

Abstract
Oncolytic virotherapy is gaining interest in the clinic as a new weapon against cancer. In vivo administration of oncolytic viruses showed important limitations that decrease their effectiveness very significantly: the antiviral immune response causes the elimination of the therapeutic effect, and the poor natural ability of oncolytic viruses to infect micrometastatic lesions significantly minimizes the effective dose of virus. This review will focus on updating the technical and scientific foundations of one of the strategies developed to overcome these limitations, ie, using cells as vehicles for oncolytic viruses. Among many candidates, a special type of adult stem cell, mesenchymal stem cells (MSCs), have already been used in the clinic as cell vehicles for oncolytic viruses, partly due to the fact that these cells are actively being evaluated for other indications. MSC carrier cells are used as Trojan horses loaded with oncoviruses, are administered systemically, and release their cargos at the right places. MSCs are equipped with an array of molecules involved in cell arrest in the capillaries (integrins and selectins), migration toward specific parenchymal locations within tissues (chemokine receptors), and invasion and degradation of the extracellular matrix (proteases). In addition to anatomical targeting capacity, MSCs have a well-recognized role in modulating immune responses by affecting cells of the innate (antigen-presenting cells, natural killer cells) and adaptive immune system (effector and regulatory lymphocytes). Therefore, carrier MSCs may also modulate the immune responses taking place after therapy, ie, the antiviral and the antitumor immune responses.
Keywords: virotherapy, mesenchymal stem cells, oncolytic adenovirus
Introduction
In recent years, we have seen an increased interest in the application of oncolytic virotherapy for the treatment of human tumors.1 These treatments have already reached the clinical arena, and the first results of clinical trials have been published. Oncolytic viruses may become a new option in the antitumor armamentarium, although there is still a long way ahead for clarifying indications, dosage, treatment guidelines, and other aspects that will determine the optimization of their use in humans. The therapeutic potential of oncolytic viruses has been repeatedly demonstrated in different in vitro models; however, the administration in experimental animals first, but especially in patients, has put in a more realistic light the therapeutic capacity of oncolytic virotherapy. There are some limitations to the clinical use of oncolytic viruses that decrease their effectiveness very significantly. On the one hand, the antiviral immune response that the body develops causes the elimination of the therapeutic effect following the first administration or even with the first dose in preimmunized patients. Moreover, the poor natural ability of oncolytic viruses to infect micrometastatic lesions significantly minimizes the effective dose of virus; increasing the dosage does not ensure a greater effect due to increased toxicity as well as due to the antiviral immune response. This problem does not occur in localized tumors, but it is significant in metastatic tumor disease, which is the leading cause of cancer death.
It is not surprising then that strategies to overcome these limitations have been pursued in order to improve the results of oncolytic virotherapy. Several groups, including ours, began to explore years ago the possibility of using cells as vehicles for avoiding those limitations.2 First, to increase the amount of viral particles that are released at metastasis areas, exploiting the ability of these cell vehicles to localize and integrate into the tumor masses after intravenous (IV) administration. Microscopic inspection of cancerous lesions identifies a series of accessory cells that are recruited during tumor growth: myeloid cells, macrophages, lymphocytes, endothelial cells, and mesenchymal cells. IV infusion of any of the aforementioned cells in animals with metastatic cancer has shown a preferential localization in tumor beds. On the basis of this fact, the cells are used as Trojan horses bearing antitumor agents in them, to be released in the right place.3 Various oncolytic viruses and tumor-infiltrating cell types have been evaluated in this strategy, using several tumor models. The common result of these experiments has been the preclinical demonstration of the feasibility of the strategy. In addition to tumor targeting capacity, the cell vehicles can facilitate the therapeutic effect of oncolytic virotherapy, extending its life by hiding the virus from recognition and attack by the immune system in the immediate time after administration. This is critical for all of the aforementioned limitations to have clinical impact.
Mesenchymal cells have arrived at the clinic as oncolytic virotherapy cell vehicles, partly due to the fact that these cells are actively being evaluated for other indications.4 In the field of cancer, there are a few early phase clinical trials using mesenchymal stem cells (MSCs). One is evaluating their capacity to home to sites of prostate cancer in men with localized adenocarcinoma of the prostate (ClinicalTrials.gov Identifier: NCT01983709). The second is assessing safety and efficacy of MSCs genetically modified to produce interleukin-12 in head and neck cancer patients (NCT02079324). Two trials are using MSCs as cell carriers for oncolytic virotherapies, with an oncolytic measles virus (NCT02068794) and with an oncolytic adenovirus (NCT01844661). A few years ago, our group began evaluating the use of mesenchymal cells as cell carriers for the administration of an oncolytic adenovirus in children with metastatic neuroblastoma.2 During this time, we have increased our clinical experience with this antitumor strategy. This review will focus on updating the fundamentals that determine their antitumor potential.
