

Abstract
The adaptive responses of a living cell to internal and external signals are controlled by networks of proteins whose interactions are so complex that the functional integration of the network cannot be comprehended by intuitive reasoning alone. Mathematical modeling, based on biochemical rate equations, provides a rigorous and reliable tool for unraveling the complexities of molecular regulatory networks. The budding yeast cell cycle is a challenging test case for this approach, because the control system is known in exquisite detail and its function is constrained by the phenotypic properties of >100 genetically engineered strains. We show that a mathematical model built on a consensus picture of this control system is largely successful in explaining the phenotypes of mutants described so far. A few inconsistencies between the model and experiments indicate aspects of the mechanism that require revision. In addition, the model allows one to frame and critique hypotheses about how the division cycle is regulated in wild-type and mutant cells, to predict the phenotypes of new mutant combinations, and to estimate the effective values of biochemical rate constants that are difficult to measure directly in vivo.
INTRODUCTION
The cell cycle is the sequence of events during which a growing cell replicates all its components and divides them more or less evenly between two daughter cells, so that each daughter contains the information and machinery necessary to repeat the process (Mitchison, 1971; Murray and Hunt, 1993). Cell proliferation underlies all biological growth, reproduction, and development, and its misregulation results in serious human diseases. As might be expected of a process so central to cell viability, the molecular machinery regulating crucial events of the cell cycle (DNA synthesis and mitosis) is highly conserved across eukaryotic organisms (Nurse, 1990). Hence, thorough genetic studies of cell cycle regulation in budding yeast (Nasmyth, 1996; Mendenhall and Hodge, 1998) and fission yeast (Nurse, 1997; Moser and Russell, 2000) have paid handsome dividends in understanding cell proliferation in multicellular plants and animals.
The cell cycle regulatory system is most fully worked out for budding yeast (Saccharomyces cerevisiae). A hypothetical molecular mechanism for regulating DNA synthesis, bud emergence, mitosis, and cell division in budding yeast is proposed in Figure 1. How does one determine whether such a hypothesis is correct? The classical approach in physical chemistry is to convert the mechanism into a set of kinetic equations (nonlinear ordinary differential equations) and to compare the solutions of these equations to the observed behavior of the chemical reaction system. If a set of rate constants can be found for which the solutions fit the observations, then the mechanism is provisionally confirmed (pending further experimental investigations). If not, inconsistencies identify aspects of the mechanism that require revision and further testing. Although a mechanism can be disproved if it is inconsistent with well-established facts, it can never be proved correct, because new observations may force modifications and additions. Hence, our intention is not to prove that the hypothesis in Figure 1 is “true” but rather only to show that the mechanism is a reasonable approximation to what is going on inside yeast cells.
Figure 1.

Consensus model of the cell cycle control mechanism in budding yeast. (For a full justification of this diagram, with references to the original literature, see our Web site at http://mpf.biol.vt.edu.) The diagram should be read from bottom left toward top right. (In the diagram, Cln2 stands for Cln1 and 2, Clb5 for Clb5 and 6, and Clb2 for Clb1 and 2; furthermore, the kinase partner of the cyclins, Cdc28, is not shown explicitly. There is an excess of Cdc28 and it combines rapidly with cyclins as soon as they are synthesized.) Newborn daughter cells must grow to a critical size to have enough Cln3 and Bck2 to activate the transcription factors MBF and SBF, which drive synthesis of two classes of cyclins, Cln2 and Clb5. Cln2 is primarily responsible for bud emergence and Clb5 for initiating DNA synthesis. Clb5-dependent kinase activity is not immediately evident because the G1-phase cell is full of cyclin-dependent kinase inhibitors (CKI; namely, Sic1 and Cdc6). After the CKIs are phosphorylated by Cln2/Cdc28, they are rapidly degraded by SCF, releasing Clb5/Cdc28 to do its job. A fourth class of “mitotic cyclins,” denoted Clb2, are out of the picture in G1 because their transcription factor Mcm1 is inactive, their degradation pathway Cdh1/APC is active, and their stoichiometric inhibitors CKI are abundant. Cln2- and Clb5-dependent kinases remove CKI and inactivate Cdh1, allowing Clb2 to accumulate, after some delay, as it activates its own transcription factor, Mcm1. Clb2/Cdc28 turns off SBF and MBF. (Clb5/Cdc28 is probably the other down-regulator of MBF.) As Clb2/Cdc28 drives the cell into mitosis, it also sets the stage for exit from mitosis by stimulating the synthesis of Cdc20 and by phosphorylating components of the APC (see text for details). Meanwhile, Cdc20/APC is kept inactive by the Mad2-dependent checkpoint signal responsive to unattached chromosomes. When the replicated chromosomes are attached, active Cdc20/APC initiates mitotic exit. First, it degrades Pds1, releasing Esp1, a protease involved in sister chromatid separation. It also degrades Clb5 and partially Clb2, lowering their potency on Cdh1 inactivation. In this model, Cdc20/APC promotes degradation of a phosphatase (PPX) that has been keeping Net1 in its unphosphorylated form, which binds with Cdc14. As the attached chromosomes are properly aligned on the metaphase spindle, Tem1 is activated, which in turn activates Cdc15 (the endpoint of the “MEN” signal-transduction pathway in the model). When Net1 gets phosphorylated by Cdc15, it releases its hold on Cdc14. Cdc14 (a phosphatase) then does battle against the cyclin-dependent kinases: activating Cdh1, stabilizing CKIs, and activating Swi5 (the transcription factor for CKIs). In this manner, Cdc14 returns the cell to G1 phase (no cyclins, abundant CKIs, and active Cdh1).
Physical chemists have used this methodology successfully to unravel molecular mechanisms that are not only complicated (many components) but also dynamically complex, involving multiple steady states, oscillations, and chaos (Field et al., 1972; Gyorgyi and Field, 1992). We have used this approach to study simpler models of cell cycle regulation in frog eggs (Novak and Tyson, 1993), fission yeast (Novak et al., 2001), and budding yeast (Chen et al., 2000). The model in Chen et al. (2000) gives an adequate description of the G1-to-S transition, but, since it was published, many more molecular details about the M-to-G1 transition (“exit from mitosis”) have come to light. By adding these details to the mechanism in Chen et al. (2000), we are able to model comprehensively and quantitatively all stages of the chromosome replication-segregation cycle in a eukaryote, based on a mechanism that is almost fully specified at the genetic level.
As explained in MATERIALS AND METHODS, we turn the mechanistic hypothesis (Figure 1) into a mathematical model (Table 1) with a preliminary set of kinetic constants (Table 2). Next, we solve these equations numerically and show, in RESULTS, that the solutions are in accord with the physiological properties of 120 mutant strains of budding yeast out of 131 studied so far (Table 3). There are 11 mutants that the model fails to account for, and these failures identify aspects of the mechanism that need further investigation. We also show how to use the model to interpret existing data, to design new experiments, and to think about the “molecular logic” of cell cycle regulation.
Table 1.
Equations
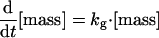 |
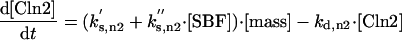 |
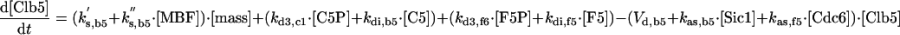 |
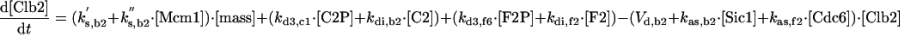 |
 |
 |
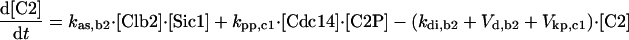 |
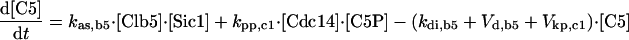 |
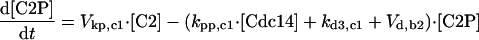 |
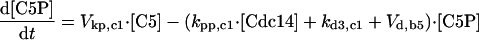 |
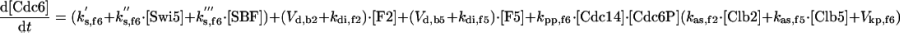 |
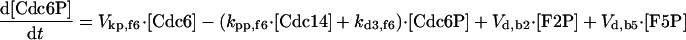 |
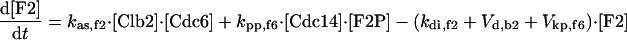 |
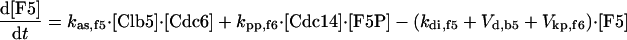 |
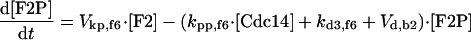 |
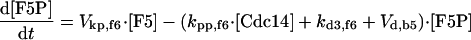 |
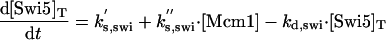 |
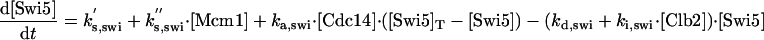 |
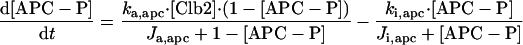 |
 |
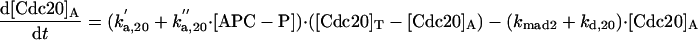 |
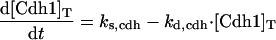 |
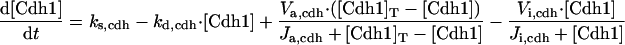 |
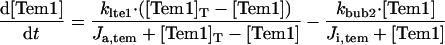 |
 |
 |
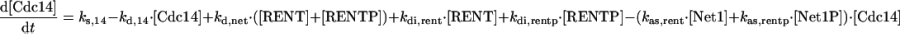 |
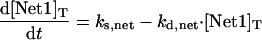 |
 |
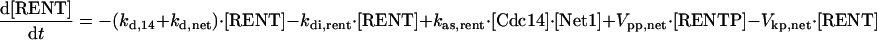 |
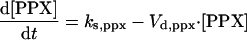 |
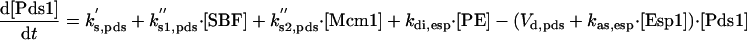 |
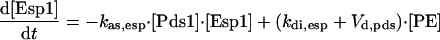 |
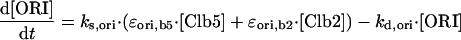 |
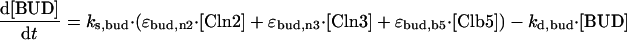 |
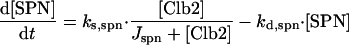 |
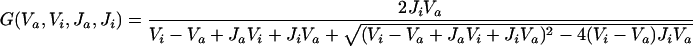 |
| [SBF] = [MBF] = G(Va,sbf, Vi,sbf, Ja,sbf, Ji,sbf) |
| [Mcm1] = G(ka,mcm · [Clb2], ki,mcm, Ja,mcm, Ji,mcm) |
| [Cln3] = C0 · Dn3 · [mass]/(Jn3 + Dn3 · [mass]) |
| [Bck2] = B0 · [mass] |
| [Clb5]T = [Clb5] + [C5] + [C5P] + [F5] + [F5P] |
| [Clb2]T = [Clb2] + [C2] + [C2P] + [F2] + [F2P] |
| [Sic1]T = [Sic1] + [Sic1P] + [C2] + [C2P] + [C5] + [C5P] |
| [Cdc6]T = [Cdc6] + [Cdc6P] + [F2] + [F2P] + [F5] + [F5P] |
| [CKI]T = [Sic1]T + [Cdc6]T |
| [RENTP] = [Cdc14]T — [RENT] — [Cdc14] |
| [Net1P] = [Net1]T — [Net1] — [Cdc14]T + [Cdc14] |
| [PE] = [Esp1]T — [Esp1] |
 |
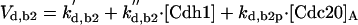 |
| Va,sbf = ka,sbf·(εsbf,n2·[Cln2] + εsbf,n3·([Cln3] + [Bck2]) + εsbf,b5·[Clb5]) |
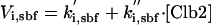 |
| Vkp,c1 = kd1,c1 + kd2,c1·(εc1,n3·[Cln3] + εc1,k2·[Bck2] + εc1,n2·[Cln2] + εc1,b5·[Clb5] + εc1,b2·[Clb2])/(Jd2,c1 + [Sic1]T) |
| Vkp,f6 = kd1,f6 + kd2,f6·(εf6,n3·[Cln3] + εf6,k2·[Bck2] + εf6,n2·[Cln2] + εf6,b5·[Clb5] + εf6,b2·[Clb2])/(Jd2,f6 + [Cdc6]T) |
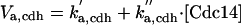 |
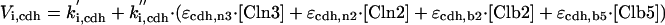 |
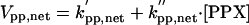 |
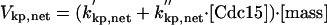 |
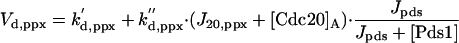 |
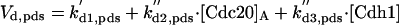 |
| Reset rules: When [Clb2] drops below Kez, we reset [BUD] and [SPN] to zero, and divide the mass between daughter cell and mother cell as follows: mass → f·mass for daughter, and mass → (1 — f)·mass for mother, with f = e—kg·D, where D = (1.026/kg) — 32 is the observed daughter cell cycle time as a function of growth rate (Lord and Wheals, 1980). When [Clb2] + [Clb5] drops below Kez2, [ORI] is reset to 0. |
| Flags: Bud emergence when [BUD] = 1, start DNA synthesis when [ORI] = 1, chromosome alignment on spindle completed when [SPN] = 1 |
Table 2.
