

Abstract
There has been much debate about the extent to which mutational epistasis, that is, the dependence of the outcome of a mutation on the genetic background, constrains evolutionary trajectories. The degree of unpredictability introduced by epistasis, due to the non‐additivity of functional effects, strongly hinders the strategies developed in protein design and engineering. While many studies have addressed this issue through systematic characterization of evolutionary trajectories within individual enzymes, the field lacks a consensus view on this matter. In this work, we performed a comprehensive analysis of epistasis by analyzing the mutational effects from nine adaptive trajectories toward new enzymatic functions. We quantified epistasis by comparing the effect of mutations occurring between two genetic backgrounds: the starting enzyme (for example, wild type) and the intermediate variant on which the mutation occurred during the trajectory. We found that most trajectories exhibit positive epistasis, in which the mutational effect is more beneficial when it occurs later in the evolutionary trajectory. Approximately half (49%) of functional mutations were neutral or negative on the wild‐type background, but became beneficial at a later stage in the trajectory, indicating that these functional mutations were not predictable from the initial starting point. While some cases of strong epistasis were associated with direct interaction between residues, many others were caused by long‐range indirect interactions between mutations. Our work highlights the prevalence of epistasis in enzyme adaptive evolution, in particular positive epistasis, and suggests the necessity of incorporating mutational epistasis in protein engineering and design to create highly efficient catalysts.
Keywords: epistasis, enzyme evolution, adaptive mutations, evolutionary constraints, directed evolution, protein engineering, rational design
Abbreviations
- DHFR
dihydrofolate reductase
- MIC
minimum inhibitory concentration
Introduction
The evolution of an enzyme toward a new function often requires the accumulation of multiple adaptive mutations. However, neutral and deleterious mutations predominate among all possible mutational steps and adaptive mutations are scarce. A strategy to efficiently identify functional mutations is pivotal for our understanding of enzyme evolution and our ability to engineer and design novel enzymes. Functional mutations can be found in the first shell (residues directly interacting with the ligand) of the active site, and are typically responsible for gain or loss of interactions between enzyme and substrate. Remote mutations, located in the second‐ (residues interacting with the first shell) and third‐ shells (residues beyond the second shell) of the active site, can also contribute to functional adaptation because of the intertwined nature of amino acids network within an enzyme.1, 2 Recent promising advances in bioinformatics and computational structural biology have rendered de novo enzyme design feasible, using computational algorithms to predict the phenotypic effects of mutations.3, 4, 5 For example, tools have been developed to rationally design enzymes through the creation of “smart” mutant libraries, where prediction guides functional “hotspots” or “islands” to explore, enabling a relatively small screening effort to isolate functional mutations.6, 7, 8, 9, 10 Despite these advances, genuine success is rare and creating de novo catalysts in the laboratory remains a challenge: only a handful of examples have been reported to yield new enzymes with high catalytic efficiency (k cat/K M> 106) in the laboratory.11, 12
On the other hand, growing evidence from molecular evolution and evolutionary biochemistry indicates that the prediction of functional mutations can be a daunting task.13, 14 Epistasis, that is, non‐additive interactions between mutations, implies that mutations exhibit different phenotypic effects, depending on the genetic background on which they occur.15, 16 For example, a functional mutation may only confer a beneficial effect upon the prior fixation of permissive mutations.17, 18, 19, 20 Conversely, the adaptive potential of a mutation can be diminished or become deleterious in the presence of other, restrictive mutations.21, 22 Thus, the accumulation of a subset of mutations that collectively provide a significant change in enzymatic function may be strongly contingent on each individual step of a mutational trajectory.23, 24 Functional mutations can be masked in the background of a starting point (for example, wild‐type enzyme) if they appear neutral or deleterious until other mutations epistatically render them beneficial on an alternative background (e.g., during adaptive trajectories). Therefore, predicting beneficial mutations may not be achievable,25 without prior knowledge of the epistatic interactions that arise upon combining mutations. Numerous studies have demonstrated that epistasis constrains the fixation of functional mutations during adaptive enzyme evolution.26, 27, 28, 29, 30 Yet, a general consensus on the role of mutational epistasis, based on a quantitative survey of its type and prevalence, remains to be established. Trends that unveil how epistatic mutations influence the accessibility of different adaptive trajectories will provide valuable insights into protein engineering and design: should we integrate epistatic interactions into the prediction and identification of functional mutations?31 And if so, how can we account for these constraints at the molecular level?
In this study, we provided a quantitative survey of epistasis during adaptive evolution. Based on nine previous studies that examined the evolution of novel functions in both natural and laboratory systems, we systematically analyzed the epistatic effects of functional mutations that were collectively responsible for large increases in new function. To investigate how epistasis hinders the predictability of functional mutations from a starting enzyme, we characterized the type and degree of epistasis occurring between two genetic backgrounds: the starting enzyme (e.g. wild type) background and the background of the intermediate variant in which the mutation was fixed during its evolutionary trajectory. We also analyzed the structural position of these functional mutations, in order to identify the physical interactions that allowed distant mutations to exhibit significant mutational epistasis. We defined several molecular mechanisms that mediate strong epistasis. Finally, we highlight the importance of integrating epistatic interactions for the identification and prediction of functional mutations in protein design and engineering.
Results
Dataset collection and analysis of mutational epistasis
We chose nine case studies from previously published work describing adaptive trajectories of enzymes towards a new function, for example, an increase in catalytic efficiency for a new substrate or a lower affinity for an inhibitor (Table 1). In these examples, the authors characterized the functional effects of adaptive mutations, that is, their dependence on the genetic background. Some explored all possible combinations (2n) of a given set of n mutations to depict an empirical fitness landscape;32, 33 others studied the mutational effects on a restricted subset of genetic backgrounds, by a posteriori engineering each individual mutation onto the wild‐type background. Out of nine examples, four represent adaptive trajectories that occurred in nature. Among these four, two examples originate from evolution observed in clinical isolates of pathogenic bacteria: TEM‐1 β‐lactamase to hydrolyze cefotaxime, a third generation cephalosporin34 and a dihydrofolate reductase (DHFR) to circumvent an anti‐malaria drug inhibitor, pyrimethamine;35 and two are reconstructed from evolutionary transitions between two extent enzymes: atrazine chlorhydrolase, AtzA, and melamine deaminase, TriA,36 and two haloalkane dehydrogenases, LinBUT and LinBMI. 37 Three examples represent adaptive trajectories created by laboratory directed evolution: Bc‐II β‐lactamase was evolved towards the hydrolysis of a first generation β‐lactam antibiotic, cephalexin.38 PTE, a phosphotriesterase, was evolved toward arylesterase activity,11 and then back to its original PTE activity.39 The last two studies represent the rational design of enzymes in the laboratory, through the improvement of enantioselectivity in ANEH epoxide hydrolase40, 41 and PAMO Baeyer−Villiger monooxygenase towards a thioether.42, 43
Table 1.
