

Abstract
Immunotherapy and oncolytic virotherapy have both shown anticancer efficacy in the clinic as monotherapies but the greatest promise lies in therapies that combine these approaches. Vesicular stomatitis virus is a prominent oncolytic virus with several features that promise synergy between oncolytic virotherapy and immunotherapy. This review will address the cytotoxicity of vesicular stomatitis virus in transformed cells and what this means for antitumor immunity and the virus’ immunogenicity, as well as how it facilitates the breaking of tolerance within the tumor, and finally, we will outline how these features can be incorporated into the rational design of new treatment strategies in combination with immunotherapy.
Keywords: virotherapy, rhabdovirus, anti-tumor immunity, t cell, natural killer cell, therapeutic vaccine
Introduction
The immunotherapy of cancer has generated widespread excitement with recent clinical findings illustrating a therapeutic modality that is capable of eliminating a wide array of cancers in various stages of development. By harnessing and augmenting a patient’s natural immune response against transformed cells, many groups have shown the potential and power of this approach for the treatment of cancer.1,2 Oncolytic viruses (OVs), which are a special subset of viruses that show preferential infection and lysis of tumor cells, have emerged as a potent tool to complement cancer immunotherapeutics. In addition to facilitating direct tumor debulking, OVs can act as potent immune stimulators that are capable of overcoming the immunosuppressive environment established by tumors.3,4 This review will focus on vesicular stomatitis virus (VSV) and its use as a flexible and potent platform for cancer immunotherapy through direct oncolysis combined with engagement of the innate and adaptive immune responses.
VSV structure and molecular biology
VSV is an enveloped virus belonging to the Rhabdoviridae family and, as such, features an 11 kb single-stranded negative-sense RNA genome. The genome serves as the template for the synthesis of five subgenomic monocistronic messenger RNAs (mRNAs) during the course of the infection cycle.5 The relative abundance of each viral protein produced during infection is determined by the location of the coding region in reference to the 3′ end of the genome (with N being the most abundant and L being the least). This is controlled by dissociation of the viral RNA-dependent RNA polymerase (composed of the L and P proteins) in the intergenic regions of the viral genome as it progresses along the genome in a 3′–5′ direction.5 Figure 1 outlines the structure and organization of the viral genome, the relative abundance of transcripts/protein products and their functions, and the structure of the mature virion.
Figure 1.
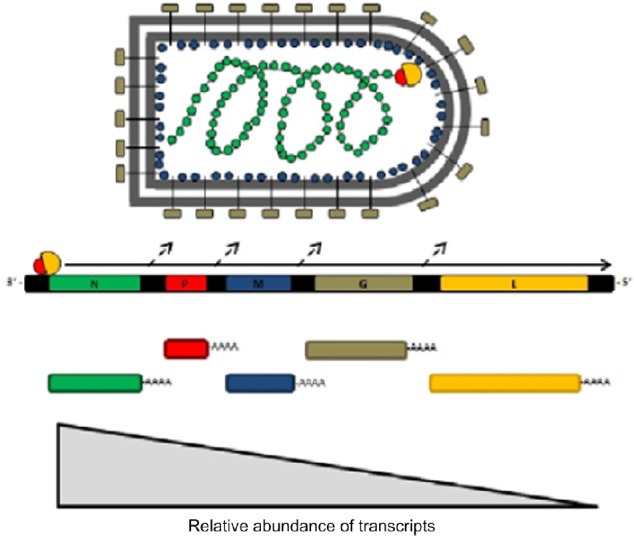
A schematic representation of the vesicular stomatitis virus virion and genome showing subgenomic mRNA and relative transcript abundance.
Notes: The negative-sense RNA genome is completely coated in the viral nucleoprotein (N protein) and is transcribed by the viral RNA-dependent RNA polymerase consisting of L and P subunits. Attachment, entry into the host cell, and membrane fusion are mediated by the glycoprotein (G protein). The matrix protein (M protein) plays a number of vital roles in the infection cycle. It is responsible for the budding of progeny virions, regulation of viral genome replication, and genome packaging, in addition to allowing VSV to shut down host cells’ innate antiviral responses through a variety of mechanisms. Dissociation of the viral polymerase in the intergenic regions of the genome controls the transcriptional abundance of five subgenomic monocistronic mRNAs. The full arrow indicates the direction of transcription of the viral genome, and the dashed arrows indicate the points at which the viral polymerase can dissociate from the viral genome.
Abbreviations: mRNA, messenger RNA; VSV, vesicular stomatitis virus.
A number of factors, such as its short replication time, the large amount of progeny generated by a single cycle of infection, a lack of preexisting humoral immunity in the general population, its broad tissue tropism, and its amenability to genetic engineering, make VSV an excellent OV. What is especially appealing about the use of VSV is its large safety window owing to its inability to induce transformation in healthy cells, as well as its sensitivity to type I interferons (IFNs).6
VSV attachment and entry
True to its originally intended purpose, the first useful feature of VSV for combination with immunotherapy is its ability to directly facilitate tumor debulking. The viral glycoprotein (G protein) is embedded in the viral envelope and facilitates attachment and entry into the host cell by binding to the low-density-lipoprotein receptor (LDLR) and its family members.7 Members of the LDLR family are ubiquitously expressed by mammalian cells; this allows VSV to infect virtually any cell type. Therefore, application of VSV in oncolytic virotherapy is not restricted by host receptor expression and can be used in an array of tumor types. In addition, this means that VSV entry is not restricted to cancer cells. However, VSV replication is still a desirable feature of the virus due to its extreme sensitivity to type I IFN (discussed in the section “Tumor-specific replication of VSV and type I IFN”).
