

Abstract
We conducted a genetic screen for mutations in myospheroid, the gene encoding the Drosophila βPS integrin subunit, and identified point mutants in all of the structural domains of the protein. Surprisingly, we find that mutations in very strongly conserved residues will often allow sufficient integrin function to support the development of adult animals, including mutations in the ADMIDAS site and in a cytoplasmic NPXY motif. Many mutations in the I-like domain reduce integrin expression specifically when βPS is combined with activating αPS2 cytoplasmic mutations, indicating that integrins in the extended conformation are unstable relative to the inactive, bent heterodimers. Interestingly, the screen has identified alleles that show gain-of-function characteristics in cell culture, but have negative effects on animal development or viability. This is illustrated by the allele mysb58; available structural models suggest that the molecular lesion of mysb58, V409>D, should promote the “open” conformation of the β subunit I-like domain. This expectation is supported by the finding that αPS2βPS (V409>D) promotes adhesion and spreading of S2 cells more effectively than does wild-type αPS2βPS, even when βPS is paired with αPS2 containing activating cytoplasmic mutations. Finally, comparisons with the sequence of human β8 suggest that evolution has targeted the “mysb58” residue as a means of affecting integrin activity.
INTRODUCTION
The integrin cell surface receptors are critical for many morphogenetic events during animal development as well as numerous physiological events in adults, including many disease-related processes (Hynes, 2002). A major goal of integrin biology is understanding the molecular structure and dynamics of the integrin αβ heterodimer. Using conformation specific antibodies, it has been clear for many years that integrins undergo significant structural changes in response to binding of ligands or signals from within the cell. These and other studies led to various molecular models for integrin structure, but only recently have high-resolution x-ray, NMR, and electron microscope studies begun to elucidate in molecular detail how the α and β subunits are organized.
A major advance in our understanding of integrin structure-function relationships was the determination of an x-ray structure for most of the extracellular part of αvβ3. This revealed a compact integrin heterodimer, bent over with the ligand-binding head domains facing back toward the plasma membrane (Xiong et al., 2001). Elegant electron microscopy studies have demonstrated that these x-ray images represent what has historically been called the inactive conformation and that activation stimulates the heterodimer to assume the extended posture where the head region is more accessible to fibrous extracellular matrix ligands (Takagi et al., 2002). Further electron microscopy experiments have shown that there are additional steps in the full activation of the heterodimer, which lead to a movement of the most distal segment of the β subunit stalk (the hybrid domain) relative to the ligand binding I-like domain (Figure 1). This pivoting of the hybrid domain is thought to be driven by a tertiary structure change in the I-like domain (Liddington et al., 2002; Luo et al., 2003a, 2004; Mould et al., 2003a, 2003b).
Figure 1.

Domain structure of integrin heterodimers, in the extended conformation. α subunit domains are in unshaded outlines. The available data suggest that ligand binding (right) stabilizes an “open” conformation, involving changes in at least two of the three darkly shaded structural domains. In this model, a change in the I-like domain tertiary structure (asterisk) drives a movement of the Hybrid domain; this may also include a dissassociation of the PSI domain from a specific binding site on the stalk. EGF 1–4, EGF-like repeats 1–4; βTD, β terminal domain; TM, transmembrane domain; Cyto, cytoplasmic domain.
Many site-directed mutagenesis studies were performed on integrin extracellular and intracellular domains before there was much of an understanding of the heterodimer structure. Residues have been identified that are required for dimer formation, ligand binding, or association with intracellular proteins, but for the most part these studies have not pointed to specific molecular interactions in the extracellular region that can explain the movements of the integrin heterodimers. The detailed structures now available are beginning to lead to more incisive site-directed mutagenesis studies in this regard (e.g., Chen et al., 2003; Luo et al., 2003a; 2004; Mould et al., 2003a, 2003b; Barton et al., 2004; Yang et al., 2004a, 2004b).
Because integrin structure is very conserved phylogenetically, invertebrate systems such as Drosophila melanogaster provide the opportunity to pursue complementary genetic approaches that are not available in vertebrate organisms (Brower, 2003). We have undertaken a forward genetics strategy in Drosophila to identify integrin β subunit mutations that alter integrin function in the context of an intact, developing animal. Here, we report the results of that screen, along with some general inferences that can be drawn from the collection. This includes data from one mutant showing that the screen has generated alleles that can shed light on the structural features that affect integrin functional states.
MATERIALS AND METHODS
Mutant Screens
To generate the mysb.. series of alleles, wa males were mutagenized with ethyl methanesulfonate (EMS;Lewis and Bacher, 1968) and crossed to CDX, y w f females at 18°C. All marker mutations and special chromosomes are described in Lindsley and Zimm (1992) or in FlyBase (http://flybase.bio.indiana.edu/). Because the mothers carried the Compound Double X chromosome, F1 males received their single X chromosome from their mutagenized fathers. Because myospheroid is on the X chromosome, any mutant myospheroid gene must therefore be able to support viability, and so null alleles were eliminated at this point. Individual wa mys? F1 males were then crossed to y mysXR04 f36a/FM7c females (mysXR04 is a weak dominant negative allele; Jannuzi et al., 2002), and the F2 progeny were raised at 28°C. If the mys?/mysXR04 females were dead, the sibling wa mys?/FM7 female and FM7 male progeny were used to make a balanced stock. The alleles were then retested for lack of complementation with the mysG1 allele, which is similar to mysXR04 but is present in a different genetic background (Jannuzi et al., 2002). myospheroid alleles generated in this way were designated mysb1-b70.
Complementation Tests
To score relative viability of various combinations of alleles, eggs were laid at the appropriate temperature, and vials were thinned to prevent overcrowding of larvae. Progeny were scored at least once a day, and if any animals from any single vial were scored, all subsequent progeny from that vial were counted, in order to guard against genotypic differences in developmental rates. In general, newly eclosed animals that were stuck in the food were scored.
Sequencing of Mutants
Adults or embryos were homogenized and genomic DNA template was isolated according to previously published procedures (Gloor et al., 1993). PCR primers were designed to yield two products covering the myospheroid coding region (exons 2–7; Yee, 1993; Zusman et al., 1993). The first fragment began 139 base pairs before the initiating AUG and the second fragment continued 62 base pairs after the UAG stop codon. The resulting PCR fragments were purified using QIAGEN′s QiaQuick PCR Purification Kit (Chatsworth, CA) and sequenced directly by the University of Arizona GATC Automated DNA Sequencing Service. The introns were not sequenced in their entirety.
Immunofluorescence
For examination of integrin expression levels in situ, all fly cultures were grown at 28°C for at least 2 days before immunostaining. Imaginal discs were dissected from late third instar larvae of the appropriate genotype and stained with the αPS2 or βPS antibodies CF.2C7 and CF.6G11 as described (Brower et al., 1984). Genotypes included hemizygous myospheroid mutant males or the same with a third chromosome containing both a UAS-αPS2-ΔCGFFN insert (Baker et al., 2002) and an enhancer trap (337; Manseau et al., 1997) that drives expression of the UAS transgene throughout the imaginal discs. Discs were mounted in VectaShield (Vector Laboratories, Burlingame, CA) and examined using a standard Zeiss immunofluorescence microscope (Thornwood, NY).
For examination of homozygous clones of mutant cells, myospheroid alleles were recombined onto a chromosome with the recombination site FRT18A, and females were generated that were heterozygous for this chromosome and an X chromosome also containing FRT18A as well as a gene encoding GFP with a nuclear localization sequence, driven by the ubiquitin promoter (Bloomington Stock Center Number 5623). A hs-FLPase insert on the second chromosome was induced by several heat shocks during larval life, which led to somatic crossing over at the FRT18A sites to produce homozygous myospheroid clones (identified by loss of GFP), and dissected discs were fixed in formaldehyde and stained using the βPS antibody. These discs were photographed using a Nikon E800 confocal microscope (Nikon, Melville, NY).
Cell Adhesion and Spreading Experiments
Drosophila S2/M3 cells transformed with integrin-expressing genes (under the regulation of the heat shock protein 70 promoter) were cultured in Shields and Sang M3 medium supplemented with 12% heat-inactivated fetal calf serum, and 2 × 10-7 M methotrexate for transformed lines (Bunch and Brower, 1992; Zavortink et al., 1993). S2/M3 cells were cotransfected with plasmids expressing the various combinations of wild-type or mutant αPS2m8 and βPS subunits and the bacterial DHFR selectable marker (Jannuzi et al., 2002). For all experiments, cells were grown in RNAi targeting the endogenous myospheroid gene, as has been described in detail (Jannuzi et al., 2002).
