nanoparticle delivery of relevant molecules, such as those of chemokine receptor CCR7, which guide DCs to lymph nodes, will enable DCs to traffic, persist in vivo, and maintain function to stimulate potent and sustained antigen-specific T-cell responses. The use of target molecules, such as neoantigens in DC immunotherapy, is also a promising strategy.
CAR T-cell immunotherapy is also an attractive immunotherapeutic mechanism, because benefit is derived through an MHC I-independent mechanism, and thus the effect of tumors downregulating MHC I to evade detection is not a concern. There are several ongoing clinical trials with genetically redirected T-cells in solid and hematologic cancers.142 However, different delivery vectors need to be tested, as do the best conditions under which to expand T-cells, as well as incorporation of specific/multiple target genes required to polarize the immune system from immunosuppressive to antitumor, and to avoid immune escape mechanisms of the tumor cells.
In the US, about a quarter of deaths each year is due to cancer. This is a very exciting and productive phase in the era of cancer-immunotherapy development, but given the complexity of immunosuppression in cancer, researchers must continue to elucidate new biomarkers for the disease and test effective immunotherapy combinations to treat this disease. Additionally, efforts need to be directed toward minimizing and managing adverse reactions to immunotherapy, and precautions need to be taken to reduce infection risks of potent immunotherapy. Finally, there is also the need for industry and health care systems to design creative ways to overcome the high cost of immunotherapy, so that this novel approach to cancer treatment can reach all who need it.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Dimberu PM, Leonhardt RM. Cancer immunotherapy takes a multifaceted approach to kick the immune system into gear. Yale J Biol Med. 2011;84(4):371–380. [PMC free article] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 4.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21(2):137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 5.Zhou J. Advances and prospects in cancer immunotherapy. N J Sci. 2014;2014:745808. [Google Scholar]
- 6.Das B, Tsuchida R, Malkin D, Koren G, Baruchel S, Yeger H. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells. 2008;26(7):1818–1830. doi: 10.1634/stemcells.2007-0724. [DOI] [PubMed] [Google Scholar]
- 7.Maitland NJ, Collins A. A tumour stem cell hypothesis for the origins of prostate cancer. BJU Int. 2005;96(9):1219–1223. doi: 10.1111/j.1464-410X.2005.05744.x. [DOI] [PubMed] [Google Scholar]
- 8.Odoux C, Fohrer H, Hoppo T, et al. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 2008;68(17):6932–6941. doi: 10.1158/0008-5472.CAN-07-5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selby P, Bizzari J, Buick R. Therapeutic implications of a stem cell model for human breast cancer: a hypothesis. Cancer Treat Rep. 1982;67(7–8):659–663. [PubMed] [Google Scholar]
- 10.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 11.Bickels J, Kollender Y, Merinsky O, Meller I. Coley’s toxin: historical perspective. Isr Med Assoc J. 2002;4(6):471–472. [PubMed] [Google Scholar]
- 12.McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. 2006;26:154–158. [PMC free article] [PubMed] [Google Scholar]
- 13.Nauts HC, Swift WE, Coley BL. The treatment of malignant tumors by bacterial toxins as developed by the late William B Coley, MD, reviewed in the light of modern research. Cancer Res. 1946;6:205–216. [PubMed] [Google Scholar]
- 14.Nelson D, Fisher S, Robinson B. The “Trojan horse” approach to tumor immunotherapy: targeting the tumor microenvironment. J Immunol Res. 2014;2014:789069. doi: 10.1155/2014/789069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 16.Lipson EJ, Drake CG. Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clin Cancer Res. 2011;17(22):6958–6962. doi: 10.1158/1078-0432.CCR-11-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prieto PA, Yang JC, Sherry RM, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res. 2012;18(7):2039–2047. doi: 10.1158/1078-0432.CCR-11-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehrlich P. Über den jetzigen stand der karzinomforschung. Ned Tijdschr Geneeskd. 1909;5:273–290. [Google Scholar]
- 19.Burnet F. The concept of immunological surveillance. Prog Exp Tumor Res. 1969;13:1–27. doi: 10.1159/000386035. [DOI] [PubMed] [Google Scholar]
- 20.Doll R, Kinlen L. Immunosurveillance and cancer: epidemiological evidence. Br Med J. 1970;4(5732):420–422. doi: 10.1136/bmj.4.5732.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamaï A, Benlalam H, Meslin F, et al. Immune surveillance of human cancer: if the cytotoxic T-lymphocytes play the music, does the tumoral system call the tune? Tissue Antigens. 2010;75(1):1–8. doi: 10.1111/j.1399-0039.2009.01401.x. [DOI] [PubMed] [Google Scholar]