Mesenchymal cells
Forty years ago, Friedenstein et al5 described the isolation of stromal cells from bone marrow by plastic adherence. These cells had a clonal capacity and the ability to support ectopic bone, stroma, and hematopoietic tissues. At the end of the last century, part of the heterogeneous stroma of bone marrow was categorized as MSCs based on their in vitro potential of trilineage differentiation (osteoblast, adipocyte, and chondrocyte) and self-renewal.6 Some studies have further reported MSC differentiation into multiple other cell types of mesodermal and nonmesodermal origin. Nevertheless, such multipotential capabilities of MSCs are not universally accepted.7 After their initial discovery in bone marrow, MSCs have been isolated and characterized from several adult and fetal tissues, including adipose, dermis, synovial fluid, umbilical cord blood, placenta, and amniotic fluid.6 These stem cells are commonly called marrow stromal stem cells, mesenchymal stromal cells, adipose-derived stromal cells, or MSCs. In 2006, the International Society for Cellular Therapy proposed the denomination of mesenchymal stromal cells and a set of minimal criteria to characterize MSCs: they must be plastic adherent and express CD105, CD73, and CD90 but not CD45, CD34, CD14 or CD11b, CD79a or CD19, and HLA-DR surface molecules. Moreover, MSCs must differentiate in vitro to osteoblasts, adipocytes, and chondroblasts.8
The use of MSCs for therapeutic applications, especially as tools in cellular therapies for clinical protocols involving immune system alterations, has been deeply explored because of their inherent ability to home to sites of inflammation following tissue injury and deploying their high immunoregulatory capacity.4,9 MSCs profoundly affect immune response via their interactions with the cellular components of the innate and adaptive immune system and through cell–cell contact and/or the secretion of soluble factors.10
Mesenchymal stem cells as carriers
More than a decade ago, Studeny et al11 reported that MSCs contribute to tumor stroma formation after IV administration. This work further demonstrated that MSCs could serve as a platform for the delivery of biological agents into tumors, by genetically overexpressing an antitumor element, IFN-beta in this report, in the carrier cells that resulted in the inhibition of the growth of malignant cells in vivo. Many other papers followed Studeny et al’s report,11 proving experimentally that MSCs targeted several models of metastatic tumors and could be used as part of a new class of medicines against cancer. Monitoring of MSCs infused into animals with tumors using different imaging techniques confirmed the preferential localization of these cells in tumor areas.12 In light of these experimental results, a clear interest emerged in deciphering the molecular basis responsible for the preferential migration of MSCs to sites where cancerous lesions are located. One of the first studies addressing this compared the gene expression profiles of MSCs cultured ex vivo with medium from either tumor cells or bone marrow.13 Several transcripts were found to be differentially expressed, including chemokines (CXCL12 and CXCL-2), CINC-2, endothelial cell specific molecule-1, fibroblast growth factor-7, nuclear factor-κB p105, and thrombomodulin. These authors applied a proteomic approach to identify soluble signaling molecules that induced MSC chemotaxis and were present in the conditioned medium of tumor cells. Cyclophilin B and hepatoma-derived growth factor were characterized and shown to promote MSC chemotaxis.14 It is interesting to note that the exposure of MSCs to cancer cell lines of different histological origins resulted in the upregulation of adhesion molecules in the MSCs that varied depending upon the cell lines used,15 indicating that the response of MSCs is not universal but modulated by tumor-derived factors.
Similarly, the process of MSC homing is compared to the migration and homing of leukocytes during inflammatory processes. It is assumed that the different steps recognized for leukocytes (tethering, rolling, firm adhesion, and diapedesis)16 occur similarly after infusing MSCs intravenously. However, extensive studies to verify whether the mediators involved during the migration and extravasation of leukocytes are the same (or what differences may exist) as those of MSCs have not yet been carried out.17 Although mechanical entrapment of MSCs has been described after IV injection,18 active arrest of MSCs within inflamed tissues has been demonstrated in studies19,20 that showed a role for molecules of the selectin and integrin families. Interfering with the adhesion molecule VLA-4 (that governs the arrest of leukocytes on activated endothelium) reduced the engraftment of MSCs in ischemic myocardium.19 In a different setting, P-selectin knockout MSCs did not slow down in postcapillary venules compared to wild-type MSCs.20 These results suggest that the engraftment of MSCs within target tissues depends on specific molecular interactions, rather than nonspecific mechanical causes.