Tumor-specific replication of VSV and type I IFN
Activation of innate immune mechanisms by type I IFN protects normal cells from VSV infection-induced lysis, while cancer cells, which commonly have defects in the type I IFN signaling pathway, are unable to induce a protective innate immune response. VSV is a potent inducer of type I IFNs and the rapid systemic dissemination of type I IFN after VSV infection selectively shields normal cells from VSV infection, thus restricting infection to cancer cells. VSV has mechanisms to inhibit type I IFN signaling, but these mechanisms are reliant on expression of the M protein to induce shutdown of gene expression within infected cells.8–10 VSV-M protein is able to block the nuclear export of host mRNA. This is achieved when M protein associates with nuclear pore complexes (NPCs) embedded in the nuclear envelope via nucleoporin Nup98 and the mRNA export factor Rae1 (Figure 2). The association of M protein to NPCs via Nup98 is mediated by amino acid residues 51–59 of the M protein, and it results in the blockade of nuclear export of mRNAs into the cytosol of the infected cell.9,11 Additionally, the M protein can inactivate DNA-dependent RNA polymerase II and stop the transcription of host antiviral response genes (Figure 2). This is accomplished via host factor-mediated inactivation of the TATA-binding protein subunit of the transcription factor IID (TFIID).8,12,13 Finally, the M protein is capable of dephosphorylating eIF4E and 4E-binding protein 1, thereby altering the structure of the eIF4F cap-binding complex, which results in inhibition of the translation of host mRNA while allowing for the translation of viral mRNA (Figure 2).10 The global suppression of host gene expression by VSV-M favors viral protein synthesis but also blunts infected cells’ ability to produce type I IFN and respond to exogenous type I IFN. This is believed to be the main mechanism for VSV’s oncolytic activity because the shutdown of host gene expression induces a cellular stress response that leads to apoptosis.
Figure 2.
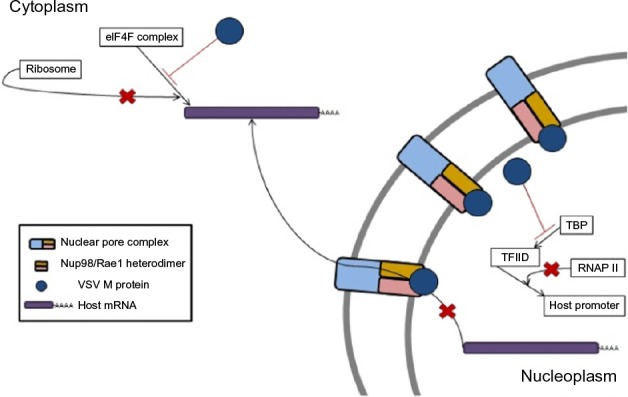
VSV M protein mediates the shutdown of host cell gene expression through three mechanisms.
Notes: Association of M protein with nuclear pore complexes inside the nucleus via binding to Nup98/Rae1 heterodimers, thereby physically blocking the export of host cell mRNA. M protein can induce the indirect inactivation of host cell DNA-dependent RNA polymerase II via inactivation of the TATA-binding subunit of TFIID, which results in the shutdown of host gene transcription. By altering the phosphorylation states of components of the eIF4F cap-binding complex, M protein acts to prevent the translation of host mRNAs.
Abbreviations: RNAP, RNA polymerase; TBP, TATA-binding protein; TFIID, transcription factor IID; VSV, vesicular stomatitis virus; mRNA, messenger RNA.
Wild-type VSV has been proven to be a powerful anticancer agent in vitro and in vivo, but the neurotropism of the virus has been linked with fatal neurotoxicity in both murine and nonhuman primate models.14,15 Given the safety concern with the use of wild-type VSV in a clinical setting, considerable effort has been invested into producing attenuated strains of the virus, which still maintain the ability to selectively kill transformed cells. Strategies have been developed to generate therapeutically effective strains of VSV with increased vulnerability to type I IFNs to increase their specificity for IFN response-defective cancer cells.16,17 The most widely used strategy of VSV attenuation was developed by John Bell’s group.18 Attenuation was accomplished via the deletion or mutation of methionine 51 in the M protein (VSV∆M51); this compromises the ability of the virus to shut down host gene expression in infected cells while retaining its oncolytic properties.18 As a result, VSV∆M51 replication is more sensitive to type I IFN signaling in the infected cell than replication of wild-type VSV.
Induction of apoptosis of infected transformed cells
The mechanisms of cell death induced by wild-type VSV and VSV∆M51 infection are different. Wild-type VSV-mediated shutdown of host expression induces signaling via the JNK/SAPK stress pathways, resulting in the formation of the mitochondrial apoptosis-induced channel (MAC) on the outer mitochondrial membrane to allow for the release of cytochrome C from mitochondria (Figure 3).19 Cytochrome C is normally associated with the inner mitochondrial membrane and functions as part of the electron transport chain during aerobic respiration; during the induction of apoptosis, it dissociates from its cardiolipin anchor, which enables it to enter the cytosol via the MAC.20
Figure 3.

Wild-type and mutant VSV induce apoptosis via distinct pathways, but cross-talk does occur.
Notes: Wild-type VSV M protein-mediated shutdown of host gene expression induces the intrinsic apoptotic pathway involving the release of cytochrome C from mitochondria into the cytosol. Formation of the apoptosome allows for the activation of caspase-9 and downstream caspases, which culminates in the death of the infected cell. VSV∆M51 induces apoptosis via the extrinsic pathway as a consequence of the detection of viral replication intermediates by double-stranded RNA-regulated protein kinase (PKR) and/or via type I interferon signaling. Downstream effects of PKR signaling induce the dimerization of FasR and Daxx to facilitate the phosphorylation of ASK-1. Phosphorylated ASK-1 activates the JNK signaling cascade, leading to the upregulation of proapoptotic factors and increased surface expression of death receptors (eg, the death-inducing signaling complex, DISC, which can activate caspase-3). Additionally, the production of viral proteins induces the endoplasmic reticulum stress response and leads to the activation of caspase-12. Blue arrows indicate points of cross–talk between the intrinsic and extrinsic pathways. The dashed arrows in the figure indicate multi-step processes.
Abbreviations: ASK, apoptosis signal-regulating kinase 1; Daxx, Fas death domain-associated-xx; dsRNA, double-stranded RNA; IFN, interferon; MAC, mitochondrial apoptosis-induced channel; MKK4, mitogen-activated protein kinase kinase 4; pASK, phosphorylated ASK; pJNK, phosphorylated JNK; PKR, RNA-regulated protein kinase; pMKK4, phosphorylated mitogen-activated protein kinase kinase 4; VSV, vesicular stomatitis virus.
