

Abstract
Oncolytic virotherapy is a new strategy to reduce tumor burden through selective virus replication in rapidly proliferating cells. Oncolytic viruses are members of at least ten virus families, each with its advantages and disadvantages. Here, I briefly review the recent advances and key challenges, as exemplified by the best-studied platforms. Recent advances include preclinical proof of feasibility, clinical evidence of tolerability and effectiveness, and the development of new strategies to improve efficacy. These include engineered tumor selectivity and expression of antitumorigenic genes that could function independently of virus replication, identification of combinatorial therapies that accelerate intratumoral virus propagation, and modification of immune responses and vascular delivery for treatment of metastatic disease. Key challenges are to select “winners” from the distinct oncolytic platforms that can stimulate anti-cancer immunity without affecting virus replication and can lyse cancer stem cells, which are most likely responsible for tumor maintenance, aggressiveness, and recurrence. Preventing the emergence of resistant tumor cells during virotherapy through the activation of multiple death pathways, the development of a better understanding of the mechanisms of cancer stem-cell lysis, and the development of more meaningful preclinical animal models are additional challenges for the next-generation of engineered viruses.
Keywords: tumor cell lysis, virus replication, tumor selectivity, programmed cell death, immune response
Introduction
Resistance is a major problem of cancer therapy. It is associated with malignant cells, known as cancer stem cells (CSCs), that retain many of the properties of normal stem cells, including the ability to differentiate into multiple cell types, self-renew, proliferate, and maintain neoplastic clonality.1 Oncolytic virotherapy is an innovative, targeted therapy with preclinical efficacy in a variety of tumors. It is designed to reduce tumor burden through selective virus replication in the tumor cells and the generation of infectious progeny that spread through the tumor mass. Oncolytic viruses kill cancer cells by (1) selectively targeting tumor cells through engineered mutations that prevent their binding to and replication in normal cells, and/or (2) expressing foreign genes that cause cell death, directly or indirectly.2 Oncolytic viruses that have already entered clinical trials are members of various families. They all have their advantages and disadvantages, which are briefly summarized in Table 1 for three of the best-studied families (oncolytic herpes simplex virus [oHSV], adenovirus, and vaccinia virus [VV]). Other platforms with notable properties include reovirus, Seneca Valley virus, and myxoma virus, which cause mild or no disease in humans, and Newcastle disease virus and vesicular stomatitis virus, which target cancer cells with loss of interferon (IFN) responses. However, available data suggest that the overall clinical efficacy of all the oncolytic virus families may be limited by relatively poor tumor penetration, induction of antiviral responses, and failure to eradicate CSCs. Here, I briefly review current achievements that underscore the promise and potential problems of this new therapeutic strategy and define areas of future research interests.
Table 1.
Properties of the three best-studied families of oncolytic viruses
| Virus type | Pros and cons | Clinical trials* |
|---|---|---|
| Adenovirus | Pros include constructed wide host-cell range and large genomic capacity. Cons include CAR variability in human cancers and expression on normal cells, preexisting antiviral immunity, hepatic adsorption, toxicity | Completed and ongoing6,55–57 |
| Vaccinia virus | Pros include wide host-cell range, large genomic capacity and established vaccine potential. Cons include potential difficulties with systemic delivery | Completed and ongoing4,60–63 |
| Herpes Simplex Virus | Pros include wide host-cell range, large genomic capacity, neurotropism, ability to evade preexisting antiviral immunity and available antiviral drugs. Cons include hepatic adsorption | Completed and ongoing3,5,65–68 |
Notes:
other viruses in clinical trials include Coxsackie virus (CVA21), measles virus (Edmonston), parvovirus, poliovirus (Sabin), retrovirus.
Abbreviation: CAR, Coxsackie adenovirus receptor.
Clinical tolerability and efficacy
The past 20 years have witnessed great progress in the development of virus-based cancer therapies with oncolytic viruses belonging to at least ten virus families and many entering the clinical arena. Overall, the oncolytic viruses have excellent clinical tolerability, and transmission from infected patients was not seen, although virus shedding was reported in urine or respiratory secretions.3 However, the ability of systemically administered virus to extravasate and spread to metastasis sites has not been fully documented. Concerns remain that (1) serious toxicities may occur at clinically effective doses and (2) the viruses may spread and mutate to regain pathogenic potential.2
Two recent phase I/II clinical trials document efficacy and provide compelling evidence of generalized treatment by intratumorally delivered virus. In the first trial, an oHSV armed with the cytokine granulocyte macrophage colony-stimulating factor (GM-CSF) known as OncoVex (and more recently as talimogene laherparepvec) that was administered by intratumoral injection to patients with metastatic melanoma caused complete regression of injected and uninjected lesions in eight of 50 treated patients.3 In the second trial, an oncolytic VV (JX594) armed with GM-CSF administered intratumorally to patients with nonresectable hepatocellular carcinoma caused objective responses in three of ten patients.4 However, it is not totally clear that direct lysis of the infected cells resulting from virus replication is an important mechanism of tumor destruction. In fact, the ability of the virus to induce programmed cell death independently of virus replication is a desirable property that needs further evaluation (see the Lysis of cancer stem cells section, below). The combined use of oncolytic viruses together with cytotoxic drugs or radiation increased clinical efficacy,5 but the exact contribution of the viruses over and above the anticancer drugs is difficult to evaluate.
Insights resulting from completed and ongoing clinical trials include the need to develop oncolytic viruses with increased tumor selectivity and intratumoral penetration, devise better methods for virus delivery to treat metastatic cancer, eg, through the engineering of viruses that avoid and/or overcome antiviral responses, and establish better animal models that retain a functional immune system. Also needed are better methods to monitor virus spread in the body, eg, through the introduction of reporter genes that enable repetitive, noninvasive determination of the location and number of virus-infected cells. Thyroidal sodium iodide symporter genes were inserted in several oncolytic viruses and used in conjunction with radioisotopes to monitor in vivo spread.6 However, recent studies have focused on the development of nonradioactive reporter gene-detection systems that can better resolve small numbers of infected cells. Improved tracking of virus spread and detection of micrometastatic tumor cells at various organ sites and in the peritoneal cavity were recently reported for an engineered adenovirus with telomerase-dependent expression of green fluorescent protein.7,8 Adenovirus vectors modified to express luciferase were also used for in vivo imaging of virus distribution9 and detection of metastatic tumors, the latter utilizing the tumor-specific mucin-1 promoter in combination with a two-step transcriptional amplification system that augments the activity of weak tissue-specific promoters.10 However, the safety of these reporter genes needs further confirmation.