A different set of molecules implicated in the homing of MSCs to specific sites, including tumors, are chemokines. They may be released from inflamed tissues, endothelial cells, or directly from tumor cells, and may promote activation of adhesion ligands, transendothelial migration, chemotaxis, and/or subsequent retention in surrounding tissue. MSCs express various chemokine receptors21,22 and respond to their corresponding ligands. A number of chemokine signaling pathways have been associated with migratory activities of MSC. The role of the CXCR4-CXCL12 (SDF-1) axis has been controversial. It was reported that only a small proportion of MSCs expressed CXCR4,23 but cytokines such as insulinlike growth factor 1 increases the expression of CXCR4 on MSC and their migratory capacity in vitro.24 The CXCR4-MIF (macrophage migration inhibitory factor) axis, and not the classic CXCR4-CXCL12, has been recently introduced as the key director of MSC migration and infiltration toward tumor cells.25 Using in vitro migration and invasion assays and an in vivo pulmonary metastasis model, the authors found that CXCR4 was the major receptor used by MIF in the homing of MSCs into the tumor environment. Genetic elimination of either CXCR4 or MIF abrogated the capacity of MSCs for homing into tumors. Human MSCs have been shown to migrate toward CCL2-containing medium derived from the primary cultures of primary cancer patient tissues,26 and in a mouse model of breast carcinoma.27 It is interesting to note that the level of CCL2 expression correlates with the level of matrix metalloproteases (MMPs; MT1-MMP/MMP14),28 for reasons discussed later. Another chemokine axis involved is CCR9-CCL25. Human MSCs express CCR9 in their surface,21 while CCL25 is expressed in multiple myeloma cells29 and has a chemotactic function for MSCs.30 All these data on chemokines as recruiters of MSCs are not unexpected since cancer and inflammation are closely related.
The final step in the migratory activity of MSCs deals with transmigration and invasion of the basement membrane of endothelium and degradation of the extracellular matrix (ECM) during chemotaxis. It is known that MSCs produce proteases for achieving this step. Genetic and pharmacologic inhibition of MMP2 in MSCs reduced transendothelial migration in vitro.31 In vitro assays of MSCs invasion through ECM-coated transwell chambers showed that downregulation of MMP-2, MT1-MMP, and TIMP-2 significantly impaired the migration of MSCs when compared with control cells.32 Using glioma cells as stimulus, functional inactivation of MMP1 abrogated the migratory potential of MSCs, while the addition of recombinant MMP1 enhanced their migratory capacity. Ectopic expression of MMP1 rendered the cells responsive to the signaling cues from the glioma cells in vivo. Finally, disrupting the interaction MMP1-PAR1 diminished the migratory ability of MSCs.33 Gelatinases are other proteases produced by MSCs during tissue invasion. Thus, MSCs possess the ability to break down endothelial basement membrane and migrate toward chemotactic factors. Such capabilities are likely a function of their responsiveness to chemotactic factors and production of ECM-degrading enzymes.
Understanding the molecular basis of the targeting ability of MSCs in the context of tumors became an important issue in the field. Being able to manipulate this capacity might open new avenues to improve the results of the delivery. One can envision the genetic manipulation of MSCs to enhance the expression of cell adhesion molecules involved in the processes previously described. An alternative way may be ex vivo selection of MSCs with improved migratory capacities, as described by Bolontrade et al.34 The authors compared in vitro adhesive capacities during MSCs isolation and then selected a specific subpopulation with the optimal highest adhesiveness, migration toward conditioned media from different cancer cell lines and fresh primary human cancer samples, and increased tumor homing toward tumor xenografts. These characteristics correlated with expression of specific integrins (integrins α2, α3, and α5) and metalloproteases production. A work by Klopp et al26 exploited the capacity of MSCs for migrating toward sites of inflammation by using low dose of irradiation on tumors in in vivo models. As early as 48 hours after irradiation, MSCs engrafted at significantly higher levels in irradiated versus unirradiated tumors. The authors characterized transforming growth factor-B1, vascular endothelial growth factor, platelet-derived growth factor-BB, and CCR2 as mediators of this effect. The results of this elegant work have clear clinical relevance, since radiotherapy is routinely used for treating many different types of cancers.