Once in the cytosol, cytochrome C is free to bind to apoptosis-activating factor 1 (Apaf-1), which results in the formation of the apoptosome; this structure cleaves inactive pro-caspase-9 into its active form in an adenosine triphosphate (ATP)-dependent manner.21 Activated caspase-9 activates a number of target initiator caspases via proteolytic cleavage – most notably, caspase-3; activated caspase-3 cleaves a number of downstream targets to initiate the death of the infected cell.19 Some cross talk between the intrinsic and extrinsic (death receptor) pathways of apoptosis can occur via capase-3-mediated cleavage of caspase-8 (Figure 3).
In contrast to the wild-type virus, which initiates apoptosis by preventing host gene expression, the M-protein-mutant strains of VSV induce apoptosis via the extrinsic “death receptor” pathway of apoptotic induction (Figure 3). A study using the infection of IFN-responsive L929 murine fibroblasts with VSV∆M51 demonstrated significantly increased surface expression of the death-receptor FasR and induction of apoptosis in a ligand-independent manner.22 Unlike canonical induction of apoptosis through the extrinsic pathway via the adaptor Fas-associated death domain (FADD), the key players in VSV-mediated apoptosis appear to be Fas death domain-associated-xx (Daxx), an alternative adaptor protein to FADD, and double-stranded RNA-regulated protein kinase (PKR).23–25 PKR is upregulated by type I IFN signaling and detects the presence of double-stranded RNA, such as the intermediates produced during VSV gene transcription/genome replication. PKR acts as a modulator of the phosphorylation of apoptosis signal-regulating kinase 1 (ASK1) to control Daxx-mediated induction of apoptosis. PKR has also been shown to actively regulate the expression of proapoptotic factors, including the Fas receptor, as well as the subcellular localization of Daxx.26,27 The Fas–Daxx interaction initiates the JNK apoptotic pathway via phosphorylation of ASK1; the phosphorylated ASK1 (pASK1) phosphorylates mitogen-activated protein kinase kinase 4 (MKK4), which leads to the phosphorylation of JNK.28 Activation of the JNK pathway then leads to upregulation of factors such as members of the proapoptotic Bcl-2 family and the activation of caspase-8.19 Precisely how caspase-8 is activated by mutant VSV infection is a matter of ongoing study. The likely mechanism is either (or both) through upregulation of death receptors leading to canonical caspase-8 activation via the death-induced signaling complex (DISC) or indirectly through endoplasmic reticulum stress-induced caspase-12 activation leading to caspase-8 activation.19
Immunostimulatory benefits of oncolytic VSV
It has long been known that tumor-specific immune attack is an important factor in the prevention of cancer.29,30 As such, transformed cells often develop mechanisms for evasion of immune-mediated clearance through the selective pressures of cancer immunoediting.31 The transformed cells that manage to survive the selective gauntlet imposed by the immune system go on to form tumors that are characterized by an immunosuppressive microenvironment. The tumor microenvironment is rich in suppressive cells, inhibitory immune checkpoint molecule expression, and immunosuppressive cytokines such as transforming growth factor (TGF)-β and interleukin-10 (IL-10).32 Collectively, these factors allow for uncontrolled tumor growth and the invariably fatal progression of untreated disease.
The immunogenicity of VSV itself can play a vital role in overcoming tumor-mediated immunosuppression through the induction of a strong innate antitumor response. Systemic type I IFN induced after VSV infection skews the inflammatory status of the tumor microenvironment from immunosuppressive to immunostimulatory. Innate immune effector cells, such as natural killer (NK) cells, are recruited and activated, which begin to facilitate tumor clearance.33 In fact, effective VSV therapy only requires a single cycle of viral gene expression to generate a potent early antitumor immune response mediated by NK cells.34
Regardless of whether tumor cell apoptosis is induced by VSV or by recruited immune cells, the end result is very much the same – tumor debulking and the stimulation of an antitumor immune response by factors released from dying tumor cells (Figure 4). The death of tumor cells results in the release of a number of molecules associated with damage-associated molecular patterns (DAMPs), such as calreticulin and high-mobility group box-1 (HMGB1), which are detected by dendritic cells (DCs) via pattern recognition receptors.35–37 Activated DCs migrate toward lymph nodes and secrete proinflammatory cytokines such as IL-1β and IL-18, which are necessary to engage adaptive immunity against tumor antigens mediated by CD8+ T-cells.38–40 Dying tumor cells also release tumor-associated antigens (including novel epitopes derived from mutated self-proteins).41 Cross-presentation of released tumor antigens on major histocompatibility complex (MHC) class I by mature DCs stimulates the activation and proliferation of tumor antigen-specific CD8+ T-cells within the lymph nodes.38,42 Cross-presentation of released tumor-associated antigens can lead to the priming of an antitumor response against multiple tumor-specific epitopes, which is advantageous when one considers the considerable heterogeneity within and between tumors of the same type.43 These immunostimulatory mechanisms of VSV allow for the treatment of advanced or aggressive tumors by overcoming tumor-mediated immunosuppression and allowing for lymphocytic infiltration of distant metastases. Both the innate and adaptive arms of the immune system are beneficial to oncolytic VSV therapy, and both need to be engaged if VSV therapy is to effectively synergize with immunotherapy in the future.
Figure 4.

VSV oncolytic virotherapy can engage antitumor immunity as a consequence of viral infection of transformed cells.
Notes: Tumor debulking is achieved via direct and indirect means. Viral replication in tumor cells will result in cytopathic effects and culminate in the death of the infected cell. Similarly, the innate antiviral response of infected cells will result in the death of tumor cells through the downstream effects of type I interferon signaling. This, coupled with the release of tumor antigens and danger-associated molecular patterns (DAMPs), stimulates the maturation of local dendritic cells. Mature dendritic cells secrete proinflammatory cytokines to recruit innate effector cells to mediate tumor killing, and they migrate to tumor-draining and distal lymph nodes to present tumor antigens to tumor-reactive CD8+ T-cells. Activated T-cells migrate to the tumor and mediate antigen-specific attack on tumor cells.