Parameter values and initial conditions
| A. Basal parameter values for the wild-type cell cycle | |||||||
|---|---|---|---|---|---|---|---|
| kg = 0.007702 | 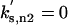 |
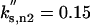 |
kd,n2 = 0.12 | 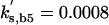 |
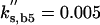 |
||
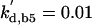 |
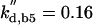 |
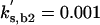 |
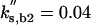 |
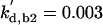 |
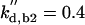 |
||
| kd,b2p = 0.15 | 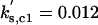 |
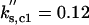 |
kd1,c1 = 0.01 | kd2,c1 = 1 | kd3,c1 = 1 | ||
| kpp,c1 = 4 | 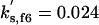 |
 |
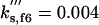 |
kd1,f6 = 0.01 | kd2,f6 = 1 | ||
| kd3,f6 = 1 | kpp,f6 = 4 | kas,b5 = 50 | kdi,b5 = 0.06 | kas,f5 = 0.01 | kdi,f5 = 0.01 | ||
| kas,b2 = 50 | kdi,b2 = 0.05 | kas,f2 = 15 | kdi,f2 = 0.5 | 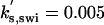 |
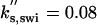 |
||
| kd,swi = 0.08 | ka,swi = 2 | ki,swi = 0.05 | ka,apc = 0.1 | ki,apc = 0.15 | 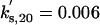 |
||
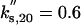 |
kd,20 = 0.3 | 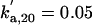 |
 |
ks,cdh = 0.01 | kd,cdh = 0.01 | ||
 |
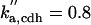 |
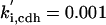 |
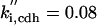 |
ks,14 = 0.2 | kd,14 = 0.1 | ||
| ks,net = 0.084 | kd,net = 0.03 | 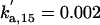 |
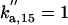 |
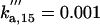 |
ki,15 = 0.5 | ||
 |
 |
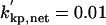 |
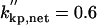 |
kas,rent = 200 | kas,rentp = 1 | ||
| kdi,rent = 1 | kdi,rentp = 2 | ks,ppx = 0.1 | 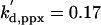 |
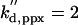 |
 |
||
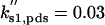 |
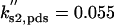 |
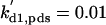 |
 |
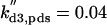 |
kas,esp = 50 | ||
| kdi,esp = 0.5 | ks,ori = 2 | kd,ori = 0.06 | ks,bud = 0.2 | kd,bud = 0.06 | ks,spn = 0.1 | ||
| kd,spn = 0.06 | ka,sbf = 0.38 | 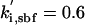 |
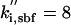 |
ka,mcm = 1 | ki,mcm = 0.15 | ||
| kmad2 = 8 (for [ORI] > 1 and [SPN] < 1) or 0.01 (otherwise) | |||||||
| kbub2 = 1 (for [ORI] > 1 and [SPN] < 1) or 0.2 (otherwise) | |||||||
| klte1 = 1 (for [SPN] > 1 and [Clb2] > Kez) or 0.1 (otherwise) | |||||||
| εsbf,n2 = 2 | εsbf,n3 = 10 | εsbf,b5 = 2 | εc1,n3 = 0.3 | εc1,n2 = 0.06 | εc1,k2 = 0.03 | ||
| εc1,b5 = 0.1 | εc1,b2 = 0.45 | εf6,n3 = 0.3 | εf6,n2 = 0.06 | εf6,k2 = 0.03 | εf6,b5 = 0.1 | ||
| εf6,b2 = 0.55 | εcdh,n3 = 0.25 | εcdh,n2 = 0.4 | εcdh,b5 = 8 | εcdh,b2 = 1.2 | εori,b5 = 0.9 | ||
| εori,b2 = 0.45 | εbud,n3 = 0.05 | εbud,n2 = 0.25 | εbud,b5 = 1 | C0 = 0.4 | Dn3 = 1 | ||
| B0 = 0.054 | [TEM1]T = 1 | [Cdc15]T = 1 | [Esp1]T = 1 | ||||
| Jd2,c1 = 0.05 | Jd2,f6 = 0.05 | Ja,apc = 0.1 | Ji,apc = 0.1 | Ja,cdh = 0.03 | Ji,cdh = 0.03 | ||
| Ja,tem = 0.1 | Ji,tem = 0.1 | Ja,sbf = 0.01 | Ji,sbf = 0.01 | Ja,mcm = 0.1 | Ji,mcm = 0.1 | ||
| Jspn = 0.14 | Jn3 = 6 | J20,ppx = 0.15 | Jpds = 0.04 | Kez = 0.3 | Kez2 = 0.2 | ||
| B. Initial conditions for a newborn, wild-type daughter cell | ||
|---|---|---|
| [mass] = 1.2060 | [F5] =7.2e-5 | [Cdc14] =0.4683 |
| [Cln2] =0.0652 | [F2P] =0.0274 | [Net1]T =2.8 |
| [Clb5] =0.0518 | [F5P] =7.9e-6 | [Net1] =0.0186 |
| [Clb2] =0.1469 | [Swi5]T =0.9765 | [RENT] =1.0495 |
| [Sic1] =0.0229 | [Swi5] =0.9562 | [PPX] =0.1232 |
| [Sic1P] =0.0064 | [APC-P] =0.1015 | [Pds1] =0.0256 |
| [C2] =0.2384 | [Cdc20]T =1.9163 | [Esp1] =0.3013 |
| [C5] =0.0701 | [Cdc20]A =0.4443 | [ORI] =0.0009 |
| [C2P] =0.0240 | [Cdh1]T =1 | [BUD] =0.0085 |
| [C5P] =0.0069 | [Cdh1] =0.9305 | [SPN] =0.0305 |
| [Cdc6] =0.1076 | [Tem1] =0.9039 | kmad2 =0.01 |
| [Cdc6P] =0.0155 | [Cdc15] =0.6565 | kbub2 =0.2 |
| [F2] =0.2361 | [Cdc14]T =2 | klte1 =0.1 |
All parameters that start with a lower case k are rate constants (min-1). All other parameters are dimensionless
Table 3.
| Wild-type |
| In glucose |
| In galactose |
| Cln mutants |
| cln1Δ cln2Δ |
| GAL-CLN2 cln1Δ cln2Δ |
| cln1Δ cln2Δ sic1Δ |
| cln1Δ cln2Δ cdh1Δ |
| GAL-CLN2 cln1Δ cln2Δ cdh1Δ |
| cln3Δ |
| GAL-CLN3 |
| Bck2 mutants |
| bck2Δ |
| Multi-copy BCK2 |
| cln1Δ cln2Δ bck2Δ |
| cln3Δ bck2Δ |
| cln3Δ bck2Δ GAL-CLN2 cln1Δ cln2Δ |
| cln3Δ bck2Δ multi-copy CLN2 |
| cln3Δ bck2Δ sic1Δ |
| cln1 cln2 cln3 strain |
| cln1Δ cln2Δ cln3Δ |
| cln1Δ cln2Δ cln3Δ GAL-CLN2 |
| cln1Δ cln2Δ cln3Δ GAL-CLN3 |
| cln1Δ cln2Δ cln3Δ sic1Δ |
| cln1Δ cln2Δ cln3Δ cdh1Δ |
| cln1Δ cln2Δ cln3Δ multi-copy CLB5 |
| cln1Δ cln2Δ cln3Δ GAL-CLB5 |
| cln1Δ cln2Δ cln3Δ multi-copy BCK2 |
| cln1Δ cln2Δ cln3Δ GAL-CLB2 |
| cln1Δ cln2Δ cln3Δ apcts |
| Cdh1, Sic1 and Cdc6 mutants |
| sic1Δ |
| GAL-SIC1 |
| GAL-SIC1-dbΔ |
| GAL-SIC1 cln1Δ cln2Δ |
| GAL-SIC1 GAL-CLN2 cln1Δ cln2Δ |
| GAL-SIC1 cln1Δ cln2Δ cdh1Δ |
| GAL-SIC1 GAL-CLN2 cln1Δ cln2Δ cdh1Δ |
| cdh1Δ(*) |
| Cdh1 constitutively active |
| sic1Δ cdh1Δ(*) |
| sic1Δ cdh1Δ GALL-CDC20 |
| cdc6Δ2-49 |
| cdc6Δ2-49 sic1Δ |
| cdc6Δ2-49 cdh1Δ(*) |
| cdc6Δ2-49 sic1Δ cdh1Δ(*) |
| cdc6Δ2-49 sic1Δ cdh1Δ GALL-CDC20 |
| swi5Δ |
| swi5Δ GAL-CLB2 |
| swi5Δ cdh1Δ(*) |
| swi5Δ cdh1Δ GAL-SIC1 |
| Clb1 Clb2 mutants |
| clb1Δ clb2Δ |
| clb2Δ CLB1(*) |
| GAL-CLB2 |
| Multi-copy GAL-CLB2 |
| clb2Δ CLB1 cdh1Δ(*) |
| clb2Δ CLB1 pds1Δ(*) |
| GAL-CLB2 sic1Δ(*) |
| GAL-CLB2 cdh1Δ |
| CLB2-dbΔ |
| CLB2-dbΔ in galactose |
| CLB2-dbΔ multi-copy SIC1 |
| CLB2-dbΔ GAL-SIC1 |
| CLB2-dbΔ multi-copy CDC6 |
| CLB2-dbΔ clb5Δ |
| CLB2-dbΔ clb5Δ in galactose |
| GAL-CLB2-dbΔ |
| Clb5 Clb6 mutants |
| clb5Δ clb6Δ |
| clb5Δ clb6Δ cln1Δ cln2Δ |
| GAL-CLB5 |
| GAL-CLB5 sic1Δ |
| GAL-CLB5 cdh1Δ |
| CLB5-dbΔ |
| CLB5-dbΔ sic1Δ |
| CLB5-dbΔ pds1Δ |
| CLB5-dbΔ pds1Δ cdc20Δ |
| GAL-CLB5-dbΔ |
| Cdc20 mutants |
| cdc20ts |
| cdc20Δ clb5Δ |
| cdc20Δ pds1Δ |
| cdc20Δ pds1Δ clb5Δ |
| GAL-CDC20 |
| cdc20ts mad2Δ |
| cdc20ts bub2Δ |
| Pds1/Esp1 interaction |
| pds1Δ(*) |
| esp1ts |
| PDS1-dbΔ |
| GAL-PDS1-dbΔ |
| GAL-PDS1-dbΔ esp1ts |
| GAL-ESP1 cdc20ts |
| MEN pathway mutants |
| tem1Δ |
| GAL-TEM1 |
| tem1ts multi-copy CDC15 |
| tem1ts GAL-CDC15 |
| tem1Δ net1ts |
| tem1Δ multi-copy CDC14 |
| cdc15Δ |
| Multi-copy CDC15 |
| cdc15ts multi-copy TEM1 |
| cdc15Δ net1ts |
| cdc15ts multi-copy CDC14 |
| Exit-of-mitosis mutants |
| net1ts |
| GAL-NET1 |
| cdc14ts |
| GAL-CDC14 |
| GAL-CDC14 GAL-NET1 |
| net1ts cdc20ts |
| cdc14ts GAL-SIC1 |
| cdc14ts then GAL-SIC1 |
| cdc14ts sic1Δ at permissive temperature |
| cdc14ts cdh1Δ at permissive temperature |
| cdc14ts GAL-CLN2 at permissive temperature |
| TAB6-1 |
| TAB6-1 cdc15ts |
| TAB6-1 clb5Δ |
| TAB6-1 clb2Δ CLB1 |
| Checkpoint mutants |
| mad2Δ |
| bub2Δ |
| mad2Δ bub2Δ |
| WTI in nocodazole |
| mad2Δ in nocodazole |
| mad2Δ GAL-TEM1 in nocodazole |
| mad2Δ pds1Δ in nocodazole |
| bub2Δ in nocodazole (*) |
| bub2Δ pds1Δ in nocodazole |
| bub2Δ mad2Δ in nocodazole |
| pds1Δ in nocodazole |
| net1ts in nocodazole |
| APC mutants |
| APC-A |
| APC-A cdh1Δ |
| APC-A cdh1Δ in galactose |
| APC-A cdh1Δ multi-copy SIC1 |
| APC-A cdh1Δ GAL-SIC1 |
| APC-A cdh1Δ multi-copy CDC6 |
| APC-A cdh1Δ GAL-CDC6 |
| APC-A cdh1Δ multi-copy CDC20 |
| APC-A sic1Δ |
| APC-A GAL-CLB2 |
indictes 11 mutants whose simulations do not agree with experimental observations
MATERIALS AND METHODS
A Quantitative Mathematical Model
The molecular mechanism in Figure 1 is a hypothetical account of the chemical reactions among the genes and proteins known to play principal roles in controlling the cell cycle of budding yeast. The mechanism summarizes information from many publications on the individual genes, their patterns of expression, and the interactions among their encoded proteins. To simplify the wiring diagram, we combine redundant cyclins (Cln2, Clb5, and Clb2 in the model refer, respectively, to Cln1 + Cln2, Clb5 + Clb6, and Clb1 + Clb2), and we ignore Clbs 3 and 4. As demonstrated by Cross et al. (2002), the kinase subunit Cdc28 that is associated with each cyclin is present in excess, so it need not be presented in the diagram or the equations.
A wiring diagram is a set of boxes (components) interconnected by arrows (reactions). An instantaneous state of the system is a specification of the current concentrations of all its components. Given a state of the system, the chemical reactions (synthesis, degradation, activation, inhibition, binding, and release) indicate how the state will change in the next moment of time. Each reaction proceeds at a rate determined by the state of the system and by kinetic parameters (e.g., rate constants and binding constants). By applying the general principles of biochemical kinetics, we can convert the mechanism in Figure 1 into a set of differential and algebraic equations (Table 1) that determine how the state of the control system evolves in time.
It is important to realize that there is no unique correspondence between a wiring diagram and a set of mathematical equations; the same mechanism can be represented by different forms of equations. For example, the modeler may choose to describe some components by differential equations and others by algebraic equations, and to describe the rates of some reactions by Michaelis-Menten kinetics and others by mass-action kinetics. These choices are somewhat arbitrary, depending on the level of detail desired in the model. Hence, there is a hierarchy of assumptions that go into a model: first, the wiring diagram, which is a hypothesis about how the components of the control system are interconnected; second, the mathematical equations, which are representations of a possible set of kinetic consequences of the mechanism; and third, the assignment of specific values to the rate constants in the equations. Once these choices are made, the equations can be solved numerically and the dynamic behavior of the control system compared with the observed properties of dividing yeast cells. If there are inconsistencies between the behavior of the model and the behavior of the cells, then one usually works backwards through the hierarchy to resolve the discrepancies. First, rate constants are adjusted to try to fit the model to the data. If that proves impossible, then one reconsiders the assumptions made in translating the diagram into equations. If modifications of those assumptions do not help, then one must reconsider the wiring diagram itself. Perhaps there are missing components or interactions that prevent the model from fitting the data; for example, see von Dassow et al. (2000). By this process of trial, error, and refinement, we have settled temporarily on the wiring diagram (Figure 1), equations (Table 1), and parameter values (Table 2) presented here as representative of the budding yeast cell cycle engine. The model will certainly continue to evolve as it is confronted with new experimental studies of the control system. In our experience, during this evolutionary process, successive models show steady improvements over earlier versions rather than wholesale replacement of one set of equations and parameters with another. We suggest that the modeling process is converging on ever better mathematical approximations of the molecular regulatory system. In the next subsection, we describe in more detail how we choose rate laws and assign values to kinetic constants.
Rate Equations and Kinetic Constants
Although the model as a whole (Figure 1) is complicated, each individual equation (Table 1) is simple. For example, Cln2-dependent kinase activity changes in time due to synthesis and degradation of the Cln2 subunit (assuming Cdc28 is in excess and binds rapidly to cyclins). The rate of Cln2 synthesis, k′s,n2 + k″s,n2 · [SBF], includes a constitutive rate, k′s,n2 (for simulation of GAL-CLN2 mutants), and a contribution dependent on the relative activity of the transcription factor SBF, controlling expression of the CLN2 gene. The activity of SBF is given by the algebraic equation [SBF] = G(Va,sbf, Vi,sbf, Ja,sbf, Ji,sbf). The “Goldbeter-Koshland function” G(Va, Vi, Ja, Ji) varies sigmoidally between 0 and 1, increasing as a function of Va and decreasing as a function of Vi (Goldbeter and Koshland, 1981). The sigmoid is steeper (more switch-like) as Ja and Ji are much <1. In the equation for SBF activity, Va,sbf = ka,sbf · (εsbf,n3 · ([Cln3] + [Bck2]) + εsbf,5 · [Clb5]) indicates that SBF is activated by Cln2/Cdc28, Cln3/Cdc28, Bck2, and Clb5/Cdc28, whereas Vi,sbf = k′i,sbf + k″i,sbf · [Clb2] indicates that SBF is inactivated by Clb2/Cdc28 (Amon et al., 1993). The coefficients εsbf express the relative efficiencies of these components in activating SBF. Our choices for the ε values (Table 2) reflect experimental data that Cln3/Cdc28 and Bck2 are about equally efficient in activating SBF because cell sizes are about the same in cln3Δ and bck2Δ mutants (Epstein and Cross, 1994), whereas Cln2/Cdc28 is much less efficient (Dirick et al., 1995; Stuart and Wittenberg, 1995). The same experiments indicate that SBF is turned on very abruptly as the cell reaches a critical size, so we choose Ja,sbf and Ji,sbf to be much <1. We have assigned a value of 2 to εsbf,b5, because there is evidence that SBF and MBF are activated by Clb5 and 6 (Schwob and Nasmyth, 1993). To make cln3Δ bck2Δ not rescued by sic1Δ or GAL-CLB5 (Wijnen and Futcher, 1999), we set εsbf,b5 < 4.
