

Abstract
Many drugs have been developed and optimized for the treatment of cancer; however, it is difficult to completely cure cancer with anticancer drugs alone. Therefore, the development of new therapeutic technologies, in addition to new anticancer drugs, is necessary for more effective oncotherapy. Oncolytic viruses are one potential new anticancer strategy. Various oncolytic viruses have been developed for safe and effective oncotherapy. Recently, Sendai virus-based oncotherapy has been reported by several groups, and attention has been drawn to its unique anticancer mechanisms, which are different from those of the conventional oncolytic viruses that kill cancer cells by cancer cell-selective replication. Here, we introduce Sendai virus-based virotherapy and its anticancer mechanisms.
Keywords: HVJ-E, cancer therapy, apoptosis, necroptosis, anticancer immunity
Introduction
It is generally thought that a virus is a health threat. When a virus infects host cells, it begins to increase its viral copies by replicating its genome and producing viral proteins for progeny viruses. Although host cells have several mechanisms to resist viral amplification, eg, innate immunity or apoptosis, some viruses can overcome these systems and lyse the infected cells when the viral copies reach a certain number.
Oncolytic virotherapy is based on the aforementioned viral features. Until now, many researchers have reported various oncolytic viruses that selectively replicate in cancer cells. Reovirus, Edmonston strain of measles virus, E1B-deficient adenovirus, and Newcastle disease virus are naturally discovered oncolytic viruses.1–3 Other oncolytic viruses have been developed by viral genome engineering. Herpes simplex virus-1 deleting some viral genes such as γ34.5, ICP6, and ICP47 has been reported as an effective virotherapeutic tool.2 Moreover, armed-type oncolytic viruses having therapeutic genes, such as granulocyte–monocyte-colony stimulating factor, are more powerful for cancer treatment.4,5
As observed earlier, various oncolytic viruses have potential as safe and effective oncotherapeutics that work in a cancer cell-selective fashion. However, the mechanism of cancer cell death induction is dependent on excess intracellular viral propagation. Sendai virus has recently received attention as a new tool for virotherapy. The oncolytic mechanism of Sendai virus differs from that of conventional oncolytic viruses. Therefore, we herein introduce Sendai virus-based virotherapy.
Sendai virus
Sendai virus is a paramyxovirus and is also known as hemagglutinating virus of Japan (HVJ). HVJ has a negative-sense single-stranded RNA genome encoding the following six viral structural proteins: nucleocapsid protein (N), phosphoprotein (P), matrix protein (M), fusion protein (F), hemagglutinin–neuraminidase (HN), and large protein (L).6 N, P, M, and L form a complex with the viral genome, and the complex is covered with a host-derived envelope (Figure 1).7 By contrast, HN and F are on the surface of the viral envelope (Figure 1).7 HN binds to sialic acid on the surface of the host cell membrane and induces the adhesion of viral particles to the host cell surface.8 F catalyzes the membrane fusion of the viral envelope with the host cell membrane and enables the viral genome to be introduced into the host cell cytoplasm for HVJ infection.9 HVJ viral proteins are translated in the infected cell to generate progeny viruses. F is originally produced as the F0 form without a catalytic function, and the F0 form is changed to the F1/F2 form, which exhibits membrane fusion activity, by the cleavage of F with a protease.10,11 Human cells do not produce the protease for F activation; therefore, HVJ is unable to amplify infectious progeny viruses in humans. Therefore, few reports have demonstrated cancer therapy using HVJ as an oncolytic virus until recently because conventional oncolytic viruses require repeated infection of cancer cells and the generation of infectious progeny for effective anticancer activity. In fact, some reports show that ultraviolet (UV)-irradiated oncolytic viruses, which lack viral reproduction potential, are unable to induce effective oncolysis.12–15 However, the generation of recombinant HVJ, which specifically infects cancer cells by viral genome engineering, has recently been reported.16,17 Moreover, UV-irradiated HVJ, which is known as HVJ-envelope (HVJ-E), effectively induces cancer cell death and stimulates antitumor immunity, even if HVJ-E lacks the ability to replicate. Since these developments were reported, Sendai virus-based oncolytic viruses have drawn attention for their potential use as cancer therapeutics. Here, we discuss attenuated HVJ and HVJ-E and explain the mechanism of HVJ-E-mediated anticancer activity, including the induction of apoptosis and necroptosis in cancer cells and the stimulation of antitumor immunity (Table 1).
Figure 1.