Abbreviations: DAMPS, danger-associated molecular patterns; DC, dendritic cell; IFN, interferon; IL, interleukin; MHC, major histocompatibility complex; NK cell, natural killer cell; TNF, tumor necrosis factor; VSV, vesicular stomatitis virus.
Engineering a more immunogenic VSV
The immune system has traditionally been viewed as a barrier to successful oncolytic virotherapy; innate and adaptive immunity were seen as factors that conspired to limit the dissemination of the oncolytic agent and the subsequent infection of transformed cells. In fact; this is especially important given the heterogeneity in the type I IFN response deficiency in tumors, which limits the ability of VSV to directly debulk some tumors.44–46 However, the discovery that the immune system plays a vital role in tumor debulking induced by OV therapy has led to a shift in this paradigm, and a number of novel strategies to effectively engage anti-tumor immunity have been developed as a result.33,34 Due to the ever-growing body of evidence that VSV can augment endogenous antitumor immunity, much attention has been focused on inserting immunostimulatory genes into the VSV genome in an effort to more effectively engage innate and adaptive immunity against tumors (Figure 5). Given the critical role of antigen-presenting cells (APCs) in mediating adaptive antitumor immune responses, numerous groups have generated VSV-encoding transgenes specifically intended to engage DCs and other APCs to break tolerance against tumor antigens.47,48 In particular, viral vectors (including VSV) encoding granulocyte-monocyte colony stimulating factor (GMCSF) or FMS-related tyrosine kinase-3 ligand (FLT3L) have shown considerable efficacy in the induction of DC maturation and subsequent antigen presentation to effector cells of the adaptive immune system.47–49
Figure 5.

The engagement of innate and adaptive immunity against tumors.
Notes: In an effort to enhance the immunogenicity of VSV, a number of groups have explored the incorporation of a transgene into the VSV genome, most commonly between the G and L genes. The expression of cytokines to enhance the antigen-presenting and co-stimulatory abilities of dendritic cells, and the expression of cytokines to enhance effector lymphocyte survival and function have shown great promise in preclinical models. Some transgenes may also improve the safety profile of the engineered virus while simultaneously enhancing the function of antigen-presenting and effector cells (eg, VSV-driven expression of type I interferons).
Abbreviations: Ag, antigen; APC, antigen-presenting cell; DC, dendritic cell; FLT3L, FMS-related tyrosine kinase-3 ligand; GM-CSF, granulocyte-monocyte colony stimulating factor; IFN, interferon; IL, interleukin; NK, natural killer cells; VSV, vesicular stomatitis virus.
It is also possible to insert transgenes that directly support T-cells and NK cells into VSV. Expression of cytokines such as IL-12 and IL-15 can provide much-needed support for effector lymphocytes by promoting proliferation, effector differentiation, and inflammatory cytokine production and can result in complete elimination of primary tumors and metastases.50–52 Some of the cytokines produced by activated lymphocytes, such as tumor necrosis factor-α, can induce apoptosis in tumor cells and cause destruction of the tumor vasculature to starve the tumor of nutrients.53,54 Others serve to stimulate tumor-reactive lymphocyte proliferation and the maintenance of a TH1-polarized immune response against the tumor.55,56
In addition to the role of these cytokines in engaging APCs and effector cells, they can facilitate the breaking of tolerance by potentiating the inflammatory response already induced by VSV infection. It is likely that recombinant VSV expressing cytokine transgenes may synergize particularly well with strategies that already act to overcome the immunosuppressive tumor microenvironment in order to enhance antitumor T-cell function.
Although immunostimulatory VSV vectors have been shown to be efficacious, a hazard remains in their use in the clinic as the antigen targeted by the induced immune response is not specific for tumor antigens. This can severely affect the efficacy of immunostimulatory VSV vectors via the priming of an adaptive immune response against off-target antigens, such as the viral proteins expressed by the VSV vector or the self-antigens shared by the tumor and healthy cells.
VSV as an oncolytic vaccine
Historically, therapeutic cancer vaccines have used replication-deficient viruses encoding tumor antigens or autologous DCs pulsed with either tumor lysate or tumor mRNA. While these approaches have been successful in generating antibody and/or cellular responses against tumor antigens, the magnitude of the responses is not sufficient to mediate complete tumor regression.57 A new paradigm is emerging with the use of “live” OVs as vaccine vectors – so-called “oncolytic vaccines.” As their name implies, oncolytic vaccines use an OV as a vaccine platform for delivery of the target antigen to the tumor bed and lymphoid organs, wherein APCs can be engaged to generate antitumor immunity.58 This is accomplished by intravenous administration of a recombinant VSV carrying a tumor-specific antigen as a transgene. In this context, the use of replication-competent OVs, such as VSV, offers several advantages. In addition to delivering the target antigen to APCs in the tumor and lymphoid organs, VSV-based oncolytic vaccines are capable of mediating direct tumor debulking as a consequence of viral replication.19,22 An advantage of a replication-competent therapeutic vaccine vector is the in situ amplification of the antigen dose due to viral replication, which can potentiate the antitumor response; this offers a benefit relative to other vaccination platforms in which the antigen dose decreases over time.
A shortfall of this system is that In addition to generating a tumor-associated-antigen-targeted response, it also induces responses against the viral vector. In this case, the antitumor response is overshadowed and outpaced by the dominance of the antiviral response because the viral target represents an exogenous antigen and the tumor response represents a self-antigen.59
With the goal of shifting the dominance of the OV vaccine-induced response toward the targeted tumor antigen and away from viral antigens, a heterologous prime-boost OV vaccination strategy has been recently developed. The principle behind this approach is to sequentially use two viral vectors that have been engineered to express a tumor antigen. The first vaccine generates immunological memory against that antigen; the second vaccine engages the memory lymphocytes generated by the primary response to facilitate a much more robust secondary response against the tumor (Figure 6).60,61
Figure 6.