In our model, SBF turns on in wild-type cells when Cln3 and Bck2 have accumulated in the nucleus to a critical level: [Cln3] + [Bck3] = k′i,sbf/(ka,sbf · εsbf,n3). Hence, the ratio k′i,sbf/ka,sbf sensitively determines cell size when SBF turns on (Dirick et al., 1995), and the ratio ka,sbf/k″i,sbf (which sets the Clb2-kinase activity required to inactivate SBF) sensitively determines the timing of Cln2 disappearance in the latter part of the cycle (Amon et al., 1993). The values given to these parameter ratios in Table 2 were chosen to fit the model to these observations.
Cln2 degradation is assumed to be a simple first-order process, kd,n2 · [Cln2] with half-life = 0.693/kd,n2 = 6 min, in reasonable accord with experiment (Salama et al., 1994; Barral et al., 1995). Actually, the Clns are degraded by the SCF-dependent proteasome pathway (Deshaies et al., 1995; Lanker et al., 1996), whose first step is phosphorylation of the protein to be degraded. We are assuming (in this version of the model) that the phosphorylation of Cln2, required for recognition by the SCF, happens uniformly throughout the cell cycle.
To fit the model to the total amount of Cln2 observed in an asynchronous culture of budding yeast cells (Cross et al., 2002), we choose k″s,n2 = 0.15 min-1.
The differential equations for [Clb5] and [Clb2] have terms for synthesis and degradation and for binding to and release from stoichiometric inhibitors Sic1 and Cdc6. The synthesis term has a transcription factor given by a Goldbeter-Koshland function, as for [Cln2]. The degradation term consists of several components describing relative contributions from different pathways: Cdc20/APC and Cdh1/APC for Clb2 (Zachariae and Nasmyth, 1999; Yeong et al., 2000), and Cdc20/APC-dependent and -independent pathways for Clb5 (Irniger and Nasmyth, 1997; Wasch and Cross, 2002). The rate constants for these pathways are estimated from the stabilities of the Clbs in various phases of the cycle when the different pathways are active. Then, the rate constants for synthesis are estimated from the data of Cross (2002) on average cyclin contents in an asynchronous culture.
By the same reasoning, one can work through every equation in the model, identifying the molecular steps involved, the assumptions made in defining the rate law, and (where relevant data are available) the numerical values assigned to rate constants. In some cases, directly relevant data are not available; for example, for the binding of Sic1 and Cdc6 to Clb2 and Clb5. Typically, we assume that binding is rapid (the rate constant for association, kas is large compared with rate constants for e.g., synthesis, degradation, and phosphorylation) and the equilibrium binding constant (kas/kdi) is large. Precise values for these parameters are not crucial to the model, with one exception. For reasons indicated in RESULTS, we assume that Cdc6 is an ineffective stoichiometric inhibitor of Clb5 kinase. This assumption is supported by the observation of Archambault et al. (2003) that Cdc6 does not complex with Clb5 in immunoprecipitation assays.
The rate constants we propose are “effective” values because they capture only the time scales of processes (note that every rate constant has a unit of minute-1). They do not capture the characteristic concentration scales of the rates. When the model was being developed, we did not know the absolute concentrations of most of the proteins in the mechanism, so we expressed the concentration of each protein in an arbitrary unit (au), which may be different for each protein. For example, for Tem1 and Cdc15, the arbitrary units are chosen so that [Tem1]T = [Cdc15]T = 1. Hence, the variables [Tem1] and [Cdc15] represent the fraction of each protein pool in the “active” form. Recently, Ghaemmaghami et al. (2003) published a comprehensive analysis of protein expression in budding yeast. Their data will allow us to assign nanomolar values to each au in a later version of our model.
Our choices of concentration units are constrained by two facts. 1) For any two proteins involved in a stoichiometric interaction (e.g., Cdc14 and Net1, Clb2 and Sic1, Pds1 and Esp1), we must use the same au for each protein of a pair. 2) Cross et al. (2002) have measured the concentrations of Clns, Clbs, Sic1, and Cdc28 in an asynchronous culture by tagging them with the same epitope. These data allow us to assign a concentration (in nanomolar) to the au used to express the concentrations of all these proteins. For example, Cross determined that there are 3000 molecules of Cln1 and Cln2 per diploid cell (averaged over the cell cycle). Given the diploid cell volume to be 100 fL, 3000 molecules corresponds to a concentration of 50 nM. (The volume of a diploid cell is calculated from the volume of a haploid cell, which was measured to be 50 fL by Nash et al., 1988 and Yaglom et al., 1995. A diploid cell is twice as big as a haploid cell (Lorincz and Carter, 1979) and presumably contains twice the amount of every protein.) From our model, the average value of [Cln2] over a full cycle is 1.25 au; therefore, 1 au of Cln2 (or Clb2 or Sic1) corresponds to a concentration of ∼40 nM, or 1200 molecules per haploid cell.
We illustrate how to relate rate constants in the model to measurements in a cell culture with a few examples. The rate constant for synthesis of Sic1 (when Swi5 is fully active) is k″s,c1 = 0.12 au/min = 4.8 mM/min. On the other hand, the rate constant for synthesis of Cdc20 (when Mcm1 is fully active), k″s,20 = 0.6 au/min, cannot be expressed in nanomolar per minute until we know the average concentration of Cdc20 in an asynchronous culture, which should correspond to 0.7 au in the model (the average value of [Cdc20]T over a full cycle). The equilibrium binding constant for Clb2 and Sic1 is kas,b2/kdi,b2 = 103 au-1 = 25 nM-1, whereas the binding constant for Cdc14 and Net1, kas,rent/kdi,rent = 200 au-1, cannot be expressed in nanomolar-1 until we know the concentration of Net1 or Cdc14 in a cell. Because Net1 and Cdc14 are expressed in the same au, the ratio [Net1]T/[Cdc14]T = 1.4 is a meaningful prediction of the model: that Net1 is in slight excess (40%) over Cdc14. On the other hand, because Tem1 and Cdc15 are expressed in different au, the ratio [Tem1]T/[Cdc15]T = 1 tells us nothing about the relative amount of these proteins in a cell.
Timing of Cell Cycle Events
Much of the quantitative data available to us refers to the timing of bud emergence, onset of DNA synthesis, and cell separation. To link the output of the model (the temporal evolution of e.g., [Cln2], [Clb5], [Clb2], [Cdc14]) to these events, we use auxiliary variables called [BUD], [ORI], and [SPN].
[BUD] represents proteins that are phosphorylated by Cdc28/cyclin dimers and subsequently initiate a new bud when the phosphorylation state reaches a threshold, [BUD] = 1. The rate of phosphorylation, ks,bud(εbud,n2[Cln2] + εbud,n3[Cln3] + εbud,b5[Clb5]), reflects the fact that bud emergence can be driven by any of the Clns or by Clb5/6 (Cvrckova and Nasmyth, 1993). The values assigned to the ε bring the timing of bud emergence in line with observations in mutants lacking various combinations of these cyclins.
In a similar manner, [ORI] = 1 signals the onset of DNA synthesis. At that time, we invoke the DNA replication/spindle assembly checkpoint by activating Mad2 and Bub2, thereby preventing activation of Cdc20 and Tem1, and keeping cells from mitotic exit.
The checkpoint is lifted when [SPN] = 1, which represents alignment of all chromosomes on the metaphase plate. We assume that exit from mitosis (telophase, cell separation) requires the elimination of Clb2-dependent kinase activity (Zachariae and Nasmyth, 1999) below a threshold value (Kez = 0.3)
At exit from mitosis, [BUD] and [SPN] are reset to zero. [ORI] is reset to zero only if [Clb2] + [Clb5] drops below another threshold (Kez2 = 0.2), which is our criterion for relicensing origins of replication (Li, 1995; Su et al., 1995).
To account for inviability of the triple-cln mutant (cln1Δ cln2Δ cln3Δ), to be described in RESULTS, we are led to assume that when cells grow too large they may not successfully initiate DNA synthesis, and they will be G1 arrested. In the model, we require that a cell must reach [ORI] = 1 before a wild-type cell in the same growth medium has divided twice; if not, the cell is considered G1 arrested, even if [ORI] = 1 sometime later.
Mass at Division
Another commonly observed property of cells is their size at division. An important variable in our model is [mass]. Typically, we record the value of [mass] at cell division in wild-type and mutant cells and compare it with observations such as “these mutant cells are roughly twice the size of wild-type cells.” The differential equation for [mass] implies that cells grow exponentially, with mass doubling time (MDT) = 0.693/kg, where kg is the specific growth rate. (MDT = 90 min on glucose medium and 150 min on galactose medium.) When a cell exits mitosis, we divide [mass] asymmetrically between mother and daughter cells, according to a rule described in Chen et al. (2000).
Notice that [mass] enters the dynamical system as a multiplier of the rates of synthesis of the cyclins. It is based on the assumption, which was made in Chen et al. (2000) as well, that cyclins are synthesized at a rate proportional to the total number of ribosomes in a cell (which is proportional to cell size) and then accumulate in a constant-volume compartment of the cell (the nucleus). Hence, [Cln3], [Cln2], [Clb5], and [Clb2] represent the concentrations of the cyclins in the nucleus. This assumption (rate of cyclin synthesis proportional to mass) is the simplest mechanism we know to coordinate the growth cycle to the chromosomal replication cycle, so that average cell size is maintained generation after generation (Futcher, 1996).
Because Sic1 and Cdc14 (Shou et al., 1999; Edgington and Futcher, 2001) are found in both nuclear and cytoplasmic compartments, their synthesis rates are not multiplied by [mass]. No additional assumptions are made on the nuclear localization of other components in the regulatory network (e.g., Swi5, Cdc20 are not assumed to be exclusively nuclear). As described in DISCUSSION, as more experimental data become available (Huh et al., 2003), we will incorporate subcellular localization of proteins in a later version of the model.
Computer Simulation
Using a computer program (http://www.math.pitt.edu/~bard/bardware/binary/winpp.zip) freely available from G. Bard Ermentrout (Department of Mathematics, University of Pittsburgh, Pittsburgh, PA), we solve the equations in Table 1, given the parameter values in Table 2 (which are appropriate for a wild-type cell), for the explicit time dependence of each variable ([Cln2](t), [Clb2](t),...). To compute a solution numerically, we also must assign initial conditions (at t = 0) to all the variables ([Cln2](0), [Clb2](0),...). Initial conditions should reasonably represent an experimental protocol, e.g., the values in Table 2 might represent conditions in a small wild-type cell selected from an elutriating rotor (newborn daughter cell with origin of replication relicensed), but their precise values are not important. The simulation was done for cells growing in culture with MDT = 90 min. Under the asymmetric-division rule of Chen et al. (2000), we give 0.4587 of the mass-at-division to the daughter, and 0.5413 to the mother; hence, the cycle time for the daughter is 101.2 min and that for the mother is 80.0 min.
To simulate each mutant, we use exactly the same equations (Table 1) and parameter values (Table 2) except for those parameter changes that are dictated by the nature of the mutation. In the simplest case (gene X is deleted), the rate of synthesis of protein X is set to zero. Overexpression is more complicated, because a gene may be overexpressed in different ways: from a plasmid or from genomically integrated copies, with its own or a foreign promoter. In the case of multiple integrated copies under control of the natural promoter, we simply multiply the appropriate k′s and k″s parameters by an integer, depending on the number of additional copies. If gene X is constitutively overexpressed (typically using the GAL promoter), then we increase k′s,x (the constant rate of synthesis of protein X), and we change the specific growth rate kg to match the new growth medium. The new value assigned to k′s,x is somewhat arbitrary, because the rates of expression of proteins from such constructs are unknown and vary for different proteins. For each GAL-construct, we choose a value of k′s,x that gives results consistent with phenotypes of all mutants containing this particular GAL-construct (e.g., all mutants containing GAL-CLB2 are assigned the same value of k′s,b2 but k′s,b5 may be assigned a different value to model GAL-CLB5).
For temperature-sensitive (ts) mutants in Table 3, we assume that the relevant catalytic activity is normal at the permissive temperature and zero at the restrictive temperature. To model “synthetic lethality” of ts alleles (where the double mutant gene1ts gene2ts is inviable at a certain temperature originally permissive to the two single mutants), we assume that the relevant rate constants for gene 1 and gene 2 are reduced to a fraction of the wild-type values at that intermediate temperature.
Assay of Model Robustness
To determine model robustness, the model was transferred to MatLab version 6.5 by using the ode23s integrator. The following rules were then applied to establish whether a given parameter set yielded a “viable” solution. First, it was required that the model executes the following events in order: origin relicensing (due to a drop in [Clb2] + [Clb5] below Kez2); origin activation (due to a subsequent rise in [Clb2] + [Clb5], causing [ORI] to increase above 1); spindle alignment (due to a rise in [Clb2], causing [SPN] to increase above 1); Esp1 activation (the [Esp1] variable going through 0.1, because of Pds1 proteolysis); and finally [Clb2] dropping below a threshold Kez to trigger division. If these “events” did not occur in this order the model was considered inviable. If division occurred in an “unbudded cell” (i.e., when [BUD] < 1), this also was considered inviable. The model also was required to meet a rough criterion for a periodic solution, that the root mean square deviation of all variables was <0.05 at subsequent divisions. Last, if the cell [mass] was >10, this was considered to be an inviable model.
Given these rules, MatLab code was written to systematically vary each of the parameters in the model up to 256-fold up and down, with 1.414-fold increments, and the maximum tolerable variation was recorded for each parameter in each direction. Importantly, only single-parameter variations were tested. Whereas formally this procedure does not establish a “volume” in parameter space, it still gives a sense of the robustness of the model. An accurate and meaningful volume determination runs into difficulties that we have not yet solved; therefore, at present, we are relying on this empirical test.
A similar test was run on several mutants: APC-A (k″a,20 = 0; or equivalently ka,apc = 0); cdh1Δ (k″d,b2 = 0.001, k″d3,pds = 0.0001; for unknown reasons, the k″d,b2 = 0 model caused an error with the Matlab integrator and so the “trivial” value of 0.001 was used instead), ckiΔ (e.g., all synthesis, association values for Sic1 and Cdc6 set to zero), cln3Δ, cln2Δ, clb5Δ (synthesis values for the relevant cyclins set to zero).
In a similar manner, the robustness of an inviable mutant (e.g., APC-A cdh1Δ) was assessed by searching for single-parameter increase or decrease that yielded a cycling model from an initially noncycling parameter set. Matlab code and complete data on parameter sensitivity runs are available on request (fcross@mail.rockefeller.edu).
RESULTS
The Model Accurately Describes the Growth and Division of Wild-Type Cells
As described in MATERIALS AND METHODS, we solve the equations in Table 1, given the parameter values and initial conditions in Table 2 for a wild-type budding yeast cell. In Figure 2, we illustrate how cell size, cyclin concentration, and other components vary during repetitive cycling of daughter cells. The computed properties of the model agree reasonably well with the observation of Brewer et al. (1984): for a wild-type diploid A364A D5 strain, growing at MDT = 90 min, the daughter cell cycle time is 97.5 min (computed value = 101.2 min), G1 length is 42 (36) min, and S/G2/M length is 57 (64) min, whereas the mother cell cycle time is 81 (80) min, G1 length is 22 (28) min, and S/G2/M is 59 (52) min.
Figure 2.

Wild-type cell cycle. Numerical solution of the differential equations in Table 1, for the parameter values in Table 2. The MDT for an asynchronous culture is 90 min. We show the cycle of a daughter cell (cycle time, 101 min; duration of G1, 36 min). The cycle time for a mother cell (not shown) is 80 min. Division is slightly asymmetric (daughter size at birth = 0.46× mother size at division). During G1 phase, Cdh1 is active and there are abundant CKIs. The G1→S transition is driven by accumulation of Cln2. The M→G1 transition is driven by activation of Cdc20. In panel 4, the left ordinate refers to [Cln2] and the right ordinate to [Cdc20] and [Cdc14].