Summary of the Nine Evolutionary Trajectories
| Enzyme | Catalytic activitya | Target moleculeb | Number of mutationsc | Most likely trajectoryd | Sourcef | References |
|---|---|---|---|---|---|---|
| TEM‐1 | β‐lactamase | Cefotaxime | 5 | G238S > E104K > A42G > M82T > (g4205a) | Clinical isolates | 34 |
| DHFR | Dihydrofolate reductase | Pyrimethamine | 4 | S117N > S58R> N50I> I73L | 35 | |
| AtzA | Atrazine chlorhydrolase | Melamine | 9 | S331C > F84L > N328D > E125D > T219P > T217I > V92L > G255W > I253L | Reconstruction of a possible trajectory between extent enzymes | 36 |
| LinBUT | Haloalkane dehydrogenase | β‐hexachloro cyclohexane | 7 | A274H > I134V > I138L > A112V > M253I > A135T > A81T | 37 | |
| ANEH | epoxide Hydrolase | (S)‐1 enantiomer | 5 | B > C > D > F > Ee | Rational design (ISM) | 40, 41 |
| PAMO | Baeyer−Villiger monooxygenase | (R) enantiomer | 4 | A442N > P440F > I67Q > L443I | 42, 43 | |
| Bc‐II | β‐lactamase | Cephalexin | 4 | G262S > L250S > N70S > V112A | Directed evolution (random) | 38 |
| PTE | Phosphotriesterase | Arylester (2NH) | 26 (for) | see ref. 39 (PTEWT‐> PTEa E) and Table S1 | 11, 39 | |
| Paraoxon | 14 (rev) | see ref. 39 (PTEa E‐> neoPTE) and Table S1 | 39 |
Catalytic activity refers to the native activity of the enzyme.
Target molecule indicates the substrate or inhibitor for which the enzyme evolved.
Number of mutations fixed during the adaptive trajectory.
Most likely trajectory extracted from the literature or reconstructed (see Mat. and Meth.). The order of fixation of mutations in the trajectory proceeds from left > right.
Source refers to the experimental framework that enabled the isolation of combinatorial mutations.
For ANEH, we used the same labelling of the mutations as ref. 40. B‐E refers to clusters of mutations co‐mutated in ANEH by ISM. See Supporting Information Table S1 for detailed clustering of mutations.
ISM iterative saturation mutagenesis, for forward, rev reverse, 2NH 2‐naphthyl hexanoate.
We then determined a mutational trajectory for each adaptive evolution, that is, the order in which mutations were accumulated in a stepwise manner during the evolutionary trajectory. For the directed evolution examples (PTE and Bc‐II), we retained the mutational order as it appeared in the evolutionary experiments. For the natural evolution examples, we either followed the plausible trajectory proposed in the original article, or we reconstructed a likely trajectory such that a mutation providing the highest improvement of the enzyme fitness among the variants would be selected at each round (Table 1). First, we observed that approximately half of adaptive trajectories exhibit diminishing returns (Supporting Information Fig. S1), a distinctive pattern of evolutionary and optimization processes, in which the enzyme fitness improvement per mutation is large in early rounds of evolution and then becomes more incremental as evolution proceeds, indicating that the evolved enzymes have approached a local fitness plateau (or local maximum).44, 45 However, in LinBUT, ANEH, PAMO and Bc‐II, the functional improvements did not decelerate but rather steadily increased which suggests that these enzymes have not reached the end of their evolutionary potential.46
Next, we quantified the extent of epistasis within each trajectory toward a new function by comparing the effect of a mutation on two different genetic backgrounds: the starting enzyme (e.g. wild type) background and the background of the intermediate variant on which the mutation was fixed during the trajectory. We calculated the fold change in enzyme fitness (F) provided by a mutation i on the wild‐type background (ΔFwt,i=Fwt+i/Fwt) and the change caused by the same mutation on the intermediate variant j in the trajectory, (ΔFj,i=Fj+i/Fj) (Supporting Information Table S1). Then, epistasis was determined by comparing the fold changes in the trajectory over the wild‐type background (ΔFj,i/ΔFwt,i). For the sake of simplicity, we considered that ≥1.5‐fold change is significant, and less than 1.5‐fold change is neutral (or nearly neutral). Whereas each experimental system exhibited different experimental errors for the enzyme fitness measurement, most studies reported error rates less than 1.5‐fold. It should be noted that the enzyme fitness represents a physicochemical property of the enzymes, such as: enzymatic catalytic efficiency (k cat/K M), enzymatic activity in crude cell lysate, inhibitory concentration by an antibiotic or inhibitor [minimum inhibitory concentration (MIC) and IC50], and enantioselectivity factors (E) (Supporting Information Table S1). Whereas these properties are not necessarily correlated to organismal fitness, they are described as selectable traits in natural or laboratory evolution. In order to circumvent the multiplicity of fitness units presented, we restricted our analysis to the fold change in enzyme fitness caused by a mutation, as measured by the ratio of the mutational effect on the trajectory over the wild type (ΔFj,i/ΔFwt,i). We classified the mutations into five different subtypes, according to their epistatic effects and single effect on each independent genetic background. i) Neutral designates mutations that show less than 1.5‐fold change in enzyme fitness on both backgrounds (0.7 < [ΔFj, i and ΔFwt,i] < 1.5). (ii ‐ v) Functional refers to non‐neutral mutations that exhibit >1.5‐fold improvement in either genetic background (ΔFj, i or ΔFwt, i > 1.5). Among functional mutations, ii) no epistasis refers to mutations that do not significantly alter the enzyme fitness depending on the background (mutations are additive, ΔFj, i/ΔFwt, i ∼ 1). (iii – iv) Positive epistasis applies to a mutation that becomes more beneficial when combined with prior substitutions on the trajectory, compared to its effect on the wild‐type background (ΔFj, i/ΔFwt, i > 1.5). It can be divided in two subclasses: iii) Positive magnitude epistasis refers to cases where the effect is neutral or positive on the wild‐type background and is further amplified on the trajectory (ΔFj, i ≫ ΔFwt, i > 0.7 or ΔFj, i ≫ ΔFwt, i > 1.5); and iv) Positive sign epistasis, which refers to mutations causing a deleterious effect on the wild‐type background but a beneficial effect on the trajectory (ΔFwt, i <0.7 and ΔFj, i > 1.5). v) Negative epistasis applies to a mutation that becomes less beneficial on the trajectory background compared to its original effect on the wild type (ΔFj, i/ΔFwt, i <0.7).