The ligand used in these assays was RBB-Tigg, a bacterial fusion protein that contains 53 amino acids of the Drosophila extracellular matrix protein Tiggrin (residues 1964–2016, including the RGD sequence and 25 amino acids upstream and downstream), fused to a histidine tag from the pTrcHisB vector (Xpress System, Invitrogen, Carlsbad, CA). This fusion protein is as active in promoting cell spreading as the previously described Tiggrin fusion protein and Tiggrin itself (Fogerty et al., 1994; T. A. Bunch, unpublished data). The fusion protein was purified by affinity chromatography on Ni-NTA agarose (QIAexpress, QIAGEN). Ninety-six well tissue culture plates or slides were coated with ligand for either 1 h at room temperature or overnight at 4°C, then blocked with 20% dried milk in phosphate-buffered saline (PBS) for 1 h at room temperature, and washed three times with PBS.
Cell spreading assays were done as described (Jannuzi et al., 2002). In brief, cells were treated with dispase/collagenase at 37°C; this heat shock also induces expression of the integrin transgenes. Cells were then allowed to spread in coated 96-well plates for 3–4 h before counting. For adhesion assays, cells were allowed to recover from the protease clearing and heat shock for 4 h, and 1–1.5 × 105 cells in 0.1 ml of M3 medium + 2 mg/ml BSA were added to ligand-coated wells. Cells were allowed to settle and attach for 20 min at 23°C, after which nonadherent cells were removed by washing with a multichannel pipetter. The adherent cells were rinsed with PBS and stained with crystal violet (0.5% in 20% methanol) for 1 min. After multiple washes with water to remove unbound dye, the crystal violet was released by the addition of 200 μl of 0.1 M citric acid to each well. Dye levels were quantified using an ELX800 Universal Microplate Reader (Bio-Tek Instruments, Burlington, VT) at 562 nm.
For both assays, dose-response curves were done to determine the level of RBB-Tigg that gave maximal cell spreading or adhesion. For the comparisons of wild-type and mysb58, RBB-Tigg concentrations were selected that gave approximately half-maximal values, in the linear range of the curves. For the spreading assays, RBB-Tigg concentrations were 32 and 8 ng/ml for the wild-type and αPS2 cytoplasmic mutant integrins; for the adhesion assays, the respective concentrations were 100 and 50 ng/ml. The average and SE from three separate experiments is given. For the adhesion assays, two wells were scored for each data point in each experiment, and background signal from wells not coated with any ligand was subtracted from the values derived from ligand coated wells.
Surface expression levels of αPS2βPS were checked by flow cytometry for each experiment (Bunch et al., 2004). In all cases, comparisons were made only between pairs of cell lines that expressed similar amounts of integrin, or slightly more wild-type relative to mysb58.
RESULTS
Screen for βPS Mutations
To screen for mutations that alter βPS function we made use of the Drosophila mutant alleles mysXR04 and mysG1, which make mild dominant negative βPS proteins (Jannuzi et al., 2002). These alleles are fully viable as heterozygotes with wild type, but they typically fail to complement weak alleles of myospheroid. This property allows one to design a screen to identify mutations that compromise βPS function without eliminating it (see MATERIALS AND METHODS). In this screen, the mutant myospheroid gene must retain sufficient βPS activity to permit the survival of an F1 male (myospheroid is on the X chromosome) after EMS treatment. The mutant is then tested for lethality when combined with a weak dominant negative allele of myospheroid. Still, we find that some relatively strong alleles have been recovered, probably because after EMS mutagenesis the F1 males can be genetically mosaic for the myospheroid mutation (Ashburner, 1989). Indeed, for three alleles (mysb23, mysb26, and mysb64) we have not seen a mutant male since the original F1 animal; each of these mutant X chromosomes can be rescued by the small mys+ duplication Tp(1;2)sn+72d, indicating that the lethality is not due to a mutation at another locus.
Point Mutations in βPS
We sequenced the coding exons and nearby splice sites for all of the new mutants from this screen as well as a number of myospheroid alleles from previous screens. Alleles generated in our screen are designated mysb.; other alleles associated with point mutations in the coding region that retain some function include mysts2 (Wright, 1968) and mysXN101 (Wieschaus et al., 1984). In all, we identified βPS point mutations in 55 hypomorphic or neomorphic alleles, which comprised 49 different molecular lesions. Included in our definition of “point” mutations are three alleles that insert one or four residues at the splice site between exons 4 and 5. Additionally, we previously described two missense alleles (mysG4 and mysG12) from another screen that have phenotypes similar to the null phenotype (Jannuzi et al., 2002).
We failed to find molecular lesions in seven alleles, including mysb5, mysb8, mysb9, and mysb36 from this screen, mysts1 and mysts3 (Wright, 1968) and mysnj42 (Costello and Thomas, 1981). In all but mysb36 (which was not mapped) the mutation has been mapped genetically to a small chromosomal region that includes myospheroid, and in some cases defects in βPS expression have been detected (unpublished data). Thus, we assume that these represent regulatory mutations.
Figure 2 shows the predicted βPS amino acid changes for all of the identified myospheroid point mutants (see Supplementary Information for a compilation of nucleotide changes). The sequences of integrin β subunits indicate that the overall structures of the proteins are strongly conserved, and the sites of almost all of the βPS changes can be easily matched to corresponding residues in human β3, for which there are x-ray structural data for most of the extracellular region (Xiong et al., 2001). We will use these correspondences as well as other data from human integrins, in making structural inferences below.
Figure 2.

Locations of βPS point mutants. The Drosophila βPS and human β3 sequences are aligned, with the locations of β3 structural domains and (for the I-like and hybrid domains) secondary structures indicated (from Xiong et al., 2001). Sites of βPS mutants are underlined with the new residue indicated below, followed by the b-series allele number for mutants generated here, or other allele names for preexisting mutants. Note that mysb70 includes two missense changes. Three alleles involve the insertion of a single residue (mysb50 and mysb62) or four amino acids (mysb69) at the splice site indicated by thick underlining between S272 and N273.
Not surprisingly, the distribution of the mutations is not uniform throughout the protein. In the I-like domain, ∼10% of the residues were mutated, whereas the hybrid domain was the least sensitive structure, with only 1% of the residues hit. The PSI domain and stalk region showed intermediate sensitivity, with 6–7% of the amino acids in each being mutated.
Genetic Characterizations
For most of the alleles we tested viability in a variety of genetic combinations, including as hemizygous males, as heterozygotes over a myospheroid null allele or the original dominant negative allele, and finally as heterozygotes over a myospheroid null in addition to being heterozygous for mew (encoding αPS1) or inflated (αPS2). (A number of the strong alleles were not tested in this way, because of the very low viability of the mutant males.) These tests were carried out at a range of temperatures from 18°C to 28°C, because myospheroid hypomorphs generally display stronger phenotypes (including lethality) at higher temperatures (Bunch et al., 1992). For strong alleles failure to complement the dominant negatives is usually complete at all temperatures, although our alleles alone or over the null allele are often viable, especially at low temperatures. Alleles designated as “weak” typically complement the dominant negative mutant at low temperatures. Heterozygosity for αPS alleles increases lethality of the βPS hemizygotes; this is especially true for αPS2. We do not see any extreme examples of α subunit specificity in viability reduction for particular βPS alleles, although weak trends could be detected for some. These viability tests were used to define alleles as strong, moderate or weak in Table 1 (see Supplementary Information for details).
Table 1.