Furthermore, the relative amounts of certain groups of proteins are in rough quantitative agreement with recent measurements by Cross et al. (2002) and Archambault et al. (2003). The ratios, for an asynchronous culture with MDT = 90 min, are as follows:
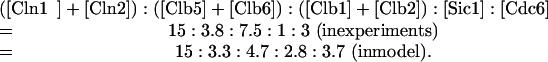 |
The predicted ratios are determined by integrating each variable over a full cycle of both a mother cell and a daughter cell, dividing these numbers by the corresponding cycle times of mother and daughter cells, and then averaging these two numbers (because there are roughly equal numbers of mother and daughter cells in rapidly growing populations).
The most significant discrepancy between the model and experiment is the ratio of [Sic1] to ([Clb1] + [Clb2] + [Clb5] + [Clb6], 1:11 in experiment and 1:3 in model. Cross's measurements indicate that rapidly growing cells have much less Sic1, on average, than would be expected from the model. By decreasing the rate of synthesis of Sic1, we could get better agreement with the experiments for wild-type cells. But the model must satisfy many other constraints implied by the phenotypes of various mutants. In particular, in the model the triple-cln deletion strain (cln1Δ cln2Δ cln3Δ) would become viable, contrary to observations, if the rate of synthesis of Sic1 were reduced by half. In our simulation of the triple-cln mutant, if Sic1 concentration is too low, then Clb5 activates its own synthesis via SBF/MBF, and the simulated cell is perfectly viable by all our criteria. We do not have any explanation for this discrepancy in Sic1 level between the model and the experiment.
The parameter values in Table 2 have been chosen taking into account the properties of wild-type and mutant cells, and they represent a compromise of many, often competing, observations. To this end we must settle on a “data set” of mutants (Table 3) that will serve as a testing ground for the sufficiency of the model. The parameter values proposed in Table 2 have been selected by a painstaking process of trial-and-error to provide a suitable fit to the full data set.
The Model Conforms to the Phenotypes of >100 Mutant Strains
The wiring diagram in Figure 1 has been composed from evidence provided by the phenotypes of dozens of budding yeast mutants that have been constructed and characterized by deleting or overexpressing each genetic component singly and in multiple combinations. It remains an informal “cartoon” of the molecular regulatory system until it is converted into a precise mathematical model and demonstrated to be consistent with most of the facts about budding yeast mitotic division. In Table 3, we list 131 mutants that have been used to test the model.
For each mutant simulation, we use exactly the same equations (Table 1) and parameter values (Table 2), and we are allowed to change only those parameters that are governed by the nature of the mutation, as described in MATERIALS AND METHODS. We compare the computed behavior of the model with the observed phenotype of the cells. For example, if the mutant is inviable, at what phase of the cell cycle is it blocked? If viable, in what subtle ways does it differ from wild-type: size at onset of DNA synthesis, size at bud emergence, size at division, and duration of G1 phase?
In most cases, 120 of 131 mutants in Table 3, the model agrees well with observations; the 11 exceptions are marked with asterisks in the table.
The Model Is Fully Described on a Web Page
At our Web site, http://mpf.biol.vt.edu, full details about the model and all simulations can be found. On this site, we summarize the basic experimental results on which the wiring diagram (Figure 1) is based. We present simulations of all mutants in Table 3, including the precise parameter values used in each case. We provide facilities for the user to repeat any simulations by using our parameter set or any new choice of parameter values. We also give instructions for how one may modify the wiring diagram, build a revised model, and run new simulations.
How the Model Represents APC Phosphorylation and the FEAR Pathway
In earlier models of cell cycle regulation in frog eggs (Novak and Tyson, 1993) and fission yeast (Novak et al., 2001), we proposed the existence of an intermediary enzyme (IE) whose purpose was to introduce a time delay between the activation of cyclin B-dependent kinase at the onset of M phase and the activation of Cdc20 (Fizzy in frog eggs and Slp1 in fission yeast) at the metaphase-to-anaphase transition. There are good reasons to suspect that IE is the APC core complex itself (Cross, 2003). First, deletion of IE would cause cells to arrest in metaphase, as is the case for most components of the APC. Rudner and Murray (2000) showed that three components of the APC core, Cdc16, Cdc23, and Cdc27, are phosphorylated by mitotic Cdc28 kinase and that only the phosphorylated forms associate with Cdc20 effectively. They also showed that APC-A mutant cells (where all the possible Cdc28-phosphorylation sites in Cdc16/23/27 are removed by substituting alanine for serine/threonine residues) are viable, with a delay in mitotic exit. This phenotype is consistent with the identification of IE with APC, if we assume that the unphosphorylated form of APC retains some intrinsic ability to activate Cdc20 (simulations not shown). Hence, in the present model, the phosphorylated, active form of IE in our earlier models (IEP) is replaced by APC-P, the phosphorylated state of APC that is necessary for full activity in conjunction with Cdc20. Notice that APC need not be phosphorylated to function in conjunction with Cdh1 (Kramer et al., 2000; Rudner and Murray, 2000). The present model is not fully correct in that IEP/APC-P is treated as if it were an enzymatic activator of Cdc20 instead of a binding partner. This problem will be corrected in future versions of the model, by revising the wiring diagram and the differential equations. These changes will undoubtedly require minor revisions of the rate constants, but we do not expect any significant changes in the conclusions reached in this article.
The sequential activation of Cdc20 and Cdh1 suggests that Cdc20/APC-P degrades, directly or indirectly, an inhibitor of Cdh1 (Morgan, 1999). The inhibitor must act before Cdc14 release from the nucleolus. If the Cdc20-sensitive inhibitor were to act downstream of Cdc14 release, then in the cdc20Δ strain, Cdc14 would be released by MEN action as soon as the spindle assembly checkpoint is satisfied, which is contrary to observation (Shirayama et al., 1999). Pds1 is an obvious candidate for this inhibitor: it is degraded by Cdc20 (Yamamoto et al., 1996b), and overexpression of nondegradable Pds1 delays Clb2 degradation for several hours (Cohen-Fix and Koshland, 1999). But how might Pds1 degradation relate to Cdc14 release? Cdc14 is released from the nucleolus in two stages (Visintin et al., 2003): an early, transient stage, via the FEAR pathway (Uhlmann et al., 1999; Stegmeier et al., 2002; Yoshida et al., 2002), involving Esp1 and Cdc5; and a late, sustained stage, involving Cdc15 and other MEN components. By binding and inhibiting Esp1, Pds1 inhibits the FEAR pathway, preventing Cdc14 release (FEAR stands for Cdc-fourteen early anaphase release).
At the time we were formulating this model, the information just cited was not known. In its place, we hypothesized a phosphatase (PPX) that inhibits Cdc14 release by opposing the action of Cdc15 on Net1. The role of Pds1 was to keep PPX level high by inhibiting its degradation by Cdc20/APC-P. Hence, in our model, Pds1 up-regulates PPX, an inhibitor of Cdc14 release. According to the FEAR story, Pds1 down-regulates activators of Cdc14 release. With appropriate choice of kinetic constants, the dynamic consequences of the two formulations should be nearly identical. In a future version of the model, we will replace PPX by a representation of the FEAR pathway.
The Model Is Based on Alternative Stable Steady States Corresponding to G1 and S/G2/M Phases of the Cell Cycle
Although the rigor and precision of the model are essential attributes in its favor, the sheer magnitude of information that comes out of the computer can overwhelm the user. To make sense of this information, we need intuitive ways to understand the model's behavior—an intuition disciplined by precise numerical simulations of the equations. We rely on the scheme in Figure 3 (described briefly in Chen et al., 2000). Fundamental to cell cycle control in budding yeast are the antagonistic relations between B-type cyclins (Clb1–6, in association with Cdc28), which promote DNA synthesis and mitosis, and G1-stabilizers (Cdh1, Sic1, and the cyclin-dependent kinase inhibitor [CKI]-role of Cdc6), which oppose cell proliferation by degrading Clbs and stoichiometrically inhibiting Clb/Cdc28 complexes (For the CKI-role of Cdc6, see Greenwood et al., 1998 and Calzada et al., 2001). We shall refer to Sic1 and Cdc6 together as the CKIs.
Figure 3.

Logic of cell cycle transitions in budding yeast. (A) Antagonistic interactions between the G1-stabilizers (Cdh1 and CKI) and the Clb/Cdc28 kinases create two coexisting stable steady states, G1 and S/G2/M. Transitions between these states are called start (G1→S) and finish (M→G1). (B) start is facilitated by Cln/Cdc28 kinases. Cell growth (“mass”) triggers accumulation of Clns. (C) finish is facilitated by Cdc20/APC. Mitotic checkpoint signals restrain the activation of Cdc20.
Because Clb-dependent kinases can inactivate Cdh1 (for review, see Zachariae and Nasmyth, 1999) and destabilize CKIs (Verma et al., 1997), these two classes of proteins are mutual antagonists (Figure 3). The model is designed to have two coexisting, self-maintaining steady states: a G1 state, when Clbs are scarce and their antagonists (Cdh1 and CKIs) are abundant; and an S/G2/M state, when the reverse is true (Nasmyth, 1996; Novak and Tyson, 1997; Novak et al., 1998; Tyson and Novak, 2001; Tyson et al., 2001). When yeast cells are proliferating, the control system is undergoing periodic transitions from the G1 state to the S/G2/M state and back again. These transitions (called start and finish) are irreversible and alternating. Once a cell has executed start, it does not normally slip back into G1 phase and does start again. Rather, it must execute a distinctly different transition (finish) to return to G1. Likewise, a cell that has executed finish does not slip back into mitosis and try to separate its chromosomes a second time. There are exceptions to these rules (endoreplication and meiosis), but they do not nullify the central role played by irreversible, alternating start and finish transitions in the cell cycle.
To a first approximation, we view the budding yeast cell cycle as an alternation between these two stable steady states generated by the antagonism between Clb kinases and G1-stabilizers. From the simulation of the wild-type cycle (Figure 2), one can see how the control mechanism shifts from one state to the other, and how the transitions are carried out.
The start transition is facilitated by Cln1,2/Cdc28 complexes, which can phosphorylate and inactivate Cdh1 and CKIs, but they themselves are unopposed by the G1-stabilizers (for reviews, see Schwob et al., 1994 and Peters, 1998). This transition occurs when the cell has grown large enough to accumulate a critical concentration of Cln3-dependent kinase in the nucleus (Miller and Cross, 2000; Cross et al., 2002). Cln3 kinase and a back-up (Bck2) activate SBF and MBF, the transcription factors for Cln1,2 and Clb5,6, so their levels increase. Clb5,6-dependent kinases are inactive due to inhibition by the CKIs, but Cln1,2-dependent kinases are not so inhibited. Cln-dependent kinases depress Cdh1 and CKIs enough to allow the Clb-dependent kinases to assert themselves, switching the control system into the stable S/G2/M state. Once the transition is made, Clb kinases by themselves are able to depress their antagonists without the help of Cln1,2 kinases. Rising activity of Clb1,2/Cdc28 turns off Cln1,2 synthesis, causing Cln-dependent kinase activity to drop. Hence, after doing their job, the start-facilitators disappear.
Cdc20/APC facilitates the finish transition. Cdc20 transcription is activated in G2/M phases by the transcription factor complex Mcm1/Fkh2/Ndd1 (Spellman et al., 1998; Zhu et al., 2000; Simon et al., 2001), which is activated in turn by Clb1,2 kinase activity. In addition, APC core proteins (Cdc16, 23, and 27) are phosphorylated by Clb1,2 kinase, which facilitates APC binding with Cdc20 to form an active complex (Rudner and Murray, 2000). Cdc20/APC-P depresses Clb kinase activity by labeling Clbs for degradation; it also initiates activation of a phosphatase, Cdc14, which reverses the inhibitory effects of Clb/Cdc28 on Cdh1 and CKIs, so the latter two can overpower the Clb kinases. As the G1-stabilizers extinguish Clb kinase activity, the transcription factor Mcm1 turns off and Cdc20 synthesis ceases. Because Cdc20 is an unstable protein, it quickly disappears, preparing the cell for the subsequent start transition.
To consider G1 and S/G2/M as two alternative steady states, however, is an oversimplification. After start, Clb-dependent Cdc28-kinase activity rises in at least two stages. First, a moderate activity, dependent primarily on Clb5,6, is responsible for initiating DNA synthesis and inactivating Cdh1. Then, a high activity, dependent primarily on Clb1,2, is responsible for driving congression of replicated chromosomes to the metaphase plate. During finish, Clb-dependent Cdc28-kinase activity drops in at least two stages. The initial drop in activity is dependent primarily on Cdc20-dependent degradation of Clb1–6, and it coincides with Pds1 degradation and subsequent separation of sister chromatids (anaphase). As mentioned in the previous section, the disappearance of Pds1 also results in the degradation of PPX (or equivalently, activation of the FEAR pathway), which results in activation of Cdh1 and a second, deeper drop in Clb1,2. The drop in Clb-dependent Cdc28-kinase activity, together with the sustained release of Cdc14-phosphatase from RENT complexes, seems to be responsible for the final stages of exit from mitosis: nuclear division (telophase), cell division, and relicensing origins of DNA replication.
S/G2/M is not a unitary state; mutants reveal states of intermediate and high activities of Clb-dependent Cdc28 kinase. For example, clb1Δ clb2Δ cells arrest in G2 phase with moderate Clb-dependent kinase activity; CLB2dbΔ cells (Clb2 protein lacks “destruction box” sequences necessary for Cdc20-mediated proteolysis) arrest in telophase with very high Clb kinase activity; and cdc14ts cells (which activate Cdc20 but not Cdh1) arrest in telophase with an intermediate activity of Cdc28 kinase, due mostly to Clb1,2.
How Can Mutant Cells Lacking start or finish Facilitators Be Rescued?
The transitions from G1 to S/G2/M and back again, which we refer to as start and finish, are induced by helper proteins Cln2 and Cdc20 as described above. The helpers are involved in negative feedback loops, i.e., the processes they induce lead to their own demise. Rising Clb2 activity after start turns off Cln2 synthesis, and falling Clb2 activity after finish turns off Cdc20 synthesis. Because Cln2 and Cdc20 are unstable proteins, they disappear rapidly after their job is done. We describe next how the inviability of various mutants lacking the helpers is related to this central scheme and how they can be rescued.
Mutations involving the facilitators of start and finish have striking phenotypes. For example, in the absence of Cln-dependent kinase activity (cln1Δ cln2Δ cln3Δ, “triple-cln” for short), start cannot occur and cells arrest in G1 phase (Wittenberg et al., 1990). In the simulation (Figure 4A), start is delayed many hours, but eventually the cell grows large enough (in the model) for Clb5-dependent kinase activity to drive [ORI] to 1. This is a serious problem for the model. To account for inviability of the triple-cln mutant, in this model we assume that a cell must reach [ORI] = 1 before a wild-type cell in the same growth medium has divided twice; if not, the cell is considered G1 arrested. Although this rule is introduced specifically to account for the phenotype of triple-cln cells, it is applied uniformly to all mutants to decide whether the simulated cells are viable or G1 arrested.
Figure 4.