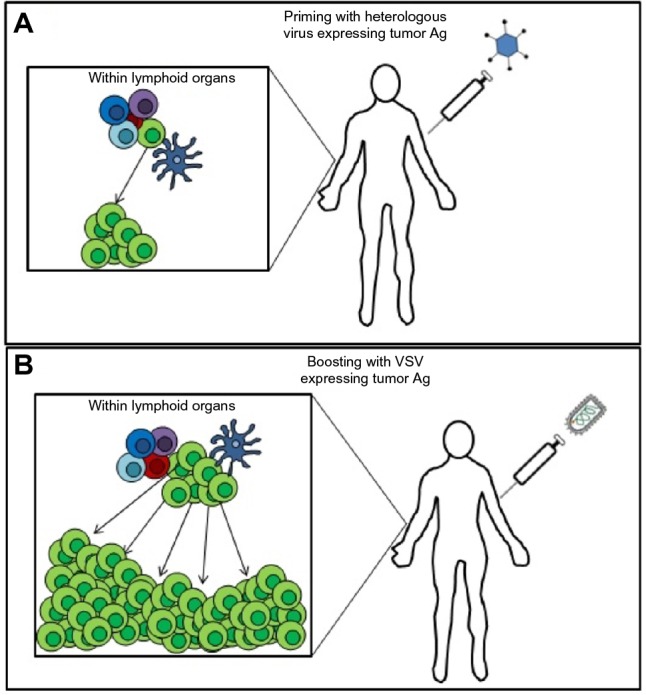
A representation of the rationale behind a heterologous prime-boost therapeutic vaccination strategy using two oncolytic viruses expressing a tumor antigen.
Notes: (A) An oncolytic virus (eg, adenovirus) expressing the target tumor antigen (Ag) is administered intravenously to the patient to engage a primary adaptive immune response and generate memory lymphocytes specific to the tumor. (B) Following the contraction of the primary immune response and the generation of memory, a booster vaccine consisting of VSV expressing the same tumor antigen is administered intravenously to engage the memory T-cells generated by the priming vaccine. This leads to a secondary response against the tumor that is much greater in magnitude than the primary response.
Abbreviations: Ag, antigen; VSV, vesicular stomatitis virus.
By using a heterologous virus in the priming phase of the treatment, the subsequent boost using VSV is not impeded by a neutralizing antibody or cellular response against the vector.62 The priming vaccine also ensures that the dominant immune response generated by the VSV boost is against the tumor antigen, and not against VSV itself. Administration of the VSV-boosting vector will have several effects; viral infection and replication within the tumor will cause immunogenic cell death in the tumor microenvironment and break tolerance to the target tumor antigen, as well as engage “better quality” (as measured by cytolytic and proliferative activity upon antigenic restimulation) CD8+ T-cells compared to those engaged by the primary vaccine vector.62,63 Although much of the work defining this paradigm was done using adenovirus as the priming vector, other viral vectors or DC vaccines are capable of priming an antitumor immune response for subsequent VSV boosting.
An interesting characteristic of VSV has recently emerged, which highlights the utility and efficacy of this OV as a boosting vector. Infection of APCs by VSV leads to the preferential engagement of central memory CD8+ T-cells (TCM) in the lymphoid organs. Trafficking of VSV to the spleen following intravenous administration results in the infection of follicular B-cells and the transfer of antigens carried by the VSV vector to MHC molecules on splenic DCs. Due to the proximity of splenic DCs to TCM, this allows efficient presentation and engagement of the TCM produced by the priming vaccine. This introduces the possibility of bypassing the need to wait for the effector T-cell-skewed primary response to subside as splenic DCs will be able to present the target antigen without lysis by effector T-cells due to their anatomical separation (Bridle et al, unpublished data, 2015).63 Once engaged, TCM reactive to the VSV transgene undergo robust secondary expansion and differentiation into effector CD8+ T-cells to mediate tumor elimination.63 In fact, it has been shown that VSV-boosted CD8+ T-cells are of higher quality, in terms of function and phenotype, compared to cells generated by the prime without any boosting. VSV-boosted TCM exhibited the ability to secrete multiple cytokines, proliferated more robustly, and had superior antitumor effect compared to the effector-memory T-cells generated by adenoviral vaccination alone.62 A limitation of this approach is that the frequency of tumor-reactive CD8+ T-cells generated by the priming vector may vary substantially from patient to patient, so the magnitude of the generated antitumor response may not be enough to mediate complete elimination of the tumor in all cases.30
Prospective VSV combination therapies
VSV oncolytic vaccination and adoptive cell therapy
After years of major obstacles, adoptive cell therapy (ACT) is becoming one of the most promising avenues for the development of future immunotherapeutic protocols. Taking into account what has been learned from heterologous prime-boost protocols, ACT is in a unique position to synergize with oncolytic vaccination with VSV. ACT involves the isolation, or generation through viral transduction, of immune effector cells (most often, CD8+ T-cells) that are reactive to a tumor antigen from a patient’s own blood, followed by ex vivo expansion to large numbers. Once expanded to an appropriate number, the cultured CD8+ T-cells are reinfused into the patient, whereupon they will migrate into the tumor to mediate elimination of antigen-positive tumor cells.64
There has been considerable effort made in improving the conditions of ex vivo culture used to expand CD8+ T-cells for adoptive transfer. New ex vivo culture protocols attempt to uncouple T-cell differentiation and proliferation to generate more desirable memory T-cell subsets.65,66 TCM are far more responsive to antigenic restimulation compared to effector-memory T-cells, as evidenced by robust secondary proliferation and effector function, and they are better able to engraft and persist in patients following transfer to provide ongoing immunosurveillance against possible relapses.67,68
Intravenous administration of VSV expressing a tumor antigen following adoptive transfer of antigen-specific TCM offers the possibility of bypassing the need for priming an endogenous antitumor response with vaccination while simultaneously providing higher-quality tumor-reactive memory T-cells for the VSV-boosting vaccine to engage compared to those generated by a priming vaccination. An additional benefit to the use of VSV is its ability to induce an acute lymphopenia via type I IFN signaling (a phenomenon to which TCM are resistant).69 Lymphodepletion is an important preparative step in ACT as it removes preexisting lymphocyte populations, which allows transferred cells better access to homeostatic cytokines and eliminates inhibitory regulatory T-cells. Lymphodepletion is usually induced with whole-body irradiation or chemotherapy treatment. In this context, VSV-induced lymphopenia opens a window for the successful engraftment of the adoptively transferred TCM without the deleterious side effects of standard lymphodepleting chemotherapy and/or radiotherapy.69 Taking the data from heterologous prime-boost studies into account, the adoptive transfer of a large number of TCM and their subsequent engagement via VSV oncolytic vaccination could produce far more potent antitumor immune responses than would be possible with heterologous vaccination alone.