Summary of βPS point mutants
| Expression
|
|||||
|---|---|---|---|---|---|
| Allele | Change | Location | Strength | Wild type α | Activated α |
| b41 | C40Y | PSI | Strong | OK (c) | |
| b33 | I50F | PSI | Weak | OK-c | |
| b3 | C58S | PSI | Moderate | OK | Reduced |
| b68 | C58Y | PSI | Strong | Reduced (c) | Much reduced |
| G4 | S196F | cation coord | Lethal | ||
| b67 | D200N | cation coord | Strong | OK-c | |
| b38, b61 | L224F | Interior | Weak | OK | OK |
| b26 | G227S | Interior | Strong | OK-c | |
| b20, b59 | P242L | α/β interface | Weak | OK | OK |
| b21 | L264F | Interior | Strong | OK-c | Gone |
| b50, b62 | 272+Q | Insertion | Weak | OK | Reduced |
| b69 | 272+SVRQ | Insertion | Strong | OK | Gone |
| b13 | E274V | Surface | Strong | OK-c | |
| b47 | A293T | Interior | Strong | OK | Gone |
| b30 | I298F | Interior | Strong | OK | Much reduced |
| b65 | A310T | Interior | Weak | OK | Much reduced |
| b45 | R312Q | Interior | Moderate | OK | |
| b25 | S317L | Interior | Weak | OK | OK |
| b48 | A325T | α/β interface | Strong | OK | Gone |
| b66 | P336S | α/β interface | Strong | ||
| b57 | G339S | Interior | Strong | OK | Gone |
| b49 | H342Y | Interior | Weak | OK | |
| ts2 | G347D | Surface | Weak | OK | |
| b42, b43 | G347S | Surface | Weak | OK | Much reduced |
| G12 | D356N | α/β interface | Lethal | ||
| b44 | I375F | Interior | Moderate | OK | |
| b53 | E387V | Surface | Weak | OK | |
| b56 | G395S | Surface | Strong | Reduced | Reduced |
| b7 | D404N | Surface | Weak | OK | |
| b23 | N407Y | Surface | Strong | OK-c | |
| b58 | V409D | “Surface” | Weak | OK-c | |
| b27 | V423E | Hybrid | Strong | OK | Gone |
| b37 | C441Y | Hybrid | Weak | OK | OK |
| b63 | G531D | EGF-1 | Weak | OK | OK |
| b31 | G541S | EGF-1 | Weak | OK | OK |
| b64 | C544Y | EGF-1 | Strong | Reduced-c | |
| b55 | R587Q | EGF-2 | Weak | OK | |
| b46 | G596S | EGF-2 | Weak | OK | |
| b52 | G596R | EGF-2 | Strong | OK | |
| b24, b28, b60 | E600K | EGF-3 | Weak | Reduced (c) | OK |
| b51 | E607K | EGF-3 | Strong | Reduced (c) | |
| XN101 | C629S | EGF-3 | Strong | OK-c | |
| b22 | R676C | EGF-4 | Weak | ||
| b39 | G679D | EGF-4 | Weak | OK | |
| b4 | C701Y | βTD | Weak | OK | |
| b34 | G707S | βTD | Weak | OK | OK |
| b29 | F743I | βTD | Weak | OK | OK |
| b32 | V763M | βTD | Weak | OK | OK |
| b40 | V775D | βTD | Weak | OK | OK |
| b1 | G792D | Transmem | Moderate | OK | OK |
| b70 | S836T, P841T | Cytoplasm | Strong | OK (c) | OK |
Allele “strength” is based on viability in various genetic tests (see text and Supplementary Information). Expression of “inactive” and “activated” proteins refers to surface protein on third instar wing imaginal disc cells with wild-type αPS2 subunits or αPS2 with a cytoplasmic CGFFN deletion. Those indicated as “-c” were determined in clones of homozygous mutant cells in the disc epithelium; those with “(c)” were scored in whole discs as well as clones. “Location” indicates the β domain or, for the I-like domain, the location of the the residue side chain; cation-coordinating, α/β interface, etc corresponds to the color coding of mutants in Figure 5. mysb21 is in the alternatively spliced fourth exon and is specific to the predominant 4A βPS isoform. Note that mysb50, mysb62, and mysb69 are insertions after residue 272, resulting from changes in splicing. The identity of these mRNAs has been confirmed by RT-PCR, and no wild-type mRNA was detected (unpublished data). mysb70 changes two nearby residues in the cytoplasmic tail. The mysb.. series alleles were generated in this screen; the null-like mysG4 and mysG12 alleles are from a screen for strong integrin mutants (Jannuzi et al., 2002); mysts2 is from a screen for hypomorphs by Wright (1968). mysXN101, from the screens of Wieschaus et al. (1984), is 100% lethal but retains some mys+ activity, based on the embryonic lethal phenotype.
In a previous small scale examination of three myospheroid hypomorphs, it was not possible to identify a discrete lethal phase or phenotype (T. Wright, personal communication). Although we have not made a comprehensive examination of all of our mutants, we also see that under conditions in which they are lethal, many of our alleles do not show well-defined phenotypes or stages of lethality. However, it does appear that embryonic development is more sensitive than most stages. For example, viability can often be enhanced significantly if animals are allowed to traverse embryogenesis at a relatively low temperature, which ameliorates the effects of integrin hypomorphs generally.
Discrete adult phenotypes also are not common for the myospheroid alleles. Some mutants can display the held-out wing phenotype that has been described previously (Wilcox, 1990), depending on temperature or genetic background, but this is not usually found in hemizygotes at low temperature, for example. Wing blisters, a clonal phenotype of null myospheroid alleles (Brower and Jaffe, 1989), are extremely rare in viable animals, with one notable exception described below. We generated clones of homozygous mutant cells via somatic recombination (Xu and Rubin, 1993) for a number of the point mutants (see Table 1). In addition to the strong alleles mysG4 and mysG12, which were originally identified based on this clonal wing blistering phenotype (Jannuzi et al., 2002), only mysb23 and mysXN101 produce wing blisters in clones. Both mysb23 and mysXN101 also show 100% embryonic lethality.
Integrin Expression in Imaginal Discs
Because we are especially interested in mutations that affect function but not expression, many of the alleles were tested for cell surface integrin levels in third instar wing imaginal discs in situ. In all cases we examined discs grown at 28°C, when mutant phenotypes are expected to be strongest (although typically the animals were allowed to traverse the sensitive embryonic period at lower temperatures to increase viability). We also examined expression in wing discs that ubiquitously expressed an αPS2 subunit containing an activating cytoplasmic mutation (deletion of the CGFFN sequence; O'Toole et al., 1994). This mutation leads to reduced heterodimer expression, and constitutive clustering on the basal surface of the disk cells (Baker et al., 2002). Because αPS2 is normally expressed only on the cells that will give rise to the ventral wing surface at this stage, staining these wing discs with an antibody against αPS2 allows one to compare expression of the βPS allele with wild-type αPS2, on the presumptive ventral epithelium, and activated αPS2 specifically on the dorsal side of the same disk (Figure 3). Not all alleles were tested for expression, in part because some were difficult to grow as hemizygotes under the conditions of the experiment. However, for some of the latter alleles expression was examined in clones of homozygous mutant cells of the wing disk epithelium (Figure 4); these clones were generated by somatic recombination in developing heterozygotes (Xu and Rubin, 1993).
Figure 3.

Expression of αPS2βPS integrins in the posterior margin of third instar larval wing imaginal discs. Typically, αPS2βPS is expressed only in the ventral (lower) half of the epithelium, but these animals are also expressing a transgenic αPS2 subunit with a cytoplasmic activating mutation (deletion of CGFFN) throughout the disk. The upper staining (arrows) is entirely from the transgenic activated αPS2 with βPS, whereas the lower immunostaining is primarily from wildtype αPS2 with βPS, which is much more stable than heterodimers containing the activating αPS2 mutation. For wild-type βPS, the dorsal staining is reduced relative to the ventral staining, and the activated heterodimers are clustered on each cell. For many βPS mutants, expression with the activated αPS2 subunits is reduced much more than for wild-type, or is even nondetectable, as shown here for mysb57. See Table 1 for a summary of the imaginal disk expression data.
Figure 4.

Examination of integrin surface expression via clonal analysis. A clone of cells homozygous for mysb60 was generated in a heterozygous mysb60/mys+ background, by somatic recombination in a cell of the early wing imaginal disk epithelium. The clone is marked by the loss of nuclear GFP staining (A), which derives from a transgene on the mys+ chromosome. The homozygous mysb60 cells show reduced surface integrin expression, as indicated by staining for βPS (B).