Engineering tumor selectivity
Oncolytic viruses reduce tumor burden through virus replication in tumor but not normal cells and the generation of infectious progeny that spreads throughout the tumor mass. They combine tumor lysis with antigen release and the generation of potentially antitumoral immune responses (Figure 1). However, virus replication in normal cells is a remaining concern, and current efforts are focused on the development of tropism-modifying strategies that can improve tumor selectivity. In one such approach, tumor-specific promoters were used to drive viral genes, as exemplified by adenovirus vectors, in which E1 gene expression is driven by the human telomerase reverse transcriptase gene (hTERT) that is robustly expressed in cancer but not normal cells.11 Another tropism-modifying approach has been to alter the virus proteins responsible for cell binding/penetration. This is exemplified by the modification of the hypervariable loop of the adenovirus hexon protein, such that the virus can no longer infect normal hepatocytes but can still infect tumor cells,12 or by the pseudotyping of vesicular stomatitis virus with the surface glycoprotein from a nonneurotropic lymphocytic choriomeningitis virus in order to reduce neurotoxicity without compromising cancer cell infection.13
Figure 1.
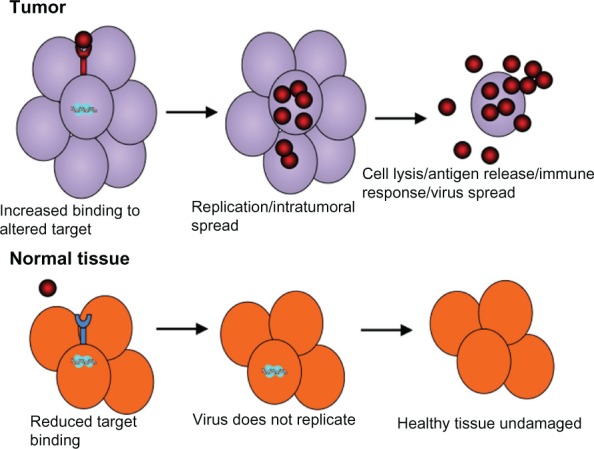
Infection and killing of tumor cells by oncolytic viruses. Oncolytic viruses are engineered to have tumor selectivity through various strategies that favor preferential infection of tumor compared to normal cells through binding of tumor-specific receptors, or preferential replication in the tumor compared to normal cells. This results in a large amount of progeny virus being released from the infected tumor cells and its spread throughout the tumor mass.
Notes: Expression of viral proteins and release of antigens eventually also leads to immune-mediated lysis of infected cells by cluster of differentiation (CD)-8+ cells. Moreover, virus gene expression and replication leads to the activation of cellular antiviral defenses that are often inactivated in tumor cells. The outcome is specific lysis of tumor but not normal cells.
Another approach has been to link virus replication to activated Ras, the levels of which are significantly higher in tumor than normal cells. In one such strategy, the expression of the HSV protein ICP4, which is required for HSV-1 replication, is driven by activated Elk, a Ras-activated transcription factor. The virus (known as SS1) preferentially replicates in cells with elevated levels of Elk, resulting in decreased cell proliferation, invasiveness, and colony formation, and increased rates of apoptosis/necrosis.14 A second such strategy took advantage of the fact that the large subunit of HSV-2 ribonucleotide reductase (R1, also known as ICP10) has a protein kinase domain that is not conserved in HSV-1, which activates the Ras signaling pathway. An HSV-2 mutant deleted in ICP10PK (known as ΔPK) preferentially replicates and lyses tumor cells through the activation of multiple death pathways.15 Finally, insertion of liver-specific micro-RNA (miRNA) binding sites into the 3′ untranslated region of the E1A gene of an oncolytic adenovirus eliminated its hepatotoxicity without compromising its anticancer activity.16 While miRNA targets can mutate during oncolysis, allowing for the formation of toxic escape variants, the risk can be reduced by dual targeting through addition of a transcriptional target (viz hTERT).17
Enhancing intratumoral penetration
A major limitation of oncolytic virotherapy is the relatively poor intratumoral penetration by the oncolytic viruses, which were modified to have a reduced replicative potential. Engineering of a cell-fusion capacity into the virus platform has been used to increase penetration. For example, a hyaluronidase-expressing oncolytic adenovirus showed improved spread and activity in a melanoma xenograft model, apparently related to improved lysis of extracellular matrix.18 Another strategy has been to inhibit antiviral proteins such as type I IFN and their receptors, which act directly against the virus and communicate with adjacent cells.19 These antiviral responses are partially inactivated in tumor cells, thereby allowing the oncolytic viruses to initiate infection, but their further inactivation is desirable. Infection with viruses that have complementing virulence may address this problem, as suggested by the finding that an IFN-sensitive oncolytic vesicular stomatitis virus replicated well in refractory tumor cells that were coinfected with an oncolytic poxvirus armed with a secreted IFN antagonist.20 This strategy carries the risk of recombination that generates a supervirus, but it is unlikely in this particular case because the viruses are RNA and DNA, respectively. Use of chemical molecules may also eliminate antiviral responses. For example, histone deacetylase inhibitors suppress the residual IFN responsiveness of tumor cells,21 and rapamycin potentiates the growth of several oncolytic viruses by disrupting the target or rapamycin complex-1-dependent production of IFN and/or the phosphatidylinositide 3-kinase pathway.22 Cyclophosphamide improves oncolytic virus efficacy by dampening the innate antiviral response, slowing the generation of neutralizing antibodies that target regulatory T cells and enhancing virus extravasation.23 However, the combination of viruses with drugs is not without risk, and it could promote off-target infections and unwanted pathology. Accordingly, the development of oncolytic viruses that inhibit the antiviral responses independent of virus replication, eg, through the induction of multiple and unrelated death programs, is particularly desirable. One such example is the HSV-2-based oncolytic virus ΔPK, which differs from the other oHSVs in that it takes advantage of the cellular programs (viz Ras) to ensure tumor selectivity, and it induces apoptosis, necroptosis, and autophagic cell death independent of its replicative potential.15
Oncolytic virus delivery and treatment of metastatic cancer
While current studies have emphasized intratumoral delivery, it is likely that systemic delivery will be required for the treatment of metastatic cancer. Important considerations in achieving this goal are to minimize virus sequestration in the liver and spleen, evade neutralization by serum factors, and increase vascular penetration through virus targeting to endothelial cells that line blood vessels and selectively enhance vessel permeability.