Positive epistasis is predominant in the adaptive trajectories of enzymes
In all trajectories, a significant fraction of mutations exhibits epistasis between the two genetic backgrounds (Figs. 1 and 2). Of 59 analyzed mutations, 20 (34%) are classified as non‐functional, while 66% (39 mutations) are functional mutations [Fig. 1(A)]. The former, which were eliminated from our analysis, may be hitchhikers‐ (bona fide neutral mutations), nearly neutral‐ (i.e. marginally beneficial or deleterious) or stability‐enhancing‐ (affecting other enzymatic properties aside from the catalysis) mutations. Seven of the functional mutations are beneficial but exhibit no or weak epistasis (i.e. their effects are additive), which represents 12% of total amount of mutations and 18% of functional mutations [Fig. 1(A)]. Thus 82% of functional mutations (54% of total mutations) are classified as “epistatic mutations”. These mutations can be further divided into 25% that exhibit negative magnitude epistasis (eight mutations, and 14% of total mutations), and 75% that cause positive epistasis (24 mutations, and 41% of total mutations). 13% of the functional mutations (∼8% of total mutations) display positive magnitude epistasis: their beneficial effect (>1.5‐fold increase) onto the wild‐type background became even more beneficial when combined in the trajectory. 33% (22% of total mutations) also belong to positive magnitude epistasis but their effects in the wild‐type were originally neutral. Moreover, ∼15% (10% of total) showed positive sign epistasis, in which the effect of the mutation was deleterious on the wild‐type background but subsequently turned beneficial as they occurred in the trajectory. Thus, 49% of functional mutations (32% of total mutations) did not appear to be beneficial in the background of the wild‐type, and then became beneficial owing to epistasis [Fig. 1(A)].
Figure 1.
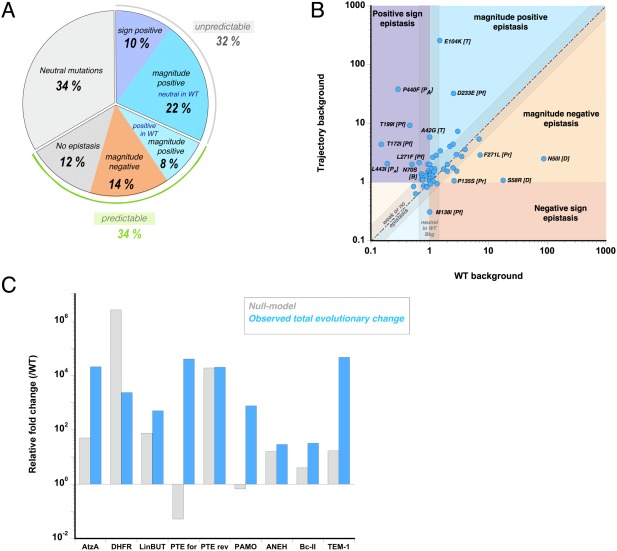
Distribution of epistatic effects between WT and trajectory backgrounds. (A) Fraction of mutations belonging to the five following types of epistasis: (I) positive sign epistasis (dark blue background), (II) Positive magnitude epistasis (light blue), (III) negative magnitude epistasis (light orange) or (IV) no epistasis and (V) Neutral or nearly neutral mutational effects on the wild‐type background are highlighted by a grey background. (B) The effects caused by each mutation are plotted on the trajectory background versus the WT background. The background of the plot is divided according to the above‐mentioned categories (I‐V). The dotted line represents neutral and additive mutations. The letter in bracket represents the enzyme, where T stands for TEM‐1, Pf for PTE‐for, Pr for PTE‐rev, D for DHFR, PA for PAMO and B for Bc‐II (C) Differences between the expected total functional change according to the Null Model (additive increase from all mutations) on the wild‐type background (grey bar) and the observed total functional change between the activity of the starting‐ and evolutionary‐ end points (blue bar).
Figure 2.
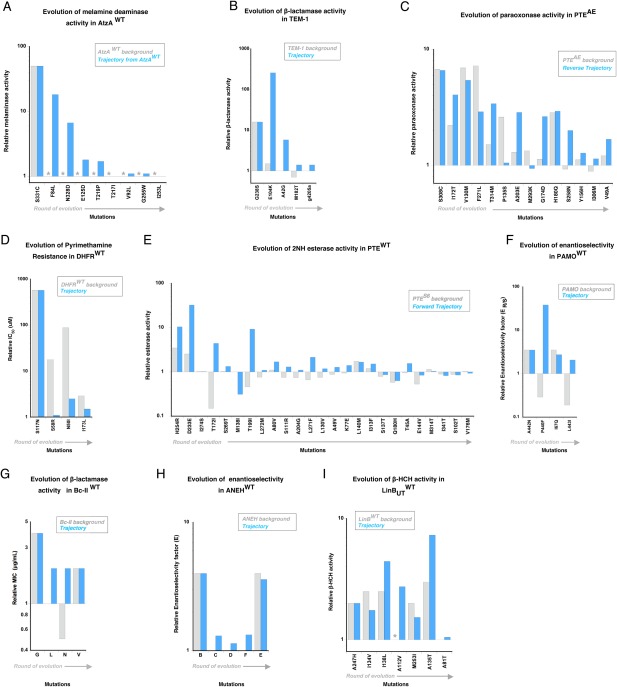
Functional changes caused by the mutations fixed within nine adaptive trajectories. The fold change in enzyme function, induced by mutations, is depicted as a bar, on the wild‐type background (grey) and as it occurs in the evolutionary trajectory (blue). Evolution of (A) melamine deaminase activity in AtzA, (B) β‐lactamase activity in TEM‐1, (C) paraoxon hydrolysis in PTEAE (reverse trajectory), (D) pyrimethamine binding in DHFR, (E) 2NH hydrolysis in PTEWT (forward trajectory), (F) enantioselectivity in PAMO (G) β‐lactamase activity in Bc‐II, (H) enantioselectivity in ANEH and (I) haloalkane dehydrogenase activity in LinBUT. Mutations are ordered according to their occurrence in the trajectory (from left to right). Detailed values are provided in Supporting Information Table S1. Non‐detectable activity/binding levels are shown as grey stars.
Positive epistasis (magnitude or sign; eight out of the nine trajectories) is the most common form of epistasis present along the evolutionary trajectories [Fig. 1(B), 2]. Positive epistasis means that an adaptive trajectory toward a new function will be constrained by the order of appearance of mutations: the beneficial effect of later mutations is contingent on the fixation of earlier ones.15, 19 AtzA represents the most drastic example: only one (S331C) out of nine mutations provides a beneficial effect on the AtzAWT background, no other mutation that became fixed at later stages yielded a detectable melaminase activity on the WT background [Fig. 2(A)]. Hence, the first mutation S331C played a permissive role for the fixation of the subsequent eight mutations that only turned beneficial in later rounds; the adaptive trajectory relied completely on the initial mutation. The evolution of TEM‐1 is also dominated by positive epistasis (three out of four mutations), whereby the first mutation G238S facilitates the fixation of the following adaptive mutations [Fig. 2(B)]. The reverse trajectory of PTE appears overall less epistatic, as 5 out of 14 mutations show positive epistasis [Fig. 2(C)]. By contrast, the trajectory of DHFR follows the opposite trend as all mutations exhibit negative magnitude epistasis: while each mutation provides a great enhancement on the wild‐type background, the beneficial effects become much smaller than expected once the first mutation is fixed [Fig. 2(D)]. Interestingly, the evolution of DHFR is the only example in which the enzyme was evolved for a loss of recognition of the target molecule (pyrimethamine inhibitor). In all other examples, the selected trait was a gain of recognition of a new molecule by increasing a promiscuous activity. Thus, the opposite trends between DHFR and other enzymes may be associated with the trait selected by evolution. Further complications of epistasis and contingency can be observed by the effect of the available, remaining, mutations at each evolutionary step along a trajectory (Supporting Information Fig. S2 and Supporting Information Table S2). However, as data are only available for a subset of the selected studies that performed combinatorial mutational analysis, we restricted our analysis to only two genetic backgrounds.