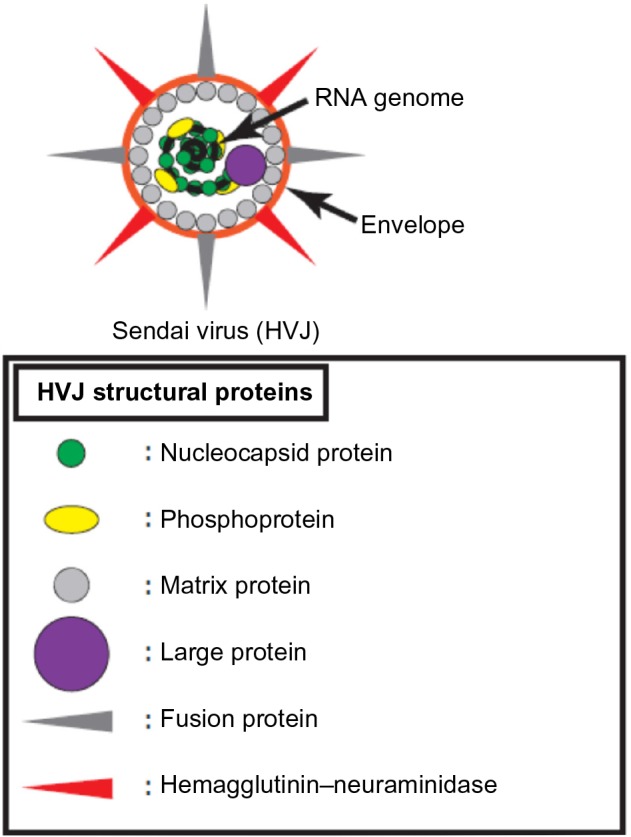
Structure of Sendai virus.
Abbreviation: HVJ, hemagglutinating virus of Japan.
Table 1.
Various oncolytic Sendai virus
| Oncolytic Sendai virus | Function |
|---|---|
| Genetic modified-Sendai virus | |
| SeV/ΔM | – Lack of production ability of infectious viral particles from infected cells |
| – Infection spread by cell–cell fusion of infected cells with uninfected cells | |
| MMP-sub SeV/ΔM | – Preferential infection to MMP-expressing cancer cells |
| uPA-sub SeV/ΔM | – Preferential infection to uPA-expressing cancer cells |
| Inactivated-Sendai virus | |
| UV-irradiated Sendai virus particle (HVJ-E) | – Induction of apoptosis in cancer cells by introduction of viral RNA genome fragments into cytoplasm |
| – Induction of necroptosis in cancer cell lack caspase-8 by HVJ-E membrane fusion | |
| – Activation of antitumor immunity (DCs stimulation, NK cells activation, Treg suppression) | |
| ΔHN-HVJ-E | – Low hemagglutinating activity |
| Tf/F-ΔHN-HVJ-E | – Transferrin receptor expressing cancer cell targeting |
| scIL12-HVJ-E | – Robust anticancer immune stimulation |
Abbreviations: ΔHN, hemagglutinin–neuraminidase-depleted; DCs, dendritic cells; HVJ-E, ultraviolet-irradiated hemagglutinating virus of Japan; MMP, matrix metalloproteinase; NK, natural killer; scIL12, single-chain interleukin-12; SeV/ΔM, recombinant hemagglutinating virus of Japan containing a viral genome lacking the M-coding region; sub, subtype; Tf/F, transferrin recombinant fusion protein; Treg, regulatory T-cell; uPA, urokinase-type plasminogen activator; UV, ultraviolet.
Genetically modified-Sendai virus
The generation of recombinant attenuated HVJ by genome engineering has recently been reported. Inoue et al generated recombinant HVJ containing a viral genome lacking the M-coding region (SeV/ΔM).18 SeV/ΔM exhibits the same infectivity as wild-type (wt) HVJ, but the production of mature viral particles is abolished in SeV/ΔM-infected cells. The SeV/ΔM infection spreads to neighboring cells by cell–cell fusion under trypsin treatment because the infected cells express F and HN. However, there is no protease for the activation of F in Homo sapiens, and thus the spread of SeV/ΔM across cancer tissues by reinfection is not expected in humans. To provide the SeV/ΔM infection with re-infectivity and cancer cell specificity, Kinoh et al generated a new SeV/ΔM containing a gene-encoding recombinant F that is cleaved by matrix metalloproteinases (MMPs) (MMP-sub SeV/ΔM).16 It has been shown that MMP expression and activity is increased in various types of human cancers.19 MMP-sub SeV/ΔM demonstrated significantly improved infectivity and decreased tumor volume compared with both in vitro and in vivo experiment with MMP-expressing cancer cells (HT1080). However, there are many cancers which lack MMP expression, and MMP-sub SeV/ΔM could not infect to such cancers. Kinoh et al generated another type of SeV/ΔM that is able to undergo fusion in the presence of urokinase-type plasminogen activator (uPA), and they reported high cytotoxicity against uPA-expressing cancer cells with the uPA subtype but not MMP-sub SeV/ΔM.17
UV-irradiated Sendai virus particles
Sendai virus particles (HVJ-E) are generated via the UV irradiation of HVJ, and the viral RNA genome is fragmented into many short RNAs by UV light.20 Therefore, HVJ-E maintains its membrane fusion activity but lacks the ability to replicate in infected cells. At first, HVJ-E was developed as a drug-delivery system for plasmids, siRNAs, proteins, and anticancer drugs due to its membrane fusion ability.21–24 However, it was recently reported that HVJ-E directly induces cancer cell death and the activation of antitumor immunity by itself; since this discovery, research has focused on the mechanism of the antitumor effects induced by HVJ-E treatment.