Minimize virus sequestration
Intravenously (IV) delivered virus is rapidly cleared from the circulation through sequestration by the mononuclear phagocytic system in the liver and spleen. Some viruses, such as adenoviruses, can bind scavenger receptors on Kupffer cells and induce proinflammatory cytokines that can result in serious dose-limiting toxicities.24 Because virus clearance is preceded by coating (opsonization) with antibodies, complement, coagulation factors, and/or other serum proteins that facilitate its recognition by splenic macrophages and hepatic Kupffer cells, strategies to minimize sequestration include chemical modification that interferes with this process. Conjugation of the viral protein coat with the biocompatible polymer polyethylene glycol (PEG), which prolongs protein and liposome circulation, was shown to slow clearance of adenovirus 5 (Ad5), increasing its oncolytic activity after IV delivery.25 The potential concern that PEGylation could reduce infectivity is addressed by the ability to restore infectivity through the reengineering of receptor-binding ligands onto the surface of the shielded particles. Oncolytic adenoviral therapy has also been combined with clodronate liposomes for depletion of Kupffer cells, and gadolinium chloride prolonged the circulatory half-life of an Ad5 vector, with a 100-fold difference in blood concentrations at 60 minutes.26 Alternative approaches to minimize virus sequestration include depletion of serum factors that bind the virus (eg, immunoglobulin M or complement proteins that bind HSV) by pretreatment with cobra venom factor or cyclophosphamide27 and saturation of scavenger receptors on mononuclear phagocytes by preconditioning with polyinosinic acid.
Protection from neutralizing serum factors
Surface coating or delivery by carrier cells also serves to protect viruses from neutralizing serum factors. Thus, conjugation with the biopolymer N-(2-hydroxypropyl)methacrylamide was shown to prolong the circulatory half-life of adenovirus.26 Also, oncolytic viruses systemically delivered by immune effector cells, including monocytes/macrophages, dendritic cells, and lymphocytes, reached the tumor even in the face of neutralizing antibodies.28 Moreover, hiding the oncolytic viruses inside carrier cells, such as mesenchymal stem cells that preferentially engraft into solid tumors, was successfully used in preclinical and small clinical trials.29,30 However, the use of stem and other cell types to deliver oncolytic viruses is still poorly evaluated.
Increase vascular penetration
Because the tumor vasculature is antigenically distinct, oncolytic viruses can be modified to target the tumor cell surface selectively, detarget sensitive tissues, or create dual-target viruses to enhance both vascular penetration and tumor infection. Adenovirus vectors encoding a fusogenic membrane glycoprotein driven by the human endothelial receptor tyrosine-kinase promoter triggered in vivo fusion between endothelial cells and epithelial cells, facilitating transendothelial virus penetration.31 Oncolytic measles virus expressing vascular targeting peptides, the N-terminal fragment of urokinase plasminogen activator, or cyclic arginine-glycine-aspartate (RGD) infected tumor vessel endothelial cells in vivo.32 Combination of vascular endothelial growth factor (VEGF) and metronomic doses of paclitaxel or cisplatin increased the vascular permeability of the tumor endothelium and improved the delivery of Sindbis virus vector to tumors.33 Also, multiple injections of VEGF165 of increased reovirus infection of proliferating tumor endothelium, thereby increasing therapeutic activity in a syngeneic B16 melanoma model.34
Immunity and virus clearance
Immunosuppressive drugs can retard immune clearance of oncolytic viruses, enhance persistence of virus infection, and prolong therapeutic efficacy,35 but a concern is toxicity. Oncolytic viruses represent a unique strategy for combining tumor lysis with potent activation of adaptive and innate immune responses, eg, by arming these viruses with proinflammatory cytokine, such as GM-CSF.3,4 Targeted infection of the tumor with viruses thus armed leads to a localized inflammatory response that facilitates immune recognition of cancer-specific neoantigens. Several groups are combining oncolytic viruses with adoptive cell immunotherapy designed to overcome the tumor-generated immunosuppressive activity, reasoning that virus-mediated tumor cell destruction should enhance the activity of the transferred cells.35 Of course, the activated immune system is likely to prematurely suppress the replication of the oncolytic virus,36 thereby further reducing the already problematic intratumoral penetration (see the Enhancing intratumoral penetration section, above). However, even limited infection of distant lymph-node metastases may lead to enhanced therapeutic benefit. In this context, oHSVs are particularly promising because they can evade immunity, and studies in animals and humans have shown that prior immunity is not an obstacle to systemic routes of administration.37
Lysis of cancer stem cells
CSCs are malignant cells that retain many of the properties of normal stem cells, including the ability to differentiate into multiple cell types, self-renew, proliferate, and maintain neoplastic clonality. They are resistant to traditional chemotherapy and radiotherapy, and have recently been identified as the cells that are most likely to be responsible for tumorigenesis, tumor maintenance, aggressiveness, and recurrence. Mechanisms used by CSCs to resist current therapies include efficient DNA-repair ability, upregulation of antiapoptotic genes, differential expression and phosphorylation of various kinases, increased expression of adenosine triphosphate-binding cassette transporters, and relative quiescence.1 A major challenge has been the ability to identify these cells and distinguish them from other tumor and normal cells in order to gain a better understanding of their biology, which would allow the development of novel targeted therapies to attack and kill CSCs. Current techniques to identify CSCs rely on an assortment of markers, including cell-surface, nuclear, or cytoplasmic proteins; transcription factors; enzymes; and/or functional attributes, notably the ability to grow in spheroid cultures, all of which offer potential strategies for targeting them.1
Direct studies of CSC lysis by oncolytic viruses are scant. Breast cancer CSCs marked by a unique phenotype are as sensitive to killing by reovirus as the total cell population.38 The oncolytic VV JX-594 killed most cells from five separate high-grade glioma cell lines grown as spheroids, which are enriched for CSCs,39 and an oncolytic VV delivered by ex vivo expanded natural killer T cells (cytokine-induced killer–oncolytic VV strain) killed murine lymphoma cells with stem-like features, including the ability to initiate tumors and resist chemotherapy and radiation.40 An adenovirus vector in which E1 expression is driven by the promoter of the antiapoptotic protein survivin effectively targeted cluster of differentiation (CD)-133+ glioma cells believed to be CSCs,41 but its therapeutic potential is unclear, because the expression of survivin in neural tumors is variable. Mahller et al42 used the rQNestin34.5 oHSV to infect and kill neuroblastoma cells, preventing tumor formation in nude mice, which presumably reflects CSC targeting. Another oHSV that carries an exogenous endo-angio fusion gene (endostatin and angiostatin are potent angiogenesis inhibitors) infected and killed glioma CSCs.43 An oHSV that produces chondroitinase adenosine triphosphate-binding cassette, a bacterial enzyme that removes the chondroitin sulfate from the tumor extracellular matrix proteoglycans, spread throughout glioma spheroids more efficiently than the unmodified virus, and had enhanced replication and antitumor activity in vivo.44 We recently found (unpublished) that the oncolytic virus ΔPK lyses breast cancer and melanoma CSCs at very low virus titers (0.1 plaque-forming units/cell) and without resistance development through autophagic cell death. Lysis is caused by calpain-mediated degradation of the selective autophagy protein p62/SQSTM1 that is implicated in tumor development.45 The importance of developing oncolytic viruses that can target death pathways with potentially CSC-specific activity, cannot be sufficiently emphasized.