The predominance of positive epistasis was also observed when we compared the overall functional increase in the trajectories (difference between starting and most evolved mutant) and the increase calculated from the null model, which assumes that the collective increase of all mutations relies on non‐epistatic cumulative effects of all single point mutations on the wild‐type background [Fig. 1(C)]. In six out of nine examples, the predicted functional increase based on the null‐model fell short compared to the actual increase obtained over the whole trajectory. These differences can be large: TEM‐1, AtzA, PTE‐for and PAMO, all exhibit more than two‐orders of magnitude difference between the predicted and observed increase. The latter two examples are particularly striking cases of unpredictability: the null model anticipates a strong negative effect upon combining 4 and 25 mutations, respectively, whereas together they epistatically contribute to more than 103‐ and 105‐fold increases in their respective trajectories.
Taken together, our observations highlight the prevalence of epistasis in adaptive trajectories. The strong tendency for positive epistasis complicates the prediction of functional mutations. In half of the cases within our analysis, mutations were identified as being beneficial in both backgrounds; hence these functional mutations are predictable, at least qualitatively. Yet, the remaining functional mutations are unpredictable, as their beneficial effects cannot be anticipated due to their negative or neutral effect on the starting background.
Indirect interactions between mutations can cause strong epistasis
We analyzed mutational sites within protein crystal structures to determine how the position of mutations relates to the degree of epistasis. First, we measured the distance between mutations and a key functional component in the active site, for example, a catalytic metal ion, a nucleophile or a bound ligand (analogue or inhibitor) (see Supporting Information Table S1 and Materials and Methods for each case study). Most functional mutations, in particular the highly beneficial ones, are located less than 10 Å away from key active site residues (Supporting Information Fig. S3), which is consistent with previous work investigating the correlation between structural position and mutational effect.47 These results indicate that most functional mutations are either directly interacting with the substrate or rather, are located within the second and third shells of the active site and indirectly affecting the enzymatic function. The same trend is observed when plotting the epistasis effect (ΔFj,i/ΔFwt,i) versus the distance from a key active site feature [Fig. 3(A)]. Mutations exhibiting strong epistasis are also clustered within the first and second shells of the active site (<10 Å from a key residue). Over the course of evolution, an increase in mutation fixation within the outer shell of the protein is observed compared to the core (Supporting Information Fig. S4). Next, we measured the distance between a mutation showing strong epistasis (arbitrarily defined as 3< ΔFj,i/ΔFwt,i, and 0.5 >ΔFj,i/ΔFwt,i) and all prior mutations accumulated in the trajectory [Fig. 3(B)]. In ∼40% of cases (five out of 13 mutations: PTE‐for‐D233E and ‐L271F, PAMO‐P440F and ‐L443I, and Bc‐II‐N70S), at least one residue located less than 4 Å away from the epistatic mutation was mutated, indicating that direct interactions contribute to epistasis. However in the remaining cases, epistasis is caused by indirect interactions between mutations (>8 Å from the closest prior mutation): TEM‐1‐E104K and DHFR‐N50I (∼8–10 Å); and PTE‐for‐T172I and ‐L271F and TEM‐1‐A42G (>10 Å). Thus, a direct interaction to either the substrate or the prior mutations is not compulsory for strong epistasis to occur. Indirect and long‐range interactions can significantly contribute to changes in mutational effects, indicating that complex network effects can affect catalysis.
Figure 3.
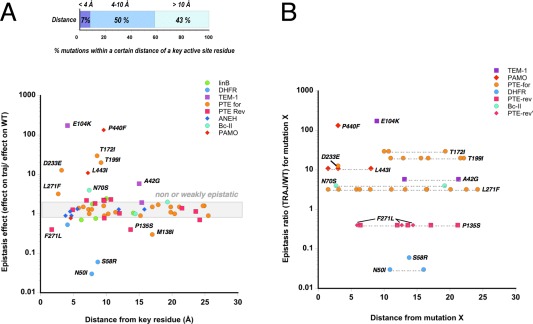
Localization of epistatic mutations on protein crystal structures. (A) Epistatic effect of mutations versus distance between these mutations and a key active site residue (the designated key residue for each enzyme is described in Supporting Information Table S1). The highlighted grey area indicates mutations for which the epistasis effects fall between 0.7 and 1.5‐fold change, that is, non‐epistatic substitutions. (B) Epistasis effect for a set of highly epistatic mutations versus distance between these mutations and others that are fixed earlier in the trajectory. PTE‐rev‐F271L and P‐135S, which have the same epistasis ratio (0.4), are depicted in pink, as triangles and squares, respectively.
Molecular basis of epistasis
A more fundamental question, and challenge, that arises from these observations is the identification of the molecular mechanisms that underlie epistasis. In several cases, the crystal structure of the evolved variant(s) was obtained and compared to that of the wild‐type enzyme. Here, we highlight these examples and discuss, in the light of previous observations, possible molecular mechanisms (Fig. 4 and Supporting Information Fig. S5). Four mechanisms can be derived from the structural observations. (i) Direct interactions between epistatic mutations, of which one mutation directly interacts with the substrate. For example, the forward evolution of PTE proceeded via two key direct interactions between the fist shell mutation H254R and subsequent mutations L271F and D233E [Fig. 4(A)]. The fixation of D233E and L271F stabilized the position of R254, which in turn improved the interaction between R254 and the substrate.11, 39 The effect of D223E and L271F increased >10‐ and 3‐fold in the trajectory, respectively, after the fixation of H254R compared to the wild‐type background. (ii) Direct interactions between mutations, but no direct interaction between the substrate and the mutations. Although there is no structure for the evolved variant, PAMOZGZ‐2 may fall into this category as the proximity of mutations P440F, A442N and L443I in the wild‐type structure suggests that they interact with each other (through hydrogen bonding interactions).48 However these mutations are unlikely to directly interact with the substrate, rather they may contribute to a functional change by altering the position of R33742 [Supporting Information Fig. S5(A)]. (iii) Indirect interaction between mutations, and direct interaction between the substrate and one of the mutations. The first mutation (S117R) in DHFR interacts with the inhibitor, pyrimethamine49 [Fig. 4(B)]. The subsequent mutations (58 and 50) are located a distance from S117R and do not interact with the substrate, but show strong negative epistasis (0.06‐fold and 0.03‐fold, respectively). Similarly, the first mutation G238S in TEM‐1 is directly interacting with the substrate (although it may also resolve a steric conflict50), and the second mutation (E104K) interacts with neither the first mutation nor the substrate [Fig. 4(C)]. Yet, the effect of E104K is altered by >100‐fold upon the fixation of G238S, which to date remains poorly understood.50 It is likely that long‐range interactions between these two residues propagate through a loop (Ω‐loop), potentially via position 240. (iv) Indirect interaction between mutations and no direct interaction between the substrate and the mutations. These examples represent the most complicated epistatic networks for enzyme function. The long‐range epistatic effects may be associated with fine‐tuning of key catalytic components in the active site and/or change in conformational dynamics. In Bc‐II, strong positive sign epistasis occurs between mutations G262S (round 1) and N70S; both of these mutations are located below the active site floor and are distant from the substrate46 [Fig. 4(D)]. While the first mutation G262S causes a 0.5–1.0 Å shift of a catalytic Zn2+, a later mutant containing N70S displays altered dynamics for several active site loops. The effect of N70S, which is deleterious in the WT background, showed strong positive sign epistasis and only became beneficial in combination with G262S.46 Repositioning of metal ions was also observed in the evolution of PTE [Supporting Information Fig. S5(B)] and a displacement of non‐mutated active site residues has been observed in several other systems such as ANEH41 and TEM‐1 [Supporting Information Fig. S5(C‐D)].