Another option for augmenting this potent combination could draw upon the success of chimeric antigen receptor (CAR) technology in the treatment of CD19+ B-cell lymphomas by including such receptors on adoptively transferred CD8+ T-cells to target multiple tumor antigens – including those not presented on MHC class I molecules.70–72 This would be especially useful given the evidence supporting the fact that many tumors show defects in antigen processing and presentation (eg, defects in TAP) and/or the surface expression of MHC class I to “mask” themselves from immune attack.31 Generation of TCM cells expressing a CAR could allow this therapy to also benefit from the potent immunostimulatory effects of oncolytic VSV vaccines.
Synergy with immune checkpoint blockade
VSV vaccination enhancement of either heterologous prime-boost oncolytic vaccination or ACT could be further augmented by inclusion of immune checkpoint–blocking antibodies to sustain T-cell activation and function within the tumor. Immune checkpoint blockade is an ideal candidate for combination with oncolytic VSV therapy due to the ease of administration and potential for overcoming tumor immunosuppression. As mentioned herein, expression of immunosuppressive cytokines such as TGF-β and IL-10 in the tumor microenvironment induces a state of immunosuppression and compromises the activity of infiltrating CD8+ T-cells. The potent early innate inflammatory response induced by VSV can help to overcome this, but this is not the only immune escape mechanism used by tumor cells. Tumors often upregulate inhibitory immune checkpoint molecules such as PD-L1, either constitutively or in response to lymphocytic attack, thereby “striking back” at infiltrating CD8+ T-cells by inducing apoptosis. In addition, lymphocytes within tumors are known to upregulate expression of the inhibitory surface marker CTLA-4, which competitively binds CD28 to inhibit co-stimulation of infiltrating lymphocytes and induce tolerance.32,73 A new class of drugs has been developed using monoclonal antibodies that target CTLA-4 and PD-L1 to block interaction of T-cells with the inhibitory checkpoint molecules and alleviate their suppressive effects.
Immune checkpoint blockade has showed remarkable efficacy in the clinic; ipilimumab, an anti-CTLA-4 monoclonal antibody, has been granted FDA approval for the treatment of metastatic melanoma.74 Treatment with the antibody resulted in improved and sustained T-cell activation within patient tumors and has been associated with complete, durable responses in some patients with advanced disease that were refractory to other treatments.75,76 While promising as a monotherapy, the combination of checkpoint blockade with VSV expressing immunostimulatory transgenes could lead to an even more potent antitumor response than antibody therapy alone. By coadministering engineered VSV and an antibody against these checkpoint molecules, the tumor microenvironment could rapidly be changed to one that greatly favors tumor destruction rather than tumor proliferation.77
Conclusion
VSV has emerged as a powerful OV with unique properties that potently synergize with immunotherapy. The existence of attenuated mutants increases the size of the therapeutic window of VSV, making it a safe option for use in the clinic. VSV offers several advantages as a treatment platform, such as high immunogenicity, the relative ease of transgene insertion into its genome, ease of administration, lack of preexisting immunity in the general population, and broad tropism, which opens up almost limitless possibilities in the rational design of new combinatorial treatment strategies.
This flexibility as a treatment platform provides unique opportunities for synergism with immunotherapy by inserting transgenes encoding tumor antigens or immunostimulatory molecules to potently engage a patient’s own immune system to eliminate tumors and metastases. These approaches make VSV’s interaction with the patient’s immune system a benefit, rather than an obstacle (as it is viewed in the context of “standard” oncolytic virotherapy). The design of future combinatorial immunotherapies would benefit strongly from the inclusion of VSV to maximize patient outcome while minimizing negative side effects. Therefore, it is likely that an increasing number of novel treatment strategies taking advantage of this powerful tool will emerge in the coming years.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12(4):278–287. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 2.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12(4):269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stojdl DF, Lichty B, Knowles S, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6(7):821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 4.Lichty BD, Power AT, Stojdl DF, Bell JC. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol Med. 2004;10(5):210–216. doi: 10.1016/j.molmed.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Barr JN, Whelan SPJ, Wertz GW. Transcriptional control of the RNA-dependent RNA polymerase of vesicular stomatitis virus. Biochim Biophys Acta. 2002;1577(2):337–353. doi: 10.1016/s0167-4781(02)00462-1. [DOI] [PubMed] [Google Scholar]
- 6.Giedlin MA, Cook DN, Dubensky TW. Vesicular stomatitis virus: an exciting new therapeutic oncolytic virus candidate for cancer or just another chapter from Field’s Virology? Cancer Cell. 2003;4(4):241–243. doi: 10.1016/s1535-6108(03)00251-4. [DOI] [PubMed] [Google Scholar]
- 7.Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci U S A. 2013;110(18):7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Black BL, Lyles DS. Vesicular stomatitis virus matrix protein inhibits host cell-directed transcription of target genes in vivo. J Virol. 1992;66(7):4058–4064. doi: 10.1128/jvi.66.7.4058-4064.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen JM, Her LS, Varvel V, Lund E, Dahlberg JE. The matrix protein of vesicular stomatitis virus inhibits nucleocytoplasmic transport when it is in the nucleus and associated with nuclear pore complexes. Mol Cell Biol. 2000;20(22):8590–8601. doi: 10.1128/mcb.20.22.8590-8601.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connor JH, Lyles DS. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1 vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephospho. J Virol. 2002;76(20):10177–10187. doi: 10.1128/JVI.76.20.10177-10187.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quan B, Seo H-S, Blobel G, Ren Y. Vesiculoviral matrix (M) protein occupies nucleic acid binding site at nucleoporin pair (Rae1•Nup98) Proc Natl Acad Sci U S A. 2014;111(M):9127–9132. doi: 10.1073/pnas.1409076111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan H, Puckett S, Lyles DS. Inhibition of host transcription by vesicular stomatitis virus involves a novel mechanism that is independent of phosphorylation of TATA-binding protein (TBP) or association of TBP with TBP-associated factor subunits. J Virol. 2001;75(9):4453–4458. doi: 10.1128/JVI.75.9.4453-4458.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan H, Yoza BK, Lyles DS. Inhibition of host RNA polymerase II-dependent transcription by vesicular stomatitis virus results from inactivation of TFIID. Virology. 1998;251(2):383–392. doi: 10.1006/viro.1998.9413. [DOI] [PubMed] [Google Scholar]