Selection of the appropriate oncolytic virus
Given the diversity of the virus families, oncolytic platforms were originally developed with the expectation that they would be differentially suited to distinct malignancy classes. However, this has proven not to be the case, and most of the oncolytic viruses show a relatively broad spectrum of antitumor activity, typically against both epithelial and hematological malignancies. When viruses are engineered to target specific cell-surface receptors or nuclear transcription factors, their use is thereafter limited to tumors that express the relevant target, but so far there has been a preference for clinical translation of oncolytic viruses with broader host-range spectra. Safety considerations must drive the choice of a virus for a given indication. Different viruses have differing toxicities, and genetic manipulation may lead to unexpected toxicities. This was the case, for example, for a murine poxvirus in which interleukin 4 insertion led to 100% lethality in prevaccinated animals.46 Important factors to be considered in the safety analysis of oncolytic viruses include natural and engineered virus tropism, virus mutability and capacity for evolution and transmissibility, immunomodulatory, antiapoptotic, and cytotoxic gene products, prevalence of population-based antiviral immunity, and the availability of drugs or antisera to eliminate unwanted or persistent infections.
State of the art: examples from studied oncolytic viruses
Virtually all the virus families have been studied for oncolytic potential, and some have already reached the clinic. Specific examples of the currently available data for three virus families that have been studied in most detail are summarized below and in Table 1.
Adenovirus
Adenovirus is a nonenveloped, nonintegrated, double-stranded DNA virus that has been extensively studied for its oncolytic potential. Oncolytic adenoviruses can be mainly classified into two groups. One group shows tumor-selective replication via deletion of certain genes, such as E1B, which are dispensable for virus growth in tumor cells. The other group possesses an E1 gene-expression cassette driven by tumor-specific promoters, and induces a tumor-specific immune response through various cytokines by directing attacks at the tumor microenvironment and angiogenesis levels.47 Mutants deleted in the immediate–early (E1A) or early (E1B) genes do not bind the cellular proteins that are required for cell proliferation, a binding which is needed for productive infection in normal cells. However, these deletions do not affect virus replication in cancer cells, which have pathway defects such as p16/retinoblastoma or p53.
Most adenovirus vectors enter cells through the Coxsackie adenovirus receptor (CAR). This is a problem, because CAR expression is highly variable (to absent) in tumor cells, while it is high on normal epithelial cells, neurons, and astrocytes.47 To overcome this problem, mutants were designed to have modifications in the fiber knob of the viral capsid, thereby altering tropism and enabling infection of cancer cells through a CAR-independent mechanism, for example via integrins. Moreover, insertion of transgene-expression cassettes into the E3 region and the region between the fiber and E4, respectively (CRAd5/11D24.TRAIL/arresten), enhanced therapeutic potential with nearly complete inhibition of glioma growth in nude mice, possibly by increased antiangiogenesis and enhanced tumor apoptosis.48
Telomerase-specific replication-selective adenovirus (TRAD; also known as telomelysin) is one of the most promising oncolytic adenoviruses that were modified to drive E1 gene expression by tumor-specific promoters. The E1 gene cassette in TRAD is driven by the hTERT promoter, achieving tumor selectivity by virtue of the preferential expression of telomerase in cancer, relative to normal cells. TRAD has undergone a phase I clinical trial with no serious adverse events, but antitumor effects were limited to only several patients.7 Efficacy was enhanced by combination therapy with anticancer agents49 or through additional modifications. For example, insertion of apoptin suppressed the growth of gastric carcinoma cells in vitro and reduced the tumor burden in xenografted nude mice.50 TRAD has the advantage that it is disseminated from the injected tumors into the systemic circulation, resulting in infection of distant tumors.51 However, this property may be a double-edged sword, because normal cells throughout the body are also likely to be infected, causing unwanted toxicities. Indeed, TRAD replicates in normal human cells, probably because low levels of E1A expression suffice in order to support virus replication.52 To overcome this limitation, Sugio et al53 incorporated a miRNA-regulated gene-expression system that can prevent TRAD replication in normal human cells. Insertion of sequences complementary to miR-199a reduced replication in normal cells by approximately 30%–50% when compared with the conventional TRAD, except for hepatocytes, in which simultaneous insertion of both miR-199a and miR-122a caused an additional reduction in virus replication.53 Among some of the other promising adenovirus-based platforms that bear mention is an oncolytic adenovirus that overexpresses the tumor suppressor p53 that had profound antitumor activity.54
Several clinical trials of oncolytic adenoviruses gave rise to promising albeit modest results. A mutant that uses the cyclooxygenase (COX)-2 promoter to drive E1A deleted in 24 base pairs within the retinoblastoma-binding site (Ad5/3-Cox2L-D24) was used to treat a 6-year-old boy with metastatic neuroblastoma resistant to several chemotherapy regimens, including autologous transplantation. A 71% regression of the primary tumor and clearance of metastatic bone marrow disease were achieved through injection of 1011 virus particles into the primary tumor bed. Side effects included mild fever, diarrhea, stomach pains, and elevated liver enzymes that resolved in 2 weeks.55 A separate study of four patients with refractory metastatic neuroblastoma used a virus that had a deletion in E1A, a substitution of the E1A promoter for E2F-responsive elements, and an RGD-4C peptide motif inserted into the fiber protein in order to enhance tumor tropism. The virus was delivered IV by autologous mesenchymal stem cells. It was tolerated well, with only fever seen in three patients and elevated liver alanine aminotransferase in one patient that resolved in 96 hours. A very good partial response was seen in one of the patients.56 In a matched case-control study of advanced cancer patients, the calcium-channel blocker verapamil significantly increased the mean titers of circulating virus without increasing the frequency or severity of adverse events, but there were no significant differences in clinical response or survival.57
Vaccinia virus
VV is a double-stranded, enveloped DNA virus that was first used as a smallpox vaccine. Oncolytic mutants are deleted in both copies of the thymidine kinase gene,58 which provides tumor selectivity because dividing cells have higher levels of thymidine triphosphate that are required for DNA synthesis. Oncolytic VVs are also deleted in the B18R gene, which counteracts type I IFNs, resulting in IFN-mediated virus inactivation in normal cells.59 Several viruses that target cancer-specific antigens and/or induce an immune response through expression of antiangiogenesis proteins or cytokines were also developed and used in clinical trials.60 The oncolytic VV GLV-1h68 had significant therapeutic potential for lymph-node metastases of human prostate carcinoma in tumor xenografts. This was associated with increased vascular permeability in lymph-node metastases, an increased number of immune cells (major histocompatibility complex Class II [MHCII]+/CD68+ macrophages and MHCII+/CDl9+ B lymphocytes), and upregulated expression of proinflammatory cytokines.61 Combination therapy with radiation improved long-term regression rates in xenografts of head and neck cancer,62 but the exact contribution of the virus over and above the radiation therapy is still a concern. Concerns also relate to the efficiency of systemic delivery of oncolytic VV platforms, but recent studies of the JX-784 oncolytic virus have shown efficient systemic delivery.63 Sequential combination of immunologically distinct viruses might enhance antitumor efficacy through the induction of tumor-specific immunity and circumvention/mitigation of antiviral immune responses. Indeed, recent findings indicate that three low doses of adenovirus followed by three low doses of VV resulted in superior antitumor efficacy (62.5% of animals becoming tumor-free) than that seen with six doses of either virus alone.64 However, this approach is subject to correct selection of the virus dose and treatment order and requires a better understanding of the mechanism of action of each one of the virus platforms. Moreover, while potentially promising, this strategy carries the inherent risk of recombination between the two viruses, leading to the generation of a supervirus and undesirable toxicities.