As summarized in Table 1, most myospheroid hypomorphs are expressed well in discs, suggesting that general destabilization of the heterodimers cannot account for the loss of viability in the screen. However, especially for the I-like domain alleles, heterodimers with a βPS mutation and the activating αPS2 alteration are often present at much lower levels, and in many cases these are virtually undetectable on the dorsal epithelium. To ask if the I-like domain point mutants cluster into any particular part of the structure, we mapped our alleles onto the x-ray trace of the human β3 subunit (Figure 5). There is no suggestion that a particular region of the I-like domain, such as the cation binding sites or the α/β interface, is especially sensitive to this type of destabilizing mutation. Alleles are found in all parts of the domain, and the collection includes residues with side chains either on the external surface or the interior of the structure (including some that simply replace an internal hydrophobic residue with a larger hydrophobic amino acid).
Figure 5.

Locations of I-like domain and hybrid domain βPS mutants, mapped onto the structure of αv(pale blue)β3(white). Categories of mutations are: α/β interface-red; side chain on surface-yellow; side chain on interior-blue; side chain that coordinates with cation-purple; location of insertions-orange; mysb58-green (see below). There is no obvious hyper-sensitive region for I-like domain mutants.
Decreased expression of activated heterodimers is not typically found for mutations in the stalk region. However, in a reversal of the I-like domain trend, a mutant (mysb60) at the boundary between the second and third EGF-like repeats (E600>K) shows reduced expression normally in discs, but is expressed at least as well as wild-type βPS when paired with the activated αPS2 subunits. Because this result was unusual, we confirmed the decreased expression with the wild-type αPS2 by making clones of homozygous mysb60 cells in a heterozygous background, where the difference in expression is clearly evident at the clone boundary (Figure 4). This region of the β stalk is not resolved in the x-ray structures, and probably undergoes significant changes when the heterodimer switches from the bent to extended conformations. The glutamate that is mutated in mysb60 is well conserved in β subunits, and our data suggest that this residue is likely to make specific interactions that stabilize the inactive conformation. These data are also consistent with recent findings that disruption of the disulfides immediately surrounding the mysb60 residue promote integrin activation (Kamata et al., 2004).
mysb58 Is a Gain of Function Allele
The allele mysb58 is unusual in that in various combinations with other integrin mutants it causes a high frequency of wing blisters. Blisters are especially common when mysb58 animals are also heterozygous for αPS mutations. Typically, wing blisters are created when patches of cells in the wing epithelium are homozygous for strong loss-of-function mutations in myospheroid (Brower and Jaffe, 1989). Blisters may also be seen in PS integrin regulatory alleles (e.g., Bloor and Brown, 1998), but they are rarely seen in the βPS point mutant hypomorphs, probably because these alleles alter integrin function more globally, and the level of function necessary to hold the wing epithelia together is less than that required for one or more essential events during development. Paradoxically, even though mysb58 causes a relatively strong wing phenotype, it is a very weak allele by all of the genetic tests for viability. One model that might explain this paradox is that mysb58 may promote integrin activation. For example, gain-of-function αPS2 mutations, if inappropriately expressed in developing wings, promote wing blistering (Baker et al., 2002).
The screen was not necessarily designed to uncover integrin gain-of-function alleles, and so we decided to further assess the properties of mysb58 more directly in cell culture. Only one nucleotide alteration was found in the coding region of mysb58 (T1226>A, where nucleotide 1 is the A of the initial AUG), which would lead to a single amino acid change (V409>D). We expressed βPS subunits containing the V409>D mutation along with αPS2 in Drosophila S2 cells and compared the activity of this mutant with wild-type βPS in two assays. Adhesion was measured by allowing cells to settle onto a substratum coated with a fragment of the αPS2βPS ligand Tiggrin (RBB-Tigg), followed by removal of nonadherent cells by washing 20 min later. Cell spreading was assessed by direct observation at 3–4 h after plating. In both assays, the mysb58 mutant cells show increased activity relative to those expressing wild-type βPS (Figure 6).
Figure 6.

(A and B) Spreading and adhesion of S2 cells expressing αPS2 and wild-type or mysb58 (V409>D) βPS. Cells were allowed to settle on plates coated with an RGD-containing fragment of the Drosophila ECM protein Tiggrin (RBB-Tigg). Spread cells (A) were defined by phase microscopy 3–4 h after plating. Adhesion (B) was defined by the number of cells remaining attached after washing 20 min after settling onto the plate. Ligand concentrations were chosen that give approximately half-maximal spreading or adhesion for each pair of bars (see MATERIALS AND METHODS), and the values are expressed as a percentage of the maximum at high RBB-Tigg concentrations; this adjusts for variations in expression or other factors between experiments. For the right pairs of bars in each histogram (labeled “act”), βPS was combined with αPS2 subunits containing a cytoplasmic-activating mutation (GFFNR>GFANA). In each pairwise comparison, spreading is significantly increased for the mysb58 allele compared with wild-type βPS, and the amount of the increase is approximately the same whether βPS is paired with wild-type or cytoplasmically activated αPS2. (C) These data are most easily explained by a two-stage model of integrin activation, in which the equilibrium between bent and extended heterodimers is regulated primarily by cellular events, and the equilibrium between the open and closed conformations of the integrin head is affected by mysb58 (protein tracings from Shimaoka and Springer, 2003). Data are averages from three experiments, and integrin expression levels in each case were generally similar or slightly higher for the wild-type βPS cells (unpublished data).
S2 cells expressing integrins with an αPS2 subunit containing an activating cytoplasmic mutation (GFFNR>GFANA) adhere and spread extraordinarily well under similar conditions even though integrin expression is typically reduced. Combining mysb58 with activating αPS2 subunits results in even greater adhesion and spreading ability than is seen for the cytoplasmic mutation alone (Figure 6), and the difference attributable to mysb58 is similar to that seen with wild-type αPS2. This suggests that these two mutations are affecting different factors that alter integrin function in these assays; this is discussed more fully below. Finally, the data presented in Figure 6 were generated using the “m8” isoform of αPS2 (Brown et al., 1989); preliminary experiments indicate that mysb58 also enhances the activity of heterodimers containing the “c” isoform of αPS2.
DISCUSSION
The results of this screen indicate that forward genetics can provide an important complement to site-directed mutagenesis for understanding complex molecular dynamics of proteins. Moreover, the scale of this mutagenic screen is such that one can begin to draw inferences based on the overall distributions of the mutants. For example, it appears that our mutations are more common in regions that are molecularly dynamic. The I-like domain, which is thought to undergo extensive tertiary structural changes, was hit most frequently (10% of all residues in the domain), whereas the neighboring hybrid domain, which is likely to function as a more solid unit, was relatively untouched (1%). The surprising number of mutations in the stalk suggests that individual domains within this region also undergo significant structural rearrangements during conformational switching; this is in keeping with the unstructured state of much of the stalk in the αvβ3 crystal (Xiong et al., 2001; see also Beglova et al., 2002).
The analysis of the mysb58 allele begs the question as to why a screen that is designed to uncover integrin loss-of-function mutations would also yield gain-of-function alleles. Proper regulation of function is essential for many integrin mediated events, and it has been documented that strongly hyperactive integrins will not necessarily support wild-type morphogenesis (or viability) of developing Drosophila (Martin-Bermudo et al., 1998). Furthermore, the screen selects for mutants that are lethal over an allele (mysXR04) that has defects in regulation (Jannuzi et al., 2002), probably including both inside-out and outside-in signaling (B. James, unpublished results). Thus, in retrospect it is not surprising that the screen can yield mutants that affect integrin function in relatively complex and unpredictable ways. Indeed, this is one of the main advantages of forward genetic approaches and especially of screens conducted in whole animals, where the full range of integrin functions required in various tissues will be sampled.
A recent study of cysteine mutants in the EGF-like repeats of the β3 stalk suggests that this region is important for the maintenance of inactive conformations (Kamata et al., 2004), and so we might expect that some of our alleles in this region will show more complex effects than simple reduction of integrin function. Comparisons between this study of αIIbβ3 and our screen point out again the complementary nature of data from cell culture and whole animals. For example, mutation C549>S of β3 gave the highest “activation index” measured by Kamata et al. (2004); mutation of the homologous residue in flies (C629>S) is 100% embryonic lethal in hemizygous males. Mutation of a PSI domain cysteine (C26>S) in αIIbβ3-expressing cells had little effect on fibrinogen binding and activation index; the homologous change in βPS (mysb3) leads to increased integrin activity in S2 cells (T. A. Bunch, unpublished results) but temperature-sensitive lethality in developing animals. Finally, the available data suggest that disruption of the long range disulfide connecting the PSI domain and stalk should be very strongly activating (Sun et al., 2002), and one might expect this change to disrupt integrin regulation sufficiently to be lethal in embryos. Although elimination of this disulfide in mysb41 is a relatively strong allele, fertile adults can be obtained, and expression of the heterodimers in imaginal discs is not significantly affected.