- 14.Johnson JE, Nasar F, Coleman JW, et al. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology. 2007;360(1):36–49. doi: 10.1016/j.virol.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sur J-H, Allende R, Doster AR. Vesicular stomatitis virus infection and neuropathogenesis in the murine model are associated with apoptosis. Vet Pathol. 2003;40(5):512–520. doi: 10.1354/vp.40-5-512. [DOI] [PubMed] [Google Scholar]
- 16.Colamonici OR, Domanski P, Platanias LC, Diaz MO. Correlation between interferon (IFN) alpha resistance and deletion of the IFN alpha/beta genes in acute leukemia cell lines suggests selection against the IFN system. Blood. 1992;80(3):744–749. [PubMed] [Google Scholar]
- 17.Wong LH, Krauer KG, Hatzinisiriou I, et al. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3γ. J Biol Chem. 1997;272(45):28779–28785. doi: 10.1074/jbc.272.45.28779. [DOI] [PubMed] [Google Scholar]
- 18.Stojdl DF, Lichty BD, ten Oever BR, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anticancer agents. Cancer Cell. 2003;4(4):263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 19.Gaddy DF, Lyles DS. Vesicular stomatitis viruses expressing wild-type or mutant M proteins activate apoptosis through distinct pathways. J Virol. 2005;79(7):4170–4179. doi: 10.1128/JVI.79.7.4170-4179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dejean LM, Martinez-Caballero S, Kinnally KW. Is MAC the knife that cuts cytochrome c from mitochondria during apoptosis? Cell Death Differ. 2006;13(8):1387–1395. doi: 10.1038/sj.cdd.4401949. [DOI] [PubMed] [Google Scholar]
- 21.Cullen SP, Martin SJ. Caspase activation pathways: some recent progress. Cell Death Differ. 2009;16(7):935–938. doi: 10.1038/cdd.2009.59. [DOI] [PubMed] [Google Scholar]
- 22.Gaddy DF, Lyles DS. Oncolytic vesicular stomatitis virus induces apoptosis via signaling through PKR, Fas, and Daxx. J Virol. 2007;81(6):2792–2804. doi: 10.1128/JVI.01760-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang X, Khosravi-Far R, Chang HY, Baltimore D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell. 1997;89(7):1067–1076. doi: 10.1016/s0092-8674(00)80294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torii S, Egan DA, Evans RA, Reed JC. Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs) EMBO J. 1999;18(21):6037–6049. doi: 10.1093/emboj/18.21.6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song JJ, Lee YJ. Daxx deletion mutant (amino acids 501-625)-induced apoptosis occurs through the JNK/p38-Bax-dependent mitochondrial pathway. J Cell Biochem. 2004;92(6):1257–1270. doi: 10.1002/jcb.20155. [DOI] [PubMed] [Google Scholar]
- 26.Takizawa T, Tatematsu C, Nakanishi Y. Double-stranded RNA-activated protein kinase interacts with apoptosis signal-regulating kinase 1: implications for apoptosis signaling pathways. Eur J Biochem. 2002;269(24):6126–6132. doi: 10.1046/j.1432-1033.2002.03325.x. [DOI] [PubMed] [Google Scholar]
- 27.Kopecky SA, Willingham MC, Lyles DS. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis V. J Virol. 2001;75(24):12169. doi: 10.1128/JVI.75.24.12169-12181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khelifi AF, D’Alcontres MS, Salomoni P. Daxx is required for stress-induced cell death and JNK activation. Cell Death Differ. 2005;12(7):724–733. doi: 10.1038/sj.cdd.4401559. [DOI] [PubMed] [Google Scholar]
- 29.Leavy O. Natural killer cells: maturation and function of NK cells. Nat Rev Immunol. 2012 Feb;12:3172. doi: 10.1038/nri3172. [DOI] [PubMed] [Google Scholar]
- 30.Lee PP, Yee C, Savage PA, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5(6):677–685. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 31.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 32.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 33.Altomonte J, Wu L, Meseck M, et al. Enhanced oncolytic potency of vesicular stomatitis virus through vector-mediated inhibition of NK and NKT cells. Cancer Gene Ther. 2009;16(3):266–278. doi: 10.1038/cgt.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galivo F, Diaz RM, Wongthida P, et al. Single-cycle viral gene expression, rather than progressive replication and oncolysis, is required for VSV therapy of B16 melanoma. Gene Ther. 2010;17(2):158–170. doi: 10.1038/gt.2009.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sukkurwala AQ, Martins I, Wang Y, et al. Immunogenic calreticulin exposure occurs through a phylogenetically conserved stress pathway involving the chemokine CXCL8. Cell Death Differ. 2014;21(1):59–68. doi: 10.1038/cdd.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang B, Sikorski R, Kirn DH, Thorne SH. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the IFN response and HMGB1. Gene Ther. 2011;18(2):164–172. doi: 10.1038/gt.2010.121. [DOI] [PubMed] [Google Scholar]