HVJ-E-induced apoptosis
Recently, many reports have shown that HVJ-E induces cancer cell apoptosis even though it is unable to replicate.25–29 Kawaguchi et al demonstrated that HVJ-E effectively induces apoptosis of castration-resistant human prostate cancer cell lines (PC3 and DU145) that express high levels of GD1a and SPG (HN receptors) on the surface but does not induce apoptosis of hormone-insensitive prostate cancer cell lines (LNCaP) that lack HN receptors.26 Therefore, it was suggested that HVJ-E-mediated cell apoptosis is dependent on the expression of HN receptors. However, despite the expression of GD1a and SPG, normal prostate epithelium (PNT2) does not undergo apoptosis following HVJ-E treatment. These results suggest that HVJ-E specifically induces apoptosis in cancer cells but not normal cells; however, the mechanism of this selectivity is unclear.
Recently, the molecular mechanism of HVJ-E-mediated cancer cell apoptosis was elucidated. Tanaka et al reported that sterile alpha motif-containing domain 9 (SAMD9) is responsible for HVJ-E-induced apoptosis in human glioblastoma cells (U251MG).25 They showed that HVJ-E treatment increases SAMD9 expression via type-I IFN and that SAMD9 upregulation induces U251 cell death. Moreover, Matsushima-Miyagi et al have reported more detailed mechanisms of HVJ-E-induced apoptosis in prostate cancer cells (Figure 2A).27 They demonstrated that HVJ-E stimulates retinoic acid-inducible gene I (RIG-I)/mitochondrial antiviral signaling protein signaling in PC3 and DU145, and this signaling enhances the expression levels of phorbol-12-myristate-13-acetate-induced protein 1 (Noxa) and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) via IRF3 and IRF 7. Noxa and TRAIL are known apoptosis inducers,30,31 and these factors induce apoptosis in HVJ-E-treated cancer cells. HVJ-E has many viral RNA fragments derived from the UV-irradiated viral genome, and the RNA fragments are introduced into the cancer cell cytoplasm by HVJ-E membrane fusion. The RNA fragments are recognized by RIG-I in cancer cells, and this recognition triggers the signaling that leads to the induction of apoptosis. Therefore, it has been suggested that RIG-I is a key factor for HVJ-E-mediated cancer cell apoptosis. In fact, HVJ-E-treated cancer cells have elevated levels of RIG-I,26,27,32 and RIG-I knockdown inhibits HVJ-E-mediated cancer cell apoptosis.27 However, HVJ-E-mediated apoptosis is not detected in normal cells because proapoptotic genes are not induced, although the RIG-I expression level of normal cells is higher than in cancer cells under both basal and stimulated conditions in various tissue-derived cells.33 Besch et al also reported that synthetic RNA induces cancer-cell-selective apoptosis via the RIG-I pathway.34 In their report, apoptotic genes such as PUMA and Noxa were activated by synthetic RNA in both melanoma cells and nonmalignant skin cells, but the antiapoptotic gene BCL-xL was upregulated by the RNA only in nonmalignant cells. Thus, the mechanism of RNA-induced apoptosis in cancer cells is controversial and remains to be further elucidated. Moreover, it remains unknown why HVJ-E specifically induces the expression of proapoptotic genes in cancer cells but not in noncancerous cells, and the elucidation of this mechanism will facilitate the understanding of the difference between cancer cells and normal cells.
Figure 2.
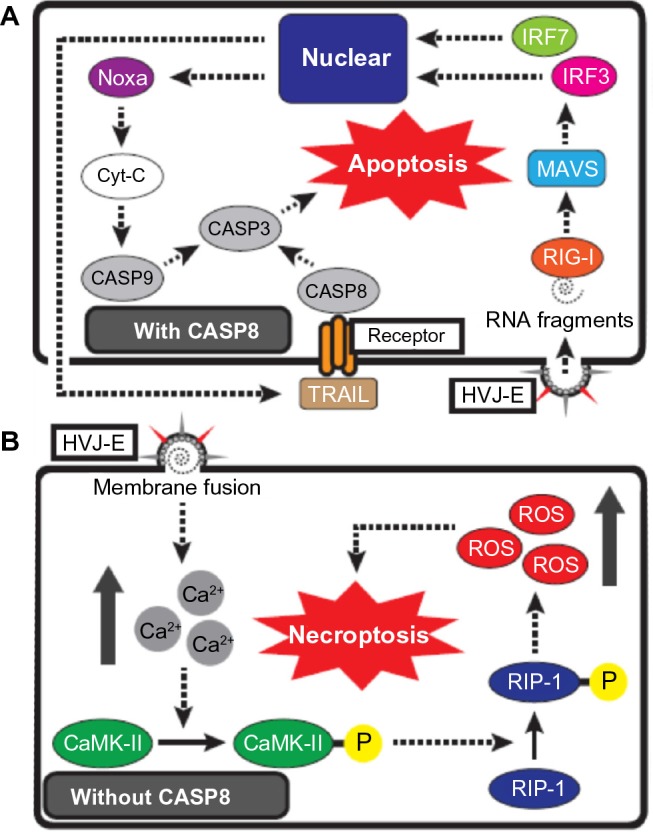
Schematic representation of HVJ-E-mediated cancer cell death.