Another feature of an unbiased genetic screen is that one routinely samples alterations that may do something other than just eliminate a functional side group. For example, the mysb67 mutation changes a conserved aspartate that coordinates with the ADMIDAS cation that appears to direct I-like domain movements (Xiong et al., 2001, 2002; Chen et al., 2003; Mould et al., 2003b). This residue might be expected to be critical for integrin function in at least one developmental context and therefore be 100% lethal; however, mysb67 routinely retains enough activity to support embryonic development through hatching into first instar larvae (well beyond the stage when null alleles die) and even can produce very rare viable adults. This could be because the asparagine that replaces the aspartate in mysb67 may not completely eliminate the ADMIDAS site function. In three other sites, the screen yielded mutations in the same residue that have different severity, depending on the nature of the change (Table 1).
Of course, because of the starting wild-type nucleotide sequence and the chemical nature of the mutagenesis, any residue is likely to be converted into only a subset of the 20 possible amino acids. However, numerous alternatives may be tested in a random EMS mutagenesis. Although G-to-A transitions are the most common mutations created by EMS, other alterations are not particularly rare. Indeed, of the missense alleles we have sequenced, 29% are caused by a nucleotide change other than G-to-A. Moreover, once a critical site has been identified by one mutation, additional changes can subsequently be sampled by more directed methods.
Requirements for Specific Amino Acids in Integrin β Subunits
The overall structure of integrins is strongly conserved, probably because of the intricate interactions between and within α and β subunits during the various conformational changes of the heterodimer, and the majority of our mutations are in residues that are at least similar in most β subunits. Phylogenetically conserved residues are most likely to be important for protein function, and so it is not surprising that changing these will have noticeable effects. However, it is remarkable that so many strongly conserved amino acids can be altered without preventing morphogenesis in all of the mutant flies. Of the mutants generated here, only mysb23 has an embryonic lethal phenotype that is somewhat similar to that of a null allele. Four alleles eliminate completely conserved cysteines but retain some adult viability. Four other alleles change residues that are 100% conserved in all of the 25 β subunits sequenced to date, and either produce some adults or reliably develop through embryogenesis and hatch as larvae. And 14 alleles that give adult animals are in residues that are conserved in 20 or more βs. (A number of these latter are conserved in all except some of the very divergent β subunits, such as β8 or the insect βν.) Not included in this group are more mutations that change the nature of residues that are similar in virtually all βs, such as small hydrophobic amino acids.
Although missense alleles with altered function were fairly easy to obtain in this screen, we previously noted that missense alleles were relatively rare among the strong myospheroid mutants generated by EMS mutagenesis, which is expected to produce point mutations primarily (Jannuzi et al., 2002). Indeed, splice site mutants were more common than missense mutations, suggesting that relatively few residues of βPS are absolutely required for integrin function, even in the context of a whole, developing animal. Taken together, the data from these screens indicate strongly that most integrin functions are driven by a set of relatively weak molecular interactions and that few contributing residues are absolutely essential.
The cytoplasmic domain is unusual in that it contains a high frequency of well conserved residues but yielded only one mutation in this screen. There are a number of possible reasons for this discordance, including the fact that the small size of this region (45–50 residues) weakens the statistical significance of the correlation. However, one possible explanation for the paucity of cytoplasmic alleles is that a relatively high percentage of the conserved residues in this domain are required in order to attain the level of integrin function required for passage of the mutagenized males through the F1 generation of the screen. It may be significant that many conserved cytoplasmic amino acids appear to contribute to specific interactions with other proteins (Liu et al., 2000), whereas most of the conserved extracellular residues are involved in integrin heterodimer structure-function. Interestingly, the one cytoplasmic allele we did find, mysb70, specifically (compared with two extracellular myospheroid hypomorphs) displays strong genetic interactions with mutations in the cytoplasmic integrin-binding protein talin (unpublished data).
Stability of Activated Heterodimers
We find that many of our β I-like domain mutants have dramatic effects on integrin expression only when paired with activating αPS2 subunits. One possibility is that all of these alleles disrupt interactions that are specific to one tertiary state of the I-like domain, but it seems unlikely that we could be so fortunate as to uncover such a high percentage of mutations with such specificity. A more plausible scenario is that most of these alleles make the β subunit slightly less stable, but in the inactive (in this case, probably bent) conformation the heterodimer is held together by an excess of molecular interactions. However, if forced into an upright conformation, the mutant heterodimers may lack sufficient stability to persist, especially in the absence of potentially stabilizing integrin-ligand interactions (Luo et al., 2003b). Consistent with this interpretation, a number of mutants were found that might be expected to “loosen up” the tertiary structure of the I-like domain, for example, by replacing an internal hydrophobic residue with a larger hydrophobic amino acid. In general, these did not significantly affect heterodimer expression levels when paired with wild-type αPS2 in imaginal discs, although they could reduce expression of the mutationally activated integrins.
α subunit cytoplasmic activating mutations often result in decreased expression, and it is not known if this is because of rapid turnover, inability to reach the cell surface, or both. We do not know which events are affected in our cells either, but wherever the instability occurs, the I-like domain mutants appear to be lowering the permissible baseline and demonstrate clearly that constitutive cytoplasmic activation does indeed decrease heterodimer stability. Our data do not directly address the issue of whether activation per se leads to a separation of the α and β heads, as has been suggested (Hantgan et al., 1999, 2001; see also Luo et al., 2003b). Most probably, observed separations seen in isolated heterodimers depend on the conditions of preparation and are not typical of integrins in cellular membranes, but in any case these structures are indicative of reduced stability that would be expected to be augmented by the destabilizing effects of I-like domain mutations.
mysb58 and Integrin “Activation”
The cell adhesion and spreading assays support the idea that the unusual wing blistering phenotype of mysb58 is actually due to a gain-of-integrin function. These assays were performed with a Tiggrin fragment. Because Tiggrin is not critical in wing morphogenesis (Bunch et al., 1998), it remains a formal possibility that mysb58 affects binding to a wing ligand specifically, although from the available structural data there is no reason to suspect that residue V409 would affect ligand-specific functions. Preliminary experiments indicate that mysb58-expressing cells function at least as well as cells expressing wild-type βPS on a fragment of an RGD-containing protein that is likely to serve as an integrin ligand in the wing, Wingblister-laminin (Graner et al., 1998; Martin et al., 1999).
In addition to the domains conserved in essentially all integrins, a number of integrins contain a ligand-binding I domain (also known as the A domain) attached to their α subunits, and for a few of these I domains x-ray and other experimental data exist for both the open (ligand binding) and closed conformations. Opening of the I domain causes a downward extension of the C terminal α7 helix (Lee et al., 1995; Emsley et al., 2000), and conversely, mutagenesis experiments have shown that promoting this movement can increase I domain ligand binding (Xiong et al., 2000; Lu et al., 2001; Yang et al., 2004b).
By analogy with α subunit I domains, it has been proposed that upon complete activation the α7 helix of the integrin β subunit I-like domain moves down, causing the observed pivoting of the hybrid domain out away from the α subunit (Takagi et al., 2002; Liddington and Ginsberg, 2002; Luo et al., 2003a). In support of this idea, mutation of a conserved hydrophobic residue at the base of the α7 helix has been demonstrated to promote the exposure of activation epitopes in the hybrid domain (Mould et al., 2003a). Most convincingly, Luo et al. (2004) have recently demonstrated that locking the α7 helix of β3 in different positions with disulfides has the predicted consequences on heterodimer affinity states, assuming that this structure functions similarly to the homologous helix of α subunit I domains. Moreover, deletion of one turn of the I-like domain α7 helix has been tied to shape changes which increase ligand binding affinities at the distal end of the domain (Yang et al., 2004a).