Figure 4.
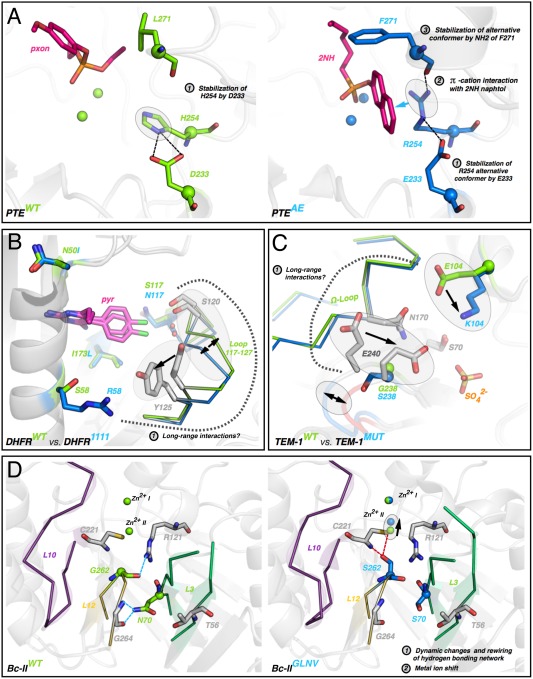
Molecular mechanisms underlying strong cases of epistasis. (A) Direct interactions between mutations in PTE. The first round mutation H254R directly interacts with the 2NH substrate (magenta sticks). Then subsequent mutations D233E/L271F directly stabilize the position of R254 in PTEAE (evolved mutant, right). (B) Indirect interaction between R58S and S117N in DHFR. R58 may cause a shift of loop 117‐127, which may reposition N117 and S120, emphasizing the steric clash with pyrimethamine (pyr, magenta sticks). (C) Indirect interactions between E104K and G238S in TEM‐1. The two mutations may interact through the Ω‐loop and the shift of E240 that is likely contacting the substrate. (D) Indirect interactions between mutations G262S and N70S originate from a rewiring of the hydrogen‐bonding network between loops L12 and L3, causing a displacement of a catalytic zinc ion.
Overall, these molecular descriptions of epistasis suggest that long‐range interactions that cause strong epistasis are rather common. The interactions could act through various ways, including loop repositioning, protein dynamics, or through displacement of active site residues or co‐factors.
Discussion
Our comprehensive characterization demonstrates that epistasis, and in particular positive epistasis, is widespread during the adaptive evolution of enzyme functions. Despite a broad range of enzyme fitness parameters used in each experiment, eight out of nine case studies are largely driven by positive epistasis within the evolutionary trajectory. Positive epistasis indicates that mutations that are fixed at early rounds of evolution play a permissive role for the beneficial effects of later mutations. Thus, evolutionary trajectories may be restricted and the accumulation of mutations limited to a certain order by positive epistasis, particularly at later stages.51 This implies that there are actually fewer available trajectories than one would predict from the total number of fixed mutations separating the wild‐type from an optimized genotype. Observations from previous studies have suggested that beneficial and adaptive mutations are very rare; 0.01–1% of single amino acid substitutions are beneficial, 30–50% are highly deleterious, 50–70% are nearly neutral or neutral,52 indicating that only a handful of mutations can increase catalytic activity. Therefore, the scarcity of available functional mutations, combined with positive epistasis restricting their fixation, indicate that only a few evolutionary trajectories may be accessible. Indeed, a number of natural and laboratory evolution studies have demonstrated that the evolution of protein function from a given starting point is repeatable, that is, only a subset of functional mutations are repeatedly fixed in parallel experimental evolution,51, 53, 54, 55 where the same substitution appears independently in distinct lineages.56, 57, 58 On the other hand, a few cases demonstrated that distinct mutational trajectories could arise, because of constraints exerted by negative sign epistasis.21, 22 Taken together, the evolution of enzymes from a given genetic background may be highly deterministic, meaning that only a few mutational trajectories may originate from a given starting point and that the evolution has to follow a certain order of occurrence to accumulate beneficial functional mutations.
It should be noted, however, that the observation of prevalent positive epistasis is only applied to mutations that collectively comprise one adaptive trajectory, but may be irrelevant to how epistasis prevails across a whole protein fitness landscape. For example, the analysis of homologous extant proteins that (neutrally) diverged in evolution suggests a lesser extent of epistasis.28, 59 In order to obtain a complete picture of a protein fitness landscape, one should ideally cover the complete landscape surrounding a starting point, within at least several mutational steps.33 Under these conditions, the size of such a library becomes extremely large, which cannot be explored by current technologies. Alternatively, exploring epistasis through the creation of multiple alternative trajectories that can be compared to each other would be highly valuable. These trajectories can be obtained from natural‐, or parallel‐ experimental evolution repeatedly starting from a unique gene,22 from various orthologous genes,29, 60 toward distinct targets21, 61, 62 or by evolving an enzyme back to its original function.39
For enzyme engineering and design, the prevalence of positive epistasis emphasizes the difficulty of predicting beneficial functional mutations. Our observations indicate that half of mutational effects are unpredictable based on the sole enzyme fitness effect conferred on the starting genetic background. Recent advances have extended the available toolkit to computationally predict functional mutations based on protein crystal structures;3, 63, 64 some of which improve their prediction capacity by integrating epistasis.65 High‐throughput screening technologies combined with deep sequencing, should enable the exploration of vast mutational sets, which may be generated by mutational scanning.66, 67 Yet, these technologies predominantly explore mutations that are located within a single or a few mutational steps away from a starting point. Thus, even if these platforms can effectively identify a number of single point functional mutations, combining these mutations may still not be able to provide the desired (and predicted) functional change. Consistent with previous studies,28, 68, 69 our analysis reveals that a significant number of strong epistatic effects stem from long‐range interactions, suggesting that epistatic networks are highly intertwined and can be mediated by conformational dynamics.13, 70, 71, 72 It is essential that we understand the molecular mechanisms that result in mutational epistasis in order to enhance our predictive ability for efficient engineering of novel enzymes. Experimental evolution strategies, when used on multiple genetic backgrounds and under different conditions, should elucidate the influence of epistasis on evolutionary outcomes, and allow us to identify genotypes that display a higher probability to adapt.