- 22.Matsueda S, Graham DY. Immunotherapy in gastric cancer. World J Gastroenterol. 2014;20(7):1657–1666. doi: 10.3748/wjg.v20.i7.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slaney CY, Rautela J, Parker BS. The emerging role of immunosurveillance in dictating metastatic spread in breast cancer. Cancer Res. 2013;73(19):5852–5857. doi: 10.1158/0008-5472.CAN-13-1642. [DOI] [PubMed] [Google Scholar]
- 24.Romero JM, Jiménez P, Cabrera T, et al. Coordinated downregulation of the antigen presentation machinery and HLA class I/β2-microglobulin complex is responsible for HLA-ABC loss in bladder cancer. Int J Cancer. 2005;113(4):605–610. doi: 10.1002/ijc.20499. [DOI] [PubMed] [Google Scholar]
- 25.Fridman WH, Galon J, Pagès F, Tartour E, Sautes-Fridman C, Kroemer G. Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res. 2011;71(17):5601–5605. doi: 10.1158/0008-5472.CAN-11-1316. [DOI] [PubMed] [Google Scholar]
- 26.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 27.Pagès F, Galon J, Dieu-Nosjean MC, Tartour E, Sautès-Fridman C, Fridman WH. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. 2010;29(8):1093–1102. doi: 10.1038/onc.2009.416. [DOI] [PubMed] [Google Scholar]
- 28.Pagès F, Kirilovsky A, Mlecnik B, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J Clin Oncol. 2009;27(35):5944–5951. doi: 10.1200/JCO.2008.19.6147. [DOI] [PubMed] [Google Scholar]
- 29.Stiff PJ, Potkul RK, Venkataraman G, Sojitra P, Drakes ML. Immune surveillance tissue antigen profiling in advanced ovarian cancer. Clin Ovarian Cancer Other Gynecol Cancer. 2012;5(2):78–86. [Google Scholar]
- 30.Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 31.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 32.Hiura T, Kagamu H, Miura S, et al. Both regulatory T cells and antitumor effector T cells are primed in the same draining lymph nodes during tumor progression. J Immunol. 2005;175(8):5058–5066. doi: 10.4049/jimmunol.175.8.5058. [DOI] [PubMed] [Google Scholar]
- 33.Manrique SZ, Correa MA, Hoelzinger DB, et al. Foxp3-positive macrophages display immunosuppressive properties and promote tumor growth. J Exp Med. 2011;208(7):1485–1499. doi: 10.1084/jem.20100730. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Miller AM, Lundberg K, Ozenci V, et al. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol. 2006;177(10):7398–7405. doi: 10.4049/jimmunol.177.10.7398. [DOI] [PubMed] [Google Scholar]
- 35.Zhang SH, Han YX, Wu JB, et al. Elevated frequencies of CD4(+) CD25(+) CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int J Cancer. 2011;129(6):1373–1381. doi: 10.1002/ijc.25791. [DOI] [PubMed] [Google Scholar]
- 36.Wolf D, Wolf AM, Rumpold H, et al. The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin Cancer Res. 2005;11(23):8326–8331. doi: 10.1158/1078-0432.CCR-05-1244. [DOI] [PubMed] [Google Scholar]
- 37.Frey DM, Droeser RA, Viehl CT, et al. High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int J Cancer. 2010;126(11):2635–2643. doi: 10.1002/ijc.24989. [DOI] [PubMed] [Google Scholar]
- 38.García-Hernández ML, Hernández-Pando R, Gariglio P, Berumen J. Interleukin-10 promotes B16-melanoma growth by inhibition of macrophage functions and induction of tumour and vascular cell proliferation. Immunology. 2002;105(2):231–243. doi: 10.1046/j.1365-2567.2002.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larmonier N, Marron M, Zeng Y, et al. Tumor-derived CD4(+)CD25(+) regulatory T cell suppression of dendritic cell function involves TGF-beta and IL-10. Cancer Immunol Immunother. 2007;56(1):48–59. doi: 10.1007/s00262-006-0160-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang ZZ, Grote DM, Ziesmer SC, et al. Soluble and membrane-bound TGF-β-mediated regulation of intratumoral T cell differentiation and function in B-cell non-Hodgkin lymphoma. PLoS One. 2013;8(3):e59456. doi: 10.1371/journal.pone.0059456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faget J, Biota C, Bachelot T, et al. Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer Res. 2011;71(19):6143–6152. doi: 10.1158/0008-5472.CAN-11-0573. [DOI] [PubMed] [Google Scholar]
- 42.Gobert M, Treilleux I, Bendriss-Vermare N, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009;69(5):2000–2009. doi: 10.1158/0008-5472.CAN-08-2360. [DOI] [PubMed] [Google Scholar]
- 43.Qin XJ SH, Deng JM, Liang QL, Jiang J, Ye ZJ. CCL22 recruits CD4-positive CD25-positive regulatory T cells into malignant pleural effusion. Clin Cancer Res. 2009;15(7):2231–2237. doi: 10.1158/1078-0432.CCR-08-2641. [DOI] [PubMed] [Google Scholar]
- 44.Jaafar F, Righi E, Lindstrom V, et al. Correlation of CXCL12 expression and FoxP3+ cell infiltration with human papillomavirus infection and clinicopathological progression of cervical cancer. Am J Pathol. 2009;175(4):1525–1535. doi: 10.2353/ajpath.2009.090295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wald O, Izhar U, Amir G, et al. CD4+CXCR4highCD69+ T cells accumulate in lung adenocarcinoma. J Immunol. 2006;177(10):6983–6990. doi: 10.4049/jimmunol.177.10.6983. [DOI] [PubMed] [Google Scholar]