- 37.Ahmed M, Mitchell LM, Puckett S, Brzoza-Lewis KL, Lyles DS, Hiltbold EM. Vesicular stomatitis virus M protein mutant stimulates maturation of Toll-like receptor 7 (TLR7)-positive dendritic cells through TLR-dependent and -independent mechanisms. J Virol. 2009;83(7):2962–2975. doi: 10.1128/JVI.02030-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baroja-Mazo A, Martín-Sánchez F, Gomez AI, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15(5 mM):1–5. doi: 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- 40.Ghiringhelli F, Apetoh L, Tesniere A, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 41.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 42.Zelenay S, Keller AM, Whitney PG, et al. The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J Clin Invest. 2012;122(5):1615–1627. doi: 10.1172/JCI60644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mcgranahan N, Swanton C. Perspective biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27(1):15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 44.Blackham AU, Northrup SA, Willingham M, D’Agostino RB, Lyles DS, Stewart JH. Variation in susceptibility of human malignant melanomas to oncolytic vesicular stomatitis virus. Surgery. 2012;153(3):333–343. doi: 10.1016/j.surg.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moerdyk-Schauwecker M, Shah NR, Murphy AM, Hastie E, Mukherjee P, Grdzelishvili VZ. Resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus: role of type I interferon signaling. Virology. 2013;436(1):221–234. doi: 10.1016/j.virol.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blackham AU, Northrup SA, Willingham M, et al. Molecular determinants of susceptibility to oncolytic vesicular stomatitis virus in pancreatic adenocarcinoma. J Surg Res. 2014;187(2):412–426. doi: 10.1016/j.jss.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergman I, Griffin JA, Gao Y, Whitaker-Dowling P. Treatment of implanted mammary tumors with recombinant vesicular stomatitis virus targeted to Her2/neu. Int J Cancer. 2007;121(2):425–430. doi: 10.1002/ijc.22680. [DOI] [PubMed] [Google Scholar]
- 48.Leveille S, Goulet M-L, Lichty BD, Hiscott J. Vesicular stomatitis virus oncolytic treatment interferes with tumor-associated dendritic cell functions and abrogates tumor antigen presentation. J Virol. 2011;85(23):12160–12169. doi: 10.1128/JVI.05703-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramsburg E, Publicover J, Buonocore L, et al. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced T-cell responses and is highly attenuated for replication in animals. J Virol. 2005;79(24):15043–15053. doi: 10.1128/JVI.79.24.15043-15053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shin EJ, Wanna GB, Choi B, et al. Interleukin-12 expression enhances vesicular stomatitis virus oncolytic therapy in murine squamous cell carcinoma. Laryngoscope. 2007;117(2):210–214. doi: 10.1097/01.mlg.0000246194.66295.d8. [DOI] [PubMed] [Google Scholar]
- 51.Stephenson KB, Barra NG, Davies E, Ashkar AA, Lichty BD. Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 2012;19(4):238–246. doi: 10.1038/cgt.2011.81. [DOI] [PubMed] [Google Scholar]
- 52.Liu RB, Engels B, Schreiber K, et al. IL-15 in tumor microenvironment causes rejection of large established tumors by T cells in a noncognate T cell receptor-dependent manner. Proc Natl Acad Sci U S A. 2013;110(20):8158–8163. doi: 10.1073/pnas.1301022110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin. 2008;29(11):1275–1288. doi: 10.1111/j.1745-7254.2008.00889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Horssen R, Ten Hagen TLM, Eggermont AMM. TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist. 2006;11(4):397–408. doi: 10.1634/theoncologist.11-4-397. [DOI] [PubMed] [Google Scholar]
- 55.Ikeda H, Old LJ, Schreiber RD. The roles of IFN?? in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002;13(2):95–109. doi: 10.1016/s1359-6101(01)00038-7. [DOI] [PubMed] [Google Scholar]
- 56.Zaidi MR, Merlino G. The two faces of interferon-γ in cancer. Clin Cancer Res. 2011;17(19):6118–6124. doi: 10.1158/1078-0432.CCR-11-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Melero I, Gaudernack G, Gerritsen W, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11(9):509–524. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- 58.Bartlett DL, Liu Z, Sathaiah M, et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. 2013;12(1):103. doi: 10.1186/1476-4598-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Larocca C, Schlom J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011;17(5):359–371. doi: 10.1097/PPO.0b013e3182325e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bridle BW, Stephenson KB, Boudreau JE, et al. Potentiating cancer immunotherapy using an oncolytic virus. Mol Ther. 2010;18(8):1430–1439. doi: 10.1038/mt.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bridle BW, Boudreau JE, Lichty BD, et al. Vesicular stomatitis virus as a novel cancer vaccine vector to prime antitumor immunity amenable to rapid boosting with adenovirus. Mol Ther. 2009;17(10):1814–1821. doi: 10.1038/mt.2009.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bridle BW, Clouthier D, Zhang L, et al. Oncolytic vesicular stomatitis virus quantitatively and qualitatively improves primary CD8(+) T-cell responses to anticancer vaccines. Oncoimmunology. 2013;2(8):e26013. doi: 10.4161/onci.26013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang L, Bridle BW, Chen L, et al. Delivery of viral-vectored vaccines by B cells represents a novel strategy to accelerate CD8(+) T-cell recall responses. Blood. 2013;121(13):2432–2439. doi: 10.1182/blood-2012-06-438481. [DOI] [PubMed] [Google Scholar]
- 64.Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257(1):56–71. doi: 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crompton JG, Clever D, Vizcardo R, Rao M, Restifo NP. Reprogramming antitumor immunity. Trends Immunol. 2014;35(4):178–185. doi: 10.1016/j.it.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hinrichs CS, Borman ZA, Gattinoni L, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2011;117(3):808–814. doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sukumar M, Liu J, Ji Y, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123(10):4479–4488. doi: 10.1172/JCI69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hinrichs CS, Spolski R, Paulos CM, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111(11):5326–5333. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bridle BW, Chen L, Lemay CG, et al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther. 2013;21(4):887–894. doi: 10.1038/mt.2012.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bagg A, June CH. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. 2011:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gilham DE, Debets R, Pule M, Hawkins RE, Abken H. CAR-T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol Med. 2012;18(7):377–384. doi: 10.1016/j.molmed.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 73.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015:1–10. doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gao Y, Whitaker-Dowling P, Griffin JA, Barmada MA, Bergman I. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther. 2009;16(1):44–52. doi: 10.1038/cgt.2008.55. [DOI] [PubMed] [Google Scholar]