Materials and Methods
Enzyme fitness measurements
To directly compare the nine evolutionary trajectories, we reported on the enzyme fitness, that is, on biophysical properties that were selected by evolution, which should not be confused with organismal fitness. While the IC50 of DHFR mutants are closely related to the organismal fitness, this may not be the case for kinetic parameters. For example, in case of Bc‐II, MIC values increase over the course of the evolution whereas k cat/K M values do not linearly correlate to fitness improvements,38 hence we used MIC values, such as for TEM‐1. For ANEH, we used the data found in the Supporting Information of reference,40 which provides E‐values for all possible combinations. Note that they slightly differ from the values presented in the main article by Reetz et al. For PAMO, the authors provided the ΔΔG ‡ reporting on the difference in the activation energy between both enantiomers, from which, to simplify, we calculated the enantiomeric ratio ER/S (preferential conversion of the enantiomer R over S) from Eqs. (1) and (2) at 298K:
| (1) |
| (2) |
For AtzA and LinBUT, we used the Michaelis‐Menten parameters (k cat/K M) provided. For DHFR, the authors supplied ln (IC 50+1) from a fit to growth rate curves,35 from which we isolated the IC 50 from Eq. (3):
| (3) |
For the PTE‐rev trajectory, we extracted crude lysate activity values from reference.39 Note that due to the complexity of both PTE trajectories, that is, having multiple mutations fixed at a given round and/or shuffling recombination, the fold change on the trajectory background were all experimentally re‐measured by the authors (calculations provided in Supporting Information Table S1), instead of dividing the catalytic activity of the mutants at round n+1/round n, as for the other seven studies (see reference 39 for more details). Furthermore, to ensure full consistency, we used the re‐measured catalytic activity values of the parent variants (R1, R2, etc…) to draw the evolutionary trajectories and perform calculations, instead of the trajectory values originally reported. We performed the same calculations for the PTE‐for trajectory, hence values were not extracted from Ref. 11, but experimentally re‐measured and provided as a personal communication from Dr. Miriam Kaltenbach (manuscript in preparation).
Trajectories
For the directed evolution examples (PTE‐for, PTE‐rev and Bc‐II), we retained the mutational order as it appeared at each round of the evolutionary experiments. In most cases, to select a unique trajectory for each enzyme, we followed the plausible trajectory suggested by the authors in the original article, whether natural (AtzA) or artificial (ANEH, PAMO). Note that for ANEH, the authors recapitulated each evolutionary step to letters A‐>F, which sometimes contain more than one mutation. In a few cases, we arbitrarily reconstructed a most likely trajectory such that each mutational step provides the highest enzyme fitness improvement possible (TEM‐1 and LinBUT). For LinBUT, we selected mutations based on the increase in the second conversion step, more relevant for the LinBMI‐type of activity, as suggested by the authors.37 For DHFR, we chose the following trajectory: S117 > S58R> N50I> I173L (labeled 0010> 0110 > 1110 > 1111 in the original papers), due to its accessibility, high frequency and common occurrence in Plasmodium falciparum 27 and Plasmodium vivax.35 This trajectory is not the favored one in P. vivax according to the authors, but this is due to the use of a multi‐parameter analysis including a combination of growth rate, drug concentration, selection and drift. To simplify the read‐out in our comparison, we selected the trajectory that provided a steady increase in IC50, and fixed all four mutations.
Structural analysis
Wild‐type and (when available) mutant structures were retrieved from the pdb database. AtzA (pdb: 4v1y) was excluded from this analysis due to the lack of detectable melaminase activity on the wild‐type background, which prevented the calculation of the epistasis ratio (ΔFj,i/ΔFwt,i). Distances were measured on the wild‐type structure using pymol,73 between the closest atoms of a key active site residue and of the residue later mutated, as follows. We used a metal ion for Bc‐II (pdb: 1bc2, Zn2+ (II)), a ligand for DHFR (pdb: 2bl9, pyrimethamine), LinBUT (pdb: 2bfn, 1,2‐dichloropropane), PTE–for (pdb: 4pcp, overlaid 2‐naphtyl hexanoate analogue from pdb: 4e3t), PTE‐rev (pdb: 4pcn, overlaid paraoxon analogue from pdb: 2r1n), ANEH (pdb: 3g0i, valpromide), TEM‐1 (pdb: 1btl, N‐ (benzyloxycarbonyl) amino] methyl] phosphate overlaid from pdb: 1axb) and another active site residue for PAMO (pdb: 2ylr, R337). When plotting the epistasis ratio versus distances, we excluded all substitutions occurring at the first round to prevent a bias in the distribution of epistatic mutation, as by definition the first occurring mutation cannot be epistatic.
Supporting information
Supporting Information
Acknowledgments
The authors thank Dave W. Anderson, Janine Copp and Rasmus A. Olesen for comments on the manuscript. N.T. is a CIHR new investigator and a Michael Smith Foundation for Health Research (MSFHR) career investigator.
Significance Statement: Epistasis, that is, non‐additive mutational effects, constrains the adaptive evolution of proteins, yet we lack a consensus view of its extent. To address this need, we performed a systematic survey of mutational epistasis occurring within nine adaptive enzyme trajectories. Of particular importance to protein engineering, our results quantify the unpredictability of functional mutations and emphasize the need to incorporate epistasis for the accurate prediction of mutational effects.
