

Abstract
The structure, function, and evolution of proteins depend on physical and genetic interactions among amino acids. Recent studies have used new strategies to explore the prevalence, biochemical mechanisms, and evolutionary implications of these interactions—called epistasis—within proteins. Here we describe an emerging picture of pervasive epistasis in which the physical and biological effects of mutations change over the course of evolution in a lineage‐specific fashion. Epistasis can restrict the trajectories available to an evolving protein or open new paths to sequences and functions that would otherwise have been inaccessible. We describe two broad classes of epistatic interactions, which arise from different physical mechanisms and have different effects on evolutionary processes. Specific epistasis—in which one mutation influences the phenotypic effect of few other mutations—is caused by direct and indirect physical interactions between mutations, which nonadditively change the protein's physical properties, such as conformation, stability, or affinity for ligands. In contrast, nonspecific epistasis describes mutations that modify the effect of many others; these typically behave additively with respect to the physical properties of a protein but exhibit epistasis because of a nonlinear relationship between the physical properties and their biological effects, such as function or fitness. Both types of interaction are rampant, but specific epistasis has stronger effects on the rate and outcomes of evolution, because it imposes stricter constraints and modulates evolutionary potential more dramatically; it therefore makes evolution more contingent on low‐probability historical events and leaves stronger marks on the sequences, structures, and functions of protein families.
Keywords: epistasis, evolutionary biochemistry, sequence‐function relationship, protein evolution, sequence space, deep mutational scanning, ancestral sequence reconstruction
Introduction
A protein's biological functions emerge from its chemical and physical properties, which in turn are determined by the interactions between its amino acid residues in three‐dimensional space. It is therefore not surprising that the functional effect of changing an amino acid often depends on the specific sequence of the protein into which the mutation is introduced. This dependency on genetic context has long been called epistasis by geneticists.1 Epistasis is invoked when the combined effect of two or more mutations deviates from that predicted by adding their individual effects.
Although studies of epistasis have traditionally focused on genetic interactions between mutations at different loci,1 recent research has begun to address epistasis within proteins—its prevalence, biochemical mechanisms, and impacts on evolution. A consensus view of these subjects has not yet emerged however. Some papers conclude that epistasis is “rampant”2 or even the “primary factor” in protein evolution,3 whereas others claim that the frequency and magnitude of epistasis is “sufficiently low” such that it does not strongly affect the patterns of substitution in evolving proteins.4 There is also no clear picture of the mechanisms that cause epistasis: many papers have focused exclusively on epistasis mediated by effects on protein stability,5, 6, 7, 8, 9 although a few have addressed effects on protein conformation, ligand binding, and allostery.10, 11, 12
These disagreements reflect, at least in part, the lack of a unified discussion of the parallels and contrasts now emerging from the diverse modes of analysis applied to epistasis and its effects on protein evolution. Here we attempt such a unified view, focusing on the following specific questions: How important a factor is epistasis in changing the effects of mutations during the course of evolutionary history? Does epistasis typically amplify or dampen the effect of individual mutations? Does most evolutionarily relevant epistasis reflect very specific interactions between mutations—for example, with only one potential “permissive” mutation that can open the path for another specific mutation—or are many‐to‐one, one‐to‐many, or many‐to‐many interactions more common? What are the molecular mechanisms of interaction that produce each form of epistasis? And how do epistatic interactions of these various types influence the pathways and outcomes of long‐term protein evolution?
Epistasis and Protein Sequence Space
The concept of sequence space provides a useful metaphor for understanding the relationship between a protein's sequence, its physical or biological properties, and its evolution. Sequence space is a multidimensional representation of all possible protein genotypes, each connected to its neighbors by edges representing changes in a single residue.13 Assigning physical or biological properties to each genotype yields a “topological map” of the sequence space, just as a topological map of a geographic landscape assigns elevations to locations defined by their latitudinal and longitudinal coordinates. Epistasis makes the topology of sequence space “rugged”,14 in that the physical or biological effect of a mutation differs in sign or magnitude depending on the sequence background into which it is introduced; similarly, on a rugged geographical landscape, the change in elevation caused by a step in some direction varies dramatically depending on the starting point.
As proteins evolve, they follow trajectories through sequence space, so this topology also determines how mutation, drift, selection, and other forces can drive genetic and functional evolution. A typical trajectory in natural or directed protein evolution consists of iterative mutational steps between functional proteins; trajectories involving strongly deleterious mutations are considered unlikely, because nonfunctional variants of biologically important proteins will usually reduce fitness and therefore be removed by natural selection.13, 15, 16, 17 In the absence of epistasis, any mutation that changes protein properties in a beneficial way can be fixed by natural selection, irrespective of the genetic background in which it occurs; the result is a large number of passable trajectories through sequence space to the functional optimum that combines all of the beneficial sequence states. When epistasis is present, however, a mutation may be beneficial in some backgrounds but deleterious (or neutral) in others; the number of passable trajectories becomes smaller, the fixation of any one mutation may be contingent on the prior occurrence of other specific mutations, and there may be multiple local optima, consisting of mutually conditional beneficial states, isolated from each other by trajectories of low fitness.