Mesenchymal stem cells and the immune responses
The immune system has a recognized role in the outcome of virotherapies.35 Natural killer cells interfere with the action of oncolytic viruses, reducing or eliminating their efficacy.36 It is also known that adaptive immunity controls viral infections.37 On the other hand, oncolysis involves tumor cell death, with the possible release of tumor-specific antigens. These antigens, coupled to danger signals associated with viral infection,38 can stimulate an antitumoral immune response, increasing the clinical effect of the virus.39 The combination of MSCs with oncolytic viruses has important implications in the development of immune responses that occur in these patients, which make them different from the virotherapy used without MSCs. These cells have a well-known role in the function of human antigen-presenting cells and in effector and regulatory leukocytes of the innate and adaptive responses.40–47 It has also been reported that autologous MSCs may function as antigen-presenting cells in animal models.48,49 Therefore, MSCs may not only act as carrier cells but might as well modulate the immune responses taking place after infusing MSCs loaded with oncolytic viruses, ie, the antiviral and the antitumor immune responses. There are technical limitations in translating the results obtained with oncolytic viruses in preclinical models (alone or in combination with MSCs) when animals are either nonpermissive for the virus (as is the case for oncolytic adenoviruses, and to some extend herpex viruses) or when the viral cycle is too fast for a successful Trojan horse transfer (ie, coxsackie virus or vesicular stomatitis virus). Also critical when studying immune aspects of oncolytic virothetapies is the fact that human cancer cell lines are implanted in immunodeficient animals. Taking into account all these limitations, authors have reported experimental data indicating that MSCs may temporarily hide the presence of the oncolytic virus from the immune system, retarding the attack and inactivation of the virus, thus allowing for a longer therapeutic window. The administration of MSCs as carriers of oncolytic measles virus in passively immunized mice with ovarian cancer resulted in increased survival compared to that of mice treated with naked virus or uninfected MSC.50 In a rat model susceptible to human adenovirus infections,51 the authors evaluated the antiadenoviral immune response following delivery of oncolytic adenoviral vectors using MSCs as carriers, and found improved delivery, enhanced dissemination, and increased persistence of viruses via suppression of the antiviral immune response. A second important aspect is the fact that MSCs may modulate their gene and protein expression profile depending upon the environment.52 Oncovirus-infected MSCs would adopt a proinflammatory MSC1 phenotype, and will arrive and lodge into tumor sites, which are “sterilized” inflamed tissues and would promote an immunomodulating MSC2 phenotype on the MSCs.53 The role of the carrier MSCs on the immune response will eventually be the result of these two opposite forces. It is not easy to decide what final phenotype would be preferable for the MSCs when carrying oncolytic viruses in cancer patients. On the one hand, inflammation may favor the benefits of virotherapies; therefore, a predominant MSC1 phenotype would be desiderable.54 However, on the other hand, MSCs less responsive to viral infection might hide the virus from recognition and attack by the patient’s innate and adaptive immune system before viral delivery at the metastasis sites, favoring better conditions for the in situ oncolytic effect. Figure 1 depicts the potential effects of MSCs as carriers for oncolytic viruses and as modulators of the antiviral and antitumor immune responses.
Figure 1.
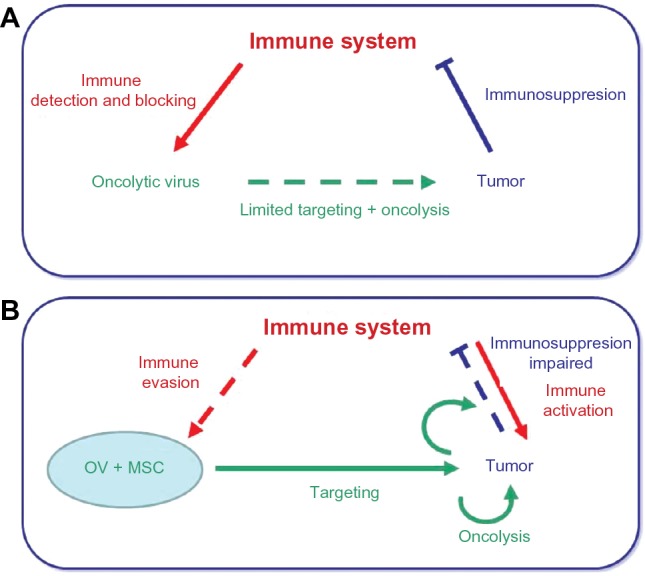
Mesenchymal stem cells, carriers for oncolytic viruses and beyond.
Notes: (A) The immune system interferes with the action of OV, reducing or eliminating their efficacy as anticancer agents. Independently, the tumor microenviroment inactivates the attack of the immune system. (B) MSCs enhance the oncolytic action of OV by protecting them from the immune system while delivering them at the tumor sites. In addition, MSCs may interfere with the mechanisms of immunosuppression developed by the tumor microenvironment, enabling an antitumor immune response. Solid lines indicate dominant action whereas dotted lines indicate impaired action. Colors identify each main character, ie, immune response (red), tumor (blue), and virotherapy (green).
Abbreviations: OV, oncolytic viruses; MSCs, mesenchymal stem cells.