References
- 1. Guthrie VB, Allen J, Camps M, Karchin R (2011) Network models of TEM beta‐lactamase mutations coevolving under antibiotic selection show modular structure and anticipate evolutionary trajectories. PLoS Comput Biol 7:e1002184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Halabi N, Rivoire O, Leibler S, Ranganathan R (2009) Protein sectors: evolutionary units of three‐dimensional structure. Cell 138:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Damborsky J, Brezovsky J (2014) Computational tools for designing and engineering enzymes. Curr Opin Chem Biol 19:8–16. [DOI] [PubMed] [Google Scholar]
- 4. Rajagopalan S, Wang C, Yu K, Kuzin AP, Richter F, Lew S, Miklos AE, Matthews ML, Seetharaman J, Su M, et al. (2014) Design of activated serine‐containing catalytic triads with atomic‐level accuracy. Nat Chem Biol 10:386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hilvert D (2013) Design of protein catalysts. Annu Rev Biochem 82:447–470. [DOI] [PubMed] [Google Scholar]
- 6. Lutz S, Patrick WM (2004) Novel methods for directed evolution of enzymes: quality, not quantity. Curr Opin Biotechnol 15:291–297. [DOI] [PubMed] [Google Scholar]
- 7. Nobili A, Tao Y, Pavlidis IV, van den Bergh T, Joosten HJ, Tan T, Bornscheuer UT (2015) Simultaneous use of in silico design and a correlated mutation network as a tool to efficiently guide enzyme engineering. Chembiochem 16:805–810. [DOI] [PubMed] [Google Scholar]
- 8. Packer MS, Liu DR (2015) Methods for the directed evolution of proteins. Nat Rev Genet 16:379–394. [DOI] [PubMed] [Google Scholar]
- 9. Reetz MT, Wang LW, Bocola M (2006) Directed evolution of enantioselective enzymes: iterative cycles of CASTing for probing protein‐sequence space. Angew Chem Int Ed Engl 45:1236–1241. [DOI] [PubMed] [Google Scholar]
- 10. Currin A, Swainston N, Day PJ, Kell DB (2015) Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently. Chem Soc Rev 44:1172–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tokuriki N, Jackson CJ, Afriat‐Jurnou L, Wyganowski KT, Tang R, Tawfik DS (2012) Diminishing returns and tradeoffs constrain the laboratory optimization of an enzyme. Nat Commun 3:1257 [DOI] [PubMed] [Google Scholar]
- 12. Meier MM, Rajendran C, Malisi C, Fox NG, Xu C, Schlee S, Barondeau DP, Hocker B, Sterner R, Raushel FM (2013) Molecular engineering of organophosphate hydrolysis activity from a weak promiscuous lactonase template. J Am Chem Soc 135:11670–11677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sykora J, Brezovsky J, Koudelakova T, Lahoda M, Fortova A, Chernovets T, Chaloupkova R, Stepankova V, Prokop Z, Smatanova IK, et al. (2014) Dynamics and hydration explain failed functional transformation in dehalogenase design. Nat Chem Biol 10:428–430. [DOI] [PubMed] [Google Scholar]
- 14. Giger L, Caner S, Obexer R, Kast P, Baker D, Ban N, Hilvert D (2013) Evolution of a designed retro‐aldolase leads to complete active site remodeling. Nat Chem Biol 9:494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Visser JA, Cooper TF, Elena SF (2011) The causes of epistasis. Proc Biol Sci 278:3617–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Phillips PC (2008) Epistasis–the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet 9:855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gong LI, Suchard MA, Bloom JD (2013) Stability‐mediated epistasis constrains the evolution of an influenza protein. Elife 2:e00631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McKeown AN, Bridgham JT, Anderson DW, Murphy MN, Ortlund EA, Thornton JW (2014) Evolution of DNA specificity in a transcription factor family produced a new gene regulatory module. Cell 159:58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaltenbach M, Tokuriki N (2014) Dynamics and constraints of enzyme evolution. J Exp Zool B Mol Dev E 322:468–487. [DOI] [PubMed] [Google Scholar]
- 20. Dean AM, Thornton JW (2007) Mechanistic approaches to the study of evolution: the functional synthesis. Nature Rev Genet 8:675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dickinson BC, Leconte AM, Allen B, Esvelt KM, Liu DR (2013) Experimental interrogation of the path dependence and stochasticity of protein evolution using phage‐assisted continuous evolution. Proc Natl Acad Sci USA 110:9007–9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salverda ML, Dellus E, Gorter FA, Debets AJ, van der Oost J, Hoekstra RF, Tawfik DS, de Visser JA (2011) Initial mutations direct alternative pathways of protein evolution. PLoS Genet 7:e1001321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harms MJ, Thornton JW (2013) Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat Rev Genet 14:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blount ZD, Borland CZ, Lenski RE (2008) Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli. Proc Natl Acad Sci USA 105:7899–7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Visser JA, Krug J (2014) Empirical fitness landscapes and the predictability of evolution. Nat Rev Genet 15:480–490. [DOI] [PubMed] [Google Scholar]
- 26. Tufts DM, Natarajan C, Revsbech IG, Projecto‐Garcia J, Hoffmann FG, Weber RE, Fago A, Moriyama H, Storz JF (2015) Epistasis constrains mutational pathways of hemoglobin adaptation in high‐altitude pikas. Mol Biol E 32:287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lozovsky ER, Chookajorn T, Brown KM, Imwong M, Shaw PJ, Kamchonwongpaisan S, Neafsey DE, Weinreich DM, Hartl DL (2009) Stepwise acquisition of pyrimethamine resistance in the malaria parasite. Proc Natl Acad Sci USA 106:12025–12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lunzer M, Golding GB, Dean AM (2010) Pervasive cryptic epistasis in molecular evolution. PLoS Genet 6:e1001162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yokoyama S, Xing J, Liu Y, Faggionato D, Altun A, Starmer WT (2014) Epistatic adaptive evolution of human color vision. PLoS Genet 10:e1004884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Podgornaia AI, Laub MT (2015) Protein evolution. Pervasive degeneracy and epistasis in a protein‐protein interface. Science 347:673–677. [DOI] [PubMed] [Google Scholar]
- 31. Weinreich DM, Lan Y, Wylie CS, Heckendorn RB (2013) Should evolutionary geneticists worry about higher‐order epistasis? Curr Opin Genet Dev 23:700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weinreich DM, Knies JL (2013) Fisher's geometric model of adaptation meets the functional synthesis: data on pairwise epistasis for fitness yields insights into the shape and size of phenotype space. Evolution 67:2957–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kogenaru M, de Vos MG, Tans SJ (2009) Revealing evolutionary pathways by fitness landscape reconstruction. Crit Rev Biochem Mol Biol 44:169–174. [DOI] [PubMed] [Google Scholar]
- 34. Weinreich DM, Delaney NF, Depristo MA, Hartl DL (2006) Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 312:111–114. [DOI] [PubMed] [Google Scholar]
- 35. Jiang PP, Corbett‐Detig RB, Hartl DL, Lozovsky ER (2013) Accessible mutational trajectories for the evolution of pyrimethamine resistance in the malaria parasite Plasmodium vivax . J Mol E 77:81–91. [DOI] [PubMed] [Google Scholar]