Herpes simplex virus
HSV is a double-stranded, enveloped DNA neurotropic virus that has shown promise in treating a variety of malignancies, including brain tumors, neuroblastoma, and sarcoma. HSV has many advantages over other viruses for cancer gene therapy. It has a broad host range and high efficiency of infection. Because a large portion (up to 30 kb) of the HSV genome is nonessential for virus replication in cancer cells, it can be substituted with foreign DNA that enhances oncolytic efficacy, eg, by restoring the neurovirulence gene under control of a tumor-specific promoter, by producing enzymes that disrupt and inhibit the tumor microenvironment, or by generating cytokines that stimulate an immune response against the tumor. The HSV genome does not integrate into the host genome, eliminating concerns of insertional mutagenesis and antiviral drugs are currently available to control infection should anything go wrong. Most oHSVs are based on HSV-1 and are typically deleted in ICP34.5 and R1. lCP34.5 is a multifunctional virulence factor that inhibits cellular antiviral responses by counteracting the ability of the double-stranded RNA-dependent protein kinase (PKR) to inhibit translation.65 Because PKR activity is reduced in tumor cells, ICP34.5 deletion confers tumor-selective growth. However, loss of ICP34.5 may contribute to relatively poor tumor penetration, as suggested by the finding that an oHSV that retains the PKR-inhibiting GADD34 domain of ICP34.5 had superior oncolytic activity in glioblastoma than an oncolytic virus in which the entire ICP34.5 was deleted.66 R1 (also known as ICP6 in HSV-1 and ICP10 in HSV-2) is the large subunit of the viral ribonucleotide reductase. Its deletion confers tumor-selective virus growth because tumor cells have high levels of ribonucleotide reductase, but the activity is required for viral DNA replication in normal and quiescent cells. Additional oHSVs are deleted in ICP47, which is responsible for immune evasion and/or are armed with a proinflammatory cytokine gene, such as interleukin 12, or GM-CSF that elicits an immune response via activation of natural killer and T cells.37
oHSVs kill a wide variety of nonneural cancers, including sarcomas, melanomas, colon, breast, lung, prostate, and hepatic tumors, and several adult human studies have demonstrated safety and antitumor effects.5 Preclinical studies in neonatal mice and New World owl monkeys suggest that engineered HSV is safe,67 and efficacy was demonstrated preclinically in gliomas, medulloblastomas, and neuroblastomas. HSV176, which is deleted in both copies of ICP34.5 and G207 and also has an insertional mutation in R1, was used safely without any dose-limiting toxicities in adult patients with recurrent glioblastoma multiforme.68 However, clinical efficacy was relatively modest, apparently due to poor intratumoral penetration caused by inefficient virus replication in the tumor cells. One approach to overcome this problem has been to establish an HSV-retargeting system that relies on the combination of two engineered viral glycoproteins, gD and gB, to mediate highly efficient HSV infection exclusively through recognition of the abundantly expressed epidermal growth-factor receptor on glioblastoma cells. Using this approach, treatment of orthotopic primary human glioblastoma xenografts demonstrated prolonged survival, with up to 73% of animals showing a complete response, as confirmed by magnetic resonance imaging.69
To increase tumor selectivity and enhance viral replication, Kambara et al70 developed rQNestin34.5, which expresses lCP34.5 under control of a synthetic nestin promoter, and HSV RQT3 was constructed to have deletions in R1 and ICP34.5 and also express the inhibitor of metalloproteinases 3. RQT3-treated neuroblastoma and peripheral nerve-sheath tumor xenografts showed delayed tumor growth, had reduced vascular density, and decreased circulating endothelial progenitors, indicating a possible antiangiogenic effect of the virus.71 However, within hours of infection, activated natural killer cells were recruited to the site of infection through ligand upregulation, and they coordinated macrophage and microglia activation within the glioblastomas, thereby substantially diminishing the therapeutic efficacy.72
The duration and extent of HSV infection in vivo are still largely dictated by host IFN activity status. A recent study designed to overcome IFN antiviral defense mechanisms used the B18R gene from the vaccinia virus, which encodes a secreted decoy receptor with a broad antagonizing effect against type I IFNs. The virus, known as Synco-B18R, largely retained its oncolytic activity in the presence of increased IFN levels in vitro. When injected intratumorally, Synco-B18R showed significantly greater oncolytic activity than the parent oncolytic virus.73 Cell-growth inhibition by pathways independent of virus replication is likely to contribute to virus oncolytic activity, likely by increasing tumor penetration. Indeed, therapeutic efficacy in mouse xenograft models of squamous cell carcinoma and ovarian and breast cancer was significantly increased by insertion of inhibitor of growth 4, associated with at least 1000-fold more infectious virus found in the tumors,74 and efficacy was also increased by combinatorial therapy with oHSVs and the alkylating agent temozolomide.75
Future directions
Over the past decade, several viruses have been translated from the laboratory to the clinic, and several more viruses are likely to be used in clinical trials in the future. Each virus has unique benefits and limitations as an oncolytic agent, which may help to determine how it is used. This review focused on some of these viruses in order to exemplify the current state of the art. While numerous engineering modifications have already been done, these have yielded limited results in terms of clinical efficacy, and strategies are still needed to improve virus delivery, tumor specificity and penetration, reduce virus clearance, and increase the tumor-directed immune response. Combination therapy with drugs, radiation, monoclonal antibodies, small-molecule inhibitors, and/or other oncolytic viruses can have increased efficacy. For example, viruses can sensitize cells to radiation, and radiation can enhance viral infection, replication, and gene expression, resulting in greater tumor cell death.76 Monoclonal antibodies and small-molecule inhibitors can complement oncolytic virotherapy by altering regulatory pathways, increasing viral replication, and enhancing the induction of apoptosis.77 However, such combinatorial approaches carry the risk of unwanted severe toxicities. Better evaluation of oncolytic potential against CSCs is needed, as is the development of oncolytic viruses that kill tumor cells and CSCs not only through virus replication but also through the induction of multiple death pathways and the stimulation of enhanced antitumorigenic immune responses. In this context, the oHSV ΔPK developed in our laboratory is particularly promising, because its oncolytic potential includes the lysis of CSCs and it involves multiple death pathways, including apoptosis, necroptosis, and autophagic cell death,11 as well as the activation of innate immunity (unpublished data). However, additional studies are needed to elucidate better the host range of the ΔPK virus and determine its ability to function in the treatment of metastatic cancer.