Practical questions
It is not currently known what oncolytic virus would be best to combine with MSCs for clinical uses. It may be anticipated that a candidate virus should possess a few characteristics before it can be considered for the Trojan horse approach using MSCs. On the one hand, the oncolytic virus must be able to infect the cellular vehicle through their own receptors (virus specific) that have to be expressed by MSCs or through modified manipulation (such as the RGD motif). Second, the lytic cycle of the virus must provide enough time for carrier cells as to home into the tumor, as described earlier. Third, virus immunogenicity is a key aspect. Fourth, the virus has to go unnoticed by the immune system as long as possible. Here, since MSCs will provide initial concealment, it seems preferable to have a virus that generates a strong local inflammatory response to trigger further leukocyte infiltration and change the immunosuppressive status of the tumor. We have used an oncolytic adenovirus in our strategy. Initially, MSCs infected with adenoviral vectors showed no major changes in phenotypic or functional characteristics,55 so all considerations commented earlier should be valid when they are infected with oncolytic adenoviruses for treating patients. MSCs have a 48–72-hour viability window following oncoviral infection,2 after which they should disappear because of the viral replication within that destroys them. Therefore, their function as vehicles should have already taken place. Studies on the kinetics of infused MSCs in animals have shown that this length of time should be enough.56,57 It is important to consider that intravenously infused MSCs may change location after their initial first-pass effect. Their function as immune modulators, either as MSC1 or MSC2, should also disappear within this time frame. In this case, their immune effect should primarily be related to the antiadenoviral response, rather than the antitumoral one. However, it has been recently reported that MSCs have a short-term memory and retain information about danger signals of the environment,58 which may allow them to participate in the antitumor immune response once they home into the metastasis sites. It is not known how MSCs, responsible of many tolerant mechanisms, may affect the tumor microenvironment during the time they are functionally viable.
The combination of MSCs and oncolytic viruses has not yet been pursued in the clinical setting by many groups. Although different sources of MSCs have been used in preclinical models, there is no data on the impact that the source (bone marrow and adipose tissue are the two most commonly used in clinic for other diseases)4 or the histocompatibility (autologous, related, or unrelated donor) of MSCs may have in clinical outcome, both interesting issues for future research. A Mayo Clinic group is leading a Phase I/II trial for assessing safety, dosage, and clinical effect of adipose tissue-derived MSCs infected with an oncolytic measles virus encoding thyroidal sodium iodide symporter (MV-NIS) in patients with relapsed ovarian cancers (ClinicalTrials.gov Identifier: NCT02068794). This trial is the continuation of preclinical experiments carried out by the investigators.57 Our group2 has been developing a strategy for the treatment of refractory and metastatic solid tumors in children for the last 10 years, based on the administration of Celyvir: autologous bone marrow–derived MSCs infected with ICOVIR-5, an oncolytic adenovirus59 designed for systemic treatment of disseminated tumors. ICOVIR-5 contains several modifications that give selective replication ability in cancer cells in which the Rb/E2F route is deregulated.60 Our strategy uses systemic infusions of Celyvir aiming at enhancing the targeted delivery of the oncolytic adenovirus to the metastasis sites based on the natural tumor tropism of the MSCs.61 Our initial clinical experience has been an already finished62 compassionate use program in children with refractory tumors, mainly neuroblastoma, and a currently open clinical trial recruiting children and adult patients with advanced cancers (EudraCT2008-000364-16; ClinicalTrials Identifier: NCT01844661). Although a final analysis of these experiences is still pending, after more than 300 doses administered to over 30 children plus six adults with cancer, the first conclusion to be drawn is that tolerance is excellent, with viral-related toxicities being very mild and self-limiting. Hematological and biochemical controls performed to patients during treatment with Celyvir have all been in the normal range. Furthermore, in addition to the oncolytic capacity of Celyvir, the antitumor immune response seems essential in the clinical course of this therapy. We will provide a thorough analysis when the trial is completed.
Conclusion and future perspectives
Delivered by MSCs, oncolytic virotherapies may add value to the intrinsic viral oncolytic capacities and immunotherapeutic effects1 (see, for instance, the recent review by Sampath and Thorne63 on oncolytic virotherapies). Several characteristics of the MSCs, related to their migratory and immune-modulation capacities, contribute to the success of the new medicinal product. Enhancing our understanding of the mechanisms governing MSCs migration and MSCs immune modulation of the antiviral and antitumor immune responses will help in designing safer and more efficacious therapies.
Footnotes
Disclosure
The authors report no conflicts of interests in this work.