Epistasis can therefore affect evolutionary processes in dramatic ways. First, it can create a strong path‐dependency in trajectories of protein evolution,18, 19, 20, 21 because the mutations that are stochastically fixed may determine which functional optimum an evolving protein ultimately occupies, and these optima may differ not only in primary sequence but also in interesting physical or biological properties. Second, epistasis can yield evolutionary “dead‐ends” in sequence space, from which a potentially beneficial mutation is not immediately accessible; in such cases, a relaxation of selection or even selection for other protein properties is necessary before a trajectory is opened to a superior optimum.10, 22, 23, 24, 25, 26, 27, 28 Third, epistasis can cause a mutation that confers or improves a function in one protein to have no effect or even be strongly deleterious in a related protein;2, 21, 29 as a result, attempts to leverage natural sequence variation or experimental observations to predict mutational effects or engineer proteins with desired properties often fail.30 These issues highlight why characterizing epistasis—including the breadth of its effect, its mechanistic underpinnings, and its evolutionary impact—is important for our basic understanding of protein biochemistry and evolution.
Prevalence and Strength of Epistasis
How prevalent is epistasis within proteins, how strongly does it modulate the effects of mutations, and to what extent does this context‐dependence affect long‐term evolution? Studies of these questions have used two primary approaches—deep mutational scanning of large numbers of mutations in individual proteins, and analyses of changes in mutational effects across long‐term trajectories of protein evolution.
Epistasis in a protein's local sequence neighborhood
A recently developed technique called deep mutational scanning makes it possible to characterize a very large library of mutant versions of some protein of interest with respect to some physical or biological property. By analyzing many or all variants that differ by one or two amino acids from a starting protein, it is possible to comprehensively characterize pairwise epistatic interactions in that protein's local sequence neighborhood.31, 32, 33, 34, 35 In the absence of epistasis, the behavior of double mutants can be predicted with perfect accuracy by adding the effects of their constituent single mutations. (That is, R 2 approaches 1 for the correlation between observed and predicted double mutant function.) In contrast, on a completely epistatic landscape, the effect of a mutation is completely independent in every background (so R 2 approaches 0). Experiments reveal an intermediate prevalence of epistasis: the properties of single mutants predict double mutant behavior moderately well (R 2 ∼ 0.65–0.75).31, 32, 33 This result indicates that strong epistasis is not all‐pervasive, pointing instead to epistasis that is pervasive and weak or relatively rare and strong. In fact, it appears that both types of interactions are important: a comprehensive study of pairwise interactions in protein G domain 1 (GB1) found strong deviations from additivity (by a factor >2) in ∼5% of all pairs of mutations, while weak epistasis (<2‐fold deviation) affected ∼30% of pairs34 (see also Ref. 35). Thus, small‐effect epistasis is very common; large‐effect epistasis is less pervasive but still affects a substantial number of mutations.
Does epistasis tend to affect protein properties in one direction more than another? In “negative epistasis” a double mutant's measured phenotype has a smaller value than expected under additivity [e.g., Fig. 1(C,D,G)], whereas in “positive epistasis” the phenotype is greater than predicted [Fig. 1(E,F,H)].1 In deep mutational scanning studies of ligand affinity and fitness effects, far more pairs exhibit negative than positive epistasis: the former group outnumbers the latter by a factor of 3–20.33, 34, 35 Most mutations have deleterious effects on these phenotypes, so negative epistasis in the majority of cases acts synergistically to make double mutants worse than either single mutant alone [Fig. 1(D)].33, 34, 35 This kind of epistasis would cause weakly deleterious mutations to become progressively less evolutionarily accessible as modifying mutations accumulate.
Figure 1.
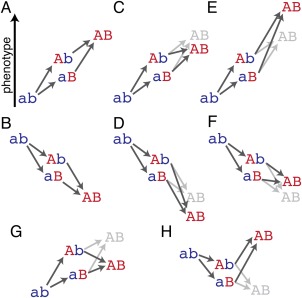
Patterns of epistasis between mutations. (A and B) Mutations a⇒A and b⇒B behave additively (non‐epistatically) with respect to the measured phenotype (e.g., stability, fitness): the phenotypic effect of a state at one site is independent of the state of the other. (C and D) The two mutations exhibit negative epistasis: the double mutant AB has a lower measured phenotype than would be expected from the effects of A and B alone, regardless of the direction of the effect.1 (E and F) The two mutations exhibit positive epistasis: the double mutant AB has a greater phenotype than would be expected from the effects of A and B alone. (G) The two mutations exhibit negative sign epistasis: the sign of the phenotypic effect of state B depends on the state at site A. (H) The two mutations exhibit reciprocal sign epistasis: the sign of the phenotypic effect of either mutation changes depends on the state at the other.
Of particular importance for evolution is positive sign epistasis [Fig. 1(H)], in which a pair of deleterious or neutral mutations becomes beneficial when combined. Although far less prevalent than negative epistasis, positive sign epistasis still appears to be widespread. In GB1, most mutations that are deleterious have at least one or more interacting mutations elsewhere in the protein that make it beneficial or neutral.34 Positive epistasis can open mutational trajectories to combinations of substitutions that would otherwise have been inaccessible. For example, in a high‐throughput screen of a mutant protein library for variants that maintained the wild‐type function, about 95% of the functional variants recovered would have been predicted to be nonfunctional from the effects of single mutations alone.36
These deep mutational scanning studies provide important insights into how epistasis might affect the first stages of an evolutionary process that begins from present‐day forms, initially closing many paths to beneficial combinations but sometimes opening new ones. But the strategy leaves untouched important questions about the effect of epistasis on long‐term historical protein evolution. There is plenty of epistasis in the local sequence neighborhood of a protein, but does this epistasis actually matter in determining proteins' historical trajectories? For example, mutational scans suggest that many mutations manifest sign epistasis in their interactions, but how frequently does the direction of a mutation's effect actually change during evolution? Is the strength and pervasiveness of epistasis in the immediate neighborhood of extant proteins similar to that in the much larger tracts of sequence space traversed by proteins evolving over hundreds of millions of years? Answering these questions requires direct analysis of epistasis across long‐term trajectories of protein evolution.