- 46.Capece D, Fischietti M, Verzella D, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int. 2013;2013:187204. doi: 10.1155/2013/187204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol. 2012;22(4):275–281. doi: 10.1016/j.semcancer.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ostrand-Rosenberg S, Sinha P, Chornoguz O, Ecker C. Regulating the suppressors: apoptosis and inflammation govern the survival of tumor-induced myeloid-derived suppressor cells (MDSC) Cancer Immunol Immunother. 2012;61(8):1319–1325. doi: 10.1007/s00262-012-1269-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 50.Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51–81. doi: 10.1016/S0065-2776(06)90002-9. [DOI] [PubMed] [Google Scholar]
- 51.Tien AH, Xu L, Helgason CD. Altered immunity accompanies disease progression in a mouse model of prostate dysplasia. Cancer Res. 2005;65(7):2947–2955. doi: 10.1158/0008-5472.CAN-04-3271. [DOI] [PubMed] [Google Scholar]
- 52.Ondondo B, Jones E, Godkin A, Gallimore A. Home sweet home: the tumor microenvironment as a haven for regulatory T cells. Front Immunol. 2013;4:197. doi: 10.3389/fimmu.2013.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4(12):941–952. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 54.Gabrilovich D, Ishida T, Oyama T, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92(11):4150–4166. [PubMed] [Google Scholar]
- 55.Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65(8):3437–3446. doi: 10.1158/0008-5472.CAN-04-4262. [DOI] [PubMed] [Google Scholar]
- 56.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11(18):6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 57.Dong H, Chen L. B7-H1 pathway and its role in the evasion of tumor immunity. Mol Med (Berl) 2003;81(5):281–287. doi: 10.1007/s00109-003-0430-2. [DOI] [PubMed] [Google Scholar]
- 58.Kryczek I, Wei S, Zhu G, et al. Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res. 2007;67(18):8900–8905. doi: 10.1158/0008-5472.CAN-07-1866. [DOI] [PubMed] [Google Scholar]
- 59.Kryczek I, Zou L, Rodriguez P, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203(4):871–881. doi: 10.1084/jem.20050930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hinrichs CS, Kaiser A, Paulos CM, et al. Type 17 CD8+ T cells display enhanced antitumor immunity. Blood. 2009;114(3):596–599. doi: 10.1182/blood-2009-02-203935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Llosa NJ, Geis AL, Orberg ET, Housseau F. Interleukin-17 and type 17 helper T cells in cancer management and research. Immunotargets Ther. 2014;2014(3):39–54. doi: 10.2147/ITT.S56529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol. 2010;10(4):248–256. doi: 10.1038/nri2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Inaba T, Ino K, Kajiyama H, et al. Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol Oncol. 2009;115(2):185–192. doi: 10.1016/j.ygyno.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 64.Malachowski WP, Metz R, Prendergast GC, Muller AJ. A new cancer immunosuppression target: indoleamine 2,3-dioxygenase (IDO). A review of the IDO mechanism, inhibition and therapeutic applications. Drugs Future. 2005;30(9):897–813. [Google Scholar]
- 65.McKenna K, Beignon AS, Bhardwaj N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol. 2005;79(1):17–27. doi: 10.1128/JVI.79.1.17-27.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sharma MD, Baban B, Chandler P, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117(9):2570–2582. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei S, Kryczek I, Zou L, et al. Plasmacytoid dendritic cells induce CD8+ regulatory T cells in human ovarian carcinoma. Cancer Res. 2005;65(12):5020–5026. doi: 10.1158/0008-5472.CAN-04-4043. [DOI] [PubMed] [Google Scholar]
- 68.McDonnell AM, Robinson BW, Currie AJ. Tumor antigen cross-presentation and the dendritic cell: where it all begins? Clin Dev Immunol. 2010;2010:539519. doi: 10.1155/2010/539519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seliger B. Different regulation of MHC class I antigen processing components in human tumors. J Immunotoxicol. 2008;5(4):361–367. doi: 10.1080/15476910802482870. [DOI] [PubMed] [Google Scholar]
- 70.Inman BA, Frigola X, Dong H, Kwon ED. Costimulation, coinhibition and cancer. Curr Cancer Drug Targets. 2007;7(1):15–30. doi: 10.2174/156800907780006878. [DOI] [PubMed] [Google Scholar]
