

Abstract
The last decade has seen a growing number of experiments aimed at systematically mapping the effects of mutations in different proteins, and of attempting to correlate their biophysical and biochemical effects with organismal fitness. While insightful, systematic laboratory measurements of fitness effects present challenges and difficulties. Here, we discuss the limitations associated with such measurements, and in particular the challenge of correlating the effects of mutations at the single protein level (“protein fitness”) with their effects on organismal fitness. A variety of experimental setups are used, with some measuring the direct effects on protein function and others monitoring the growth rate of a model organism carrying the protein mutants. The manners by which fitness effects are calculated and presented also vary, and the conclusions, including the derived distributions of fitness effects of mutations, vary accordingly. The comparison of the effects of mutations in the laboratory to the natural protein diversity, namely to amino acid changes that have fixed in the course of millions of years of evolution, is also debatable. The results of laboratory experiments may, therefore, be less relevant to understanding long‐term inter‐species variations yet insightful with regard to short‐term polymorphism, for example, in the study of the effects of human SNPs.
Keywords: protein evolution, mutational scan, systematic mutational mapping, SNP, polymorphism
Introduction
Systematic mappings of the effects of protein mutations have increased in popularity. By now, such mappings have been conducted in well over a dozen proteins, and for a few proteins, several independent datasets are available (for a recent review, see Ref. 1). Systematic mappings are clearly insightful and valuable, for example, in the context of protein engineering, in predicting the effects of protein mutations in general. Such mappings also promote a better understanding of the linkages between genotype, phenotype, and fitness. These linkages are far from trivial, with the correlation between the biophysical and biochemical effects of mutations and their effects on physiology and organismal fitness often being a key missing link.2
Systematic mappings examine either a large set of mutations, typically by saturation mutagenesis at a given set of positions (e.g., using NNS codons to diversify individual sites), or exhaustively, by random mutagenesis of the encoding gene (for early examples see Refs. 3, 4, 5). The resulting gene repertoires are subsequently subjected to a functional screen, or a selection, with the aim of quantifying the effects of individual mutations (in the simplest manner, delineating deleterious mutations from neutral and beneficial ones). Although systematic mutational mappings are routinely performed, there remain technical challenges and open conceptual questions. For example, how do the results of laboratory experimental mappings relate to natural protein evolution? The focus of this perspective is on the utility as well as the limitations of systematic mappings in measuring the fitness effects of mutations. However, a comprehensive description of these approaches is beyond the scope of this review. We thus discuss a specific aspect of these experiments—fitness values. What do laboratory fitness values mean? What are the limitations when measuring fitness values in the laboratory? How do laboratory measured fitness values compare to fitness values in living organisms?
Fitness Is a Multidimensional Parameter
Fitness, usually denoted as W, relates to the reproduction potential of competing alleles, or genotypes. Given a competition between two alleles, to take the simplest case, their ratio in a growing population will change in accordance with their individual fitness values. One allele is typically assigned as the reference, or wild‐type, with its fitness defined as 1. The other allele(s) would have a fitness value (W′) that may be equal to, less than, or greater than W. The difference in fitness between the alleles is defined as the selection coefficient, or the fitness effect, and is denoted as s:
whereby W for the reference (wild‐type) allele is defined as 1, and s > 0 denotes beneficial mutations.
In an exponentially growing population, the fraction of an allele (f) competing against wild‐type would then be:
| (1) |
whereby f0 is the fraction of the competing allele at generation zero, s is its selection coefficient, and n is the number of generations.
However, despite an unambiguous mathematical definition, fitness is not a simplistic parameter, particularly in natural populations.6 Selection can act equally strongly, sometimes even stronger, on survival rather than growth (e.g., during stationary phase in bacteria or in host‐pathogen arms races). The effects of mutations are often pleiotropic, and thus the fitness effect of a mutation typically depends on the environment and growth phase. For example, tradeoffs between growth and survival commonly exist, such that a given mutation may have a deleterious fitness effect during growth under standard conditions yet become beneficial for survival under stress.7 Fitness is also an elusive parameter when it comes to laboratory measurements, specifically in relation to the fitness effects of protein mutations. The results of essentially all systematic mappings are presented as fitness values under a certain experimental condition. This is largely justified, especially when the distributions of fitness effects (DFEs) of mutations are sought (addressed later in a section devoted to DFEs). However, as discussed below, the derived fitness values relate to different experimental setups and/or were calculated in different ways, and may thus have a different meaning and relevance. The authors' own works are not an exception in this respect.