Epistasis in long‐term protein evolution
One way to gain insight into epistasis in real protein evolution is to compare the effects of some mutation on physical or biological properties when it is introduced into different proteins related by evolutionary descent (homologs). Some studies have addressed this question experimentally, while others have used computational approaches to indirectly infer the prevalence and strength of epistasis during long‐term evolution.
Experimental comparisons of mutational effects between homologs
Manipulative experiments on protein homologs point to both strong and pervasive effects of epistasis that cause the functional effects of mutations to differ between related proteins. One study tested the functional effect of 168 amino acid differences that separate orthologs enzymes that have maintained the same function in two bacterial species.2 Each individual residue from one ortholog was introduced into the other, and about one third of these “sequence swaps” severely decreased enzyme activity. This result indicates that permissive epistatic interactions made the residue tolerable in its native background, that restrictive epistatic mutations made it intolerable in the other, or both. A similar study examined all combinations of nine variable residues that differ between closely related orthologs proteins and statistically determined both the average effects of each residue on catalytic activity, as well as the variance of its effect across different combinations.37 The standard deviation of every mutation's effect was at least 45% of its average effect (up to 75% in the most extreme case), indicating significant epistasis among the sequence differences between the proteins.
These studies demonstrate widespread epistasis, but they do not trace the accumulation of epistatically interacting mutations over time. A rapid study addressed this question using the rapid evolution of influenza nucleoprotein.7 The mutational trajectory of the protein over the last 39 years was reconstructed. Each of the substitutions that occurred during this trajectory was assessed for its effects on viral RNA transcription when introduced into the sequence context in which it occurred historically and into the sequence from an extinct strain that closely resembles an ancestral version of the protein. Every substitution was neutral in the background in which it occurred, but three were radically deleterious with respect to both function and fitness in the ancestral background, indicating relatively rare but extremely strong epistasis that allowed these mutations to be tolerated later.
The above examples illuminate the variability in mutational effect for states that were actually incorporated into diverging proteins during evolution. But what is the impact of epistasis on the effects of all mutations, including those that are never observed because they are deleterious? A recent study compared site‐specific mutational preferences between two influenza nucleoprotein orthologs by assessing the effect on viral fitness of all 19 possible single‐amino‐acid replacement mutations at every site in the two proteins, whether or not they changed during evolution.4 The two proteins differ at only 6% of sites, but significant differences in site‐specific amino acid preference were found at 3–15% percent of sites (depending on the statistical method used to evaluate differences). Thus, on average, each substitution during the evolution of these two closely related proteins modulated the amino acid preferences at one or two other sites.
Strong epistasis is also apparent in laboratory evolution studies. One study placed a protein under strong selective pressure to evolve a new activity and then reimposed selection for the original activity, a trajectory that involved 28 amino acid changes in all.21 The “ancestral” amino acid state at each of these 28 sites was then introduced singly into the “derived” protein, and the derived states were each introduced into the ancestral protein to test for context‐dependence. Almost half of the substitutions were deleterious when swapped into the other background, pointing to widespread epistatic interactions among the sites and states that were substituted during the laboratory evolutionary process.
Comparative sequence analysis
Computational analyses of protein sequence data have investigated epistasis by seeking evidence that the effects of mutations differ among phylogenetic lineages. These studies have detected several major signatures (Fig. 2) that point to a strong and pervasive effect of epistasis on protein evolution.
Figure 2.

Evidence for epistasis in extant sequence data. (A) A mutation from F to V at a given site is known to cause disease in humans, but V is observed as the wild‐type state in mouse, implying permissive mutations in mouse or restrictive mutations in human. (B) Amino acid usage for a site in a given clade (ua or ub) only represents a fraction of the total amino acid usage observed for the site over its long‐term evolution (uT), suggesting different epistatic constraints between clades. (C) Phylogenetic analysis is used to infer historical amino acid substitutions (indicated by vertical bars; additional sequences used to polarize changes leading to bottom two sequences are not shown). The number of paired substitutions to the same amino acid state (convergent substitutions, Nc) decreases with increasing evolutionary distance, relative to the number of paired substitutions to different amino acid states (divergent substitutions, Nd), suggesting lineage‐specific constraints on acceptable states. (D) Pairs of sites that exhibit significant covariation correspond to protein structural contacts.
First, amino acid states that cause disease in one lineage frequently correspond to a wild‐type state in the orthologous protein from other species [Fig. 2(A)].38, 39, 40, 41, 42, 43, 44 These states do not cause disease in the lineages in which they have fixed, so other lineage‐specific substitutions must have modulated their effects. Remarkably, of the sequence differences between orthologous proteins in humans and other vertebrates, about 1% of the states from other species are known to cause disease in humans;38, 40 when the incomplete nature of databases of pathogenic mutations is taken into account, it is estimated that up to 10% of differences between orthologouss might correspond to epistatically modified pathogenic states.