- 36. Noor S, Taylor MC, Russell RJ, Jermiin LS, Jackson CJ, Oakeshott JG, Scott C (2012) Intramolecular epistasis and the evolution of a new enzymatic function. PLoS One 7:e39822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moriuchi R, Tanaka H, Nikawadori Y, Ishitsuka M, Ito M, Ohtsubo Y, Tsuda M, Damborsky J, Prokop Z, Nagata Y (2014) Stepwise enhancement of catalytic performance of haloalkane dehalogenase LinB towards beta‐hexachlorocyclohexane. AMB Express 4:72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meini MR, Tomatis PE, Weinreich DM, Vila AJ (2015) Quantitative description of a protein fitness landscape based on molecular features. Mol Biol E 32:1774–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaltenbach M, Jackson CJ, Campbell EC, Hollfelder F, Tokuriki N (2015) Reverse evolution leads to genotypic incompatibility despite functional and active site convergence. Elife 4:e.06492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reetz MT, Sanchis J (2008) Constructing and analyzing the fitness landscape of an experimental evolutionary process. Chembiochem 9:2260–2267. [DOI] [PubMed] [Google Scholar]
- 41. Reetz MT, Bocola M, Wang LW, Sanchis J, Cronin A, Arand M, Zou J, Archelas A, Bottalla AL, Naworyta A, et al. (2009) Directed evolution of an enantioselective epoxide hydrolase: uncovering the source of enantioselectivity at each evolutionary stage. J Am Chem Soc 131:7334–7343. [DOI] [PubMed] [Google Scholar]
- 42. Zhang ZG, Lonsdale R, Sanchis J, Reetz MT (2014) Extreme synergistic mutational effects in the directed evolution of a baeyer‐villiger monooxygenase as catalyst for asymmetric sulfoxidation. J Am Chem Soc 136:17262–17272. [DOI] [PubMed] [Google Scholar]
- 43. Reetz MT, Wu S (2009) Laboratory evolution of robust and enantioselective Baeyer‐Villiger monooxygenases for asymmetric catalysis. J Am Chem Soc 131:15424–15432. [DOI] [PubMed] [Google Scholar]
- 44. Stebbins J (1944) The law of diminishing returns. Science 99:267–271. [DOI] [PubMed] [Google Scholar]
- 45. Hartl DL, Dykhuizen DE, Dean AM (1985) Limits of adaptation: the evolution of selective neutrality. Genetics 111:655–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tomatis PE, Fabiane SM, Simona F, Carloni P, Sutton BJ, Vila AJ (2008) Adaptive protein evolution grants organismal fitness by improving catalysis and flexibility. Proc Natl Acad Sci USA 105:20605–20610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morley KL, Kazlauskas RJ (2005) Improving enzyme properties: when are closer mutations better? Trends Biotechnol 23:231–237. [DOI] [PubMed] [Google Scholar]
- 48. Malito E, Alfieri A, Fraaije MW, Mattevi A (2004) Crystal structure of a Baeyer‐Villiger monooxygenase. Proc Natl Acad Sci USA 101:13157–13162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kongsaeree P, Khongsuk P, Leartsakulpanich U, Chitnumsub P, Tarnchompoo B, Walkinshaw MD, Yuthavong Y (2005) Crystal structure of dihydrofolate reductase from Plasmodium vivax: pyrimethamine displacement linked with mutation‐induced resistance. Proc Natl Acad Sci USA 102:13046–13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salverda ML, De Visser JA, Barlow M (2010) Natural evolution of TEM‐1 beta‐lactamase: experimental reconstruction and clinical relevance. FEMS Microbiol Rev 34:1015–1036. [DOI] [PubMed] [Google Scholar]
- 51. Draghi JA, Plotkin JB (2013) Selection biases the prevalence and type of epistasis along adaptive trajectories. Evolution 67:3120–3131. [DOI] [PubMed] [Google Scholar]
- 52. Romero PA, Arnold FH (2009) Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol 10:866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lobkovsky AE, Koonin EV (2012) Replaying the tape of life: quantification of the predictability of evolution. Front Genet 3:246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. da Silva J, Coetzer M, Nedellec R, Pastore C, Mosier DE (2010) Fitness epistasis and constraints on adaptation in a human immunodeficiency virus type 1 protein region. Genetics 185:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Christin PA, Salamin N, Savolainen V, Duvall MR, Besnard G (2007) C4 Photosynthesis evolved in grasses via parallel adaptive genetic changes. Curr Biol 17:1241–1247. [DOI] [PubMed] [Google Scholar]
- 56. Barlow M, Hall BG (2002) Predicting evolutionary potential: in vitro evolution accurately reproduces natural evolution of the tem beta‐lactamase. Genetics 160:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Counago R, Chen S, Shamoo Y (2006) In vivo molecular evolution reveals biophysical origins of organismal fitness. Mol Cell 22:441–449. [DOI] [PubMed] [Google Scholar]
- 58. Meyer JR, Dobias DT, Weitz JS, Barrick JE, Quick RT, Lenski RE (2012) Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 335:428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ashenberg O, Gong LI, Bloom JD (2013) Mutational effects on stability are largely conserved during protein evolution. Proc Natl Acad Sci USA 110:21071–21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Khanal A, Yu McLoughlin S, Kershner JP, Copley SD (2015) Differential effects of a mutation on the normal and promiscuous activities of orthologs: implications for natural and directed evolution. Mol Biol E 32:100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Palmer AC, Toprak E, Baym M, Kim S, Veres A, Bershtein S, Kishony R (2015) Delayed commitment to evolutionary fate in antibiotic resistance fitness landscapes. Nat Commun 6:7385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schenk MF, Witte S, Salverda ML, Koopmanschap B, Krug J, de Visser JA (2015) Role of pleiotropy during adaptation of TEM‐1 beta‐lactamase to two novel antibiotics. Evol Appl 8:248–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bolon DN, Voigt CA, Mayo SL (2002) De novo design of biocatalysts. Curr Opin Chem Biol 6:125–129. [DOI] [PubMed] [Google Scholar]
- 64. Das R, Baker D (2008) Macromolecular modeling with rosetta. Annu Rev Biochem 77:363–382. [DOI] [PubMed] [Google Scholar]
- 65. Wilkins AD, Venner E, Marciano DC, Erdin S, Atri B, Lua RC, Lichtarge O (2013) Accounting for epistatic interactions improves the functional analysis of protein structures. Bioinformatics 29:2714–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fowler DM, Fields S (2014) Deep mutational scanning: a new style of protein science. Nat Methods 11:801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McLaughlin RN, Jr. , Poelwijk FJ, Raman A, Gosal WS, Ranganathan R (2012) The spatial architecture of protein function and adaptation. Nature 491:138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. LiCata VJ, Ackers GK (1995) Long‐range, small magnitude nonadditivity of mutational effects in proteins. Biochemistry 34:3133–3139. [DOI] [PubMed] [Google Scholar]
- 69. Natarajan C, Inoguchi N, Weber RE, Fago A, Moriyama H, Storz JF (2013) Epistasis among adaptive mutations in deer mouse hemoglobin. Science 340:1324–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Perica T, Kondo Y, Tiwari SP, McLaughlin SH, Kemplen KR, Zhang X, Steward A, Reuter N, Clarke J, Teichmann SA (2014) Evolution of oligomeric state through allosteric pathways that mimic ligand binding. Science 346:1254346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jimenez‐Oses G, Osuna S, Gao X, Sawaya MR, Gilson L, Collier SJ, Huisman GW, Yeates TO, Tang Y, Houk KN (2014) The role of distant mutations and allosteric regulation on LovD active site dynamics. Nat Chem Biol 10:431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dellus‐Gur E, Elias M, Caselli E, Prati F, Salverda ML, de Visser JA, Fraser JS, Tawfik DS (2015) Negative epistasis and evolvability in TEM‐1 beta‐lactamase–the thin line between an enzyme's conformational freedom and disorder. J Mol Biol 427:2396–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information