- 71.Krempski J, Karyampudi L, Behrens MD, et al. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol. 2011;186(12):6905–6913. doi: 10.4049/jimmunol.1100274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11(2):155–164. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- 73.Gelao L, Criscitiello C, Esposito A, Goldhirsch A, Curigliano G. Immune checkpoint blockade in cancer treatment: a double-edged sword cross-targeting the host as an “innocent bystander”. Toxins. 2014;6(3):914–933. doi: 10.3390/toxins6030914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lesterhuis WJ, Salmons J, Nowak AK, et al. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS One. 2013;8(4):e61895. doi: 10.1371/journal.pone.0061895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Galili U, Clark MR, Shohet SB, Buehler J, Macher BA. Evolutionary relationship between the natural anti-Gal antibody and the Gal α1→3Gal epitope in primates. Proc Natl Acad Sci U S A. 1987;84(5):1369–1373. doi: 10.1073/pnas.84.5.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gunturu KS, Rossi GR, Saif MW. Immunotherapy updates in pancreatic cancer: are we there yet? Ther Adv Med Oncol. 2012;5(1):81–89. doi: 10.1177/1758834012462463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rossi GR, Mautino MR, Awwad DZ, et al. Allogeneic melanoma vaccine expressing αGal epitopes induces antitumor immunity to autologous antigens in mice without signs of toxicity. J Immunother. 2008;31(6):545–554. doi: 10.1097/CJI.0b013e31817d2f45. [DOI] [PubMed] [Google Scholar]
- 78.Hanna MG., Jr Immunotherapy with autologous tumor cell vaccines for treatment of occult disease in early stage colon cancer. Hum Vaccin Immunother. 2012;8(8):1156–1160. doi: 10.4161/hv.20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Murshid A, Gong J, Stevenson MA, Calderwood SK. Heat shock proteins and cancer vaccines: developments in the past decade and chaperoning in the decade to come. Expert Rev Vaccines. 2011;10(11):1553–1568. doi: 10.1586/erv.11.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Calderwood SK, Stevenson MA, Murshid A. Heat shock proteins, autoimmunity, and cancer treatment. Autoimmune Dis. 2012;2012:486069. doi: 10.1155/2012/486069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Solit DB, Rosen N. Hsp90: a novel target for cancer therapy. Curr Top Med Chem. 2006;6(11):1205–1214. doi: 10.2174/156802606777812068. [DOI] [PubMed] [Google Scholar]
- 82.Antonarakis ES. Combining active immunotherapy with immune checkpoint blockade for the treatment of advanced prostate cancer. Asian J Androl. 2012;14(4):520–521. doi: 10.1038/aja.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Madan RA, Mohebtash M, Arlen PM, et al. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(5):501–508. doi: 10.1016/S1470-2045(12)70006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Larocca C, Schlom J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011;17(5):359–371. doi: 10.1097/PPO.0b013e3182325e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bartlett DL, Liu Z, Sathaiah M, et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. 2013;12(1):103. doi: 10.1186/1476-4598-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cerullo V, Koski A, Vähä-Koskela M, Hemminki A. Chapter eight – Oncolytic adenoviruses for cancer immunotherapy: data from mice, hamsters, and humans. Adv Cancer Res. 2012;115:265–318. doi: 10.1016/B978-0-12-398342-8.00008-2. [DOI] [PubMed] [Google Scholar]
- 87.Guo ZS, Thorne SH, Bartlett DL. Oncolytic virotherapy: molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta. 2008;1785(2):217–231. doi: 10.1016/j.bbcan.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Patel MR, Kratzke RA. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Transl Res. 2013;161(4):355–364. doi: 10.1016/j.trsl.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 89.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang MH, You SL, Chen CJ, et al. Decreased incidence of hepatocellular carcinoma in hepatitis B vaccinees: a 20-year follow-up study. J Natl Cancer Inst. 2009;101(19):1348–1355. doi: 10.1093/jnci/djp288. [DOI] [PubMed] [Google Scholar]
- 91.Olsson SE, Villa LL, Costa RL, et al. Induction of immune memory following administration of a prophylactic quadrivalent human papillomavirus (HPV) types 6/11/16/18 L1 virus-like particle (VLP) vaccine. Vaccine. 2007;25(26):4931–4939. doi: 10.1016/j.vaccine.2007.03.049. [DOI] [PubMed] [Google Scholar]
- 92.Van Kriekinge G, Castellsague X, Cibula D, Demarteau N. Estimation of the potential overall impact of human papillomavirus vaccination on cervical cancer cases and deaths. Vaccine. 2014;32(6):733–739. doi: 10.1016/j.vaccine.2013.11.049. [DOI] [PubMed] [Google Scholar]
- 93.Villa LL, Ault KA, Giuliano AR, et al. Immunologic responses following administration of a vaccine targeting human papillomavirus types 6, 11, 16, and 18. Vaccine. 2006;24(27):5571–5583. doi: 10.1016/j.vaccine.2006.04.068. [DOI] [PubMed] [Google Scholar]
- 94.Bagarazzi ML, Yan J, Morrow MP, et al. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci Transl Med. 2012;4(155):155ra138. doi: 10.1126/scitranslmed.3004414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maldonado L, Teague JE, Morrow MP, et al. Intramuscular therapeutic vaccination targeting HPV16 induces T cell responses that localize in mucosal lesions. Sci Transl Med. 2014;6(221):221ra213. doi: 10.1126/scitranslmed.3007323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bogdanov KV, Frolova OI, Marinets OV, Ogorodnikova IuS, Afanas’ev BV, Zaritskiĭ AIu. [The effectiveness of interferon-alpha therapy in Ph-positive chronic myeloid leukemia] Vopr Onkol. 2003;49(2):189–192. Russian. [PubMed] [Google Scholar]