Second, genome‐scale alignments of orthologs with conserved functions from distant taxa point to extensive changes in the tolerability of specific mutations over the long term. For example, the amino acid states found in clades of related species represent only a small subset of the states that occur across its long‐term evolution [Fig. 2(B)],3 pointing to lineage‐specific epistatic constraints. Other studies have found that the rate of convergent substitutions between orthologs declines as sequence divergence increases, as expected if the site‐specific tolerability of each amino acid changes in a lineage‐specific manner [Fig. 2(C)].45, 46, 47, 48 By analyzing these data with an evolutionary model that incorporates epistatic interactions, one study found that an average amino acid substitution switches the tolerability of five other potential mutations, making deleterious mutations at other sites nondeleterious, or vice versa.49 Although the patterns—and particularly the quantitative estimates of the extent of epistasis—identified in these large‐scale computational studies depend on assumptions and statistical models,50 their congruence with experimental studies of specific proteins suggests that epistasis is indeed likely to be pervasive during long‐term protein evolution.
Finally, sequence signatures of covariation provide circumstantial evidence for pervasive epistasis in long‐term evolution [Fig. 2(D)]. Epistasis between residues causes sites within a protein to constrain each other's evolution, so the state present at one site in a sequence should provide information about the state at an interacting site. Such signatures of covariation in sequence alignments have been used to correctly predict protein folds from sequence and to engineer novel sequences that fold in a desired conformation.51, 52, 53, 54, 55, 56 They have also been used to predict and engineer protein–protein interactions,57, 58, 59, 60 to predict the functional effects of mutations,61, 62 and to understand other sequence‐structure‐function relations.63, 64, 65 Although the physical basis for the signal of covariation in these analyses has not been established, some of the strongest pairwise covariation terms have been experimentally validated as strong epistatic interactions.61, 62 Further, efforts to design and predict protein structure and function have been far more successful when mutual information among residues is included in the analysis than when only site‐specific amino acid frequency profiles are used, suggesting that covariation signatures have indeed captured distinct and biologically meaningful dependencies among sites.51, 61, 62 That such analyses can capture enough of the relevant details about protein architecture to do this kind of practical work suggests that epistasis is likely to be a strong determinant of protein structure and function.
In silico evolutionary simulations
Epistasis has a similarly pervasive role in computational simulations of neutral5, 6 and adaptive66 protein evolution. In these studies, some ancestral protein with a defined structure is allowed to evolve in silico under defined population genetic conditions. For example, a recent study simulated the evolution of argT for replicate evolutionary trajectories 30 substitutions long; purifying selection was imposed to maintain a stable fold, implemented by applying a stability prediction algorithm that uses the protein's known crystallographic structure and a very simple function that relates stability to fitness.6 Each substitution that occurred during a trajectory was then evaluated for its predicted effects on stability and fitness when introduced into every protein sequence that existed at some point during that trajectory. The vast majority of substitutions had different predicted effects on stability and fitness at the time they occurred than they would have if introduced at a different time. Specifically, most substitutions were neutral at the time of their fixation but would have been deleterious at earlier points in the trajectory, reflecting an important role for permissive mutations in the turnover of evolutionarily viable mutations. Epistasis also caused substantial irreversibility in the evolutionary process: once a substitution enabled by an earlier permissive substitution at some other sequence site occurs, then the ancestral state at the permissive site becomes deleterious, making reversion to that state unlikely.
Relationship to epistasis in the local sequence network
How does the epistasis observed in long‐term evolution compare with the mixture of large‐ and small‐effect epistasis observed in the local sequence neighborhoods of various proteins? Two kinds of analyses bear on this problem but do not clearly resolve it. Computational simulations suggest that both small‐ and large‐effect epistasis have a pervasive influence on proteins' evolutionary trajectories6; however, the models used in these simulations are highly simplified, so the real‐world relevance of their quantitative conclusions is unclear.67 Experimental analyses of the effects of mutations introduced into homologous proteins have detected extensive large‐effect epistasis,2, 7, 21, 37 demonstrating its relevance in real‐world, historical evolutionary trajectories. The pervasive small‐effect epistasis visible in local sequence networks, however, has not been generally observed in these kinds of experiments. It could be that small‐effect epistasis does not meaningfully impact natural evolution, but we cannot rule out ascertainment and reporting bias as alternative explanations: statistically establishing small deviations from additivity is considerably more difficult than establishing large ones, and a general view that only large‐effect epistasis is worth reporting may be at play, as well.
Taken together, analyses to date point to extensive large‐effect epistasis in the local sequence neighborhoods of present day proteins and in the substitutions that become fixed during evolutionary trajectories. Small‐effect epistasis is clearly present in sequence neighborhoods; the extent to which it affects and is incorporated into real‐world evolutionary trajectories remains unresolved.
Specificity of Epistasis and Causal Mechanisms
The above examples demonstrate that epistasis is pervasive and often strong during the course of long‐term evolution. But a complete description of epistasis in protein evolution requires more detailed attention to the nature of the interactions, including their specificity, their effects on evolutionary trajectories, and the molecular mechanisms that produce them.
The biological properties of a protein are ultimately determined by its physical properties (such as conformation, stability, ligand affinity, or dynamics), which in turn are determined by protein sequence (Fig. 3). Nonlinearity in either mapping—from protein sequence to physical property, or physical property to biological property—results in epistasis at the level of function or fitness. We can distinguish two broad classes of epistatic interaction—specific and nonspecific epistasis68—which refer to whether the epistasis involves mutations that modify the effects of few or many other potential mutations. The difference in specificity arises from a difference in the biophysical mechanisms that produce each class of epistasis. The two classes also differ, in turn, in the mapping the interaction affects—from sequence to physical property or from physical property to biological characteristic—and in their implications for evolutionary processes.