Summary
Oncolytic virotherapy is a new strategy to reduce tumor burden through selective virus replication in rapidly proliferating cells.
Oncolytic viruses belong to various families. They have undergone different engineered modifications, which are designed to enhance tumor selectivity, improve intratumoral replication, modify immune responses, enhance vascular delivery, and express antitumorigenic genes that function independently of virus replication.
Recent advances include preclinical proof of feasibility, the development of strategies to monitor virus spread and detect micrometastatic tumor cells, and clinical trials that document tolerability and effectiveness.
Current efforts have favored intratumoral delivery, but systemic delivery is likely required for the treatment of metastatic disease. Strategies to increase specific tumor penetration, minimize virus sequestration in the liver and spleen, evade neutralization by serum factors, and enhance vascular penetration are under investigation.
In addition to developing more meaningful animal models, key challenges are to select “winners” from the distinct oncolytic platforms that can stimulate anticancer immunity without affecting virus replication, lyse CSCs, and prevent the emergence of resistant tumor cells.
Footnotes
Disclosure
The author reports no conflicts of interest in this work.
References
- 1.Visvader LE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Zeyaullah M, Patro M, Ahmad I, et al. Oncolytic viruses in the treatment of cancer: a review of current strategies. Pathol Oncol Res. 2012;18:771–781. doi: 10.1007/s12253-012-9548-2. [DOI] [PubMed] [Google Scholar]
- 3.Senzer NN, Kaufman HL, Amatruda T, et al. Phase II clinical trial of a granulocyte-macrophage Colony- stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27:5763–5771. doi: 10.1200/JCO.2009.24.3675. [DOI] [PubMed] [Google Scholar]
- 4.Park BH, Hwang T, Liu TC, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9:533–542. doi: 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- 5.Harrington KJ, Hingorani M, Tanay MA, et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res. 2010;16:4005–4015. doi: 10.1158/1078-0432.CCR-10-0196. [DOI] [PubMed] [Google Scholar]
- 6.Barton KN, Stricker H, Brown SL, et al. Phase I study of noninvasive imaging of adenovirus-mediated gene expression in the human prostate. Mol Ther. 2008;16:1761–1769. doi: 10.1038/mt.2008.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kishimoto H, Urata Y, Tanaka N, Fujiwara T, Hoffman RM. Selective metastatic tumor labeling with green fluorescent protein and killing by systemic administration of telomerase-dependent adenoviruses. Mol Cancer Ther. 2009;8:3001–3008. doi: 10.1158/1535-7163.MCT-09-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kishimoto H, Zhao M, Hayashi K, et al. In vivo internal tumor illumination by telomerase-dependent adenoviral GFP for precise surgical navigation. Proc Natl Acad Sci U S A. 2009;106:14514–14517. doi: 10.1073/pnas.0906388106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guse K, Dias JD, Bauerschmitz GJ, et al. Luciferase imaging for evaluation of oncolytic adenovirus replication in vivo. Gene Ther. 2007;14:902–911. doi: 10.1038/sj.gt.3302949. [DOI] [PubMed] [Google Scholar]
- 10.Huyn ST, Burton JB, Sato M, Carey M, Gambhir SS, Wu L. A potent, imaging adenoviral vector driven by the cancer-selective mucin-1 promoter that targets breast cancer metastasis. Clin Cancer Res. 2009;15:3126–3134. doi: 10.1158/1078-0432.CCR-08-2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nemunaitis I, Tong AW, Nemunaitis M, et al. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol Ther. 2010;18:429–434. doi: 10.1038/mt.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shashkova EV, May SM, Doronin K, Barry MA. Expanded anticancer therapeutic window of hexon-modified oncolytic adenovirus. Mol Ther. 2009;17:2121–2130. doi: 10.1038/mt.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muik A, Kneiske I, Werbizki M, et al. Pseudotyping vesicular stomatitis virus with lymphocytic choriomeningitis virus glycoproteins enhances infectivity for glioma cells and minimizes neurotropism. J Virol. 2011;85:5679–5684. doi: 10.1128/JVI.02511-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan W, Bodempudi V, Esfandyari T, Farassati F. Utilizing ras signaling pathway to direct selective replication of herpes simplex virus-1. PLoS One. 2009;4:e6514. doi: 10.1371/journal.pone.0006514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colunga AG, Laing M, Aurelian L. The HSV-2 mutant DeltaPK induces melanoma oncolysis through nonredundant death programs and associated with autophagy and pyroptosis proteins. Gene Ther. 2010;17:315–327. doi: 10.1038/gt.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cawood R, Chen HH, Carroll F, Bazan-Peregrino M, van Rooijen N, Seymour LW. Use of tissue-specific microRNA to control pathology of wild-type adenovirus without attenuation of its ability to kill cancer cells. PLoS Pathog. 2009;5:e1000440. doi: 10.1371/journal.ppat.1000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugio K, Sakurai F, Katayama K, et al. Enhanced safety profiles of the telomerase-specific replication-competent adenovirus by incorporation of normal cell-specific microRNA-targeted sequences. Clin Cancer Res. 2011;17:2807–2818. doi: 10.1158/1078-0432.CCR-10-2008. [DOI] [PubMed] [Google Scholar]
- 18.Guedan S, Rojas JJ, Gros A, Mercade E, Cascallo M, Alemany R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol Ther. 2010;18:1275–1283. doi: 10.1038/mt.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schoggins JW, Wilson SJ, Panis M, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Boeuf F, Diallo JS, McCart JA, et al. Synergistic interaction between oncolytic viruses augments tumor killing. Mol Ther. 2010;18:888–895. doi: 10.1038/mt.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyên TL, Abdelbary H, Arguello M, et al. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc Natl Acad Sci U S A. 2008;105:14981–14986. doi: 10.1073/pnas.0803988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alain T, Lun X, Martineua Y, et al. Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc Natl Acad Sci U S A. 2010;107:1576–1581. doi: 10.1073/pnas.0912344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiao J, Wang H, Kottke T, et al. Cyclophospharnide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin Cancer Res. 2008;14:259–269. doi: 10.1158/1078-0432.CCR-07-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haisma HJ, Boesjes M, Beerens AM, et al. Scavenger receptor A: a new route for adenovirus 5. Mol Pharm. 2009;6:366–374. doi: 10.1021/mp8000974. [DOI] [PubMed] [Google Scholar]