Laboratory Fitness Measurements of Protein Alleles
A detailed description of the many different approaches taken to systematically measure the effects of mutations is beyond the scope of this perspective. Indeed, a whole variety of experimental setups have been employed primarily depending on the protein in question. Alternative approaches include parallel analyses of many individual clones, whereby each clone carries a protein variant with a different mutation. Clones are then pooled according to their levels of function and the pools are subjected to deep sequencing. Alternatively, a competition mode is applied whereby all clones are grown in bulk, and deep sequencing of the entire population reveals the frequency and hence the relative fitness of each mutation in the repertoire. In the context of our discussion here, laboratory experiments generally follow two different modes: (i) direct measurements of the effects of mutations on “protein fitness”; or, (ii) measurements of the effects of mutations on organismal fitness under a defined condition. In the first mode, protein fitness effects, the selection or screen is applied directly to the biochemical or biophysical properties of the protein under study, for example, measuring fluorescence levels to map the effects of GFP mutations. In the second mode, organismal fitness effects, the protein under study impacts growth rate under the conditions of the experiments and the selection is accordingly applied for organismal growth and/or survival—for example, systematically mapping the effects of mutations in ubiquitin on yeast growth rates.8
While we schematically categorized two measurement modes: direct, protein fitness versus organismal fitness, certain experiments fall somewhere in between these categories. For example, measuring the levels of antibiotics resistance conferred by mutants of a resistance enzyme formally addresses organismal growth rates. However, in effect, this setup directly measures the enzyme's functional capacity as defined above (e.g., Refs. 9 and 10).
When directly measuring protein fitness effects or protein fitness landscapes,11 protein fitness, or W P, actually relates to the functional capacity of the protein in question. The latter relates to both the levels of soluble, functional protein and the protein's specific activity (e.g., k cat or k cat /K M for an enzyme). Protein fitness may, therefore, be addressed, certainly in the case of enzymes, as the multiplication of these two parameters (W P α [E] k cat /K M, or [E] k cat, depending on cellular substrate concentrations). However, the relationship between the functional capacity of a given protein (W P) and organismal fitness (W) is highly complex. Even in relatively simple cases, the correlation is almost always nonlinear (detailed below). And thus, the functional capacity of protein alleles may not linearly, or even directly, correlate with allele frequencies in living organisms carrying these protein variants.
Buffering the Effects of Mutations
In both the direct, protein fitness mode and the organism mode, laboratory experiments generally buffer the effects of mutations. Buffering may be due to experimental design, such as altering the expression of the protein under study using multiple‐copy plasmids and/or variable‐strength promoters.12 Proteins are often expressed with fusion tags that may “float” mutants with impaired stability. Mutants with impaired stability may also be masked by expression at sub‐challenging temperatures—for example, E. coli's optimal growth temperature in rich media is 42°C, but 37°C or even lower temperatures are often applied. Indeed, most destabilizing mutations cause misfolding and loss of protein function at higher temperature. Whatever the specific reason(s) may be, it is clear that mildly deleterious effects are often masked, certainly when the effects of single mutations are measured. Indeed, when the cumulative effects of mutations were measured in a mode of a prolonged drift, namely, by iterative rounds of mutagenesis and selection, a much higher fraction of deleterious mutations was observed (>80% of all possible mutations).13
However, buffering is not a phenomenon limited to laboratory setups, as it is also widely observed in the natural context (i.e., chromosomal genes, endogenous promoters, etc.). This is primarily because the relationship between the functional output of a protein which mutations affect directly (W P), and organismal fitness (W) is non‐linear. Buffering, therefore, applies also to the mode of organismal fitness effects. In a typical, although still simplified scenario, this relationships follows a saturation curve that can be described by14, 15:
| (2) |
whereby W P is the protein's functional output, or protein fitness (for the wild‐type protein, W P = 1), and B is the buffering coefficient, or mid‐value, that is, the protein's functional output level that results in W = 0.5; note that for W P = 1, organismal fitness, W, is also equal to 1 (Fig. 1).