Figure 3.
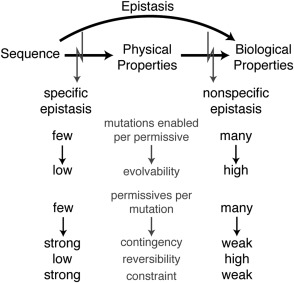
Mechanisms of epistasis and their evolutionary implications. Biological properties (e.g., function, fitness) depend on the physical properties of protein molecules (e.g., stability, solubility, affinity for ligand), which in turn depend on the peptide sequence. Epistasis is defined as nonadditivity somewhere in the mapping from sequence to biological properties. Specific epistasis causes nonadditivity in the mapping from sequence to physical properties because of physical interactions between sites. Nonspecific epistasis arises from an intrinsic nonlinear relationship between physical and biological properties. Specific permissive mutations enable fewer mutations than nonspecific permissive mutations and therefore have a less dramatic impact on protein evolvability. Similarly, mutations that require a specific permissive mutation to be tolerated have fewer possible permissive mutations than a mutation that can be enabled through a nonspecific effect. This causes specific epistasis to underlie stronger historical contingency, lower reversibility, and stronger long‐term evolutionary constraints.
Specific epistasis
In specific epistasis, a mutation modulates the effects of a small number of other mutations. Specific epistasis is typically mediated by physical interactions among residues; these may involve direct interactions between amino acid side chains,20, 21, 69 mutual interaction with other side chains70 or ligands,26, 71, 72 or a dependence of one mutation on a structural change caused by another.10, 21, 73, 74 Because of these physical interactions, two specifically epistatic mutations affect a physical property of the protein—such as stability, affinity, catalysis, or dynamic motions—in a nonadditive fashion [Fig. 4(B)].
Figure 4.
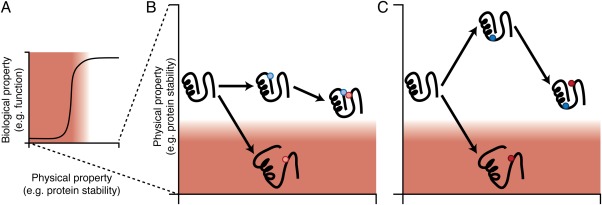
Examples of specific and nonspecific epistasis. (A) The relationship between stability and function is often modeled by a sigmoidal function as expected due to the nonlinear relation between stability and the fraction of protein folded at a given temperature. This relationship is projected onto the y‐ and z‐axes of the mutational reaction coordinates of (B) and (C) as a gradient from white (functional) to red (nonfunctional). (B) A graphical example of specific epistasis. The red mutation in the parental background is destabilizing, resulting in a large functional defect. However, a blue mutation, which by itself does not alter stability, interacts with the red mutation to reduce its effect on stability and, in turn, on function. (C) An example of nonspecific epistasis. The red mutation in the parental background is destabilizing, resulting in a large functional defect. The blue mutation is stabilizing and by itself has little impact on function. However, in this stability‐buffered background, the red mutation (the destabilizing effect of which is unchanged) can occur with no functional defect.
A comprehensive case study on specific epistasis has emerged from investigations on vertebrate steroid receptors using ancestral protein reconstruction and experimental analysis. This work traced the evolution of specificity in the glucocorticoid receptor for its ligand—the steroid hormone cortisol—from a promiscuous ancestral protein that was activated by both cortisol and a structurally related class of steroids called mineralocorticoids. Seven historical substitutions, when introduced into the ancestral protein, are sufficient to fully recapitulate the evolution of cortisol specificity. Introducing five of these substitutions, however, yields a completely nonfunctional receptor unless the other two “permissive” substitutions, which themselves have no effect on function, are in place first.10, 22 Structural analysis suggested a direct conformational mechanism for specific epistasis: the function‐switching substitutions dramatically shifted the position of a helix that lines the ligand pocket, destabilizing key elements of the active conformation but also allowing formation of a new cortisol‐specific hydrogen bond. The two permissive substitutions appeared to directly compensate, generating new physical interactions that stabilized the same structural elements destabilized by the function‐switching mutations, thereby permitting the evolution of a receptor that could be activated only by cortisol.
To determine whether the permissive substitutions were truly specific, a follow‐up experiment assessed how many other epistatically acting mutations might have been available that could have permitted the function‐switching substitutions.75 A library of thousands of variants of the ancestral receptor was prepared, each of which contained the function‐switching substitutions but neither of the permissive substitutions; this library was then screened for epistatic mutations that could rescue cortisol activation. In addition to the historical permissive substitutions, this screen uncovered three new compensatory mutations. However, none of these could have been permissive during evolution, because each dramatically compromised the ancestral receptor's function when introduced on its own. Thus, the epistatic interaction between the historical permissive and function‐switching substitutions was extremely specific, with alternate permissive mutations in the sequence neighborhood of the ancestral receptor being extremely rare. This genetic specificity arose directly from the biophysical basis of the interaction: both the permissive and compensatory mutations acted locally, restoring contacts to the structural elements destabilized by the function‐switching mutations, but only the historical permissive substitutions were also compatible with both the ancestral and derived ensembles of conformations that contribute to ligand‐induced activation. Thus, the direct, local relationship between permissive and function‐switching substitutions caused them to interact nonlinearly in their effects on physical properties (ligand‐activation and structure), giving rise to a very specific, few‐to‐few genetic interaction.