- 97.Lutz D. [Interferon-alpha therapy in chronic myeloid leukemia] Wien Med Wochenschr. 1993;143(16–17):416–419. German. [PubMed] [Google Scholar]
- 98.Mocellin S, Pasquali S, Rossi CR, Nitti D. Interferon alpha adjuvant therapy in patients with high-risk melanoma: a systematic review and meta-analysis. J Natl Cancer Inst. 2010;102(7):493–501. doi: 10.1093/jnci/djq009. [DOI] [PubMed] [Google Scholar]
- 99.von Wussow P, Block B, Hartmann F, Deicher H. Intralesional interferon-alpha therapy in advanced malignant melanoma. Cancer. 1988;61(6):1071–1074. doi: 10.1002/1097-0142(19880315)61:6<1071::aid-cncr2820610603>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 100.Elias EG, Zapas JL, Beam SL, Brown SD. GM-CSF and IL-2 combination as adjuvant therapy in cutaneous melanoma: early results of a phase II clinical trial. Oncology. 2005;19(4 Suppl 2):15–18. [PubMed] [Google Scholar]
- 101.Fateh S, Schell TD, Gingrich R, Neves RI, Drabick JJ. Unsuccessful high dose IL-2 therapy followed immediately by near continuous low dose temozolomide can result in rapid durable complete and near-complete remissions in metastatic melanoma. Cancer Biol Ther. 2010;10(11):1091–1097. doi: 10.4161/cbt.10.11.13452. [DOI] [PubMed] [Google Scholar]
- 102.Borrello IM, Levitsky HI, Stock W, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting cellular immunotherapy in combination with autologous stem cell transplantation (ASCT) as postremission therapy for acute myeloid leukemia (AML) Blood. 2009;114(9):1736–1745. doi: 10.1182/blood-2009-02-205278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sorkin P, Abu-Abid S, Lev D, et al. Systemic leakage and side effects of tumor necrosis factor alpha administered via isolated limb perfusion can be manipulated by flow rate adjustment. Arch Surg. 1995;130(10):1079–1084. doi: 10.1001/archsurg.1995.01430100057012. [DOI] [PubMed] [Google Scholar]
- 104.Vom Berg J, Vrohlings M, Haller S, et al. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J Exp Med. 2013;210(13):2803–2811. doi: 10.1084/jem.20130678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang K, Grivennikov SI, Karin M. Implications of anti-cytokine therapy in colorectal cancer and autoimmune diseases. Ann Rheum Dis. 2013;72(Suppl 2):ii100–ii103. doi: 10.1136/annrheumdis-2012-202201. [DOI] [PubMed] [Google Scholar]
- 106.Chao MP, Weissman IL, Majeti R. The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24(2):225–232. doi: 10.1016/j.coi.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Edris B, Weiskopf K, Volkmer AK, et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc Natl Acad Sci U S A. 2012;109(17):6656–6661. doi: 10.1073/pnas.1121629109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jaiswal S, Chao MP, Majeti R, Weissman IL. Macrophages as mediators of tumor immunosurveillance. Trends Immunol. 2010;31(6):212–219. doi: 10.1016/j.it.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sick E, Jeanne A, Schneider C, Dedieu S, Takeda K, Martiny L. CD47 update: a multifaceted actor in the tumour microenvironment of potential therapeutic interest. Br J Pharmacol. 2012;167(7):1415–1430. doi: 10.1111/j.1476-5381.2012.02099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tseng D, Volkmer JP, Willingham SB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A. 2013;110(27):11103–11108. doi: 10.1073/pnas.1305569110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rech AJ, Mick R, Martin S, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 2012;4(134):134ra162. doi: 10.1126/scitranslmed.3003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rech AJ, Vonderheide RH. Clinical use of anti-CD25 antibody daclizumab to enhance immune responses to tumor antigen vaccination by targeting regulatory T cells. Ann N Y Acad Sci. 2009;1174:99–106. doi: 10.1111/j.1749-6632.2009.04939.x. [DOI] [PubMed] [Google Scholar]
- 113.Rasku MA, Clem AL, Telang S, et al. Transient T cell depletion causes regression of melanoma metastases. J Transl Med. 2008;6:12. doi: 10.1186/1479-5876-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ghiringhelli F, Larmonier N, Schmitt E, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34(2):336–344. doi: 10.1002/eji.200324181. [DOI] [PubMed] [Google Scholar]
- 115.Ghiringhelli F, Menard C, Puig PE, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56(5):641–648. doi: 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schuetze SM, Zhao L, Chugh R, et al. Results of a phase II study of sirolimus and cyclophosphamide in patients with advanced sarcoma. Eur J Cancer. 2012;48(9):1347–1353. doi: 10.1016/j.ejca.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 117.Teng MW, Ngiow SF, von Scheidt B, McLaughlin N, Sparwasser T, Smyth MJ. Conditional regulatory T-cell depletion releases adaptive immunity preventing carcinogenesis and suppressing established tumor growth. Cancer Res. 2010;70(20):7800–7809. doi: 10.1158/0008-5472.CAN-10-1681. [DOI] [PubMed] [Google Scholar]