Figure 1.
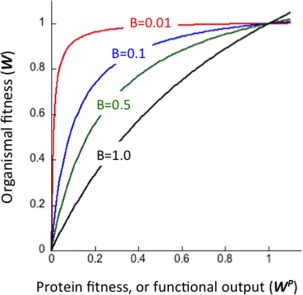
The relationship between organismal fitness (W) and a protein's functional output, or protein fitness (W P), that is, the level of soluble, functional protein in the cell multiplied by its specific activity (e.g., enzymatic k cat /K M, or k cat). Both fitness values are given as relative, that is, taking the value of 1 for the wild‐type protein and organism. The simulated graphs follow Eq. (2) with the buffering coefficient (B) taking the values of 0.01 up to 1.
The sensitivity of organismal fitness to changes in protein fitness (as reflected in the B value) varies. For one thing, per given growth condition, knockouts of the majority of genes have no effect on organismal fitness (i.e., B = 0; for E. coli data see Ref. 16). However, even when under a selective growth condition, essential proteins may exhibit very different B values. For example, for E. coli growing on lactose as the sole carbon source, lactose permease exhibits high sensitivity (i.e., mutations that mildly affect its functional transport capacity have large effects on E. coli growth rates). In contrast, for β‐galactosidase, an enzyme that is as essential for lactose utilization (and belongs to the same pathway and operon), the functional output could be reduced by nearly an order of magnitude relative to wild‐type with nearly no effect on organismal fitness.14
The underlying reasons for this widely observed buffering are largely unknown. It could be that strong buffering (i.e., low B values) is typical to laboratory growth conditions, or even, to the specific laboratory condition applied in a given experiment. The dependency of growth rate (organismal fitness) upon the functional output of a given enzyme and, accordingly, its metabolic control15 are bound to vary from rich versus minimal media or from one temperature to another. In natural environments, B will also differ, possibly even more dramatically. However, given that the evolutionary history of an organism encompasses an entire range of different environments, there would be certain environments or conditions with no buffering. Buffering may also relate to cellular mechanisms of rescuing impaired protein mutants, such as chaperones. In most likelihood, the traits of natural proteins were shaped in response to most relevant environment, that is, under the most demanding conditions with respect to a given protein. These conditions may relate to the distant past17 and are largely unknown, and would thus be extremely difficult to reproduced in the laboratory. Regardless of its origins, buffering results in “robustness” to mutations—namely, mutations show no experimentally measureable effects on organismal phenotype unless they drastically decrease the protein's functional output.
Finally, we note that in reality, Eq. (2) is obviously an over‐simplification. There would be cases, for example, where an increase in a protein's functional capacity (W P) would result in a decrease in organismal fitness (e.g., higher enzyme levels and/or k cat/K M may result in metabolic imbalance, and may thus decrease organismal fitness).
Limits in Detection of Fitness Effects
Laboratory fitness measurements have an additional limitation, even when assuming a native‐like context, such as measuring the effects of mutations for an endogenous protein expressed from a chromosomal copy under its original promoter, and with the organism grown under challenging conditions (such an experimental setup has been so far implemented for measuring the fitness effects of a given set of mutations (e.g., Ref. 18) but not for systematic mappings). This limitation relates to the low sensitivity of detection for fitness differences. In the protein fitness mode, sensitivity is limited by the magnitude of error in the applied measurement, say GFP fluorescence levels as measured by FACS. Whilst every experiment has its own error range, the error range is considerable. Very small fitness differences that may drive the purging or fixation of mutations in nature are usually obscured in such laboratory measurements.