Nonspecific epistasis
In nonspecific epistasis, a mutation modulates the effects of a relatively large number of other mutations. Nonspecific epistasis occurs when two mutations interact nonadditively with respect to some biological property, despite contributing additively at the level of physical protein properties. The epistasis arises because of a nonlinearity in the mapping from the physical property to the biological property [Fig. 4(A)], so a mutation with the same biophysical effect size has a different impact on function or fitness, depending on the current position of the parental protein on this property's topological landscape [Fig. 4(C)].
Nonspecific epistasis has been most thoroughly studied for mutations that independently affect the stability of the protein's native fold but exhibit epistasis in the protein's functionality or contribution to fitness.7, 76, 77, 78, 79, 80, 81, 82 Nonspecific epistasis has also been observed for mutations that are additive with respect to other physical properties (folding, ligand‐binding affinity, and enzyme activity) but are non‐additive with respect to surface expression, transcriptional activity, or fitness, simply because of the nonlinear mapping between the two types of property.27, 28, 83
Mutations that interact epistatically in this way manifest low specificity in their coupling to each other. Every mutation that affects a physical property that maps nonlinearly to biological function or fitness will epistatically interact with every other mutation that affects the same property. For example, a mutation that increases stability and is therefore permissive for another mutation that reduces stability should be permissive for any mutation that reduces stability by a similar amount; further, its permissive effect could be replaced by that of any other mutation that has a similar positive effect on stability. Because the effects on the protein's physical property are independent of each other, the mechanisms that produce nonspecific epistasis typically involve no physical interaction, direct or indirect, between the relevant residues.
For example, in the evolutionary trajectory of influenza nucleoprotein discussed above, each of the three cases of epistasis involves a destabilizing mutation that required a counterbalancing stabilizing mutation to be tolerated.7 Any single stabilizing mutation can rescue any one of the individual destabilizing mutations, highlighting the nonspecific nature of this coupling. The permissive substitutions do not substantially alter the destabilizing effect of the interacting mutations; rather, they simply increase the overall stability of the protein so that the double mutants are not substantially destabilized relative to the parent. Despite additivity at the level of protein stability, these pairs of mutations exhibit strong epistasis at the level of protein function and viral fitness.
This kind of epistasis fits a model by which stability maps nonlinearly to function and fitness: in the simplest case, suppose that the biological function of a protein scales linearly with the quantity of folded protein in the cell.84 Two mutations that independently affect protein stability will epistatically affect the quantity of folded protein (and thus function), simply because of the sigmoidal shape of the Boltzmann distribution that relates changes in the energy of a conformational state to changes in the probability that the state is occupied. Many proteins are only marginally stable, existing slightly above the steep part of the curve that relates stability to fraction folded, so this particular type of nonspecific epistasis through stability could be to be a strong and common factor in protein evolution.
Specific and nonspecific epistasis are not mutually exclusive. Mutations may interact nonadditively at the level of a physical property, and a nonlinear mapping from that property to function or fitness may further amplify72 or buffer84 the interaction. For example, mutations in ancestral transcription factors exhibited pervasive specific epistasis for DNA‐binding affinity,72 and there is also a nonlinear relationship between DNA‐binding affinity and transcriptional activation. Together, these two nonlinearities yield dramatic epistasis in the effects of mutations in the protein or DNA on transcriptional activation; as a result, mutations in the transcription factor can allow the DNA to tolerate affinity‐reducing mutations, and subsequent mutations in the DNA can exert a permissive effect on the protein, opening up mutational pathways that lead to a new regulatory complex with entirely new specificity. In contrast, if two mutations interact to increase each other's effect on stability, but the protein is already far above the stability threshold, neither the individual mutations nor their combination will strongly affect the protein's function or fitness.84
The distinction between two types of epistasis highlights the need for researchers to be transparent and cognizant of where epistasis comes from in their data. Some studies set a threshold to distinguish between putatively functional and nonfunctional (or fit and unfit) genotypes and, in turn, to classify evolutionary pathways through sequence space as passable or not.36, 60, 72 Imposing this kind of nonlinearity will necessarily lead to apparent epistasis and determine a study's conclusions about the availability of mutational trajectories. If the threshold does not have a sound biological motivation—or if its role in determining a study's conclusions is not explored—spurious inferences about epistasis and evolution may result.
Specific positive epistasis versus nonspecific negative epistasis
What is the relative prevalence of specific and nonspecific epistasis and what is the nature of their effects? Mutational scanning studies have identified “hotspot” residues, that interact epistatically with dozens or hundreds of other mutations, indicating a nonspecific effect.32, 33, 34, 35 As expected, some of these hotspots contain stabilizing mutations that can permit many different destabilizing mutations to be tolerated,32 or destabilizing mutations that interact negatively with many other mildly destabilizing mutations.33, 34 The epistasis arises because the functions being assayed—fitness, or affinity for a binding partner—depend on the fraction of protein folded, which is nonlinearly related to stability.