Mutations that interfere with the start and finish transitions. (A) Deletion of all three CLN genes arrests cells in G1 because the start-facilitators are missing. (B and C) The triple-cln mutant is rescued by further deletion of SIC1, but not by deletion of CDH1.(D–F) CDC20 mutations (deletion or temperature-sensitive lethal) block cells in metaphase (simulation not shown) can be rescued by deleting both PDS1 and CLB5, but not by deleting either gene alone. (G, H) Deletion of all G1-stabilizers makes inviable cells, and they can be rescued by GALL-CDC20. *In reality, the mutant in panel G is not telophase arrested as predicted by the model. See text for a description of its phenotype and possible modifications of the model to account for the discrepancy.
In the model, deletion of either SIC1 or CDH1 allows triple-cln mutant cells to undergo start (Figure 4, B and C) before transgressing the just mentioned rule about G1 arrest. The former construct is viable in the model and in reality (Tyers, 1996). The latter, though able to replicate its DNA (just barely, according to the “rule”), gets stuck in telophase. Schwab et al. (1997) studied the response of cdh1Δ cells to pheromone treatment (analogous to triple-cln deletion). Although the cells were sensitive to pheromone (i.e., they did not form colonies), they did not arrest uniformly in G1. A fraction of the cell population proceeded through S phase and arrested with 2C DNA content. Considering that the simulated cells just barely satisfy our condition for entering S phase, we might expect, in a population of real cells that vary around the mean simulated behavior, some cells will arrest with 1C DNA content and some with 2C DNA content, as observed.
In triple-cln sic1Δ cells, what molecules initiate the start transition? These cells contain modest amounts of Clb5 in G1 phase ([Clb5] = k′s,b5/k′d,b5 = 0.08 au). Because Sic1 is missing and Cdc6, we assume, is a poor inhibitor of Clb5/Cdc28, Clb5-dependent kinase activity is not inhibited. Together with Bck2, Clb5 activates SBF and MBF when cells are large enough. At this point, Clb5-kinase activity rises sharply and initiates DNA synthesis. Clb5 also inhibits Cdh1, enabling Clb2 to accumulate and the division cycle to progress normally.
In the case of cln1Δ cln2Δ clb5Δ clb6Δ, all the possible helpers for start (except for Cln3) are eliminated, and the cell arrests in G1 (Schwob and Nasmyth, 1993). In the model, this mutant can be rescued only be restoring CLN2 or CLB5. Deletion of Sic1 (or CKI altogether) is not enough for rescue, because Cdh1 is active, and it keeps the cell arrested in G1. Deletion of Cdh1 (or addition of GAL-CLB2) allows DNA synthesis to initiate and exit of mitosis to occur, but these quintuple mutant cells are predicted to be inviable because they do not form buds (See Figure S1 in Online Supplement A).
Exit from mitosis is initiated by Cdc20, and, as expected, cdc20Δ is lethal (Lim et al., 1998; Shirayama et al., 1998), with cells arrested in metaphase. Cdc20 plays a role in the degradation of Clb2, Clb5, Pds1 (Yamamoto et al., 1996a; Shirayama et al., 1999; Yeong et al., 2000) and our hypothetical PPX. The double mutants cdc20Δ pds1Δ and cdc20Δ clb5Δ arrest in telophase and metaphase, respectively (Figure 4, D and E), but the triple mutant cdc20Δ pds1Δ clb5Δ is viable (Shirayama et al., 1999; Figure 4F).
What is the helper for the finish transition of the triple mutant cdc20Δ pds1Δ clb5Δ? Shirayama et al. (1999) have shown that APC/Cdh1 activity is essential for the viability of this mutant. In the model, the negative feedback loop responsible for exiting mitosis is the following. Clb2 activates chromosome alignment on the mitotic spindle (SPN). When SPN = 1, Bub2 is inhibited and Cdc15 is activated, which in turn activates Cdc14 and Cdh1, causing Clb2 level to decrease. The CKIs are restored, and the cell returns to G1. This interpretation predicts that bub2Δ would render cdc20Δ pds1Δ clb5Δ inviable: because the negative feedback is broken, the quadruple mutant would arrest in the G1 phase.
How Can Mutant Cells Lacking G1-Stabilizers Be Rescued?
Removing G1-stabilizers (Cdh1 and/or CKIs) causes problems at start and finish.
Although cdh1Δ and sic1Δ are both viable, they show synthetic lethality with viable mutations that have higher than normal Clb2-kinase activity. For example, both cdh1Δ and sic1Δ show synthetic lethality with cdc14ts at 29°C (Yuste-Rojas and Cross, 2000).
Deletion of all three G1-stabilizers (sic1Δ cdc6Δ2-49 cdh1Δ, “triple-antagonist” for short) abolishes the stable G1 steady state. cdc6Δ2-49 encodes a truncated Cdc6 protein that has lost its role as a CKI but retains its role as a DNA licensing factor. In simulations, the triple-antagonist arrests in telophase, with intermediate activity of Clb2-kinase (Figure 4G). However, in reality, although these cells are inviable (Cross, 2003), they are not arrested in telophase (Archambault et al., 2003). In the latter report, the authors showed that these cells have an unusual phenotype, they are able to undergo DNA synthesis and nuclear division but not cell division, and arrest as a “four-cell body object” with 4C DNA content. (We will describe in more detail below the problems of our model with this mutant.)
Although the model is not able to describe the phenotype of the triple-antagonist correctly, it is able to simulate the rescue of this mutant by overproduction of Cdc20 (with GALL-CDC20) as reported by Cross (2003) (their Supplementary Figure 4). In this case, Cdc20 serves as the G1-stabilizer. Because it is synthesized constitutively, Cdc20 does not disappear from these cells as Clb2 is degraded during finish (Figure 4H). SBF and MBF turn on as usual, but Clb5 does not accumulate because it continues to be degraded by Cdc20. The cell is delayed in G1 until Clb5 kinase eventually triggers DNA synthesis. At that point, Cdc20 is inactivated by Mad2, allowing Clb2 to rise and drive the cell into mitosis. When chromosomes are aligned, [SPN] = 1, the inhibition on Cdc20 is removed, and Clb2 is degraded, returning the cell to G1. This interpretation predicts that mad2Δ would render sic1Δ cdc6Δ2-49 cdh1Δ GALL-CDC20 inviable.
The Model Predicts Phenotypes of Novel Mutants
As we have already pointed out with regard to (cln1Δ cln2Δ clb5Δ clb6Δ cdh1Δ), (cdc20Δ pds1Δ clb5Δ bub2Δ), and (sic1Δ cdc6Δ2-49 cdh1Δ GALL-CDC20 mad2Δ), we can use the model to predict phenotypes of mutants that have not yet been investigated experimentally, to test crucial features of the model.
For example, we assume that the Cdc6 is a much weaker inhibitor of Clb5,6 kinases than is Sic1 and that Clb5,6 kinases are not able to phosphorylate and destabilize Cdc6 as efficiently as Clb1,2 kinases. The first assumption that Cdc6 is a weak inhibitor of Clb5 kinase is based on the following observations. 1) sic1Δ cells have very short G1 period (Schneider et al., 1996), initiating DNA synthesis before SBF/MBF activation and budding. For DNA replication to occur early in those mutant cells, the high concentration of Cdc6 in G1 must not be able to inhibit even the low level of Clb5,6 kinases generated by MBF-independent transcription. Hence, Cdc6 cannot be a strong inhibitor of Clb5,6. 2) GAL-CLB5 cells do not show advancement in DNA synthesis (Schwob et al., 1994), indicating that Clb5/Cdc28 must be effectively inhibited by Sic1. This assumption was made independently of experiments published recently by Archambault et al. (2003), who showed that Sic1 coimmunoprecipitates with Clb5 but Cdc6 does not.
The second assumption, that Clb5 kinase is less able to phosphorylate and inactivate Cdc6, is inferred from the large size of cln1Δ cln2Δ cln3Δ sic1Δ cells. As described in 1), Clb5,6 kinases are able to initiate DNA synthesis early and to inactivate Cdh1 in the quadruple mutant, but Clb2 is still inhibited by Cdc6. The cell has to grow to very large size to accumulate enough Clb5 to inactivate Cdc6. Only then can Clb2 kinase activity rise, driving the cell into mitosis.
If it is true that Cdc6 is a weaker inhibitor to Clb5 kinase than is Sic1, then whenever the activity of Clb5 kinase is important, sic1Δ will have very different effects than cdc6Δ2-49. For example, triple-cln sic1Δ is viable (Tyers, 1996), but the model predicts that triple-cln cdc6Δ2-49 will remain arrested in G1. Similarly, the model predicts (Table 4, row i) that the inviability of GAL-CLB5-dbΔ (Jacobson et al., 2000) can be rescued by overproducing Sic1 but not Cdc6. On the other hand, because Sic1 and Cdc6 are both strong inhibitors of Clb2, the inviability of Clb2-dbΔ (Wasch and Cross, 2002) should be rescued by overproduction of either Sic1 or Cdc6 (Table 4, row ii), as confirmed recently by Archambault et al. (2003).
Table 4.
Prediction of mutant phenotypes
| Genotype | Phenotype | |
|---|---|---|
| i | GAL-CLB5-dbΔ | Inviable, inefficient origin licensing (Jacobson et al., 2000) |
| GAL-CLB5-dbΔ 2×SIC1 | Viable, size similar to WT (prediction) (Figure S1 a) | |
| GAL-CLB5-dbΔ GAL-SIC1 | Viable, size bigger than GAL-CLB5-dbΔ 2×SIC1 (prediction) | |
| GAL-CLB5-dbΔ GAL-CDC6 | Still inviable (prediction) | |
| ii | CLB2-dbΔ | Arrests in telophase (Wasch and Cross, 2002) |
| CLB2-dbΔ GAL-SIC1 | Viable (Cross, 2003) | |
| CLB2-dbΔ GAL-CDC6 | Viable (prediction) | |
| iii | cdc20Δ pds1Δ clb5Δ | Viable (Shirayama et al., 1999) |
| triple-deletion + 1.5×NET1 (or 0.5×CDC15, or 0.5×CDC14) | Arrests in telophase (prediction) | |
| triple-deletion + bub2Δ | Arrests in G1 (prediction) | |
| triple-deletion + sic1Δ (or cdc6Δ2-49, or both) | Viable (prediction) (Figure S1 b) | |
| iv | cdc20Δ pds1Δ | Arrests in telophase (Shirayama et al., 1999) |
| cdc20Δ pds1Δ TAB6-1 | Viable (prediction) (Figure S1 c) | |
| cdc20Δ pds1Δ 2×CDC15 | Viable (prediction) | |
| v | APC-A cdh1Δ | Inviable in glucose (Cross, 2003) |
| APC-A sic1Δ | Viable (Cross, 2003) | |
| APC-A sic1Δ cdc6Δ 2-49 | Viable (prediction) | |
| vi | sic1Δ cdc6Δ2-49 cdh1Δ GALL-CDC20 | Viable (Archambault et al., 2003) |
| quadruple mutant + mad2Δ | Inviable, G2 arrest (prediction) | |
| vii | net1ts in nocodazole | Loses viability (Visintin et al., 1999) |
| net1ts 2×CLB5 (or 2×CLB2) in nocodazole | Retains viability (prediction) (Figure S1 d) | |
| net1ts 2×CLN2 (or 2×CLN3) in nocodazole | Loses viability (prediction) | |
| net1ts sic1Δ (or cdc6Δ2-49) in nocodazole | Loses viability (prediction) (Figure S1 e) | |
| viii | cln1Δ cln2Δ clb5Δ clb6Δ | Inviable, arrests in G1 (Schwob and Nasmyth, 1993) |
| quadruple mutant + cdh1Δ (or + GAL-CLB2) | Able to do DNA synthesis and mitosis, becoming dinucleate because no bud formation, similar to cdc24ts (prediction) (Figure S1 f) |
Simulations of some of these mutants are shown in Online Supplement A, Figure S1.
Viability of the triple mutant cdc20Δ pds1Δ clb5Δ (Shirayama et al., 1999) depends on a feedback loop that activates Cdh1 at the end of the cycle. If we perturb elements in this loop (Table 4, row iii), we are likely to render cdc20Δ pds1Δ clb5Δ inviable. On the other hand, the viability of cdc20Δ pds1Δ clb5Δ does not depend on CKIs.
As shown in Figure 3, Cdc20 helps the finish transition by degrading Clb kinases and by activating the CKIs and Cdh1 (through Cdc14). Hence, in row iv (Table 4), the model predicts that inviability of the double mutant cdc20Δ pds1Δ (telophase arrest, with Cdc14 released from the nucleolus; Shirayama et al., 1999) could be rescued by an extra copy of genomic CDC15. The double mutant also can be rescued by TAB6-1 (a dominant Cdc14 mutation where Cdc14 binding to NET1 is reduced; Shou et al., 2001). In TAB6-1 cells, less Cdc14 is sequestered in the nucleolus, making it easier for the triple mutant (cdc20Δ pds1Δ TAB6-1) to exit from mitosis even in the presence of Clb5.
Row v (Table 4) shows when Cdc20 is crippled, as in APC-A mutants, Cdh1 is more important than the CKIs in getting out of M phase. Row vi (Table 4) suggests that ability of Cdc20 overexpression to rescue the lethality of the triple-antagonist critically depends on the checkpoint mechanism. Row vii (Table 4) indicates that net1ts can retain viability in nocodazole if Clbs are overexpressed but not if CKIs are deleted.
The Model Predicts Effective Values of Rate Constants
Table 2 makes ∼100 predictions about the rates of component biochemical processes involved in cell cycle control, e.g., synthesis and degradation rates, phosphorylation and dephosphorylation rates, relative activities of cyclins with overlapping substrate specificities. Some of these rates are well established experimentally (e.g., protein half-lives are easily measured), whereas most others were completely unknown. As biochemists seek to measure these rate constants and binding constants directly, our estimates will serve as landmarks for relating in vitro measurements to in vivo activities. Some of these predictions are described in detail in Online Supplement A, along with a few supporting observations.
Inconsistencies between Simulations and Observations Identify Problems in the Model
Although the model gives a quantitatively accurate representation of many aspects of the budding yeast cell cycle, it fails to account for certain properties of 11 mutants in our test series (Table 3). Although these failures (Table 5) may reflect faulty parameter settings (Table 2) or mistranslations of the mechanism into equations (Table 1), careful consideration of the inconsistencies (see Online Supplement B) indicates that there are problems in the wiring diagram itself (Figure 1). Identifying these problems suggests ways to improve the model in the future.