Errors also stem from the fact that in systematic mappings, the mutational frequencies and the corresponding fitness effects are typically derived by deep sequencing. Although many datasets do not provide an explicit estimate of the error range, the cumulative noise level of such experiments is considerable. One method to estimate experimental noise is to use the frequency across synonymous mutations as the error range, based on the assumption that their fitness effects are negligible relative to those of amino acid exchanges. Applying this criterion, the error range in allele frequencies was found to be ≤2.5% in a recent study.10 However, this error range relates to wild‐type whose frequency is relatively high. In fact, for most alleles, the error range is much higher (15–40% in Ref. 10). The error ranges also relate to the fact that the allele frequencies are far from being equally distributed in both NNS libraries (due to synthesis biases) and error‐prone ones (due to base and position biases especially).13 The degree of sampling also affects the error range and, therefore, the ability to detect small fitness differences.19 The standard deviation (SD) for the distribution of fitness effects of all synonymous mutations indicates a much higher error range. We found, for example, these SD values to be in the range of 0.1.13 Applying a standard cutoff of 2xSD means that fitness effects (i.e., s values per round of selection) that are smaller than 0.2 could not be reliably measured.13 However, these error ranges relate to a particular experimental setup that is not applicable to others.
The sensitivity of determining fitness effects is also limited by the number of generations that can be applied in the laboratory. Allele frequency differences are exponentially amplified along generations [Eq. (1)], and thus, after many generations, comparatively small fitness effects (small s values) result in significant changes in allele frequencies. In the laboratory, this amplification is typically achieved by serial passages. However, there exists a tangible danger of genomic mutations biasing the effects of the measured alleles (background, or hitchhiking mutations).20 When plasmid encoded genes are utilized, recloning and retransformation can be performed between passages in order to minimize any biases due to background genomic mutations. However, even in this case, the total number of generations that can be applied in the laboratory is limited and, thereby, so are the fitness effects that can be discerned. In effect, s values on the order of 10−4 or even lower are highly relevant in natural populations, certainly for microorganisms (the effect of population size is discussed below). However, such fitness effects cannot be resolved by laboratory experiments, as this would not only demand selection to continuously act for ∼104 generations with no hitchhiking, background mutations, but also a markedly low error range in the measurement of allele frequencies. The above caveats are obviously much more dominant in systematic “multiplexing” experiments compared to “binary” competitions (competing two, or a few alleles) in which relatively small s values can be measured (for example Ref. 21).
Distribution of Fitness Effects of Protein Mutations
The results of systematic mapping experiments are often provided in the form of a distribution of fitness effect (DFE). The published data indicate quite different distributions, not only per protein but also per experiment or lab. The differences are evident even when sampling the results of just three mappings, two of which come from the authors' labs (Fig. 2).
Figure 2.
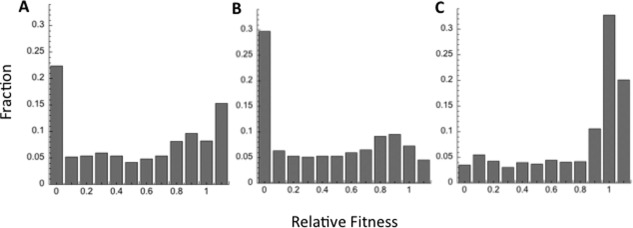
The variation in laboratory measured DFEs of mutations of three different proteins. A. The DFE of TEM‐1 β‐lactamase measured by saturation mutagenesis and deep sequencing.10 B. The DFE for a bacterial DNA methyltransferase, M.HaeIII, derived by a prolonged drift (17 rounds of random mutagenesis and selection) and deep sequencing.13 C. The DFE for ubiquitin obtained by systematic mapping using saturation mutagenesis and deep sequencing.8 The presented DFEs address nonsynonymous mutations only. The fitness values were normalized (wild‐type = 1) and sorted to 0.1 bins to allow their histogram presentations on the same scale. Note that the 1.1 bin encompasses all mutations with fitness values ≥1.1. The dramatic differences between these DFEs (e.g., the fraction of the most deleterious mutations, fitness < 0.1 versus the fraction of beneficial mutations, fitness > 1) relate primarily to the experimental mode and the manner by which fitness effects were measured and calculated, and less so to the different nature of these three proteins. For example, the highest fraction of deleterious mutations is observed for M.HaeIII. This is due to the cumulative load of mutations under a prolonged drift that resulted in strong purging, possibly at a level seen in natural evolution.13