- 118.Vermeij R, Leffers N, Hoogeboom BN, et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: a single-arm phase II study. Int J Cancer. 2012;131(5):E670–E680. doi: 10.1002/ijc.27388. [DOI] [PubMed] [Google Scholar]
- 119.Fonsatti E, Maio M, Altomonte M, Hersey P. Biology and clinical applications of CD40 in cancer treatment. Semin Oncol. 2010;37(5):517–523. doi: 10.1053/j.seminoncol.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 120.Zhang M, Yao Z, Dubois S, Ju W, Muller JR, Waldmann TA. Interleukin-15 combined with an anti-CD40 antibody provides enhanced therapeutic efficacy for murine models of colon cancer. Proc Natl Acad Sci U S A. 2009;106(18):7513–7518. doi: 10.1073/pnas.0902637106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sznol M, Hodi F, Margolin K, et al. Phase I study of BMS-663513, a fully human anti-CD137 agonist monoclonal antibody, in patients (pts) with advanced cancer (CA) J Clin Oncol. 2008;26(Suppl 15):3007. [Google Scholar]
- 122.Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. 2007;56(5):739–745. doi: 10.1007/s00262-006-0272-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Flies DB, Sandler BJ, Sznol M, Chen L. Blockade of the B7-H1/PD-1 pathway for cancer immunotherapy. Yale J Biol Med. 2011;84(4):409–421. [PMC free article] [PubMed] [Google Scholar]
- 124.Hirano F, Kaneko K, Tamura H, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65(3):1089–1096. [PubMed] [Google Scholar]
- 125.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1 (PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24(2):207–212. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Genova C, Rijavec E, Barletta G, et al. Ipilimumab (MDX-010) in the treatment of non-small cell lung cancer. Expert Opin Biol Ther. 2012;12(7):939–948. doi: 10.1517/14712598.2012.681371. [DOI] [PubMed] [Google Scholar]
- 127.Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828–833. doi: 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Slovin SF, Higano CS, Hamid O, et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: results from an open-label, multicenter phase I/II study. Ann Oncol. 2013;24(7):1813–1821. doi: 10.1093/annonc/mdt107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yang JC, Hughes M, Kammula U, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. 2007;30(8):825–830. doi: 10.1097/CJI.0b013e318156e47e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hurwitz AA, Yu TF, Leach DR, Allison JP. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Natl Acad Sci U S A. 1998;95(17):10067–10071. doi: 10.1073/pnas.95.17.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fecher LA, Agarwala SS, Hodi FS, Weber JS. Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist. 2013;18(6):733–743. doi: 10.1634/theoncologist.2012-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15(17):5591–5598. doi: 10.1158/1078-0432.CCR-09-1024. [DOI] [PubMed] [Google Scholar]
- 133.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma – are we there yet? Neuro Oncol. 2013;15(1):4–27. doi: 10.1093/neuonc/nos273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Grauer OM, Nierkens S, Bennink E, et al. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int J Cancer. 2007;121(1):95–105. doi: 10.1002/ijc.22607. [DOI] [PubMed] [Google Scholar]
- 137.Jacobs JF, Idema AJ, Bol KF, et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol. 2010;225(1–2):195–199. doi: 10.1016/j.jneuroim.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 138.Selby MJ, Engelhardt JJ, Quigley M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1(1):32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 139.Knutson KL, Wagner W, Disis ML. Adoptive T cell therapy of solid cancers. Cancer Immunol Immunother. 2006;55(1):96–103. doi: 10.1007/s00262-005-0706-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jenq RR, Van den Brink MR. Allogeneic haematopoietic stem cell transplantation: individualized stem cell and immune therapy of cancer. Nat Rev Cancer. 2010;10(3):213–221. doi: 10.1038/nrc2804. [DOI] [PubMed] [Google Scholar]
- 141.Goff SL, Smith FO, Klapper JA, et al. Tumor infiltrating lymphocyte therapy for metastatic melanoma: analysis of tumors resected for TIL. J Immunother. 2010;33(8):840–847. doi: 10.1097/CJI.0b013e3181f05b91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer. 2013;13(8):525–541. doi: 10.1038/nrc3565. [DOI] [PubMed] [Google Scholar]
- 143.Billingham R. Dendritic cells. J Anat. 1948;82(Pt 1–2):93–109. [PMC free article] [PubMed] [Google Scholar]