Table 5.
nconsistencies between simulations and observations
| Genotype | Observed phenotype | Simulated phenotype | Notesa |
|---|---|---|---|
| cdh1Δ | Viable, grows slowly, reduced size at birth, reduced size at budding (Jorgensen et al., 2002; Wasch and Cross, 2002) | Viable, normal growth rate, small at birth, normal size at budding | Cdh1 promotes proteolysis of some activator of start? |
| cdh1Δ cdc6Δ2-49 | Viable, large size, extended G2 phase (Calzada et al., 2001) | Viable, size between WT and cdh1Δ, slightly shorter G2 | |
| CLB1 clb2Δ | Viable (Richardson et al., 1992) | Inviable. Sister chromatid separation occurs 1.5 min after nuclear division | Faulty parameter settings, or model criterion for cell viability too strict |
| cdh1Δ clb2Δ CLB1 | Inviable in glucose, viable in galactose (Cross, 2003) | Viable in both glucose and galactose | Clb1 may not be a perfect backup of Clb2. Clb2 may be essential for activation of APC/Cdc20 |
| GAL-CLB2 sic1Δ | Inviable, telophase arrest (Toyn et al., 1997) | Inviable. Division occurs in an unbudded cell | A delay in the onset of mitosis by the morphogenesis checkpoint may cause telophase arrest |
| cdh1Δ sic1Δ | Inviable. Most cells are able to exit mitosis after 1st cycle and arrest in the 2nd cycle as large budded cells with replicated DNA (Schwab et al., 1997; Visintin et al., 1997; Wasch and Cross, 2002; Archambault et al., 2003) | Completes 1st cycle, but no bud in the 2nd cycle. If budding problem is ignored, then the mutant would be viable | See discussion of the triple-antagonist strain in the text |
| cdh1Δ sic1Δ cdc6Δ2-49 (triple-antagonist) | Inviable. The triple-antagonist strain has a cytokinesis defect, arresting as a 4-cell body objects with 2 short spindles and 4C DNA content (Archambault et al., 2003) | Inviable, telphase arrest with 2C DNA content | Cdc14 may trigger mitotic exit in the presence of high Clb2 kinase activity. See text for details |
| swi5Δ cdh1Δ | Inviable, similar to cdh1Δ sic1Δ cdc6Δ2-49 (Archambault et al., 2003) | Similar to cdh1Δ sic1Δ cdc6Δ2-49 | Same as cdh1Δ sic1Δ cdc6Δ2-49 |
| pds1Δ | Viable (Yamamoto et al., 1996a) | Inviable, Esp1 active before chromosome alignment | Model neglects role of Cdc5 in activating the substrate Scc1 for Esp1 |
| pds1Δ clb2Δ CLB1 | Inviable (Shirayama et al., 1999) | Inviable for the wrong reasons. If both single mutants are viable then the double mutant would be viable as well | New role of Clb2 in inactivating Esp1 |
| bub2Δ in nocodazole | Exits from mitosis after many hours in nocodazole (Hoyt et al., 1991; Alexandru et al., 1999) | Tightly arrests in metaphase in nocodazole | New cross talk from the BUB2 pathway to the MAD2 pathway? |
More detailed analysis of the problem mutants and how to resolve these inconsistencies are described in Online Supplement B
The most troublesome mutants for the model are the triple mutant cdh1Δ sic1Δ cdc6Δ2-49 and the double mutant swi5Δ cdh1Δ strains, which have very similar phenotypes (Table 5).
The triple-antagonist strain (cdh1Δ sic1Δ cdc6Δ2-49, where all three G1-stabilizers are deleted) is inviable (Archambault et al., 2003), but the cells are not telophase arrested. When a triple-antagonist GAL-SIC1 (integrated) strain is transferred from galactose to glucose medium, the mutant cells reproducibly accumulate as four-cell body objects that are resistant to sonication. The cells are able to undergo DNA synthesis and nuclear division but not cell division, and they arrest as binucleate cells with 4C DNA content. In our simulation, this mutant would be unbudded, arrested in telophase with 2C DNA content.
We do not know how to interpret the phenotype of the triple-antagonist and how to model it properly. There are three problems. First, how can the mutant exit from mitosis? What drives Clb2 activity below the threshold for nuclear division? Degradation by Cdc20 is not enough, because cdc14Δ (which would be equivalent to the triple-antagonist) arrests in telophase. The observed phenotype of the triple-antagonist suggests that our requirement for nuclear division is incorrect and that Cdc14 must have additional roles besides activating CKI and Cdh1.
Perhaps it is the rise in Cdc14 phosphatase activity, rather than (or in addition to) the fall of Clb2 kinase activity, that triggers nuclear division (see Online Supplement C). Suppose that nuclear division is triggered when the phosphorylation state of a target protein drops below a critical value. If the target protein is phosphorylated by Clb2 kinase and dephosphorylated by Cdc14 phosphatase, then the triple-antagonist cells, which have high Cdc14 activity, could exit from mitosis. However, there are other problems.
Even if they could execute nuclear division, they would have trouble forming a bud in the next cell cycle. The persistent high Clb2 kinase activity after nuclear division would prevent SBF activation and Cln2 accumulation, hence in simulation [BUD]max = 0.25 (it never reaches 1). However, triple-antagonist cells do form buds. It may be that Clb2 kinases are able to trigger bud formation, albeit at very low efficiencies compared with Cln2 kinases.
A third problem for the triple-antagonist cells is that high Clb2 kinase activity after the first nuclear division would presumably prevent reactivation of the licensing factors. Surprisingly, triple-antagonist cells are able to replicate their DNA but block in G2/M for some unknown reason. Maybe high Clb activity is not able to block origin relicensing totally, allowing these mutants to enter S phase from a few licensed origins. In this case, S phase may not be properly completed, causing cells to arrest in a G2/M state with seemingly replicated DNA.
In Online Supplement C, we simulate the model with a target protein (as described above). Ignoring the bud problem, we can find a basal parameter set that describes the phenotypes of triple-antagonist cells and the double mutant swi5Δ cdh1Δ (except their deficiency in cytokinesis). The same basal parameter set can explain the phenotype of cdh1Δ sic1Δ cells as well, without causing additional problems for the other mutants in Table 3.
A drawback of this “target hypothesis” is its lack of robustness; it is very sensitive to small changes of certain parameter values. However, as described in Online Supplement C, if proper checkpoint mechanisms are added, then the extended model is expected to be suitably robust.
Is the Model Robust?
The eukaryotic cell cycle engine must satisfy two basic conditions. 1) It must alternate between phases of DNA replication and sister chromatid segregation (Botchan, 1996). 2) It must coordinate the DNA replication-segregation cycle with overall cell growth, so that the cycle times of mother and daughter cells (P and D) and the mass doubling time of the culture (MDT = ln2/kg) satisfy the relationship e-kgD + e-kgP = 1 (Hartwell and Unger, 1977; Fantes and Nurse, 1981). Furthermore, it must be robust in the sense that these basic requirements are satisfied over a broad range of parameter values, so that cell viability is not delicately dependent on enzymatic activities. For a budding yeast cell, we have an operational definition of exactly how robust is its cell cycle engine, because the large set of phenotypically characterized mutants probes the parameter space of the control system. The viability/inviability of these mutants tells us exactly how large the region of robust control is in the budding yeast parameter space. Because the model is consistent with most mutant phenotypes, we can say that it is neither more nor less robust than warranted by observations.
We also have tested the robustness of the model systematically, as in Cross (2003), by changing the parameters in the basal (wild-type) parameter set one at a time to determine the range over which cyclic behavior persists (see MATERIALS AND METHODS for the requirements of viability in simulations). We find that 72% of the parameters can be changed at least 10-fold in either direction (Figure 5). In Online Supplement D and Table S2, those 35 parameters that do not have this flexibility are described. They identify fragile parts of the model. It remains to be determined whether these fragilities are all real features of the control system. Some may be artifacts of the model that need to be corrected.
Figure 5.

Robustness assay of the wild-type and six mutant strains, cdh1Δ, ckiΔ, APC-A, cln2Δ, cln3Δ, and clb5Δ. For each strain, simulations were run with systematic variations in each parameter to determine the maximum increase or decrease of that particular parameter the model could still tolerate to give a viable cell according to the criteria described in MATERIALS AND METHODS. Maximum variation tested is 256-fold up and 1/256-fold down. The cumulative distribution of parameters exhibiting a tolerance for a given fold of increase or decrease is plotted for each strain. Wild-type and cln3Δ mutant are most robust to parameter changes, whereas cln2Δ and cdh1Δ are least robust.
For example, measurements of Ghaemmaghami et al. (2003) and Huh et al. (2003) suggest that [Net1]T/[Cdc14]T = 0.2, which lies outside the range required by the model, 1 < [Net1]T/[Cdc14]T < 2.8. Perhaps the model is incorrect in assuming 1:1 stoichiometric inhibition of Cdc14 by Net1. Furthermore, the model does not include the fact that Cdc14 and Cdc5 are involved in a homeostatic negative feedback loop: Cdc5 activates Cdc14, which activates Cdh1, which in turn degrades Cdc5 (Charles et al., 1998; Shou et al., 1999; Pereira et al., 2002; Stegmeier et al., 2002; Visintin et al., 2003). This loop may buffer fluctuations in Cdc14 amount, making the model more robust to [Cdc14]T. And the requirement of Cdc14 phosphorylation by Cdc5 for its release from the nucleolus and activation, as reported recently by Visintin et al. (2003), may help to explain how cells can have higher [Cdc14]T than [Net1]T.
The Phenotypes of Some Mutants Are Not as Robust as Wild Type
One would expect that most mutants will be more sensitive to parameter changes than wild-type cells. Figure 5 also shows robustness analyses of six mutants: cdh1Δ, ckiΔ (= sic1Δ cdc6Δ2-49), APC-A, cln2Δ, clb5Δ, and cln3Δ. The first five mutants are all less robust than wild type.
For example, in cdh1Δ, because SBF is turned off earlier due to higher Clb2 level, less Cln2 is made, and the cell is compromised in its ability to bud. Changes in other parameters that further decrease Cln2 activity causes the cdh1Δ mutant to fail our viability criteria by failing to bud.
The other example is the APC-A mutant (Cross, 2003). Because this mutant is compromised in its degradation of Clb2 by Cdc20/APC-P and depends on Cdh1 activity for viability (APC-A cdh1Δ telophase arrest), parameter changes that further decrease Cdh1 activity tend to make APC-A inviable.
On the other hand, the ckiΔ mutant (or the single sic1Δ mutant) seems to be very robust to parameter changes in our analysis (less robust than wild type, but more than cdh1Δ or APC-A). This may not be true in reality. Because we have not taken into account the fact that sicΔ mutant cells have poor viability and show increased genome instability (Nugroho and Mendenhall, 1994; Lengronne and Schwob, 2002), the physiology of the mutant is not properly described by the model.
DISCUSSION
In all eukaryotes studied thoroughly to date, cyclin B-dependent kinase drives cells into mitosis. When chromosomes are properly aligned on the mitotic spindle, cyclin B-dependent kinase then activates the anaphase promoting complex (APC-P/Cdc20), which degrades securin (Pds1), which permits separase (Esp1) to degrade cohesin (Scc1), which permits sister chromatids to be pulled apart by spindle microtubules (for review, see Zachariae and Nasmyth, 1999). Cdc20/APC-P is also responsible for significant degradation of B-type cyclins, as cells exit mitosis. The rest of the story, as cells divide and reenter G1 phase, varies considerably from organism to organism (for review, see Bardin and Amon, 2001). In budding yeast cells, Cdc20 needs help from Cdc14, Cdc15, and other MEN-components to remove B-type cyclins completely and establish a stable G1 state (high levels of Cdh1, Sic1, and Cdc6). In fission yeast, the Cdc20 homolog (Slp1) is solely responsible for guiding cells from metaphase back to G1 (Kim et al., 1998). Fission yeast homologues of MEN-components function in the septum initiation network (SIN), which is down stream of cyclin B removal (McCollum and Gould, 2001). Similarly, during early embryonic cell cycles, full exit from mitosis is accomplished by Cdc20 alone (known as fizzy in frog and fruit fly; Sigrist et al., 1995; Lorca et al., 1998). In somatic cells of higher eukaryotes, several homologous proteins in the MEN/SIN pathways are known (Hirota et al., 2000; Hudson et al., 2001), but it is not yet known what specific roles these proteins play in mitotic exit and cell separation.
To contribute to a general understanding of cell cycle regulation, especially as cells exit mitosis and reenter G1 phase, we have created a computational model of the full molecular mechanism of the cell cycle engine in budding yeast S. cerevisiae. We have shown that the model is consistent with the observed phenotypes of >100 mutants of budding yeast. A similar, comprehensive model of cell cycle controls in fission yeast, including the septum initiation network, is in preparation. We expect that these thorough, quantitative models of yeast cell cycle regulation will provide a solid foundation for developing models of the regulatory properties of mammalian cell proliferation.
Assessing the Basic Principles of the Model
Our models of budding yeast, fission yeast, and frog eggs suggest some new ways to look at cell cycle control. We view the classical phases of the cell cycle (G1, S/G2, and M) as alternative stable steady states of the underlying control system (Novak and Tyson, 1993; Nasmyth, 1996; Tyson et al., 1996; Chen et al., 2000). A cell progresses irreversibly from G1 to S/G2 to M and back to G1 because cell growth and division drive the control system out of the region of attraction of one state into a region of attraction of the next (Tyson et al., 2001; Tyson and Novak, 2001). In this point of view, checkpoints are thought of as signals that delay these transitions by stabilizing a stage of the cycle (Novak et al., 2002; Tyson et al., 2002; Ciliberto et al., 2003).
In our budding yeast model, this view of cell cycle control depends crucially on the antagonism between Clb-dependent kinases and G1-stabilizers (Cdh1, Sic1, and Cdc6), which creates alternative steady states of low and high Clb-dependent Cdc28 kinase activity. Transitions between these states are driven by Cln-dependent phosphorylation of the G1-stabilizers and by APC/Cdc20-dependent proteolysis of Clbs. In the model, activation of Cln1,2-kinases is intimately connected to cell growth by our assumption that Cln3 accumulates in the nucleus as a cell grows (Chen et al., 2000). When the intranuclear concentration of Cln3/Cdc28 crosses a critical threshold, a sequence of molecular events is initiated that rapidly switches the cell from G1 (Clb-kinase activity low) to S/G2/M (Clb-kinase activity high). Sustained high level of Clb kinase activates APC/Cdc20, which initiates the reverse transition to come back to G1.
In the model, the transition facilitators, Cln1,2 and Cdc20, are involved in negative feedback loops with Clb1,2. Cln1,2/Cdc28 removes Sic1 and inactivates Cdh1, allowing Clb1,2/Cdc28 activity to rise, which turns off transcription of the CLN1,2 gene. Clb1,2/Cdc28 activates transcription of CDC20 and phosphorylates APC components, thereby promoting formation of active APC/Cdc20 complex, which then degrades Clb1,2.
Specific aspects of this theoretical picture of the budding yeast cell cycle have been tested. First, mutants probe the basic wiring diagram of the model, and the success ratio of the model (120/131 = 92%) indicates that the fundamental antagonistic interactions and negative feedback loops are most likely correct. Second, the notion of “bistability” (alternative stable steady states) has been examined in budding yeast by Cross et al. (2002). They constructed a strain (cln1Δ cln2Δ cln3Δ GAL-CLN3 cdc14ts) that, when growing on glucose at 37°C, lacks both start-facilitators and finish-facilitators. Under these conditions, cells can arrest stably in either a low Clb-kinase state or a high Clb-kinase state, depending on which state the cell is in when it is shifted to glucose at 37°C. (In the “high” state, Cdc20 is active but Cdc14 is not, so the cell arrests with moderate Clb2 level characteristic of telophase arrested cells.) Bistability also has been confirmed recently for interphase and M-phase states of frog egg extracts (Pomerening et al., 2003; Sha et al., 2003). Third, our crucial assumption that cell size is monitored by Cln3 accumulation in the nucleus has received careful attention. The average size of yeast cells is quite sensitive to changes in CLN3 dosage (Cross et al., 2002). Results of Miller and Cross (2001) by using mislocalized Cln3 are in qualitative agreement with the idea that Cln3 nuclear accumulation is important for size regulation, although the quantitative relationship is somewhat unclear (discussed in Miller and Cross 2001). Qualitative agreement also is observed by Edgington and Futcher (2001) in similar experiments.