Although nonspecific epistasis accounts for a large number of interactions, specific epistasis is also very important. In one mutational scanning study, the most densely connected hotspots—the most nonspecifically coupled mutations—accounted for less than 20% of all epistatic interactions between mutations.33 As expected, the more specific couplings typically involve direct interactions that modulate the effects of two sequence changes on a protein's physical properties. For example, two individually destabilizing mutations to cysteine yield a stabilizing disulfide bond when combined.34
Nonspecific mechanisms seem to be associated strongly with negative epistasis, while specific mechanisms are associated with positive interactions. In the mutational scan of the GB1 protein, nearly all of the strong, negatively epistatic pairs in GB1 involve combinations of two destabilizing mutations, and these pairs are distributed relatively uniformly in three‐dimensional space on the protein structure.34 In contrast, most of the positively interacting pairs are in close structural proximity (Cβ distance <10 Å), and many affect hydrogen bond networks—suggesting direct and specific physical interactions34 (see also Ref. 33). The association of nonspecific interactions with negative epistasis makes sense, because most mutations are destabilizing and most proteins have only a small “stability reservoir” above the critical threshold, below which the proportion of folded protein drops off precipitously [Fig. 4(A)]. A slightly destabilizing mutation—if it does not exhaust the stability reservoir—will therefore typically have only a weak effect on folding and function, but combining two such mutations may be strongly deleterious.
Specific and nonspecific epistasis in long‐term evolution
Although both positive specific epistasis and negative nonspecific epistasis are prevalent in mutational scanning studies, this might not be true in long‐term evolution. Deleterious combinations of mutations will usually be removed by purifying selection, so positive interactions—such as permissive epistasis—might be expected to dominate the record of long‐term sequence evolution. Does specific epistasis therefore dominate, as well? Case‐studies of the historical evolution of individual proteins have uncovered both specific10, 75 and nonspecific7 mechanisms of permissive interactions, but such studies are insufficient to determine the general prevalence of the two classes of epistasis in protein evolution.
Several larger‐scale studies do suggest that specific positive epistasis is pervasive. In the in silico evolution of argT discussed above, most substitutions were tolerated at the time of their fixation only because of prior permissive substitutions6; that is, they were deleterious in at least some sequence backgrounds that existed earlier in the evolutionary trajectory. Further, these substitutions had smaller predicted stability effects when they occurred than they did at earlier times, pointing to a specific epistatic effect of permissive mutations on the mapping from sequence to stability. The average epistatic effect with respect to stability was small (predicted ΔΔΔG = 0.5 kcal/mol), suggesting moderate epistatic effects with respect to a protein's physical properties can have a meaningful impact on fitness and evolution.
Structural analyses of compensated mutations that are pathogenic in humans but fixed in other species support a similar conclusion. Most pathogenic mutations are predicted to be destabilizing when introduced into the human structure in silico. However, when they are introduced into structures in which the nearby residues have the amino acids found in the species in which they have fixed, they are predicted to be less destabilizing, again by about half a kcal/mol.41 This points to a general role for specific epistasis in this mode of permissive evolution, but it does not rule out an additional role for nonspecific, structurally distant epistatic modifiers.
Is the widespread specific epistasis for stability suggested by these in silico predictions actually observed when epistasis is experimentally characterized in real proteins? Two studies have experimentally measured the effects on folding stability of a handful of mutations when introduced into divergent orthologs, which differ at up to 28% and 43% of sites.8, 9 Both studies found that it is rare for mutations that are destabilizing in one ortholog to become stabilizing in the other, or vice versa. But the magnitude of many mutations' effects on stability does change notably, with the correlation of a residue's stability effects in the two orthologs degrading as sequence distance increases, reaching R 2 = 0.8 among the most divergent orthologs. One study observed that the stability effects of a mutation when introduced into different orthologs frequently differ by more than 0.5 kcal/mol,9 consistent with computational predictions.
Taken together, these studies point to a pervasive effect of specific, positive epistasis in evolution that is retained in protein sequences over long periods of time. They do not rule out an additional role for nonspecific epistasis. Although combinations of amino acids that negatively interact are typically removed from the sequence record by purifying selection, this does not mean they are not important during historical evolution, because determining the paths that evolution does not take is as important in evolutionary outcomes as shaping those it may pass through.
Evolutionary Implications of Epistasis
We have discussed a body of research that suggests frequent epistasis among substitutions that fix along evolutionary trajectories. How does this epistasis impact the evolutionary process? Further, how do specific and nonspecific epistasis differentially affect these processes?
Evolvability and robustness
A protein's evolvability is determined by the accessibility of mutations that confer new functions; its robustness is determined by the set of available neutral mutations. Positive epistasis—permissive substitutions of either the specific or nonspecific type—increases both evolvability and robustness, because it opens some mutational trajectories that would otherwise have involved deleterious steps. A nonspecific permissive mutation, however, has the potential to open many more evolutionary pathways than specific epistatic mutations do. For example, when stabilizing permissive mutations are introduced into a cytochrome P450 enzyme, it can tolerate a wider range of mutations that confer new functions but are moderately destabilizing.79 Similarly, in the antibiotic resistance gene TEM‐1 β‐lactamase, a “global suppressor” mutation M182T stabilizes the protein, relieving the otherwise deleterious effect of many other mutations that reduce protein stability, including many that enhance the protein's activity on new antibiotics.76, 78, 82 Permissive mutations that globally buffer other physical properties should promote similar increases in evolvability and robustness, though this remains to be demonstrated.