- 144.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137(5):1142–1162. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 146.Hart DN. Dendritic cells: unique leukocyte populations which control the primary immune response. Blood. 1997;90(9):3245–3287. [PubMed] [Google Scholar]
- 147.Liu YJ. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell. 2001;106(3):259–262. doi: 10.1016/s0092-8674(01)00456-1. [DOI] [PubMed] [Google Scholar]
- 148.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5(4):296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 149.Harada Y, Yonemitsu Y. Recent developments in patented DC-based immunotherapy for various malignancies. Recent Pat Regen Med. 2011;1(1):72–87. [Google Scholar]
- 150.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Graddis TJ, McMahan CJ, Tamman J, Page KJ, Trager JB. Prostatic acid phosphatase expression in human tissues. Int J Clin Exp Pathol. 2011;4(3):295–306. [PMC free article] [PubMed] [Google Scholar]
- 152.Fong L, Ruegg CL, Brockstedt D, Engleman EG, Laus R. Induction of tissue-specific autoimmune prostatitis with prostatic acid phosphatase immunization: implications for immunotherapy of prostate cancer. J Immunol. 1997;159(7):3113–3117. [PubMed] [Google Scholar]
- 153.Thakur A, Vaishampayan U, Lum LG. Immunotherapy and immune evasion in prostate cancer. Cancers (Basel) 2013;5(2):569–590. doi: 10.3390/cancers5020569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Okada H, Kalinski P, Ueda R, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymeth-ylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29(3):330–336. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lee JJ, Foon KA, Mailliard RB, Muthuswamy R, Kalinski P. Type 1- polarized dendritic cells loaded with autologous tumor are a potent immunogen against chronic lymphocytic leukemia. J Leukoc Biol. 2008;84(1):319–325. doi: 10.1189/jlb.1107737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mailliard RB, Wankowicz-Kalinska A, Cai Q, et al. α-Type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res. 2004;64(17):5934–5937. doi: 10.1158/0008-5472.CAN-04-1261. [DOI] [PubMed] [Google Scholar]
- 157.Zhu X, Fallert-Junecko BA, Fujita M, et al. Poly-ICLC promotes the infiltration of effector T cells into intracranial gliomas via induction of CXCL10 in IFN-α and IFN-γ dependent manners. Cancer Immunol Immunother. 2010;59(9):1401–1409. doi: 10.1007/s00262-010-0876-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhu X, Nishimura F, Sasaki K, et al. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med. 2007;5(1):10. doi: 10.1186/1479-5876-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Eyrich M, Rachor J, Schreiber SC, Wölfl M, Schlegel PG. Dendritic cell vaccination in pediatric gliomas: lessons learnt and future perspectives. Front Pediatr. 2013;1:12. doi: 10.3389/fped.2013.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Fong L, Brockstedt D, Benike C, Wu L, Engleman EG. Dendritic cells injected via different routes induce immunity in cancer patients. J Immunol. 2001;166(6):4254–4259. doi: 10.4049/jimmunol.166.6.4254. [DOI] [PubMed] [Google Scholar]
- 161.Lesterhuis WJ, de Vries IJ, Schreibelt G, et al. Route of administration modulates the induction of dendritic cell vaccine-induced antigen-specific T cells in advanced melanoma patients. Clin Cancer Res. 2011;17(17):5725–5735. doi: 10.1158/1078-0432.CCR-11-1261. [DOI] [PubMed] [Google Scholar]
- 162.Schuler-Thurner B, Schultz ES, Berger TG, et al. Rapid induction of tumor-specific type 1 T helper cells in metastatic melanoma patients by vaccination with mature, cryopreserved, peptide-loaded monocyte-derived dendritic cells. J Exp Med. 2002;195(10):1279–1288. doi: 10.1084/jem.20012100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gilboa E, Vieweg J. Cancer immunotherapy with mRNA-transfected dendritic cells. Immunol Rev. 2004;199:251–263. doi: 10.1111/j.0105-2896.2004.00139.x. [DOI] [PubMed] [Google Scholar]
- 164.Thumann P, Moc I, Humrich J, et al. Antigen loading of dendritic cells with whole tumor cell preparations. J Immunol Methods. 2003;277(1–2):1–16. doi: 10.1016/s0022-1759(03)00102-9. [DOI] [PubMed] [Google Scholar]
- 165.Whiteside TL. Evaluation of dendritic cell products generated for human therapy and post-treatment immune monitoring. Biopharm Int. 2008;21(3):42–67. [Google Scholar]
- 166.Brossart P, Wirths S, Stuhler G, Reichardt VL, Kanz L, Brugger W. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood. 2000;96(9):3102–3108. [PubMed] [Google Scholar]
- 167.Gnjatic S, Nishikawa H, Jungbluth AA, et al. NY-ESO-1: review of an immunogenic tumor antigen. Adv Cancer Res. 2006;95:1–30. doi: 10.1016/S0065-230X(06)95001-5. [DOI] [PubMed] [Google Scholar]
- 168.Odunsi K, Jungbluth AA, Stockert E, et al. NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res. 2003;63(18):6076–6083. [PubMed] [Google Scholar]