The derived fitness effects in all systematic mapping experiments are not fitness values per se, but rather, relative fitness values at a scale that is experiment specific. The snag is that, although these distributions are all relative, that is, normalized to wild‐type, they were acquired via very different experimental modes. One critical point is that the fitness effects are usually provided per experiment, or per round of selection or screening. However, fitness effects (s values) are defined per generation (for a normalization method, see Ref. 22). The variability, however, goes far beyond the number of generations. Additionally, as discussed above, some experiments directly measured protein effects whilst others measured organismal effects. But there are many other factors that vary between experiments. Even when assuming the same selection mode, for example, Ampicillin resistance, the growth and selection regimes can vary. In most experiments, the effects of single mutations within a wild‐type background are measured. This strategy has clear advantages, foremost because mutations can interact such that their contributions to fitness are inter‐dependent or epistatic. However, a potential disadvantage of measuring the effects of single mutations is the buffering of mildly deleterious effects13—a tendency that is further intensified when the protein under study is expressed at high levels.12
The manner of calculating the fitness effects also varies between experiments. Finally, the comparison between experiments is further complicated by limited data availability—raw data are not available in most cases, certainly not in a format amenable to downloading and further processing. Overall, the differences between laboratory‐measured DFEs are large. However, these differences relate primarily to differences in experimental setups rather than to differences in the biophysical and functional properties of the analyzed proteins. For example, although in the laboratory ubiquitin appears to be more permissive to mutations [Fig. 2(C)] compared to a bacterial β‐lactamase or a DNA methyltransferase [Fig. 2(A,B)], in nature, it is the most conserved protein of the three examples shown here.
Laboratory Mappings Compared with Natural Divergence
Compared to laboratory mapping experiments, divergence in natural populations almost always occurs over much longer time‐scales and in more genetically diverse populations. These distinctions in timescale (or number of generations) and population structure impact the interpretations and comparisons that can be made between sequences changes seen under natural versus laboratory evolution.
The appearance of mutations and their fixation across an entire population are a fundamentally important process in evolution. Work led by Wright, Fisher, Gillespie, Ohta, Kimura, and others developed the foundational theory that describes the probability and time to fixation of a new mutation based on its selective advantage.23 Population genetics theory provides lessons that are increasingly valuable across all fields of biology, including protein science, because of the explosion in our knowledge of sequence diversity driven by next‐generation sequencing. Here, we will highlight a few of the key features of population genetics theory and how they relate to sequence diversity in nature and in the laboratory.
Because evolution is heavily influenced by stochastic events, not all beneficial mutations (W′ > W) that initially appear within a population become fixed. The role of stochastic events in evolution and our ability to model them is a key aspect of population genetics that can seem counter‐intuitive. Why do not all adaptive mutations fix? And conversely, why are not all fixed mutations adaptive? When any new mutation, including those that are adaptive, arises in a population, its initial frequency is so low that the likelihood that it will survive in the population is heavily influenced by stochastic processes (e.g., the individual originally harboring the mutation must survive to reproduce, which is never a certainty). Stochastic processes act on the reproductive fitness of each individual in each generation.
If stochastic events influence evolution, then what is meant by selective advantage? Selective advantages, or positive fitness effects, are averages over a large number of individuals and generations such that the stochastic components are minimized or hidden. This is analogous to a measure of the bulk property of a population of cells or molecules, which does not provide a detailed description of the variation among individuals. For example, in an experimental fitness competition, the selective advantage of a mutation is measured as the average growth rate increase, whereby the latter comprises an average per millions of cells harboring that mutation.