The mysb58 mutation alters a residue (βPS V409) which, by homology to β3, sits near the top of the α7 helix of the I-like domain. This residue is hydrophobic in 24 of the 25 integrin β subunits sequenced to date, and in the original x-ray structure of αvβ3 (Xiong et al., 2001) the closest side chain neighbors to the residue homologous to βPS V409 (β3 L341) are two conserved hydrophobic residues of the α1′ helix. mysb58 inserts an aspartic acid residue into this hydrophobic interface and would be expected to disrupt the association between these structures. Indeed, in the x-ray structure of αvβ3 in combination with an RGD peptide ligand (Xiong et al., 2002) the top of the α7 helix dissolves and β3 L341 (homologous to βPS V409) is rotated away from the α1′ helix, becoming exposed on the exterior of the I-like domain (Figure 7). Previous data have indicated that ligand binding induces movements of the α1-α1′ helices (Xiong et al., 2002; Mould et al., 2002), and the data from mysb58 point to mechanisms whereby this change could be coordinated with movements of the α7 helix (Luo et al., 2004).
Figure 7.

Structural basis for the activating effects of mysb58. The movement of the β3 residue (L341) homologous to βPS V409, in the unliganded I-like domain (A) and in similar crystals infused with a peptide ligand (B) (structures from Xiong et al., 2001, 2002). In the “liganded” structure, the L341 side chain (red) rotates away from the neighboring hydrophobic side chains of the α1′ helix (purple), toward the solvent. This would facilitate a proposed downward movement of the α7 helix (green) and the rightward rotation of the hybrid domain (blue arrow), although these latter movements are not seen in these crystals, which are still in the “bent” conformation (see Figure 6).
Although there is no significant downward movement of the α7 helix in the αvβ3-RGD peptide x-ray structure (Xiong et al., 2002), this is likely due to constraints imposed on the bent conformation of the integrins in the crystal lattice. The increase in activity seen for mysb58 even in the presence of a strongly activating cytoplasmic α subunit is most easily explained by a two-stage model of integrin activation (Takagi et al., 2002; Figure 6C), in which the mysb58 mutation preferentially promotes structural changes after the transition to the upright conformation. In this regard, it is noteworthy that artificially locking the β3 subunit α7 helix in the “high-affinity” position did not by itself drive heterodimers into an extended conformation (Luo et al., 2004). We agree with Takagi et al. (2002) that cellular regulation is probably mediated primarily by adjusting the bent-extended equilibrium. Extended heterodimers can then sample the open and closed conformations, and the stabilization of the open state by ligand binding can trigger integrin clustering, cellular signaling, and other downstream effectors. In any case, our data provide genetic support for the idea that hydrophobic interactions involving the α7 and α1-α1′ helices of the β subunit I-like domain regulate integrin activity, in a manner analogous to that shown for α subunit I domains (e.g., Xiong et al., 2000).
Finally, the important complementary nature of forward genetics and site-directed mutagenesis is illustrated by a recent article reporting an alanine-scanning mutagenesis of the β1 I-like domain helices α1-α1′ and α7 (Barton et al., 2004). Although these authors did not check the residue corresponding to our mysb58 allele, they did alter the two hydrophobic residues in the α1 helix region that make a partially exposed pocket for the mysb58 homologue of β1 in the closed conformation. These changes had little affect on activation in their assay system, but we would predict that changing these residues to polar amino acids (instead of alanines) would likely result in a more active integrin.
β8: The Exception that Proves the Rule?
When comparing 25 β subunit sequences from sponges to humans, only vertebrate β8 does not have a hydrophobic residue in the position homologous to that mutated in mysb58 (Moyle et al., 1991); in β8 this residue is asparagine. β8 integrins have been reported to have broad ligand-binding capabilities and appear to function in the nervous system, in vascular morphogenesis, and regulation of cell growth (Nishimura et al., 1994, 1998; Venstrom and Reichardt, 1995; Cambier et al., 2000; Zhu et al., 2002), but little is known about the regulation of β8 integrins in situ.
The presence of a highly polar residue in the mysb58 position led us to take a closer look at the rest of the β8 sequence. Xiong et al. (2003) have hypothesized that a loop in the membrane proximal “β terminal domain” of β subunits interacts with the I–like domain and serves as a “deadbolt” to stabilize the bent conformation of inactive heterodimers. Sequence comparisons reveal that the deadbolt structure is specifically deleted in β8 (Figure 8), and like the presence of a polar residue in the mysb58 position, β8 is unique among the 25 reported β subunits for this deletion. (Additionally, β8 is missing the otherwise completely conserved aspartates that coordinate the ADMIDAS cation [Xiong et al., 2001, 2002], which apparently mediates tertiary structural changes of the I-like domain [Chen et al., 2003; Mould et al., 2003b]). The mysb58-like polymorphism and lack of a deadbolt lead to the prediction that β8 containing integrins are activated very easily, perhaps constitutively. In any case, the comparisons indicate that the mysb58 mutation has identified an important side chain with physiological relevance in regulating integrin function.
Figure 8.
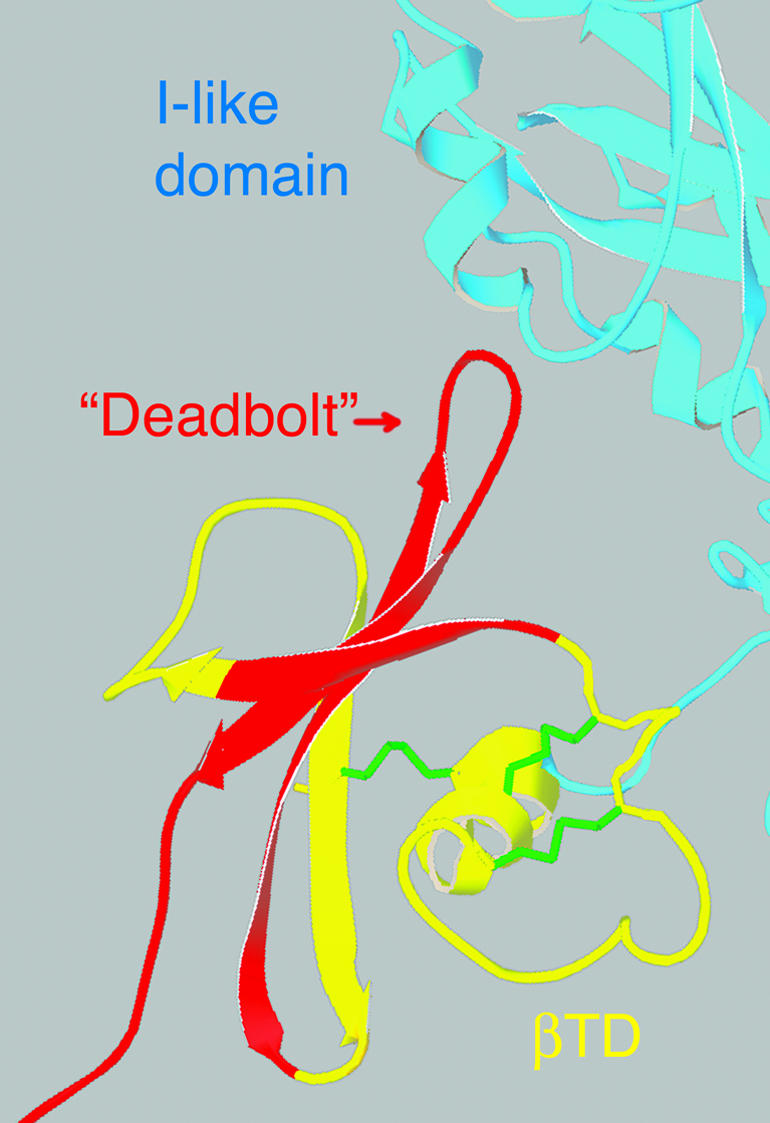
Ribbon diagram of the β-terminal domain of β3 (from Xiong et al., 2001), showing structures expected to be missing in β8 (red). The deleted sequences include those that comprise the “deadbolt” that has been hypothesized to interact with the I-like domain and stabilized the bent, inactive conformation (Xiong et al., 2003). Stabilizing disulfides that are retained in β8 are indicated in green.