Notes: (A) HVJ-E-mediated apoptosis signaling in cancer cells with CASP8. RNA fragments, which are introduced by HVJ-E membrane fusion, are recognized by RIG-I, and the signaling induces the transcription of TRAIL and Noxa by the activation of IRF7 and IRF3 via the RIG-I/MAVS pathway. TRAIL activates CASP8 via TRAIL receptor binding, and Noxa activates CASP9 via Cyt-C release from mitochondria. Activation of CASP8 and CASP9 results in cancer cell apoptosis via the subsequent activation of CASP3. (B) HVJ-E-mediated necroptosis signaling in cancer cells without CASP8. HVJ-E membrane fusion elevates the cytoplasmic Ca2+ level, and Ca2+ induces the phosphorylation of CaMK II. CaMK-II-P phosphorylates RIP1, and RIP1-P induces cancer cell necroptosis by elevating the levels of intracellular ROS.
Abbreviations: CaMK-II-P, phosphorylated CaMK II; CASP, caspase; Cyt-C, cytochrome-C; HVJ-E, ultraviolet-irradiated hemagglutinating virus of Japan; RIP1-P, phosphorylated RIP1; ROS, reactive oxygen species; TRIAL, TNF-related apoptosis-inducing ligand.
HVJ-E-induced necroptosis
As described earlier, various HVJ-E-treated cancer cells undergo apoptosis following the activation of apoptosis-related caspases (caspase-8, -9, and -3) by the recognition of RNA fragments. However, HVJ-E was recently reported to induce an entirely different cell death system from the conventional HVJ-E-mediated apoptosis.
Nomura et al reported that HVJ-E induced cell death by a mechanism other than apoptosis in human neuroblastoma cells (SK-N-SH and SK-N-AS) (Figure 2B).35 First, they revealed that HVJ-E-treated SK-N-SH cells underwent cell death that was not blocked by treatment with an apoptosis inhibitor (a pan-caspase inhibitor). SK-N-SH cells are unable to recognize HVJ-E RNA fragments because they lack RIG-I expression. Thus, the authors next focused their attention on the elevation of cytoplasmic Ca2+ levels following HVJ-E membrane fusion.36 The Ca2+ increase induced the phosphorylation of RIP1 via CaMK II activation, and cell death was subsequently induced by reactive oxygen species production. Moreover, HVJ-E-mediated SK-N-SH cell death was inhibited by necrostatin-1, which is a necroptosis inhibitor. Necroptosis is a cell death process that occurs during caspase-8 inactivation, and the phosphorylation of RIP1 and RIP3 is involved in this system.37 SK-N-SH does not express caspase-8, suggesting that HVJ-E can induce necroptosis in cancer cells lacking caspase-8 expression. Caspase-8 is a key factor for apoptosis, and many anticancer drugs induce cancer cell apoptosis by activating caspase-8.38 However, some cancer cells acquire drug resistance through the absence of caspase-8 expression.39 Therefore, it is possible that HVJ-E will be a promising treatment tool against drug-resistant cancer cells without caspase-8 expression.
HVJ-E-mediated activation of anticancer immunity
HVJ-E induces not only cancer cell apoptosis and necroptosis but also activates anticancer immunity. Kurooka and Kaneda reported that the intratumoral administration of HVJ-E dramatically decreased the tumor volume of murine colon carcinoma (CT26) growing in the backs of mice.40 Moreover, HVJ-E treatment of CT26 in the right back flank of mice significantly inhibited the engraftment of transplanted CT26 into the left back flank, suggesting that the intratumoral administration of HVJ-E activates a tumor-specific antitumor immunity. The researchers demonstrated that HVJ-E promotes the maturation of dendritic cells (DCs) and induces the production of various cytokines, namely IFN-α and -β, TNF-α and interleukin (IL)-6, by mature DCs. In particular, IL-6 was an important factor for the activation of HVJ-E-mediated antitumor immunity, and IL-6 enhanced the proliferation and activation of cytotoxic T-lymphocytes (CTLs) by suppressing the activity of regulatory T-cells (Tregs).41 When an RNA virus is detected by DCs, the viral RNA genome is recognized by Toll-like receptor (TLR)-7 and -8 in the endosome42–44 or RIG-I in the cytoplasm.45 However, Suzuki et al showed that IL-6 secretion by DCs is activated by reconstituted particles that contain HVJ-E components but lack an RNA genome,46 suggesting that HVJ-E-mediated stimulation of DCs is independent from the recognition of RNA fragments by TLR-7, TLR-8, and RIG-I. Moreover, they revealed that F is responsible for the induction of IL-6 secretion from DCs. However, the DC receptor that recognizes F is unknown.