The primary parameter that determines the balance between stochastic drift and selection in evolution is the effective population size, Ne.11, 24 The larger the effective, or reproductive population, the smaller the influence of random events. By analogy, the more times that you flip a coin, the more likely it is that the observed distributions of heads and tails accurately represents the underlying probability of the coin flip. In evolving populations, the relative influence of stochastic processes or drift compared to selection depends on the effective population size. In considering the fixation of a mutation, drift and selection are balanced when Eq. (3) is satisfied.
| (3) |
Whereby s is the selection coefficient and Ne is the effective population size of a haploid organism.
A key prediction of this equation is that organisms with smaller population sizes will be more heavily influenced by drift, resulting in proteins that have been less stringently selected. It is important to note that effective population sizes in nature vary by over four orders of magnitude, so the stringency of selection acting on different organisms is tremendously varied. For example, the effective population size estimate for humans is about 104, 26 which represents the most recent bottleneck in human evolution, while microbial effective population sizes can be as high as 10.10 For this reason, the sequences of human proteins have not been as stringently selected as those in microbes with larger effective population sizes. This idea is supported by observations that the fidelity of genetic replication correlates with effective population size,27 and is low for humans relative to species with larger Ne, including flies, worms, and most microbes.
However, even in organisms with relatively small effective population sizes, such as humans, mutations with adaptive benefits beyond the limit of detection in most experimental evolution studies can be subject to positive natural selection. This makes it challenging to utilize experimental results to distinguish mutations that may have been adaptive in natural evolution from those that were non‐adaptive or nearly neutral. The detection limits of experiments also make it difficult to know if a mutation that is experimentally indistinguishable from wild‐type would be neutral, beneficial, or deleterious in nature.
This article focuses on describing why experimental mutational scanning results should be interpreted with care, and in particular with regards to natural evolution. The issues discussed above are of importance in the face of a rapidly expanding technology. However, there are many valuable lessons about natural evolution that experimental mutational scans have and can continue to provide. It is worth noting that not all adaptive mutations are of such small effects that they cannot be observed using experimental mappings approaches. Indeed, the resolution limit of experimental mappings continues to improve, and many experiments have identified mutations that are adaptive to specific environmental conditions.28, 29 In addition, by performing mutational mappings of both protein fitness and organism fitness, links between biochemistry (e.g., enzyme proficiency) biophysics (e.g., protein stability) and physiology (e.g., growth rate) can be revealed that are valuable for developing quantitative models (e.g., see Refs. 2, 8, 9, 30, and 31).
Experimental mutational mappings also have a tremendous potential in helping us understand polymorphism and genetic disease (Fig. 3). Mutations that are deleterious enough to be identified as such in mutational scanning experiments are almost certainly deleterious relative to 1/Ne, which means that they will be deleterious in natural populations.
Figure 3.
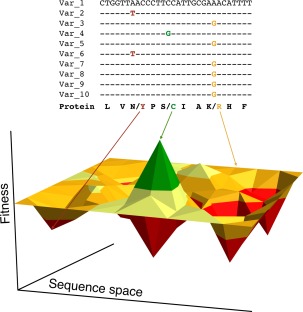
Potential use of experimental fitness maps to interpret polymorphisms observed in natural populations. The top panel illustrates potential DNA sequences from 10 individuals in a population. In these 10 theoretical sequences there are three polymorphisms that cause amino acid changes in the encoded protein. Interpreting the potential impact of polymorphism on fitness and health is a major challenge in the genomic era. Fitness maps from mutational scanning as theoretically illustrated in the bottom panel could be a powerful approach to interpret the impacts of polymorphisms in humans and other natural populations.
As more individuals of a species are sequenced, we learn about the diversity of genetic polymorphism, but polymorphism data alone do not reveal which polymorphisms are deleterious or contribute to disease. Mutational mappings approaches have recently been adapted to mammalian cells in culture32 and have been utilized to identify drug resistant mutations in oncogenic bRAF, the principle driver of melanoma.33 Further, the strongest drug‐resistant mutation seen in the laboratory mapping was subsequently observed in samples from patients undergoing drug treatment.34 In addition to identifying the sequence determinants of resistance, the ability to screen mutations under multiple conditions can pinpoint the specific function(s) that mutations are impacting, thus providing insights into the mechanism underlying the disease state. As recently reviewed,35 mutational mapping approaches provide a potential approach to rapidly evaluate the disease propensity of human polymorphisms, and could thereby be influential in the transition to personalized medicine.