- 25.Doronin K, Shashkova EV, May SM, Hofherr SE, Barry MA. Chemical modification with high molecular weight polyethylene glycol reduces transduction of hepatocytes and increases efficacy of intravenously delivered oncolytic adenovirus. Hum Gene Ther. 2009;20:975–988. doi: 10.1089/hum.2009.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green NK, Herbert CW, Hale SJ, et al. Extended plasma circulation time and decreased toxicity of polymer-coated adenovirus. Gene Ther. 2004;11:1256–1263. doi: 10.1038/sj.gt.3302295. [DOI] [PubMed] [Google Scholar]
- 27.Ikeda K, Wakimoto H, Ichikawa T, et al. Complement depletion facilitates the infection of multiple brain tumors by an intravascular, replication-conditional herpes simplex virus mutant. J Virol. 2000;74:4765–4775. doi: 10.1128/jvi.74.10.4765-4775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dwyer RM, Khan S, Barry FP, O’Brien T, Kerin MJ. Advances in mesenchymal stem cell-mediated gene therapy for cancer. Stem Cell Res Ther. 2010;1:25. doi: 10.1186/scrt25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ilett EJ, Barcena M, Errington-Mais F, et al. Internalization of oncolytic reovirus by human dendritic cell carriers protects the virus from neutralization. Clin Cancer Res. 2011;17:2767–2776. doi: 10.1158/1078-0432.CCR-10-3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iankov ID, Blechacz B, Liu C, et al. Infected cell carriers: a new strategy for systemic delivery of oncolytic measles viruses in cancer virotherapy. Mol Ther. 2007;15:114–122. doi: 10.1038/sj.mt.6300020. [DOI] [PubMed] [Google Scholar]
- 31.Chen HH, Cawood R, El-Sherbini Y, et al. Active adenoviral vascular penetration by targeted formation of heterocellular endothelial-epithelial syncytia. Mol Ther. 2011;19:67–75. doi: 10.1038/mt.2010.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ong HT, Trejo TR, Pham LD, Oberg AL, Russell SJ, Peng KW. Intravascularly administered RGD-displaying measles viruses bind to and infect neovessel endothelial cells in vivo. Mol Ther. 2009;17:1012–1021. doi: 10.1038/mt.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tseng JC, Granot T, DiGiacomo V, Levin B, Meruelo D. Enhanced specific delivery and targeting of oncolytic Sindbis viral vectors by modulating vascular leakiness in tumor. Cancer Gene Ther. 2010;17:244–255. doi: 10.1038/cgt.2009.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kottke T, Hall G, Pulido J, et al. Antiangiogenic cancer therapy combined with oncolytic virotherapy leads to regression of established tumors in mice. Clin Invest. 2010;120:1551–1560. doi: 10.1172/JCI41431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. 2011;6:1008–1016. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diaz RM, Galivo F, Kottke T, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- 37.Todo T. Active immunotherapy: oncolytic virus therapy using HSV-1. Adv Exp Med. 2012;746:178–186. doi: 10.1007/978-1-4614-3146-6_14. [DOI] [PubMed] [Google Scholar]
- 38.Marcato P, Dean CA, Giacomantonio CA, Lee PW. Oncolytic reovirus effectively targets breast cancer stem cells. Mol Ther. 2009;17:972–979. doi: 10.1038/mt.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lun X, Chan I, Zhou H, et al. Efficacy and safety/toxicity study of recombinant vaccinia virus JX-594 in two immunocompetent animal models of glioma. Mol Ther. 2010;18:1927–1936. doi: 10.1038/mt.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Contag CH, Sikorski R, Negrín RS, et al. Definition of an enhanced immune cell therapy in mice that can target stem-like lymphoma cells. Cancer Res. 2010;70:9837–9845. doi: 10.1158/0008-5472.CAN-10-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nandi S, Ulasov IV, Tyler MA, et al. Low-dose radiation enhances survivin-mediated virotherapy against malignant glioma stem cells. Cancer Res. 2008;68:5778–5784. doi: 10.1158/0008-5472.CAN-07-6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahller YY, Williams IP, Baird WH, et al. Neuroblastoma cell lines contain pluripotent tumor initiating cells that are susceptible to a targeted oncolytic virus. PLoS One. 2009;4:e4235. doi: 10.1371/journal.pone.0004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu G, Su W, Jin G, et al. Glioma stem cells targeted by oncolytic virus carrying endostatin-angiostatin fusion gene and the expression of its exogenous gene in vitro. Brain Res. 2011;1390:59–69. doi: 10.1016/j.brainres.2011.03.050. [DOI] [PubMed] [Google Scholar]
- 44.Dmitrieva N, Yu L, Viapiano M, et al. Chondroitinase ABC I- mediated enhancement of oncolytic virus spread and antitumor efficacy. Clin Cancer Res. 2011;17:1362–1372. doi: 10.1158/1078-0432.CCR-10-2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inoue D, Suzuki T, Mitsuishi Y, et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012;103:760–766. doi: 10.1111/j.1349-7006.2012.02216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen N, Bellone CJ, Schriewer J, et al. Poxvirus interleukin-4 expression overcomes inherent resistance and vaccine-induced immunity: pathogenesis, prophylaxis, and antiviral therapy. Virology. 2011;409:328–337. doi: 10.1016/j.virol.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Short H, Curiel DT. Oncolytic adenoviruses targeted to cancer stem cells. Mol Cancer Ther. 2009;8:2096–2102. doi: 10.1158/1535-7163.MCT-09-0367. [DOI] [PubMed] [Google Scholar]
- 48.Li X, Mao O, Wang D, ZiieirieI W, Xia H. A fiber chimeric CRAd vector Ad5/11-D24 double-armed with TRAIL and arresten for enhanced glioblastoma therapy. Hum Gene Ther. 2012;23:589–596. doi: 10.1089/hum.2011.130. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe T, Hioki M, Fujiwara T, et al. Histone deacetylase inhibitor FR901228 enhances the antitumor effect of telomerase-specific replication-selective adenoviral agent OBP-301 in human lung cancer cells. Exp Cell Res. 2006;312:256–265. doi: 10.1016/j.yexcr.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 50.Liu L, Wu W, Zhu G, et al. Therapeutic efficacy of an hTERT promoter-driven oncolytic adenovirus that expresses apoptin in gastric carcinoma. Int J Mol Med. 2012;30:747–754. doi: 10.3892/ijmm.2012.1077. [DOI] [PubMed] [Google Scholar]