It has been reported that HVJ-E induces the activation of not only CTLs but also natural killer (NK) cells. Fujihara et al reported that HVJ-E elicits antitumor activity against murine renal cell carcinoma (Renca) via NK cell activation.47 They demonstrated that intratumoral HVJ-E administration induces C-X-C motif chemokine (CXCL)-10 secretion and inhibits Renca tumor growth in the backs of the mice. The HVJ-E-mediated antitumor effect against Renca was also revealed in SCID mice lacking T- and B-cells, and the effect was impaired by inhibiting NK cells with an NK cell-neutralizing antibody. CXCL10 is a chemokine for monocytes/macrophages, DCs, T-cells, and NK cells,48 suggesting that HVJ-E treatment enhances the recruitment of activated NK cells by CXCL-10. HVJ-E-treated DCs, but not Renca, produce CXCL-10, and HVJ-E treatment of DCs and Renca co cultures synergistically enhance CXCL-10 secretion. Although the mechanism of the synergistic effect of CXCL-10 secretion is unknown, it is possible that HVJ-E treatment specifically induces the accumulation of active NK cells in tumor tissues but not in normal tissues.
Modified HVJ-E
Recently, to enhance the antitumor effect and to decrease the side effects of HVJ-E, various modifications of HVJ-E were attempted. GD1a and SPG, which are receptors for HN, are expressed by nearly all cell types; therefore, HVJ-E does not have the specificity to attack only cancer cells. Moreover, red blood cells present high levels of these HN receptors on their surface, and HVJ-E induces hemagglutination in the blood. Therefore, HVJ-E could acquire cancer cell specificity and low-hemagglutinating activity for effective cancer treatment via systemic administration. To inhibit the nonspecificity and the hemagglutinating activity of HVJ-E, HN-depleted HVJ-E (ΔHN-HVJ-E) was generated using siRNA against HN.49 In comparison with the wt-HVJ-E, ΔHN-HVJ-E exhibits low hemagglutinating activity and decreased cell adhesion, suggesting that ΔHN-HVJ-E also has a decreased affinity for cancer cells. Shimbo et al increased the tumor-targeting efficiency of ΔHN-HVJ-E by altering the virus to bind to the transferrin (Tf) receptor.50 Tf is a carrier protein of ferric ion, and cancer cells express high levels of the Tf receptor for ferric ion uptake because ferric ion is required for DNA synthesis during active cell division.51 The researchers constructed a recombinant F of Tf and F (Tf/F) and succeeded in the presentation of Tf/F on ΔHN-HVJ-E (Tf/F-ΔHN-HVJ-E). After intravenous administration, Tf/F-ΔHN-HVJ-E, but not ΔHN-HVJ-E, accumulated in uterocervical cancer (HeLa) cells, expressing high levels of the Tf receptor. Moreover, Kawachi et al succeeded in the presentation of a single-chain antibody (scFv) on HVJ-E.52 Many previous reports have described various tumor-associated antigens.53–56 Therefore, the generation of HVJ-E showing effective cancer specificity by the presentation of scFv against tumor-associated antigens is anticipated.
The presentation of an immune-stimulatory factor on the surface of HVJ-E has been reported.57 As described earlier, HVJ-E induces the production of various cytokines by DCs and enhances antitumor immunity via the downregulation of Tregs.40 However, HVJ-E is unable to effectively induce Th1-inducing cytokines (eg, IL-12 and IFN-γ). IL-12 and IFN-γ are important factors for the activation of antitumor immunity. IL-12 induces Th1 differentiation and IFN-γ secretion, and IFN-γ enhances Th1 differentiation.58 Saga et al reported that HVJ-E dramatically enhances IL-12 activity and IFN-γ secretion from murine splenocytes, much more than IL-12 alone, although HVJ-E cannot induce IFN-γ secretion from these cells.57 To enhance the activation effect of HVJ-E-mediated antitumor immunity, the researchers generated IL-12-presenting ΔHN-HVJ-E (IL12-HVJ-E).57 Compared with wt-HVJ-E, IL12-HVJ-E exhibited a robust ability to induce IFN-γ secretion from splenocytes in vitro and significantly enhanced the antitumor immunity against mouse malignant melanomas (F10 melanoma) via intratumoral administration in vivo. Moreover, compared with wt-HVJ-E, IL12-HVJ-E reduced the lung metastatic foci of F10 melanoma via intravenous administration, suggesting the possibility of applying HVJ-E as a therapy against metastatic cancer.
Conclusion
Here, we have described the oncolytic properties of Sendai virus, especially HVJ-E. The HVJ-E-mediated oncolytic mechanism differs from that of conventional oncolytic viruses. Recently, many reports have discussed cancer stem cells (CSCs), which are closely related to cancer malignancy.59–61 CSCs are a rare population of cancer tissue and exhibit the phenotypes of drug resistance and cell growth dormancy.62 Conventional oncolytic virus-mediated cell lysis requires a high proliferation of cancer cells for viral replication; therefore, conventional virotherapy might exhibit a poor effect against CSCs. However, the mechanism of HVJ-E-mediated cancer cell death is not related to cancer proliferation; consequently, it is expected that HVJ-E treatment will be effective for CSCs.
Systemic administration of HVJ-E is an attractive application for cancer therapy. To provide effective anticancer reaction by systemic HVJ-E administration, HVJ-E should be accumulated in cancer tissue. However ΔHN-HVJ-E, which has low hemagglutinating activity, specifically accumulates in lung by intravenous administration thereof.57 Therefore, intravenous administration of ΔHN-HVJ-E is likely to show effective therapeutic outcome against lung cancer and pulmonary metastasis but not against cancers of the other tissues. In order to effectively treat various cancers by systemic HVJ-E administration, a new highly-efficient HVJ-E delivery system to cancers has to be developed. Recently, systemic administration of platelets incorporating HVJ-E has demonstrated effective accumulation of HVJ-E to intradermal melanoma and induced robust anticancer therapeutic effect.63 Development of a cancer-specific delivery system of HVJ-E will improve its therapeutic effect against cancers throughout a body by systemic HVJ-E administration.