Concluding Remarks
Systematic measurements of mutational effects can provide broad insights spanning from protein evolution to protein engineering and personalized medicine. However, several technical and conceptual issues must be kept in mind. The current experimental mappings clearly have a relatively high threshold for acceptance of mutations, such that mutations that would usually be purged in nature (subject to population size considerations as discussed above) exhibit no measurable experimental effects. The modes of laboratory measurements, and the manner by which fitness effects are measured and calculated, vary, presently, to an extent that renders a reliable comparison between these measurements, and between different proteins, a daunting challenge.
Further discussion of the above issues, and in particular, standardization of the manner by which fitness effects are defined and calculated,22 as well as standardization of datasets and their public availability, would greatly increase the long‐term value of systematic mappings of protein mutations.
Acknowledgments
DST is the Nella and Leon Benoziyo Professor of Biochemistry. The authors thank Marc Ostermeier for providing the fitness data for TEM‐1 and for his insightful comments regarding this manuscript.
References
- 1. Fowler DM, Fields S (2014) Deep mutational scanning: a new style of protein science. Nat Methods 11:801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bershtein S, Choi JM, Bhattacharyya S, Budnik B, Shakhnovich E (2015) Systems‐level response to point mutations in a core metabolic enzyme modulates genotype‐phenotype relationship. Cell Rep 11:645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang W, Petrosino J, Hirsch M, Shenkin PS, Palzkill T (1996) Amino acid sequence determinants of beta‐lactamase structure and activity. J Mol Biol 258:688–703. [DOI] [PubMed] [Google Scholar]
- 4. Loeb DD, Swanstrom R, Everitt L, Manchester M, Stamper SE, Hutchison CAIII (1989) Complete mutagenesis of the HIV‐1 protease. Nature 340:397–400. [DOI] [PubMed] [Google Scholar]
- 5. Rennell D, Bouvier SE, Hardy LW, Poteete AR (1991) Systematic mutation of bacteriophage T4 lysozyme. J Mol Biol 222:67–88. [DOI] [PubMed] [Google Scholar]
- 6. Orr HA (2009) Fitness and its role in evolutionary genetics. Nat Rev Genet 10:531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dessau M, Goldhill D, McBride R, Turner PE, Modis Y (2012) Selective pressure causes an RNA virus to trade reproductive fitness for increased structural and thermal stability of a viral enzyme. PLoS Genet 8:e1003102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roscoe BP, Thayer KM, Zeldovich KB, Fushman D, Bolon DN (2013) Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J Mol Biol 425:1363–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS (2006) Robustness‐epistasis link shapes the fitness landscape of a randomly drifting protein. Nature 444:929–932. [DOI] [PubMed] [Google Scholar]
- 10. Firnberg E, Labonte JW, Gray JJ, Ostermeier M (2014) A comprehensive, high‐resolution map of a gene's fitness landscape. Mol Biol Evol 31:1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang L, Mishra P, Hietpas RT, Zeldovich KB, Bolon DN (2013) Latent effects of Hsp90 mutants revealed at reduced expression levels. PLoS Genet 9:e1003600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rockah‐Shmuel L, Toth‐Petroczy A, Tawfik DS (2015) Systematic mapping of protein mutational space by prolonged drift reveals the deleterious effects of seemingly neutral mutations. PLoS Comput Biol 11:e1004421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dykhuizen DE, Dean AM, Hartl DL (1987) Metabolic flux and fitness. Genetics 115:25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kacser H, Burns JA (1981) The molecular basis of dominance. Genetics 97:639–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H (2006) Construction of Escherichia coli K‐12 in‐frame, single‐gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Newton MS, Arcus VL, Patrick WM (2015) Rapid