References
- 1.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14(8):559–567. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 2.García-Castro J, Alemany R, Cascalló M, et al. Treatment of metastatic neuroblastoma with systemic oncolytic virotherapy delivered by autologous mesenchymal stem cells: an exploratory study. Cancer Gene Ther. 2010;17(7):476–483. doi: 10.1038/cgt.2010.4. [DOI] [PubMed] [Google Scholar]
- 3.Willmon C, Harrington K, Kottke T, Prestwich R, Melcher A, Vile R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol Ther. 2009;17(10):1667–1676. doi: 10.1038/mt.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.García-Gómez I, Elvira G, Zapata AG, et al. Mesenchymal stem cells: biological properties and clinical applications. Expert Opin Biol Ther. 2010;10(10):1453–1468. doi: 10.1517/14712598.2010.519333. [DOI] [PubMed] [Google Scholar]
- 5.Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. 1968;6(2):230–247. [PubMed] [Google Scholar]
- 6.Murphy MB, Moncivais K, Caplan AI. Mesenchymal stem cells: environmentally responsive therapeutics for regenerative medicine. Exp Mol Med. 2013;45:e54. doi: 10.1038/emm.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nombela-Arrieta C, Ritz J, Silberstein LE. The elusive nature and function of mesenchymal stem cells. Nat Rev Mol Cell Biol. 2011;12(2):126–131. doi: 10.1038/nrm3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 9.Salem HK, Thiemermann C. Mesenchymal stromal cells: current understanding and clinical status. Stem Cells. 2010;28(3):585–596. doi: 10.1002/stem.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castro-Manrreza ME, Montesinos JJ. Immunoregulation by mesenchymal stem cells: biological aspects and clinical applications. J Immunol Res. 2015;2015:394917. doi: 10.1155/2015/394917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62(13):3603–3608. [PubMed] [Google Scholar]
- 12.Reagan MR, Kaplan DL. Concise review: mesenchymal stem cell tumor-homing: detection methods in disease model systems. Stem Cells. 2011;29(6):920–927. doi: 10.1002/stem.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menon LG, Picinich S, Koneru R, et al. Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem Cells. 2007;25(2):520–528. doi: 10.1634/stemcells.2006-0257. [DOI] [PubMed] [Google Scholar]
- 14.Lin SY, Yang J, Everett AD, et al. The isolation of novel mesenchymal stromal cell chemotactic factors from the conditioned medium of tumor cells. Exp Cell Res. 2008;314(17):3107–3117. doi: 10.1016/j.yexcr.2008.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15(10):730–738. doi: 10.1038/gt.2008.39. [DOI] [PubMed] [Google Scholar]
- 16.Simon SI, Green CE. Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu Rev Biomed Eng. 2005;7:151–185. doi: 10.1146/annurev.bioeng.7.060804.100423. [DOI] [PubMed] [Google Scholar]
- 17.Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4(3):206–216. doi: 10.1016/j.stem.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Barbash IM, Chouraqui P, Baron J, et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: feasibility, cell migration, and body distribution. Circulation. 2003;108(7):863–868. doi: 10.1161/01.CIR.0000084828.50310.6A. [DOI] [PubMed] [Google Scholar]
- 19.Ip JE, Wu Y, Huang J, Zhang L, Pratt RE, Dzau VJ. Mesenchymal stem cells use integrin beta1 not CXC chemokine receptor 4 for myocardial migration and engraftment. Mol Biol Cell. 2007;18(8):2873–2882. doi: 10.1091/mbc.E07-02-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rüster B, Göttig S, Ludwig RJ, et al. Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood. 2006;108(12):3938–3944. doi: 10.1182/blood-2006-05-025098. [DOI] [PubMed] [Google Scholar]
- 21.Chamberlain G, Wright K, Rot A, Ashton B, Middleton J. Murine mesenchymal stem cells exhibit a restricted repertoire of functional chemokine receptors: comparison with human. PLoS One. 2008;3:e2934. doi: 10.1371/journal.pone.0002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Y, Zhao RC. The role of chemokines in mesenchymal stem cell homing to myocardium. Stem Cell Rev. 2012;8(1):243–250. doi: 10.1007/s12015-011-9293-z. [DOI] [PubMed] [Google Scholar]
- 23.Wynn RF, Hart CA, Corradi-Perini C, et al. A small proportion of mesenchymal stem cells strongly expresses functionally active CXCR4 receptor capable of promoting migration to bone marrow. Blood. 2004;104(9):2643–2645. doi: 10.1182/blood-2004-02-0526. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Yu X, Lin S, Li X, Zhang S, Song YH. Insulin-like growth factor 1 enhances the migratory capacity of mesenchymal stem cells. Biochem Biophys Res Commun. 2007;356:780–784. doi: 10.1016/j.bbrc.2007.03.049. [DOI] [PubMed] [Google Scholar]