HVJ-E-mediated cytotoxic mechanisms differ depending on the cancer cell type. Therefore, it is believed that there are unknown mechanisms of HVJ-E-mediated cancer cell death, and this process needs to be studied in greater detail. However, it is a clear that HVJ-E is a robust anticancer agent that can induce both direct cytotoxicity and antitumor immunity. Moreover, HVJ-E-mediated oncolytic reactions do not need viral replication; there is no side effect of uncontrollable viral expansion which is feared in conventional oncolytic virus. Additionally, to enhance anticancer activity, armed-type HVJ-E can be easily constructed by incorporating therapeutic genes into the envelope.20,64–66 Therefore, HVJ-E has a great potential for clinical application. Now, clinical trials of HVJ-E against melanoma and prostate cancer are ongoing in Japan. It is anticipated that HVJ-E will eventually be employed as a robust therapeutic tool against cancer.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vähä-Koskela MJ, Heikkilä JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett. 2007;254(2):178–216. doi: 10.1016/j.canlet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bischoff JR, Kirn DH, Williams A, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274(5286):373–376. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 4.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14(8):559–567. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 5.Bell J, McFadden G. Viruses for tumor therapy. Cell Host Microbe. 2014;15(3):260–265. doi: 10.1016/j.chom.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curran J, Kolakofsky D. Replication of paramyxoviruses. Adv Virus Res. 1999;54:403–422. doi: 10.1016/s0065-3527(08)60373-5. [DOI] [PubMed] [Google Scholar]
- 7.Sakaguchi T, Kato A, Kiyotani K, Yoshida T, Nagai Y. Studies on the paramyxovirus accessory genes by reverse genetics in the Sendai virus-mouse system. Proc Jpn Acad Ser B Phys Biol Sci. 2008;84(10):439–451. doi: 10.2183/pjab/84.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takimoto T, Taylor GL, Connaris HC, Crennell SJ, Portner A. Role of the hemagglutinin-neuraminidase protein in the mechanism of paramyxovirus-cell membrane fusion. J Virol. 2002;76(24):13028–13033. doi: 10.1128/JVI.76.24.13028-13033.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asano A, Asano K. Viral proteins in cell fusion. Tokai J Exp Clin Med. 1982;7(Suppl):193–196. [PubMed] [Google Scholar]
- 10.Gotoh B, Ogasawara T, Toyoda T, Inocencio NM, Hamaguchi M, Nagai Y. An endoprotease homologous to the blood clotting factor X as a determinant of viral tropism in chick embryo. EMBO J. 1990;9(12):4189–4195. doi: 10.1002/j.1460-2075.1990.tb07643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kido H, Yokogoshi Y, Sakai K, et al. Isolation and characterization of a novel trypsin-like protease found in rat bronchiolar epithelial Clara cells. A possible activator of the viral fusion glycoprotein. J Biol Chem. 1992;267(19):13573–13579. [PubMed] [Google Scholar]
- 12.Lun XQ, Zhou H, Alain T, et al. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007;67(18):8818–8827. doi: 10.1158/0008-5472.CAN-07-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alain T, Hirasawa K, Pon KJ, et al. Reovirus therapy of lymphoid malignancies. Blood. 2002;100(12):4146–4153. doi: 10.1182/blood-2002-02-0503. [DOI] [PubMed] [Google Scholar]
- 14.Cinatl J, Jr, Cinatl J, Michaelis M, et al. Potent oncolytic activity of multimutated herpes simplex virus G207 in combination with vincristine against human rhabdomyosarcoma. Cancer Res. 2003;63(7):1508–1514. [PubMed] [Google Scholar]
- 15.Fueyo J, Gomez-Manzano C, Alemany R, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19(1):2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 16.Kinoh H, Inoue M, Washizawa K, et al. Generation of a recombinant Sendai virus that is selectively activated and lyses human tumor cells expressing matrix metalloproteinases. Gene Ther. 2004;11(14):1137–1145. doi: 10.1038/sj.gt.3302272. [DOI] [PubMed] [Google Scholar]
- 17.Kinoh H, Inoue M, Komaru A, Ueda Y, Hasegawa M, Yonemitsu Y. Generation of optimized and urokinase-targeted oncolytic Sendai virus vectors applicable for various human malignancies. Gene Ther. 2009;16(3):392–403. doi: 10.1038/gt.2008.167. [DOI] [PubMed] [Google Scholar]
- 18.Inoue M, Tokusumi Y, Ban H, et al. A new Sendai virus vector deficient in the matrix gene does not form virus particles and shows extensive cell-to-cell spreading. J Virol. 2003;77(11):6419–6429. doi: 10.1128/JVI.77.11.6419-6429.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 20.Kaneda Y, Nakajima T, Nishikawa T, et al. Hemagglutinating virus of Japan (HVJ) envelope vector as a versatile gene delivery system. Mol Ther. 2002;6(2):219–226. doi: 10.1006/mthe.2002.0647. [DOI] [PubMed] [Google Scholar]