Recently, Cross (2003) tested specifically the interplay between Cdc20 and the G1-stabilizers Cdh1 and Sic1 as cells exit from mitosis and return to G1. Genetic results were interpreted in terms of two oscillatory mechanisms: a negative feedback oscillator (Clb1,2 activating Cdc20, which then degrades Clb1,2) and a relaxation oscillator (alternating low Clb/high Sic1,Cdh1 and high Clb/low Sic1,Cdh1, with switches between the two states triggered by Clns and Cdc14). In this view, the two oscillatory mechanisms are coupled in wild-type cells, although each can apparently function in isolation with appropriate genetic manipulations (Cross, 2003). For example, in the model Clb2 proteolysis is initiated by Cdc20/APC, which removes the bulk of Clb2 (but not all), and at mitotic exit Clb2 proteolysis is `handed off' to Cdh1/APC, essentially as shown by Yeong et al. (2000). Compromising the ability of either Cdc20 or Cdh1 to remove Clb2 (APC-A or cdh1Δ mutations) is predicted to not block the cell cycle because the other activity is able to compensate, whereas compromising both abilities at once is predicted to block the cell cycle with massive Clb2 accumulation, as shown experimentally by Cross (2003).
The present model introduces many improvements to the previous models (Chen et al., 2000; Cross, 2003). It not only is consistent with the genetic results in Cross (2003) but also it accounts for some facts that are awkward for the two-oscillator idea. For example, although the double mutant (APC-A cdh1Δ) is inviable in glucose medium, spores show 8% viability in galactose (Cross, 2003), i.e., the double mutant with both oscillators compromised can survive at low growth rates. In our present model, the double mutant is inviable for mass-doubling-time <150 min but viable for slower growth rates (see Supplementary Material F).
Parameter Identification
The preceding discussion suggests that accurate and comprehensive explanations of the properties of budding yeast growth and division will require careful attention to mechanistic details and modeling issues (equations and parameter values). For complex, interconnected networks like Figure 1, it is impossible to anticipate all the consequences of multiple mutations by undisciplined, “hand-waving” explanations. To be certain of the sufficiency and consistency of the mechanism, we must create a well-defined mathematical representation of the molecular interactions and demonstrate that the model fits all (or most) of the relevant data, as we have done in this article.
With >100 parameters at our disposal, is it any surprise that we can fit the model to the phenotypes of lots of mutants? After all, with four parameters, one is supposed to be able to fit an elephant. That is true, if the model is elephant shaped to begin with. But if the model is yeast shaped, it will not fit any particular elephant and vice versa. Hence, it is essential to prove that the model in Figure 1 is yeast shaped by displaying a particular parameter set that brings the model into agreement with the observed properties of yeast cell growth and division. In our experience, many reasonable assumptions about the wiring diagram must be rejected because no amount of parameter “twiddling” can bring the model into agreement with the phenotypes of all (or nearly all) the mutants in Table 3. Parameter changes that “rescue” a model with respect to one mutant usually have unintended and unanticipated negative effects on other mutants that were fitted just fine by the original parameter set. A mathematical model is the only way to keep track of the subtle interactions among genes and proteins in regulatory mechanisms of such complexity.
Our success in simulating most of the mutants in Table 3 indicates that the mechanism in Figure 1 is a reasonable facsimile of the Cdc28-cyclin control system in budding yeast. But a model of this sort (wiring diagram + equations + parameter values) is not a static, finished product. We have already pointed out that the model is faulty in its account of several mutant phenotypes, and lacking important components like Cdc5 and the FEAR pathway. The model also begs to be extended to related phenomena and to new experimental observations.
Future Developments
In the present model, we have neglected subcellular localization of proteins except cyclins, which we assume accumulate in the nucleus. A recent global study (Huh et al., 2003) reveals the spatial distributions of nearly all proteins within the budding yeast cell. This information provides a basis for building a more realistic model of a compartmentalized yeast cell. In particular, the model's treatment of Cln2 as a nuclear protein needs to be revised, because there is considerable evidence that Cln2 is present in both nucleus and cytoplasm (Miller and Cross, 2000; Edgington and Futcher, 2001; Miller and Cross, 2001; Huh et al., 2003). To account for this fact, we should remove “[mass]” from the differential equation for d[Cln2]/dt in Table 1. With some modification of the rate constant k″s,n2 and the efficiencies of Cln2 kinase on its substrates, we expect to recover all the results of the present model.
In a companion study (Ciliberto et al., 2003), we have developed a model of the morphogenesis checkpoint in budding yeast, which involves the phosphorylation and dephosphorylation of Cdc28 by Swe1 and Mih1, respectively. In the future, we plan to graft this checkpoint mechanism smoothly onto Figure 1, much like one would slid together two large sections of a jigsaw puzzle. The grafting process will require adjustments of some parameter values to bring the extended model into accord with the mutant phenotypes in Table 3 and in Ciliberto's Table 1.
While this manuscript was under review, Thornton and Toczyski (2003) reported a viable mutant strain that lacks essential components of the APC. The usual requirement for APC activity was circumvented by simultaneously deleting the PDS1 and CLB5 genes and by inserting multiple copies of genomic SIC1. In this mutant strain (apc2Δ apc11Δ cdh1Δ cdc20Δ pds1Δ clb5Δ SIC110×), denoted “Apc-” for short, the obligatory oscillation of Clb2/Cdc28 activity is driven not by the synthesis and degradation of Clb2, but by the rise and fall of its inhibitor Sic1. We modified our basal parameter set slightly to account for this mutant phenotype (Thornton et al., 2004) as well as 27 other related strains characterized by Thornton and Toczyski. (The model equations and parameter set are available for download from our Web site at http://mpf.biol.vt.edu.) Although most of the mutants in Table 1 are correctly derived from the basal parameter set for the Apc- mutants, a few are not. To fit the 131 mutants in Table 1 with the 13 mutants in Ciliberto's collection and the 28 mutants in Thornton's experiments, the parameter set will need further adjustments.
As the model becomes more comprehensive, the problem of parameter identification becomes more daunting. Thus, we are developing computational tools for comparing simulations to experimental data and for automatic parameter optimization.
“Problem-Solving Environments”
The model reported here has been “hand crafted,” in the sense that we used computers only to simulate the governing differential equations for given parameter values. Comparison of the simulations to experimental data was done by visual inspection, and parameter adjustments, to align the model with the data, were all done by hand. Whenever the basal parameter set (Table 2) is changed, all 131 simulations must be recomputed and compared again to the data. To make kinetic modeling of this sort available to molecular cell biologists and to extend the model to new levels of complexity, the scientific community needs efficient, reliable, powerful, user-friendly software tools. These tools should take the drudgery out of the modeling process, enabling the user to apply his or her considerable intuitive understanding of life's devices to a disciplined mathematical representation of molecular control systems. Computer scientists call such tools problem-solving environments (PSEs).
In conjunction with computer scientists, we are developing a PSE, called JigCell (Allen et al., 2003), containing tools for model construction, simulation management, and comparison of simulations to experiments. Tools for data management and automatic parameter estimation are under development. For more information, consult our Web site.
JigCell is part of the BioSPICE consortium (http://www.biospice.org/), whose goal is to develop publicly available, open-source software for modeling intracellular regulatory networks. Other PSEs that may prove useful to molecular cell biologists are the Virtual Cell (http://www.nrcam.uchc.edu/vcellR3/login/login.jsp), the System Biology Workbench (http://www.sbw-sbml.org/index.html), and Gepasi (http://www.gepasi.org/gepasi.html). These PSEs are all cutting-edge technologies that are still under development. Experimentalists should not expect that making a mathematical model of a molecular control system will ever be as simple as BLASTing a sequence, but work is underway to build software tools that will eventually bring sophisticated modeling within the grasp of motivated bench scientists.
Summary
The molecular machinery of eukaryotic cell cycle control is known in more detail for budding yeast than for any other organism. Molecular biologists have painstakingly dissected and characterized the genes and protein interactions that underlie the regulatory network. By formulating this network in differential equations and computing their solutions numerically, we have shown that a consensus mechanism successfully reproduces the behavior of wild-type and mutant cells in quantitative detail. Hence, we conclude that our present understanding of the control system, properly interpreted, is accurate and adequate. The model also helps to organize information in a logical, comprehensive and predictive manner, and it is freely available for these purposes on our Web site at http://mpf.biol.vt.edu.
Acknowledgments
K.C. and L.C. are primarily responsible for developing the model. This research was supported by grants from the National Science Foundation (MCB-0078920) and the Defense Advanced Research Project Agency (AFRL #F30602-02-0572). We thank Masaki Shirayama, Orna Cohen-Fix, and Wolfgang Zachariae for valuable discussions of the model, and Jason Zwolak for help in automating simulations of the set of mutants. The article was written in part while in residence at the Collegium Budapest, supported by a grant from the Volkswagen Stiftung.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E03-11-0794. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E03-11-0794.
Abbreviations used: CKI, cyclin-dependent kinase inhibitor; MDT, mass doubling time.
Online version of this article contains supporting material. Online version is available at www.molbiolcell.org.
References
- Alexandru, G., Zachariae, W., Schleiffer, A., and Nasmyth, K. (1999). Sister chromatid separation and chromosome reduplication are regulated by different mechanisms in response to spindle damage. EMBO J. 18, 2707-2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, N.A., et al. (2003). Modeling regulatory networks at Virginia Tech. Omics 7, 285-299. [DOI] [PubMed] [Google Scholar]
- Amon, A., Tyers, M., Futcher, B., and Nasmyth, K. (1993). Mechanisms that help the yeast cell cycle clock tick: G2 cyclins transcriptionally activate G2 cyclins and repress G1 cyclins. Cell 74, 993-1007. [DOI] [PubMed] [Google Scholar]
- Archambault, V., Li, C.X., Tackett, A.J., Wasch, R., Chait, B.T., Rout, M.P., and Cross, F.R. (2003). Genetic and biochemical evaluation of the importance of Cdc6 in regulating mitotic exit. Mol. Biol. Cell 14, 4592-4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardin, A.J., and Amon, A. (2001). MEN and SIN: what's the difference? Nat. Rev. Mol. Cell. Biol. 2, 815-826. [DOI] [PubMed] [Google Scholar]
- Barral, Y., Jentsch, S., and Mann, C. (1995). G1 cyclin turnover and nutrient uptake are controlled by a common pathway in yeast. Genes Dev. 9, 399-409. [DOI] [PubMed] [Google Scholar]
- Botchan, M. (1996). Coordinating DNA replication with cell division: current status of the licensing concept. Proc. Natl. Acad. Sci. USA 93, 9997-10000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer, B.J., Chlebowicz-Sledziewska, E., and Fangman, W.L. (1984). Cell cycle phases in the unequal mother/daughter cell cycles of Saccharomyces cerevisiae. Mol. Cell. Biol. 4, 2529-2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzada, A., Sacristan, M., Sanchez, E., and Bueno, A. (2001). Cdc6 cooperates with Sic1 and Hct1 to inactivate mitotic cyclin-dependent kinases. Nature 412, 355-358. [DOI] [PubMed] [Google Scholar]
- Charles, J.F., Jaspersen, S.L., Tinker-Kulberg, R.L., Hwang, L., Szidon, A., and Morgan, D.O. (1998). The Polo-related kinase Cdc5 activates and is destroyed by the mitotic cyclin destruction machinery in S. cerevisiae. Curr. Biol. 8, 497-507. [DOI] [PubMed] [Google Scholar]
- Chen, K.C., Csikasz-Nagy, A., Gyorffy, B., Val, J., Novak, B., and Tyson, J.J. (2000). Kinetic analysis of a molecular model of the budding yeast cell cycle. Mol. Biol. Cell 11, 369-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciliberto, A., Novak, B., and Tyson, J.J. (2003). Mathematical model of the morphogenesis checkpoint in budding yeast. J. Cell Biol. 163, 1243-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Fix, O., and Koshland, D. (1999). Pds1p of budding yeast has dual roles: inhibition of anaphase initiation and regulation of mitotic exit. Genes Dev. 13, 1950-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross, F.R. (2003). Two redundant oscillatory mechanisms in the yeast cell cycle. Dev. Cell 4, 741-752. [DOI] [PubMed] [Google Scholar]
- Cross, F.R., Archambault, V., Miller, M., and Klovstad, M. (2002). Testing a mathematical model for the yeast cell cycle. Mol. Biol. Cell 13, 52-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvrckova, F., and Nasmyth, K. (1993). Yeast G1 cyclins CLN 1 and CLN 2 and a GAP-like protein have a role in bud formation. EMBO J. 12, 5277-55286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies, R., Chau, V., and Kirschner, M. (1995). Ubiquitination of the G1 cyclin Cln2p by a Cdc34-dependent pathway. EMBO J. 14, 303-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirick, L., Bohm, T., and Nasmyth, K. (1995). Roles and regulation of Cln/Cdc28 kinases at the start of the cell cycle of Saccharomyces cerevisiae. EMBO J. 14, 4803-4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington, N.P., and Futcher, B. (2001). Relationship between the function and the location of G1 cyclins in S. cerevisiae. J. Cell Sci. 114, 4599-4611. [DOI] [PubMed] [Google Scholar]
- Epstein, C.B., and Cross, F.R. (1994). Genes that can bypass the CLN requirement for Saccharomyces cerevisiae cell cycle START. Mol. Cell. Biol. 14, 2041-2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantes, P.A., and Nurse, P. (1981). Division timing: controls, models and mechanisms. In: The Cell Cycle, ed. P.C.L. John, Cambridge, United Kingdom: Cambridge University Press, 11-33.