- 25.Lourenco S, Teixeira VH, Kalber T, Jose RJ, Floto RA, Janes SM. Macrophage migration inhibitory factor – CXCR4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors. J Immunol. 2015;194(7):3463–3474. doi: 10.4049/jimmunol.1402097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klopp AH, Spaeth EL, Dembinski JL, et al. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res. 2007;67(24):11687–11695. doi: 10.1158/0008-5472.CAN-07-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dwyer RM, Potter-Beirne SM, Harrington KA, et al. Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin Cancer Res. 2007;13(17):5020–5027. doi: 10.1158/1078-0432.CCR-07-0731. [DOI] [PubMed] [Google Scholar]
- 28.Saji H, Koike M, Yamori T, et al. Significant correlation of monocyte chemoattractant protein-1 expression with neovascularization and progression of breast carcinoma. Cancer. 2001;92(5):1085–1091. doi: 10.1002/1097-0142(20010901)92:5<1085::aid-cncr1424>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 29.Xu S, Menu E, De Becker A, Van Camp B, Vanderkerken K, Van Riet I. Bone marrow-derived mesenchymal stromal cells are attracted by multiple myeloma cell-produced chemokine CCL25 and favor myeloma cell growth in vitro and in vivo. Stem Cells. 2012;30(2):266–279. doi: 10.1002/stem.787. [DOI] [PubMed] [Google Scholar]
- 30.Binger T, Stich S, Andreas K, et al. Migration potential and gene expression profile of human mesenchymal stem cells induced by CCL25. Exp Cell Res. 2009;315(8):1468–1479. doi: 10.1016/j.yexcr.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 31.De Becker A, Van Hummelen P, Bakkus M, et al. Migration of culture-expanded human mesenchymal stem cells through bone marrow endothelium is regulated by matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-3. Haematologica. 2007;92(4):440–449. doi: 10.3324/haematol.10475. [DOI] [PubMed] [Google Scholar]
- 32.Ries C, Egea V, Karow M, Kolb H, Jochum M, Neth P. MMP-2, MT1-MMP, and TIMP-2 are essential for the invasive capacity of human mesenchymal stem cells: differential regulation by inflammatory cytokines. Blood. 2007;109(9):4055–4063. doi: 10.1182/blood-2006-10-051060. [DOI] [PubMed] [Google Scholar]
- 33.Ho IA, Chan KY, Ng WH, et al. Matrix metalloproteinase 1 is necessary for the migration of human bone marrow-derived mesenchymal stem cells toward human glioma. Stem Cells. 2009;27(6):1366–1375. doi: 10.1002/stem.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolontrade MF, Sganga L, Piaggio E, et al. A specific subpopulation of mesenchymal stromal cell carriers overrides melanoma resistance to an oncolytic adenovirus. Stem Cells Dev. 2012;21(14):2689–2702. doi: 10.1089/scd.2011.0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. 2011;19(6):1008–1016. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alvarez-Breckenridge CA, Yu J, Price R, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med. 2012;18(12):1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78(11):5535–5545. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 39.Qiao J, Kottke T, Willmon C, et al. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat Med. 2008;14(1):37–44. doi: 10.1038/nm1681. [DOI] [PubMed] [Google Scholar]
- 40.Beyth S, Borovsky Z, Mevorach D, et al. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. 2005;105(5):2214–2219. doi: 10.1182/blood-2004-07-2921. [DOI] [PubMed] [Google Scholar]
- 41.Jiang XX, Zhang Y, Liu B, et al. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood. 2005;105(10):4120–4126. doi: 10.1182/blood-2004-02-0586. [DOI] [PubMed] [Google Scholar]
- 42.Li YP, Paczesny S, Lauret E, et al. Human mesenchymal stem cells license adult CD34+ hemopoietic progenitor cells to differentiate into regulatory dendritic cells through activation of the Notch pathway. J Immunol. 2008;180(3):1598–1608. doi: 10.4049/jimmunol.180.3.1598. [DOI] [PubMed] [Google Scholar]
- 43.Zhang B, Liu R, Shi D, et al. Mesenchymal stem cells induce mature dendritic cells into a novel Jagged-2-dependent regulatory dendritic cell population. Blood. 2009;113(1):46–57. doi: 10.1182/blood-2008-04-154138. [DOI] [PubMed] [Google Scholar]
- 44.Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood. 2008;111(3):1327–1333. doi: 10.1182/blood-2007-02-074997. [DOI] [PubMed] [Google Scholar]