- 21.Kim YD, Park KG, Morishita R, et al. Liver-directed gene therapy of diabetic rats using an HVJ-E vector containing EBV plasmids expressing insulin and GLUT 2 transporter. Gene Ther. 2006;13(3):216–224. doi: 10.1038/sj.gt.3302644. [DOI] [PubMed] [Google Scholar]
- 22.Shimamura M, Morishita R, Endoh M, et al. HVJ-envelope vector for gene transfer into central nervous system. Biochem Biophys Res Commun. 2003;300(2):464–471. doi: 10.1016/s0006-291x(02)02807-3. [DOI] [PubMed] [Google Scholar]
- 23.Oshima K, Shimamura M, Mizuno S, et al. Intrathecal injection of HVJ-E containing HGF gene to cerebrospinal fluid can prevent and ameliorate hearing impairment in rats. FASEB J. 2004;18(1):212–214. doi: 10.1096/fj.03-0567fje. [DOI] [PubMed] [Google Scholar]
- 24.Kawano H, Komaba S, Kanamori T, Kaneda Y. A new therapy for highly effective tumor eradication using HVJ-E combined with chemotherapy. BMC Med. 2007;5:28. doi: 10.1186/1741-7015-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaka M, Shimbo T, Kikuchi Y, Matsuda M, Kaneda Y. Sterile alpha motif containing domain 9 is involved in death signaling of malignant glioma treated with inactivated Sendai virus particle (HVJ-E) or type I interferon. Int J Cancer. 2010;126(8):1982–1991. doi: 10.1002/ijc.24965. [DOI] [PubMed] [Google Scholar]
- 26.Kawaguchi Y, Miyamoto Y, Inoue T, Kaneda Y. Efficient eradication of hormone-resistant human prostate cancers by inactivated Sendai virus particle. Int J Cancer. 2009;124(10):2478–2487. doi: 10.1002/ijc.24234. [DOI] [PubMed] [Google Scholar]
- 27.Matsushima-Miyagi T, Hatano K, Nomura M, et al. TRAIL and Noxa are selectively upregulated in prostate cancer cells downstream of the RIG-I/MAVS signaling pathway by nonreplicating Sendai virus particles. Clin Cancer Res. 2012;18(22):6271–6283. doi: 10.1158/1078-0432.CCR-12-1595. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Q, Xu X, Yuan Y, Gong X, Chen Z, Xu X. IPS-1 plays a dual function to directly induce apoptosis in murine melanoma cells by inactivated Sendai virus. Int J Cancer. 2014;134(1):224–234. doi: 10.1002/ijc.28340. [DOI] [PubMed] [Google Scholar]
- 29.Shi LY, Han Z, Li XX, et al. Inactivated Sendai virus strain Tianjin induces apoptosis in breast cancer MCF-7 cells by promoting caspase activation and Fas/FasL expression. Cancer Biother Radiopharm. 2015;30(1):33–40. doi: 10.1089/cbr.2014.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ploner C, Kofler R, Villunger A. Noxa: at the tip of the balance between life and death. Oncogene. 2008;27(Suppl 1):S84–S92. doi: 10.1038/onc.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holoch PA, Griffith TS. TNF-related apoptosis-inducing ligand (TRAIL): a new path to anti-cancer therapies. Eur J Pharmacol. 2009;625(1–3):63–72. doi: 10.1016/j.ejphar.2009.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao H, Gong XC, Chen ZD, Xu XS, Zhang Q, Xu XM. Induction of apoptosis in hormone-resistant human prostate cancer PC3 cells by inactivated Sendai virus. Biomed Environ Sci. 2014;27(7):506–514. doi: 10.3967/bes2014.082. [DOI] [PubMed] [Google Scholar]
- 33.Su ZZ, Sarkar D, Emdad L, Barral PM, Fisher PB. Central role of interferon regulatory factor-1 (IRF-1) in controlling retinoic acid inducible gene-I (RIG-I) expression. J Cell Physiol. 2007;213(2):502–510. doi: 10.1002/jcp.21128. [DOI] [PubMed] [Google Scholar]
- 34.Besch R, Poeck H, Hohenauer T, et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest. 2009;119(8):2399–2411. doi: 10.1172/JCI37155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nomura M, Ueno A, Saga K, Fukuzawa M, Kaneda Y. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res. 2014;74(4):1056–1066. doi: 10.1158/0008-5472.CAN-13-1283. [DOI] [PubMed] [Google Scholar]
- 36.Hallett MB, Fuchs P, Campbell AK. Sendai virus causes a rise in intracellular free Ca2+ before cell fusion. Biochem J. 1982;206(3):671–674. doi: 10.1042/bj2060671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 38.Kim PK, Mahidhara R, Seol DW. The role of caspase-8 in resistance to cancer chemotherapy. Drug Resist Updat. 2001;4(5):293–296. doi: 10.1054/drup.2001.0223. [DOI] [PubMed] [Google Scholar]
- 39.Fulda S, Küfer MU, Meyer E, van Valen F, Dockhorn-Dworniczak B, Debatin KM. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene. 2001;20(41):5865–5877. doi: 10.1038/sj.onc.1204750. [DOI] [PubMed] [Google Scholar]