- Field, R.J., Koros, E., and Noyes, R.M. (1972). Oscillations in chemical systems. II. Thorough analysis of temporal oscillation in the bromate-cerium-malonic acid system. J. Am. Chem. Soc. 94, 8649-8664. [Google Scholar]
- Futcher, B. (1996). Cyclins and the wiring of the yeast cell cycle. Yeast 12, 1635-1646. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami, S., Huh, W.-K., Bower, K., Howson, R.W., Belle, A., Dephoure, N., O'Shea, E.K., and Weissman, J.S. (2003). Global analysis of protein expression in yeast. Nature 425, 737-741. [DOI] [PubMed] [Google Scholar]
- Goldbeter, A., and Koshland, Jr., D.E. (1981). An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl. Acad. Sci. USA 78, 6840-6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood, E., Nishitan, H., and Nurse, P. (1998). Cdc18p can block mitosis by two independent mechanisms. J. Cell Sci. 111, 3101-3108. [DOI] [PubMed] [Google Scholar]
- Gyorgyi, L., and Field, R.J. (1992). A three-variable model of deterministic chaos in the Belousov-Zhabotinsky reaction. Nature 355, 808-810. [Google Scholar]
- Hartwell, L.H., and Unger, M.W. (1977). Unequal division in Saccharomyces cerevisiae and its implications for the control of cell division. J. Cell Biol. 75, 422-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota, T., Morisaki, T., Nishiyama, Y., Marumoto, T., Tada, K., Hara, T., Masuko, N., Inagaki, M., Hatakeyama, K., and Saya, H. (2000). Zyxin, a regulator of actin filament assembly, targets the mitotic apparatus by interacting with h-warts/LAST1 tumor suppressor. J. Cell Biol. 149, 1073-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt, M.A., Trotis, L., and Roberts, B.T. (1991). Saccharomyces cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 66, 507-517. [DOI] [PubMed] [Google Scholar]
- Hudson, J.W., Kozarova, A., Cheung, P., Macmillan, J.C., Swallow, C.J., Cross, J.C., and Dennis, J.W. (2001). Late mitotic failure in mice lacking Sak, a polo-like kinase. Curr. Biol. 11, 441-446. [DOI] [PubMed] [Google Scholar]
- Huh, W.-K., Falvo, J.V., Gerke, L.C., Carroll, A.S., Howson, R.W., Weissman, J.S., and O'Shea, E.K. (2003). Global analysis of protein localization in budding yeast. Nature 425, 686-691. [DOI] [PubMed] [Google Scholar]
- Irniger, S., and Nasmyth, K. (1997). The anaphase-promoting complex is required in G1 arrested yeast cells to inhibit B-type cyclin accumulation and to prevent uncontrolled entry into S-phase. J. Cell Sci. 110, 1523-1531. [DOI] [PubMed] [Google Scholar]
- Jacobson, M.D., Gray, S., Yuste-Rojas, M., and Cross, F.R. (2000). Testing cyclin specificity in the exit from mitosis. Mol. Cell. Biol. 20, 4483-4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, P., Nishikawa, J.L., Breitkreutz, B.-J., and Tyers, M. (2002). Systematic identification of pathways that couple cell growth and division in yeast. Science 297, 395-400. [DOI] [PubMed] [Google Scholar]
- Kim, S.H., Lin, D.P., Matsumoto, S., Kitazono, A., and Matsumoto, T. (1998). Fission yeast Slp1, an effector of the Mad2-dependent spindle checkpoint. Science 279, 1045-1047. [DOI] [PubMed] [Google Scholar]
- Kramer, E.R., Scheuringer, N., Podtelejnikov, A.V., Mann, M., and Peters, J.M. (2000). Mitotic regulation of APC activator proteins CDC20 and CDH1. Mol. Biol. Cell 11, 1555-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanker, S., Valdivieso, M.H., and Wittenberg, C. (1996). Rapid degradation of the G1 cyclin Cln2 induced by CDK-dependent phosphorylation. Science 271, 1597-1601. [DOI] [PubMed] [Google Scholar]
- Lengronne, A., and Schwob, E. (2002). The yeast CDK inhibitor Sic1 prevents genomic instability by promoting replication origin licensing in late G1. Mol. Cell 9, 1067-1078. [DOI] [PubMed] [Google Scholar]
- Li, J.J. (1995). Once, and only once. Curr. Biol. 5, 472-475. [DOI] [PubMed] [Google Scholar]
- Lim, H.H., Goh, P.-Y., and Surana, U. (1998). Cdc20 is essential for the cyclosome-mediated proteolysis of both Pds1 and Clb2 during M phase in budding yeast. Curr. Biol. 8, 231-234. [DOI] [PubMed] [Google Scholar]
- Lorca, T., Castro, A., Martinez, A.M., Vigneron, S., Morin, N., Sigrist, S., Lehner, C., Doree, M., and Labbe, J.C. (1998). Fizzy is required for activation of the APC/cyclosome in Xenopus egg extract. EMBO J. 17, 3565-3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord, P.G., and Wheals, A.E. (1980). Asymmetrical division of Saccharomyces cerevisiae. J. Bacteriol. 142, 808-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz, A., and Carter, B.L.A. (1979). Control of cell size at bud initiation in Saccharomyces cerevisiae. J. Gen. Microbiol. 113, 287-295. [Google Scholar]
- McCollum, D., and Gould, K.L. (2001). Timing is everything: regulation of mitotic exit and cytokinesis by the MEN and SIN. Trends Cell Biol. 11, 89-95. [DOI] [PubMed] [Google Scholar]
- Mendenhall, M.D., and Hodge, A.E. (1998). Regulation of Cdc28 cyclin-dependent protein kinase activity during the cell cycle of the yeast Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 62, 1191-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M.E., and Cross, F.R. (2000). Distinct subcellular localization patterns contribute to functional specificity of Cln2 and Cln3 cyclins of Saccharomyces cerevisiae. Mol. Cell. Biol. 20, 542-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M.E., and Cross, F.R. (2001). Mechanisms controlling subcellular localization of the G1 cyclins Cln2p and Cln3p in budding yeast. Mol. Cell. Biol. 21, 6292-6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison, J.M. (1971). The Biology of the Cell Cycle, Cambridge, United Kingdom: Cambridge University Press.
- Morgan, D.O. (1999). Regulation of the APC and the exit from mitosis. Nat. Cell Biol. 1, E47-E53. [DOI] [PubMed] [Google Scholar]
- Moser, B.A., and Russell, P. (2000). Cell cycle regulation in Schizosaccharomyces pombe. Curr. Opin. Microbiol. 3, 631-638. [DOI] [PubMed] [Google Scholar]
- Murray, A., and Hunt, T. (1993). The Cell Cycle. An introduction, New York, W.H. Freeman & Co.
- Nash, R., Tokiwa, G., Anand, S., Erickson, K., and Futcher, A.B. (1988). The WHI1+ gene of Saccharomyces cerevisiae tethers cell division to cell size and is a cyclin homolog. EMBO J. 7, 4335-4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth, K. (1996). At the heart of the budding yeast cell cycle. Trends Genet. 12, 405-412. [DOI] [PubMed] [Google Scholar]
- Novak, B., Csikasz-Nagy, A., Gyorffy, B., Nasmyth, K., and Tyson, J.J. (1998). Model scenarios for evolution of the eukaryotic cell cycle. Phil. Trans. R. Soc. Lond. B 353, 2063-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak, B., Pataki, Z., Ciliberto, A., and Tyson, J.J. (2001). Mathematical model of the cell division cycle of fission yeast. Chaos 11, 277-286. [DOI] [PubMed] [Google Scholar]
- Novak, B., Sible, J.C., and Tyson, J.J. (2002). Checkpoints in the cell cycle. Encyclopedia of Life Sci. www.els.net, 1-8.
- Novak, B., and Tyson, J.J. (1993). Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos. J. Cell Sci. 106, 1153-1168. [DOI] [PubMed] [Google Scholar]
- Novak, B., and Tyson, J.J. (1997). Modeling the control of DNA replication in fission yeast. Proc. Natl. Acad. Sci. USA 94, 9147-9152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugroho, T.T., and Mendenhall, M.D. (1994). An inhibitor of yeast cyclin-dependent protein kinase plays an important role in ensuring the genomic integrity of daughter cells. Mol. Cell. Biol. 14, 3320-3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse, P. (1990). Universal control mechanism regulating onset of M-phase. Nature 344, 503-508. [DOI] [PubMed] [Google Scholar]
- Nurse, P. (1997). The Josef Steiner Lecture: CDKs and cell-cycle control in fission yeast: relevance to other eukaryotes and cancer. Int. J. Cancer 71, 507-508. [DOI] [PubMed] [Google Scholar]
- Pereira, G., Manson, C., Grindlay, J., and Schiebel, E. (2002). Regulation of the Bfa1p-Bub2p complex at spindle pole bodies by the cell cycle phosphatase Cdc14p. J. Cell Biol. 157, 367-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, J.-M. (1998). SCF and APC: the Yin and Yang of cell cycle regulated proteolysis. Curr. Opin. Cell Biol. 10, 759-768. [DOI] [PubMed] [Google Scholar]
- Pomerening, J.R., Sontag, E.D., and Ferrell, Jr., J.E. (2003). Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nat. Cell Biol. 5, 346-351. [DOI] [PubMed] [Google Scholar]
- Richardson, H., Lew, D.J., Henze, M., Sugimoto, K., and Reed, S.I. (1992). Cyclin-B homologs in Saccharomyces cerevisiae function in S phase and in G2. Genes Dev. 6, 2021-2034. [DOI] [PubMed] [Google Scholar]
- Rudner, A., and Murray, A. (2000). Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase promoting complex. J. Cell Biol. 149, 1377-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama, S.R., Hendricks, K.B., and Thorner, J. (1994). G1 cyclin degradation: the PEST motif of yeast Cln2 is necessary, but not sufficient, for rapid protein turnover. Mol. Cell. Biol. 14, 7953-7966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, B.L., Yang, Q.-H., and Futcher, B. (1996). Linkage of replication to Start by the Cdk inhibitor Sic1. Science 272, 560-562. [DOI] [PubMed] [Google Scholar]
- Schwab, M., Lutum, A.S., and Seufert, W. (1997). Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell 90, 683-693. [DOI] [PubMed] [Google Scholar]
- Schwob, E., Bohm, T., Mendenhall, M.D., and Nasmyth, K. (1994). The B-type cyclin kinase inhibitor p40sic1 controls the G1 to S transition in S. cerevisiae. Cell 79, 233-244. [DOI] [PubMed] [Google Scholar]
- Schwob, E., and Nasmyth, K. (1993). CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev. 7, 1160-1175. [DOI] [PubMed] [Google Scholar]
- Sha, W., Moore, J., Chen, K., Lassaletta, A.D., Yi, C.-S., Tyson, J.J., and Sible, J.C. (2003). Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts. Proc. Natl. Acad. Sci. USA 100, 975-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama, M., Toth, A., Galova, M., and Nasmyth, K. (1999). APC (CDC20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature 402, 203-207. [DOI] [PubMed] [Google Scholar]
- Shirayama, M., Zachariae, W., Ciosk, R., and Nasmyth, K. (1998). The Pololike kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J. 17, 1336-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou, W., et al. (2001). Net1 stimulates RNA polymerase I transcription and regulates nucleolar structure independently of controlling mitotic exit. Mol. Cell 8, 45-55. [DOI] [PubMed] [Google Scholar]
- Shou, W., Seol, J.H., Shevchenko, A., Baskerville, C., Moazed, D., Chen, Z.W.S., Jang, J., Shevchenko, A., Charbonneau, H., and Deshaies, R.J. (1999). Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell 97, 233-244. [DOI] [PubMed] [Google Scholar]
- Sigrist, S., Jacobs, H., Stratmann, R., and Lehner, C.F. (1995). Exit from mitosis is regulated by Drosophila fizzy and the sequential destruction of cyclins A, B, and B3. EMBO J. 14, 4827-4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, I., et al. (2001). Serial regulation of transcriptional regulators in the yeast cell cycle. Cell 106, 697-708. [DOI] [PubMed] [Google Scholar]
- Spellman, P.T., Sherlock, G., Zhang, M.Q., Iyer, V.R., Anders, K., Eisen, M.B., Brown, P.O., Botstein, D., and Futcher, B. (1998). Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 9, 3273-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier, F., Visintin, R., and Amon, A. (2002). Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell 107, 207-220. [DOI] [PubMed] [Google Scholar]
- Stuart, D., and Wittenberg, C. (1995). CLN3, not positive feedback, determines the timing of CLN2 transcription in cycling cells. Genes Dev. 9, 2780-2794. [DOI] [PubMed] [Google Scholar]
- Su, T.T., Follette, P.J., and O'Farrell, P.H. (1995). Qualifying for the license to replicate. Cell 81, 825-828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, B.R., Chen, K.C., Cross, F.R., Tyson, J.J., and Toczyski, D.P. (2004). Cycling without the cyclosome: modeling a yeast strain lacking the APC. Cell Cycle 3, 629-633. [PubMed] [Google Scholar]
- Thornton, B.R., and Toczyski, D.P. (2003). Securin and B-cyclin/CDK are the only essential targets of the APC. Nat. Cell Biol. 5, 1090-1094. [DOI] [PubMed] [Google Scholar]
- Toyn, J.H., Johnson, A.L., Donovan, J.D., Toone, W.M., and Johnston, L.H. (1997). The Swi5 transcription factor of Saccharomyces cerevisiae has a role in exit from mitosis through induction of the cdk-inhibitor Sic1 in telophase. Genetics 45, 85-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyers, M. (1996). The cyclin-dependent kinase inhibitor p40sic1 imposes the requirement for Cln G1 cyclin function at Start. Proc. Natl. Acad. Sci. USA 93, 7772-7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson, J.J., Chen, K., and Novak, B. (2001). Network dynamics and cell physiology. Nat. Rev. Mol. Cell. Biol. 2, 908-916. [DOI] [PubMed] [Google Scholar]
- Tyson, J.J., Csikasz-Nagy, A., and Novak, B. (2002). The dynamics of cell cycle regulation. Bioessays 24, 1095-1109. [DOI] [PubMed] [Google Scholar]
- Tyson, J.J., and Novak, B. (2001). Regulation of the eukaryotic cell cycle: molecular antagonism, hysteresis and irreversible transitions. J. Theor. Biol. 210, 249-263. [DOI] [PubMed] [Google Scholar]
- Tyson, J.J., Novak, B., Odell, G.M., Chen, K., and Thron, C.D. (1996). Chemical kinetic theory as a tool for understanding the regulation of M-phase promoting factor in the cell cycle. Trends Biochem. Sci. 21, 89-96. [PubMed] [Google Scholar]
- Uhlmann, F., Lottspeich, F., and Nasmyth, K. (1999). Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 400, 37-42. [DOI] [PubMed] [Google Scholar]
- Verma, R., Feldman, R.M.R., and Deshaies, R.J. (1997). Sic1 is ubiquitinated in vitro by a pathway that requires CDC4, CDC34 and Cyclin/CDK activities. Mol. Biol. Cell 8, 1427-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin, R., Hwang, E.S., and Amon, A. (1999). Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus. Nature 398, 818-823. [DOI] [PubMed] [Google Scholar]
- Visintin, R., Prinz, S., and Amon, A. (1997). CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science 278, 460-463. [DOI] [PubMed] [Google Scholar]
- Visintin, R., Stegmeier, F., and Amon, A. (2003). The role of the polo kinase Cdc5 in controlling Cdc14 localization. Mol. Biol. Cell 14, 4486-4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Dassow, G., Meir, E., Munro, E.M., and Odell, G.M. (2000). The segment polarity network is a robust developmental module. Nature 406, 188-192. [DOI] [PubMed] [Google Scholar]
- Wasch, R., and Cross, F. (2002). APC-dependent proteolysis of the mitotic cyclin Clb2 is essential for mitotic exit. Nature 418, 556-562. [DOI] [PubMed] [Google Scholar]
- Wijnen, H., and Futcher, B. (1999). Genetic analysis of the shared role of CLN3 and BCK2 at the G1-S transition in Saccharomyces cerevisiae. Genetics 153, 1131-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenberg, C., Sugimoto, K., and Reed, S.I. (1990). G1-specific cyclins of S. cerevisiae: cell cycle periodicity, regulation by mating pheromone, and association with p34CDC28 protein kinase. Cell 62, 225-237. [DOI] [PubMed] [Google Scholar]
- Yaglom, J., Linskens, H.K., Rubin, D.M., Futcher, B., and Finley, D. (1995). p34Cdc28-mediated control of Cln3 cyclin degradation. Mol. Cell. Biol. 15, 731-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, A., Guacci, V., and Koshland, D. (1996a). Pds1p is required for faithful execution of anaphase in the yeast Saccharomyces cerevisiae. J. Cell Biol. 133, 85-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, A., Guacci, V., and Koshland, D. (1996b). Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s). J. Cell Biol. 133, 99-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeong, F.M., Lim, H.H., Padmashree, C.G., and Surana, U. (2000). Exit of mitosis in budding yeast: biphasic inactivation of the Cdc28-Clb2 mitotic kinase and the role of Cdc20. Mol. Cell 5, 501-511. [DOI] [PubMed] [Google Scholar]
- Yoshida, S., Asakawa, K., and Toh-e, A. (2002). Mitotic exit network controls the localization of Cdc14 to the spindle pole body in Saccharomyces cerevisiae. Curr. Biol. 12, 944-950. [DOI] [PubMed] [Google Scholar]
- Yuste-Rojas, M., and Cross, F.R. (2000). Mutations in CDC14 result in high sensitivity to cyclin gene dosage in Saccharomyces cerevisiae. Mol. Gen. Genet. 263, 60-72. [DOI] [PubMed] [Google Scholar]
- Zachariae, W., and Nasmyth, K. (1999). Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev. 13, 2039-2058. [DOI] [PubMed] [Google Scholar]
- Zhu, G., Spellman, P.T., Volpe, T., Brown, P.O., Botstein, D., Davis, T.N., and Futcher, B. (2000). Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature 406, 90-94. [DOI] [PubMed] [Google Scholar]