- 51.Taki M, Kagawa S, Nishizaki M, et al. Enhanced oncolysis by a tropism-modified telomerase-specific replication-selective adenoviral agent (‘Telomelysin-RGD’) Oncogene. 2005;24:3130–3140. doi: 10.1038/sj.onc.1208460. [DOI] [PubMed] [Google Scholar]
- 52.Hitt MM, Graham FL. Adenovirus E1A under the control of heterologous promoters: wide variation in E1A expression levels has little effect on virus replication. Virology. 1990;179:667–678. doi: 10.1016/0042-6822(90)90134-d. [DOI] [PubMed] [Google Scholar]
- 53.Sugio K, Sakurai F, Katavama K, et al. Enhanced safety profiles of the telomerase-specific replication-competent adenovirus by incorporation of normal cell-specific microRNA-targeted sequences. Clin Cancer Res. 2011;17:2807–2818. doi: 10.1158/1078-0432.CCR-10-2008. [DOI] [PubMed] [Google Scholar]
- 54.Yamasaki Y, Tazawa H, Hashimoto Y, et al. A novel apoptotic mechanism of genetically engineered adenovirus-mediated tumour-specific p53 overexpression through E1A-dependent p21 and MDM2 suppression. Eur J Cancer. 2012;48:2282–2291. doi: 10.1016/j.ejca.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 55.Pesonen S, Helin H, Nokisalmi P, et al. Oncolytic adenovirus treatment of a patient with refractory neuroblastoma. Acta Oncol. 2010;49:117–119. doi: 10.3109/02841860903071369. [DOI] [PubMed] [Google Scholar]
- 56.García-Castro J, Alemany R, Cascallo M, et al. Treatment of metastatic neuroblastoma with systemic oncolytic virotherapy delivered by autologous mesenchymal stem cells: an exploratory study. Cancer Gene Ther. 2010;17:476–483. doi: 10.1038/cgt.2010.4. [DOI] [PubMed] [Google Scholar]
- 57.Koski A, Raki M, Nokisalmi P, et al. Verapamil results in increased blood levels of oncolytic adenovirus in treatment of patients with advanced cancer. Mol Ther. 2012;20:221–229. doi: 10.1038/mt.2011.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCart JA, Ward JM, Lee J, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61:875l–8757. [PubMed] [Google Scholar]
- 59.Kirn DH, Wang Y, Le Boeuf F, Bell I, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4:e353. doi: 10.1371/journal.pmed.0040353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guse K, Cerullo V, Hemminki A. Oncolytic vaccinia virus for the treatment of cancer. Expert Opin Biol Ther. 2011;11:595–608. doi: 10.1517/14712598.2011.558838. [DOI] [PubMed] [Google Scholar]
- 61.Donat U, Weibel S, Hess M, et al. Preferential colonization of metastases by oncolytic vaccinia virus strain GLV-1h68 in a human PC-3 prostate cancer model in nude mice. PLoS One. 2012;7:e45942. doi: 10.1371/journal.pone.0045942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mansfield D, Pencavel T, Kyula JN, et al. Oncolytic vaccinia virus and radiotherapy in head and neck cancer. Oral Oncol. 2013;49:108–118. doi: 10.1016/j.oraloncology.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 63.Breitbach CJ, Burke J, Jonker D, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99–102. doi: 10.1038/nature10358. [DOI] [PubMed] [Google Scholar]
- 64.Tysome JR, Li X, Wang S, et al. A novel therapeutic regimen to eradicate established solid tumor with an effective induction of tumor-specific immunity. Clin Cancer Res. 2012;18:6679–6689. doi: 10.1158/1078-0432.CCR-12-0979. [DOI] [PubMed] [Google Scholar]
- 65.Orvedahl A, Alexander D, Tallóczy Z, et al. HSV-1 ICP345 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 66.Kanai R, Zaupa C, Sgubin D, et al. Effect of γ34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J Virol. 2012;86:4420–4431. doi: 10.1128/JVI.00017-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hunter WD, Martuza RL, Feigenbaum F, et al. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation of intracerebral injection in nonhuman primates. J Virol. 1999;73:6319–6326. doi: 10.1128/jvi.73.8.6319-6326.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rampling R, Cruickshank G, Papanastassiou V, et al. Toxicity evaluation of replication-competent herpes simplex virus ICP 34.5 null mutant 1716 in patients with recurrent malignant glioma. Gene Ther. 2000;7:859–866. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 69.Uchida H, Marzulli M, Nakano K, et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol Ther. 2013;21:561–569. doi: 10.1038/mt.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kambara H, Okano H, Chiocca EA, Saeki Y. An oncolytic HSV-1 mutant expressing ICP345 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005;65:2832–2839. doi: 10.1158/0008-5472.CAN-04-3227. [DOI] [PubMed] [Google Scholar]
- 71.Mahller YY, Vaikunth SS, Ripberger MC, et al. Tissue inhibitor of metalloproteinase-3 via oncolytic herpesvirus inhibits tumor growth and vascular progenitors. Cancer Res. 2008;68:1170–1179. doi: 10.1158/0008-5472.CAN-07-2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alvarez-Breckenridge CA, Yu I, Price R, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med. 2012;18:1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fu X, Rivera A, Tao L, Zhang X. Incorporation of the B18R gene of vaccinia virus into an oncolytic herpes simplex virus improves antitumor activity. Mol Ther. 2012;20:1871–1881. doi: 10.1038/mt.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Conner I, Braidwood L. Expression of inhibitor of growth 4 by HSV1716 improves oncolytic potency and enhances efficacy. Cancer Gene Ther. 2012;19:499–507. doi: 10.1038/cgt.2012.24. [DOI] [PubMed] [Google Scholar]
- 75.Kanai R, Rabkin SD, Yin S, et al. Oncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cells. J Natl Cancer Inst. 2012;104:42–55. doi: 10.1093/jnci/djr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Touchefeu Y, Vassaux G, Harrington KI. Oncolytic viruses in radiation oncology. Radiother Oncol. 2011;99:262–270. doi: 10.1016/j.radonc.2011.05.078. [DOI] [PubMed] [Google Scholar]
- 77.Nguyen TL, Tumilasci VF, Singhroy D, Arguello M, Hiscott I. The emergence of combinatorial strategies in the development of RNA oncolytic virus therapies. Cell Microbiol. 2009;11:889–897. doi: 10.1111/j.1462-5822.2009.01317.x. [DOI] [PubMed] [Google Scholar]