- 40.Kurooka M, Kaneda Y. Inactivated Sendai virus particles eradicate tumors by inducing immune responses through blocking regulatory T cells. Cancer Res. 2007;67(1):227–236. doi: 10.1158/0008-5472.CAN-06-1615. [DOI] [PubMed] [Google Scholar]
- 41.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299(5609):1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 42.Lund JM, Alexopoulou L, Sato A, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101(15):5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heil F, Ahmad-Nejad P, Hemmi H, et al. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur J Immunol. 2003;33(11):2987–2997. doi: 10.1002/eji.200324238. [DOI] [PubMed] [Google Scholar]
- 44.Lee J, Chuang T-H, Redecke V, et al. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc Natl Acad Sci U S A. 2003;100(11):6646–6651. doi: 10.1073/pnas.0631696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki H, Kurooka M, Hiroaki Y, Fujiyoshi Y, Kaneda Y. Sendai virus F glycoprotein induces IL-6 production in dendritic cells in a fusion-independent manner. FEBS Lett. 2008;582(9):1325–1329. doi: 10.1016/j.febslet.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 47.Fujihara A, Kurooka M, Miki T, Kaneda Y. Intratumoral injection of inactivated Sendai virus particles elicits strong antitumor activity by enhancing local CXCL10 expression and systemic NK cell activation. Cancer Immunol Immunother. 2008;57(1):73–84. doi: 10.1007/s00262-007-0351-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu M, Guo S, Stiles JK. The emerging role of CXCL10 in cancer (Review) Oncol Lett. 2011;2(4):583–589. doi: 10.3892/ol.2011.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saga K, Tamai K, Kawachi M, et al. Functional modification of Sendai virus by siRNA. J Biotechnol. 2008;133(3):386–394. doi: 10.1016/j.jbiotec.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 50.Shimbo T, Kawachi M, Saga K, et al. Development of a transferrin receptor-targeting HVJ-E vector. Biochem Biophys Res Commun. 2007;364(3):423–428. doi: 10.1016/j.bbrc.2007.09.135. [DOI] [PubMed] [Google Scholar]
- 51.Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121(2):144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 52.Kawachi M, Tamai K, Saga K, et al. Development of tissue-targeting hemagglutinating virus of Japan envelope vector for successful delivery of therapeutic gene to mouse skin. Hum Gene Ther. 2007;18(10):881–894. doi: 10.1089/hum.2007.046. [DOI] [PubMed] [Google Scholar]
- 53.Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GA. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature. 1990;343(6260):774–778. doi: 10.1038/343774a0. [DOI] [PubMed] [Google Scholar]
- 54.Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60(3):509–520. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 55.van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254(5038):1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 56.Wang RF. Human tumor antigens: implications for cancer vaccine development. J Mol Med. 1999;77(9):640–655. doi: 10.1007/s001099900042. [DOI] [PubMed] [Google Scholar]
- 57.Saga K, Tamai K, Yamazaki T, Kaneda Y. Systemic administration of a novel immune-stimulatory pseudovirion suppresses lung metastatic melanoma by regionally enhancing IFN-γ production. Clin Cancer Res. 2013;19(3):668–679. doi: 10.1158/1078-0432.CCR-12-1947. [DOI] [PubMed] [Google Scholar]
- 58.Sykes L, MacIntyre DA, Yap XJ, Teoh TG, Bennett PR. The Th1:Th2 dichotomy of pregnancy and preterm labour. Mediators Inflamm. 2012;2012:967629. doi: 10.1155/2012/967629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–5828. [PubMed] [Google Scholar]
- 60.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 61.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 63.Nishikawa T, Tung LY, Kaneda Y. Systemic administration of platelets incorporating inactivated sendai virus eradicates melanoma in mice. Mol Ther. 2014;22(12):2046–2055. doi: 10.1038/mt.2014.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kiyohara E, Tamai K, Katayama I, Kaneda Y. The combination of chemotherapy with HVJ-E containing Rad51 siRNA elicited diverse anti-tumor effects and synergistically suppressed melanoma. Gene Ther. 2012;19(7):734–741. doi: 10.1038/gt.2011.123. [DOI] [PubMed] [Google Scholar]
- 65.Matsuda M, Nimura K, Shimbo T, et al. Immunogene therapy using immunomodulating HVJ-E vector augments anti-tumor effects in murine malignant glioma. J Neurooncol. 2011;103(1):19–31. doi: 10.1007/s11060-010-0355-x. [DOI] [PubMed] [Google Scholar]
- 66.Matsuda M, Yamamoto T, Matsumura A, Kaneda Y. Highly efficient eradication of intracranial glioblastoma using Eg5 siRNA combined with HVJ envelope. Gene Ther. 2009;16(12):1465–1476. doi: 10.1038/gt.2009.99. [DOI] [PubMed] [Google Scholar]