bursts and slow declines: on the possible evolutionary trajectories of enzymes. J R Soc Interface 12: 2015036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bershtein S, Mu W, Shakhnovich EI (2012) Soluble oligomerization provides a beneficial fitness effect on destabilizing mutations. Proc Natl Acad Sci USA 109:4857–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boucher JI, Cote P, Flynn J, Jiang L, Laban A, Mishra P, Roscoe BP, Bolon DNA (2014) Viewing protein fitness landscapes through a next‐gen lens. Genetics 198:461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hughes TR, Roberts CJ, Dai H, Jones AR, Meyer MR, Slade D, Burchard J, Dow S, Ward TR, Kidd MJ, Friend SH, Marton MJ (2000) Widespread aneuploidy revealed by DNA microarray expression profiling. Nat Genet 25:333–337. [DOI] [PubMed] [Google Scholar]
- 21. Geiler‐Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, Drummond DA (2011) Misfolded proteins impose a dosage‐dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci USA 108:680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kowalsky CA, Klesmith JR, Stapleton JA, Kelly V, Reichkitzer N, Whitehead TA (2015) High‐resolution sequence‐function mapping of full‐length proteins. PLoS One 10:e0118193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mayr E, Provine WB (1998) The evolutionary synthesis: perspectives on the unification of biology. Harvard University Press, Cambridge, MA. [Google Scholar]
- 24. Otto SP, Whitlock MC (1997) The probability of fixation in populations of changing size. Genetics 146:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buri P (1956) Gene frequency in small populations of mutant Drosophila. Evolution 10:367–402. [Google Scholar]
- 26. Hayes BJ, Visscher PM, McPartlan HC, Goddard ME (2003) Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res 13:635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M (2012) Drift‐barrier hypothesis and mutation‐rate evolution. Proc Natl Acad Sci USA 109:18488–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hietpas RT, Jensen JD, Bolon DN (2011) Experimental illumination of a fitness landscape. Proc Natl Acad Sci USA 108:7896–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jiang L, Liu P, Bank C, Renzette N, Prachanronarong K, Yilmaz LS, Caffrey DR, Zeldovich KB, Schiffer CA, Kowalik TF, Jensen JD, Finberg RW, Wang JP, Bolon DNA (2016) A balance between inhibitor binding and substrate processing confers influenza drug resistance. J Mol Biol 428:538–553. [DOI] [PubMed] [Google Scholar]
- 30. Bershtein S, Mu W, Serohijos AW, Zhou J, Shakhnovich EI (2013) Protein quality control acts on folding intermediates to shape the effects of mutations on organismal fitness. Mol Cell 49:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lunzer M, Golding GB, Dean AM (2010) Pervasive cryptic epistasis in molecular evolution. PLoS Genet 6:e1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosenberg AB, Patwardhan RP, Shendure J, Seelig G (2015) Learning the sequence determinants of alternative splicing from millions of random sequences. Cell 163:698–711. [DOI] [PubMed] [Google Scholar]
- 33. Wagenaar TR, Ma L, Roscoe B, Park SM, Bolon DN, Green MR (2014) Resistance to vemurafenib resulting from a novel mutation in the BRAFV600E kinase domain. Pigment Cell Melanoma Res 27:124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hoogstraat M, Gadellaa‐van Hooijdonk CG, Ubink I, Besselink NJM, Pieterse M, Veldhuis W, van Stralen M, Meijer EFJ, Willems SM, Hadders MA, Kuilman T, Krijgsman O, Peeper DS, Koudijs MJ, Cuppen E, Voest EE, Lolkema MP (2015) Detailed imaging and genetic analysis reveal a secondary BRAF(L505H) resistance mutation and extensive intrapatient heterogeneity in metastatic BRAF mutant melanoma patients treated with vemurafenib. Pigment Cell Melanoma Res 28:318–323. [DOI] [PubMed] [Google Scholar]
- 35. Young DL, Fields S (2015) The role of functional data in interpreting the effects of genetic variation. Mol Biol Cell 26:3904–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]