Acknowledgments
We are extraordinarily indebted to many individuals who helped with the screens and other genetics and sequencing of mutants, including Irene Alvarez, Emily Brower, Melissa Davis, Marty Glansberg, Stefanie Hager, Wyatt Ho, Augustine Lau, Kirsten Metz, Ryan Reese, Michael Rhee, Jeff Rosenberger, Rick Salatino, Priyanka Sundareshan, Stephanie Tang, and especially Michael Zavortink. This work was supported by grants from the National Institutes of Health and the Arizona Disease Control Research Commission. We give thanks to the sequencing facility of the GATC at the University of Arizona, to Carl Boswell for help with the confocal microscopy, to Mark Ginsberg for help in design and construction of cytoplasmic mutants and for comments on the manuscript, to Anne Cress for help with the adhesion assays, to Marc Brabant for advice, and to Ted Wright for sharing unpublished data. Those who contributed fly stocks or other reagents include Eric Wieschaus, Ted Wright, Norbert Perrimon, Nick Brown, Mani Ramaswami and the Bloomington Stock Center. The authors have no competing financial interests.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E04-02-0085. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-02-0085.
Online version of this article contains supporting material. Online version is available at www.molbiolcell.org.
References
- Ashburner, M. (1989). Drosophila: A Laboratory Handbook. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Baker, S.E., Lorenzen, J.A., Miller, S.W., Bunch, T.A., Jannuzi, A.L., Ginsberg, M.H., Perkins, L.A., and Brower, D.L. (2002). Genetic interaction between integrins and moleskin, a gene encoding a Drosophila homologue of Importin-7 (DIM-7). Genetics 162, 285-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, S.J., Travis, M.A., Askari, J.A., Buckley, P.A., Craig, S.E., Humphries, M.J., and Mould, A.P. (2004). Novel activating and inactivating mutations in the integrin β1 subunit A domain. Biochem. J. 380, 401-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beglova, N., Blacklow, S.C., Takagi, J., and Springer, T.A. (2002). Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat. Struct. Biol. 9, 282-287. [DOI] [PubMed] [Google Scholar]
- Bloor, J.W., and Brown, N.H. (1998). Genetic analysis of the Drosophila αPS2 integrin subunit reveals discrete adhesive, morphogenetic and sarcomeric functions. Genetics 148, 1127-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower, D.L., and Jaffe, S.M. (1989). Requirement for integrins during Drosophila wing development. Nature 342, 285-287. [DOI] [PubMed] [Google Scholar]
- Brower, D.L., Wilcox, M., Piovant, M., Smith, R.J., and Reger, L.A. (1984). Related cell-surface antigens expressed with positional specificity in Drosophila imaginal discs. Proc. Natl. Acad. Sci. USA 81, 7485-7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower, D.L. (2003). Platelets with wings: the maturation of Drosophila integrin biology. Curr. Opin. Cell Biol. 15, 607-613. [DOI] [PubMed] [Google Scholar]
- Brown, N.H., King, D.L., Wilcox, M., and Kafatos, F.C. (1989). Developmentally regulated alternative splicing of Drosophila integrin PS2 α transcripts. Cell 59, 185-195. [DOI] [PubMed] [Google Scholar]
- Bunch, T.A., and Brower, D.L. (1992). Drosophila PS2 integrin mediates RGD dependent cell-matrix interactions. Development 116, 239-247. [DOI] [PubMed] [Google Scholar]
- Bunch, T.A., Graner, M.W., Fessler, L.I., Fessler, J.H., Schneider, K.D., Kerschen, A., Choy, L.P., Burgess, B.W., and Brower, D.L. (1998). The PS2 integrin ligand tiggrin is required for proper muscle function in Drosophila. Development 125, 1679-1689. [DOI] [PubMed] [Google Scholar]
- Bunch, T.A., Miller, S.W., and Brower, D.L. (2004). Analysis of the Drosophila βPS subunit indicates that regulation of integrin activity is a primal function of the C8–C9 loop. Exp. Cell Res. 294, 118-129. [DOI] [PubMed] [Google Scholar]
- Bunch, T.A., Salatino, R., Engelsgjerd, M.C., Mukai, L., West, R.F., and Brower, D.L. (1992). Characterization of mutant alleles of myospheroid, the gene encoding the β subunit of the Drosophila PS integrins. Genetics 132, 519-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier, S., Mu, D.Z., O'Connell, D., Boylen, K., Travis, W., Liu, W.H., Broaddus, V.C., and Nishimura, S.L. (2000). A role for the integrin αvβ8 in the negative regulation of epithelial cell growth. Cancer Res. 60, 7084-7093. [PubMed] [Google Scholar]
- Chen, J., Salas, A., and Springer, T.A. (2003). Bistable regulation of integrin adhesiveness by a bipolar metal ion cluster. Nat. Struct. Biol. 10, 995-1001. [DOI] [PubMed] [Google Scholar]
- Costello, W.J., and Thomas, J.B. (1981). Development of thoracic muscles in muscle-specific mutant and normal Drosophila melanogaster. Soc. Neurosci. Abstr. 7, 543. [Google Scholar]
- Emsley, J., Knight, C.G., Farndale, R.W., Barnes, M.J., and Liddington, R.C. (2000). Structural basis of collagen recognition by integrin α2β1. Cell 101, 47-56. [DOI] [PubMed] [Google Scholar]
- Fogerty, F.J., Fessler, L.I., Bunch, T.A., Yaron, Y., Parker, C.G., Nelson, R.E., Brower, D.L., and Fessler, J.H. (1994). Tiggrin, a novel Drosophila extracellular matrix protein that functions as a ligand for αPS2βPS integrins. Development 120, 1747-1758. [DOI] [PubMed] [Google Scholar]
- Gloor, G.B., Preston, C.R., Johnson-Schlitz, D.M., Nassif, N.A., Phillis, R.W., Benz, W.K., Robertson, H.M., and Engels, W.R. (1993). Type I repressors of P element mobility. Genetics 135, 81-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graner, M.W., Bunch, T.A., Baumgartner, S., Kerschen, A., and Brower, D.L. (1998). Splice variants of the Drosophila PS2 integrins differentially interact with RGD-containing fragments of the extracellular proteins tiggrin, ten-m, and D-laminin 2. J. Biol. Chem. 273, 18235-18241. [DOI] [PubMed] [Google Scholar]
- Hantgan, R.R., Paumi, C., Rocco, M., and Weisel, J.W. (1999). Effects of ligand-mimetic peptides arg-gly-asp-X (X = phe, trp, ser) on αIIbβ3 integrin conformation and oligomerization. Biochemistry 38, 14461-14474. [DOI] [PubMed] [Google Scholar]
- Hantgan, R.R., Rocco, M., Nagaswami, N., and Weisel, J.W. (2001). Binding of a fibrinogen mimetic stabilizes integrin αIIbβ3's open conformation. Protein Sci. 10, 1614-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes, R.O. (2002). Integrins: bidirectional allosteric signaling machines. Cell 110, 673-687. [DOI] [PubMed] [Google Scholar]
- Jannuzi, A.L., Bunch, T.A., Brabant, M.C., Miller, S.W., Mukai, L. Zavortink, M., and Brower, D.L. (2002). Disruption of C-Terminal cytoplasmic domain of βPS integrin subunit has dominant negative properties in developing Drosophila. Mol. Biol. Cell 4, 1352-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata, T., Ambo, H., Puzon-McLaughlin, W., Tieu, K.K., Handa, M., Ikeda, Y., and Takada, Y. (2004). Critical cys residues for regulation on integrin αIIbβ3 are clustered in the EGF domains of the β3 subunit. Biochem. J. 378, 1079-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.O., Rieu, P., Arnaout, M.A., and Liddington, R. (1995). Crystal structure of the A domain from the subunit of integrin CR3 (CD11b/CD18). Cell 80, 631-638. [DOI] [PubMed] [Google Scholar]
- Lewis, E.B., and Bacher, F. (1968). Methods of feeding ethyl methane sulfonate (EMS) to Drosophila males. Dros. Inf. Serv. 43, 193. [Google Scholar]
- Liddington, R.C., and Ginsberg, M.H. (2002). Integrin activation takes shape. J. Cell Biol. 158, 833-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley, D.L., and Zimm, G.G. (1992). The Genome of Drosophila melanogaster. San Diego, CA: Academic Press.