- 45.Duffy MM, Ritter T, Ceredig R, Griffin MD. Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Res Ther. 2011;2(4):34. doi: 10.1186/scrt75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi M, Liu ZW, Wang FS. Immunomodulatory properties and therapeutic application of mesenchymal stem cells. Clin Exp Immunol. 2011;164(1):1–8. doi: 10.1111/j.1365-2249.2011.04327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tabera S, Pérez-Simón JA, Díez-Campelo M, et al. The effect of mesenchymal stem cells on the viability, proliferation and differentiation of B-lymphocytes. Haematologica. 2008;93(9):1301–1309. doi: 10.3324/haematol.12857. [DOI] [PubMed] [Google Scholar]
- 48.Chan JL, Tang KC, Patel AP, et al. Antigen-presenting property of mesenchymal stem cells occurs during a narrow window at low levels of interferon-gamma. Blood. 2006;107(12):4817–4824. doi: 10.1182/blood-2006-01-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stagg J, Pommey S, Eliopoulos N, Galipeau J. Interferon-gamma-stimulated marrow stromal cells: a new type of nonhematopoietic antigen-presenting cell. Blood. 2006;107(6):2570–2577. doi: 10.1182/blood-2005-07-2793. [DOI] [PubMed] [Google Scholar]
- 50.Mader EK, Maeyama Y, Lin Y, et al. Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin Cancer Res. 2009;(23):7246–7255. doi: 10.1158/1078-0432.CCR-09-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahmed AU, Rolle CE, Tyler MA, et al. Bone marrow mesenchymal stem cells loaded with an oncolytic adenovirus suppress the anti-adenoviral immune response in the cotton rat model. Mol Ther. 2010;18(10):1846–1856. doi: 10.1038/mt.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13(4):392–402. doi: 10.1016/j.stem.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 53.Waterman RS, Tomchuck SL, Henkle SL, Betancourt AM. A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an immunosuppressive MSC2 phenotype. PLoS One. 2010;5(4):e10088. doi: 10.1371/journal.pone.0010088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waterman RS, Henkle SL, Betancourt AM. Mesenchymal stem cell 1 (MSC1)-based therapy attenuates tumor growth whereas MSC2-treatment promotes tumor growth and metastasis. PLoS One. 2012;7(9):e45590. doi: 10.1371/journal.pone.0045590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Treacy O, Ryan AE, Heinzl T, et al. Adenoviral transduction of mesenchymal stem cells: in vitro responses and in vivo immune responses after cell transplantation. PLoS One. 2012;7(8):e42662. doi: 10.1371/journal.pone.0042662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao J, Dennis JE, Muzic RF, Lundberg M, Caplan AI. The dynamic in vivo distribution of bone marrow-derived mesenchymal stem cells after infusion. Cells Tissues Organs. 2001;169(1):12–20. doi: 10.1159/000047856. [DOI] [PubMed] [Google Scholar]
- 57.Mader EK, Butler G, Dowdy SC, et al. Optimizing patient derived mesenchymal stem cells as virus carriers for a phase I clinical trial in ovarian cancer. J Transl Med. 2013;11:20. doi: 10.1186/1479-5876-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu GY, Liu Y, Lu Y, et al. Short-term memory of danger signals or environmental stimuli in mesenchymal stem cells: implications for therapeutic potential. Cell Mol Immunol. 2015 Mar 16; doi: 10.1038/cmi.2015.11. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cascallo M, Alonso MM, Rojas JJ, Perez-Gimenez A, Fueyo J, Alemany R. Systemic toxicity-efficacy profile of ICOVIR-5, a potent and selective oncolytic adenovirus based on the pRB pathway. Mol Ther. 2007;15(9):1607–1615. doi: 10.1038/sj.mt.6300239. [DOI] [PubMed] [Google Scholar]
- 60.Alonso MM, Cascallo M, Gomez-Manzano C, et al. ICOVIR-5 shows E2F1 addiction and potent antiglioma effect in vivo. Cancer Res. 2007;67(17):8255–8263. doi: 10.1158/0008-5472.CAN-06-4675. [DOI] [PubMed] [Google Scholar]
- 61.Cussó L, Mirones I, Peña-Zalbidea S, García-Vázquez V, García-Castro J, Desco M. Combination of single-photon emission computed tomography and magnetic resonance imaging to track 111 in-oxine-labeled human mesenchymal stem cells in neuroblastoma-bearing mice. Mol Imaging. 2014;13 doi: 10.2310/7290.2014.00033. [DOI] [PubMed] [Google Scholar]
- 62.Ramírez M, García-Castro J, Alemany R, et al. Virotherapy delivered by autologous mesenchymal stem cells for children with metastatic and refractory neuroblastoma: results of a trial of compassionate use [abstract] Pediatr Blood Cancer. 2014;61(Suppl 2):S107. [Google Scholar]
- 63.Sampath P, Thorne SH. Novel therapeutic strategies in human malignancy: combining immunotherapy and oncolytic virotherapy. Oncolytic Virother. 2015;4:75–82. doi: 10.2147/OV.S54738. [DOI] [PMC free article] [PubMed] [Google Scholar]