- 169.Sabado RL, Bhardwaj N. Directing dendritic cell immunotherapy towards successful cancer treatment. Immunotherapy. 2010;2(1):37–56. doi: 10.2217/imt.09.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vlad AM, Kettel JC, Alajez NM, Carlos CA, Finn OJ. MUC1 immunobiology: from discovery to clinical applications. Adv Immunol. 2004;82:249–293. doi: 10.1016/S0065-2776(04)82006-6. [DOI] [PubMed] [Google Scholar]
- 171.Jenne L, Schuler G, Steinkasserer A. Viral vectors for dendritic cell-based immunotherapy. Trends Immunol. 2001;22(2):102–107. doi: 10.1016/s1471-4906(00)01813-5. [DOI] [PubMed] [Google Scholar]
- 172.Skoberne M, Yewdall A, Bahjat KS, et al. KBMA Listeria monocytogenes is an effective vector for DC-mediated induction of antitumor immunity. J Clin Invest. 2008;118(12):3990–4001. doi: 10.1172/JCI31350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Veron P, Allo V, Rivière C, Bernard J, Douar AM, Masurier C. Major subsets of human dendritic cells are efficiently transduced by self-complementary adeno-associated virus vectors 1 and 2. J Virol. 2007;81(10):5385–5394. doi: 10.1128/JVI.02516-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hacohen N1, Fritsch EF, Carter TA, Lander ES, Wu CJ. Getting personal with neoantigen-based therapeutic cancer vaccines. Cancer Immunol Res. 2013;1(1):11–15. doi: 10.1158/2326-6066.CIR-13-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.van Rooij N, van Buuren MM, Philips D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31(32):e439–e442. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Passweg JR, Halter J, Bucher C, et al. Hematopoietic stem cell transplantation: a review and recommendations for follow-up care for the general practitioner. Swiss Med Wkly. 2012;142:w13696. doi: 10.4414/smw.2012.13696. [DOI] [PubMed] [Google Scholar]
- 177.Eapen M, Horowitz MM, Klein JP, et al. Higher mortality after allogeneic peripheral-blood transplantation compared with bone marrow in children and adolescents: the Histocompatibility and Alternate Stem Cell Source Working Committee of the International Bone Marrow Transplant Registry. J Clin Oncol. 2004;22(24):4872–4880. doi: 10.1200/JCO.2004.02.189. [DOI] [PubMed] [Google Scholar]
- 178.Barker J, Weisdorf D, Wagner J. Creation of a double chimera after the transplantation of umbilical-cord blood from two partially matched unrelated donors. N Engl J Med. 2001;344(24):1870–1871. doi: 10.1056/NEJM200106143442417. [DOI] [PubMed] [Google Scholar]
- 179.Barker JN, Weisdorf DJ, DeFor TE, et al. Transplantation of 2 partially HLA-matched umbilical cord blood units to enhance engraftment in adults with hematologic malignancy. Blood. 2005;105(3):1343–1347. doi: 10.1182/blood-2004-07-2717. [DOI] [PubMed] [Google Scholar]
- 180.Gluckman E, Broxmeyer H, Auerbach AD, et al. Hematopoietic reconstitution in a patient with Fanconi’s anemia by means of umbilical-cord blood from an HLA-identical sibling. N Engl J Med. 1989;321(17):1174–1178. doi: 10.1056/NEJM198910263211707. [DOI] [PubMed] [Google Scholar]
- 181.Cieri N, Mastaglio S, Oliveira G, Casucci M, Bondanza A, Bonini C. Adoptive immunotherapy with genetically modified lymphocytes in allogeneic stem cell transplantation. Immunol Rev. 2014;257(1):165–180. doi: 10.1111/imr.12130. [DOI] [PubMed] [Google Scholar]
- 182.Knudtzon S. In vitro growth of granulocytic colonies from circulating cells in human cord blood. Blood. 1974;43(3):357–361. [PubMed] [Google Scholar]
- 183.Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med. 2002;195(9):1145–1154. doi: 10.1084/jem.20011284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Berenson R, Andrews R, Bensinger W, et al. Antigen CD34+ marrow cells engraft lethally irradiated baboons. J Clin Invest. 1988;81(3):951–955. doi: 10.1172/JCI113409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.DiPersio JF, Stadtmauer EA, Nademanee A, et al. Plerixafor and G-CSF versus placebo and G-CSF to mobilize hematopoietic stem cells for autologous stem cell transplantation in patients with multiple myeloma. Blood. 2009;113(23):5720–5726. doi: 10.1182/blood-2008-08-174946. [DOI] [PubMed] [Google Scholar]
- 186.Rosenblatt J, Avivi I, Vasir B, et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin Cancer Res. 2013;19(13):3640–3648. doi: 10.1158/1078-0432.CCR-13-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Dudley ME, Gross CA, Somerville RP, et al. Randomized selection design trial evaluating CD8+-enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. J Clin Oncol. 2013;31(17):2152–2159. doi: 10.1200/JCO.2012.46.6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12(4):269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8(4):299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med. 2012;14(6):405–415. doi: 10.1002/jgm.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Han EQ, Li XL, Wang CR, Li TF, Han SY. Chimeric antigen receptor-engineered T cells for cancer immunotherapy: progress and challenges. J Hematol Oncol. 2013;6:47. doi: 10.1186/1756-8722-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]