- Liu, S., Calderwood, D.A., and Ginsberg, M.H. (2000). Integrin cytoplasmic domain binding proteins. J. Cell Sci. 113, 3563-3571. [DOI] [PubMed] [Google Scholar]
- Lu, C., Shimaoka, M., Ferzly, M., Oxvig, C., Takagi, J., and Springer, T.A. (2001). An isolated, surface-expressed I domain of the integrin L2 is sufficient for strong adhesive function when locked in the open conformation with a disulfide. Proc. Natl Acad. Sci. USA 98, 2387-2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, B.H., Springer, T.A., and Takagi, J. (2003a). Stabilizing the open conformation of the integrin headpiece with a glycan wedge increases affinity for ligand. Proc. Natl. Acad. Sci. USA 100, 2403-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, B.-H., Springer, T.A., and Takagi, J. (2003b). High affinity ligand binding by integrins does not involve head separation. J. Biol. Chem. 278, 17185-17189. [DOI] [PubMed] [Google Scholar]
- Luo, B.-H., Takagi, J., and Springer, T.A. (2004). Locking the β3 integrin I-like domain into high and low affinity conformations with disulfides. J. Biol. Chem. 279, 10215-10221. [DOI] [PubMed] [Google Scholar]
- Manseau, L. et al. (1997). GAL4 enhancer traps expressed in the embryo, larval brain, imaginal discs and ovary of Drosophila. Dev. Dynam. 209, 1-13. [DOI] [PubMed] [Google Scholar]
- Martin-Bermudo, M.D., Dunin-Borkowski, O.M., and Brown, N.H. (1998). Modulation of integrin activity is vital for morphogenesis. J. Cell Biol. 141, 1073-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D., Zusman, S., Li, X., Williams, E.L., Khare, N., DaRocha, S., Chiquet-Ehrismann, R., and Baumgartner, S. (1999). Wing blister, a new Drosophila laminin α chain required for cell adhesion and migration during embryonic and imaginal development. J. Cell Biol. 145, 191-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould, A.P., Askari, J.A., Barton, S., Kline, A.D., McEwan, P.A., Craig, S.E., and Humphries, M.J. (2002). Integrin activation involves a conformational change in the α1 helix of the β subunit A-domain. J. Biol. Chem. 277, 19800-19805. [DOI] [PubMed] [Google Scholar]
- Mould, A.P., Barton, S.J., Askari, J.A., McEwan, P.A., Buckley, P.A., Craig, S.E., and Humphries, M.J. (2003a). Conformational changes in the integrin βA domain provide a mechanism for signal transduction via hybrid domain movement. J. Biol. Chem. 278, 17028-17035. [DOI] [PubMed] [Google Scholar]
- Mould, A.P., Barton, S.J., Askari, J.A., Craig, S.E., and Humphries, M.J. (2003b). Role of ADMIDAS cation-binding site in ligand recognition by integrin α5β1. J. Biol. Chem. 278, 51622-51629. [DOI] [PubMed] [Google Scholar]
- Moyle, M., Napier, M.A., and McLean, J.W. (1991). Cloning and expression of a divergent integrin subunit β8. J. Biol. Chem. 266, 19650-19658. [PubMed] [Google Scholar]
- Nishimura, S.L., Boylen, K.P., Einheber, S., Milner, T.A., Ramos, D.M., and Pytela, R. (1998). Synaptic and glial localization of the integrin αvβ8 in mouse and rat brain. Brain Res. 791, 271-282. [DOI] [PubMed] [Google Scholar]
- Nishimura, S.L., Sheppard, D., and Pytela, R. (1994). Integrin αvβ8; interaction with vitronectin and functional divergence of the β8 cytoplasmic domain. J. Biol. Chem. 269, 28708-28715. [PubMed] [Google Scholar]
- O'Toole, T.E., Katagiri, Y., Faull, R.J., Peter, K., Tamura, R., Quaranta, V., Loftus, J.C., Shattil, S.J., and Ginsberg, M.H. (1994). Integrin cytoplasmic domains mediate inside-out signal transduction. J. Cell Biol. 124, 1047-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimaoka, M., and Springer, T.A. (2003). Therapeutic antagonists and conformational regulation of integrin function. Nat. Rev. Drug Discov. 2, 703-716. [DOI] [PubMed] [Google Scholar]
- Sun, Q.H., Liu, C.Y., Wang, R., Paddock, C., and Newman, P.J. (2002). Disruption of the long-range GPIIIa Cys5-Cys435 disulfide bond results in the production of constitutively active GPIIb-IIIa (αIIb-β3) integrin complexes. Blood 100, 2094-2101. [DOI] [PubMed] [Google Scholar]
- Takagi, J., Petre, B.M., Walz, T., and Springer, T.A. (2002). Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110, 599-611. [DOI] [PubMed] [Google Scholar]
- Venstrom, K., and Reichardt, L. (1995). β8 integrins mediate interactions of chick sensory neurons with laminin-1, collage IV, and fibronectin. Mol. Cell. Biol. 6, 419-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieschaus, E.C., Nusslein-Volhard, C., and Jurgens, G. (1984). Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster III. Zygotic loci on the X chromosome. Roux's Arch. Dev. Biol. 193, 296-307. [DOI] [PubMed] [Google Scholar]
- Wilcox, M. (1990). Genetic analysis of the Drosophila PS integrins. Cell Diff. Dev. 32, 391-400. [DOI] [PubMed] [Google Scholar]
- Wright, T.R.F. (1968). Phenogenetics of temperature sensitive alleles of lethal myospheroid in Drosophila. Proc. 12th Int. Congr. Genet. 1, 41. [Google Scholar]
- Xiong, J.P., Li, R., Essafi, M., Stehle, T., and Arnaout, M.A. (2000). An isoleucine-based allosteric switch controls affinity and shape shifting in integrin CD11b A-domain. J. Biol. Chem. 275, 38762-38767. [DOI] [PubMed] [Google Scholar]
- Xiong, J.-P., Stehle, T., Diefenbach, B., Zhang, R., Dunker, R., Scott, D.L., Joachimiak, A., Goodman, S.L., and Arnaout, M.A. (2001). Crystal structure of the extracellular segment of integrin αvB3. Science 294, 339-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, J.-P., Stehle, T., Goodman, S.L., and Arnaout, M.A. (2003). New insights into the structural basis of integrin activation. Blood 102, 1155-1159. [DOI] [PubMed] [Google Scholar]
- Xiong, J.-P., Stehle, T., Zhang, R., Joachimiak, A., Frech, M., Goodman, S.L., and Arnaout, M.A. (2002). Crystal structure of the extracellular segment of integrin αvβ3 in complex with an arg-gly-asp ligand. Science 296, 151-155. [DOI] [PubMed] [Google Scholar]
- Xu, T., and Rubin, G.M. (1993). Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117, 1223-1237. [DOI] [PubMed] [Google Scholar]
- Yang, W., Shimaoka, M., Chen, J.-F., and Springer, T.A. (2004a). Activation of integrin β subunit I-like domains by one-turn C-terminal helix deletions. Proc. Natl. Acad. Sci. USA 101, 2333-2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, W., Shimaoka, M., Salas, A., Takagi, J., and Springer, T.A. (2004b). Intersubunit signal transmission in integrins by a receptor-like interaction with a pull string. Proc. Natl Acad. Sci. USA 101, 2906-2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee, G.H. (1993). Identification and characterization of integrin receptor subunits in Drosophila melanogaster. PhD Thesis, Cambridge: Massachusettts Institute of Technology.
- Zavortink, M., Bunch, T.A., and Brower, D.L. (1993). Functional properties of alternatively spliced forms of the Drosophila PS2 integrin α subunit. Cell Adhes. Commun. 1, 251-264. [DOI] [PubMed] [Google Scholar]
- Zhu, J., Motejlek, K., Wang, D., Zang, K., Schmidt, A., and Reichardt, L.F. (2002). β8 integrins are required for vascular morphogenesis in mouse embryos. Development 129, 2891-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zusman, S., Grinblat, Y., Yee, G., Kafatos, F.C., and Hynes, R.O. (1993). Analyses of PS integrin functions during Drosophila development. Development 118, 737-750. [DOI] [PubMed] [Google Scholar]