In contrast, a permissive substitution of specific effect can influence the evolutionary potential of, at most, the subset of residues with which it is physically coupled. Assessing cases in which specific epistatic interactions have narrow effects on evolvability and robustness is challenging, because it requires a robust, negative result to demonstrate a few‐to‐few relationship between mutations. Mutational scanning studies have met this challenge to some extent, identifying interactions that increase evolvability and robustness at specifically coupled positions, without as global an impact on evolvability as nonspecifically permissive mutations.32, 33, 34 Further case studies will be required to assess the generality of this result and assess the effects of nonspecific and specific epistasis on these properties during historical evolution.
Historical contingency
When positive epistasis is highly specific, then the outcomes of evolution will be contingent on low‐probability chance events. Permissive substitutions are a prerequisite for the function‐switching changes, but their acquisition cannot be driven by selection for the new function; specific permissive substitutions are rare, so the chance that one will fix by mutation and drift alone is very small.75 In such cases, evolutionary processes exhibit strong stochasticity in their outcomes: parallel populations evolving under the same dynamics will reach different endpoints in response to some selective pressure, because the permissive substitutions that happen to fix will generally be different in each population, and each set of permissive substitutions in turn will open trajectories to distinct functional optima.19, 20
In contrast, evolutionary contingency should be much weaker when nonspecific permissive epistasis is at play. In such cases, a large number of possible permissive mutations could permit any particular function‐switching mutation; across parallel evolving populations, the probability is reasonably high that one of these permissive mutations would occur eventually, opening paths to similar or identical outcomes. Indeed, in the evolution of drug resistance in TEM‐1 β‐lactamase85 and influenza neuraminidase86 or immune escape in influenza nucleoprotein,7 many different mutations were discovered that permit a particular function‐enhancing mutation through nonspecific buffering of properties such as folding, stability, and expression. Thus, nonspecific epistasis appears to be associated with much less stochasticity in the outcomes of evolutionary trajectories.
Reversibility
The reversibility of evolution has long been a topic of interest to evolutionary biologists, because irreversibility implies that the accessibility of some genetic or phenotypic state—the ancestral one—depends on the moment in the genetic history of the organism when it occurs. Specific and nonspecific epistasis appear to differentially affect the reversibility of evolution.
Specific epistasis contributes to evolutionary irreversibility. In several cases, some time after a substitution that affects function has taken place, restrictive epistatic substitutions have occurred, making the ancestral state at the first site deleterious. In each case, specific steric clashes have been involved: the restrictive amino acid is compatible with the derived state but not the ancestral state at the first site, either because of a direct clash between side chains or because of conformational changes that produce conflicts at other sites.21, 73, 87 The physical interaction between these residues affects in a nonadditive fashion the protein's propensity to fold into its functional conformations, making a reversal strongly deleterious—and thus very unlikely—once the restrictive substitution has occurred.
In contrast, nonspecific epistatic substitutions appear to be reversible over long‐term evolution. Several studies have shown that destabilizing substitutions that were initially permitted by a stabilizing substitution can revert, even after relatively long evolutionary intervals.7, 8, 9 In contrast to the specific examples above, the relative stability of the ancestral state remains unchanged, and reversion is freely accessible in the subsequent evolution of the protein.
Long‐term evolutionary constraints
Specific and nonspecific epistasis also imprint themselves differently in the constraints that leave marks on modern‐day sequences. The rate of evolution at a site reflects the strength of selective constraint that acts on that sequence position. A mutation can tighten or relax the selective constraints at a site it interacts with, slowing or accelerating its rate and changing the set of amino acid states that it tolerates. These dynamics lead to signatures of amino acid covariation in extant sequence data, which reflect the extent and nature of epistatic constraints among sites.
Nonspecific epistasis—irrespective of its prevalence and strength—should leave only sparse signals of covariation in the evolutionary record. Consider a destabilizing substitution that was initially permitted by a prior stabilizing substitution, leading to a temporary epistatic dependence: any subsequent stabilizing substitution at the same or another site could relieve the constraint that this coupling creates. The association between the two originally dependent states would then break down.8
In contrast, specific epistasis permanently changes the effects of interacting mutations, altering the native preference of sites for particular amino acid states. These types of interactions should thus generate strong, precise signals of covariation, as restraints of co‐occurrence are not easily reduced with subsequent evolutionary change. The fact that signals of covariation in sequence alignments are strongly related to the colocalization of amino acid pairs in protein's three‐dimensional structures supports the notion that specific epistasis underlies most retained signals of epistasis in the evolutionary record. Indeed, across a large number of protein families, the majority of the sites with the strongest signal of covariation are in direct structural contact.53, 54
Conclusions and Future Directions
The studies we have discussed paint a picture of pervasive epistasis among the sites and states that change during protein evolution. Mutational scanning indicates that both specific and nonspecific couplings between residues contribute strongly to nonadditivity in mutational effects at any moment in time. Over long‐term evolution, permissive substitutions—either specific or nonspecific—play a particularly critical role in opening evolutionary trajectories. The specific epistatic interactions, however, appear to most profoundly affect the long‐term outcomes of evolution.
We emphasize that this picture is emerging, not complete. Many more case studies, particularly of historical evolution—over various time scales, using proteins with different functions and architectures—are required to understand the full range of biophysical mechanisms and evolutionary implications of epistatic interactions within proteins. Several specific questions and approaches seem particularly ripe to explore.
First, combining high‐throughput mutational scanning techniques with ancestral protein reconstruction would provide insight unavailable to either technique alone. Library‐based explorations of sequence space around some extant protein tell us how epistasis shapes the pathways that evolution could follow from the present, but it does not tell us anything about the history that produced that protein. Conversely, mechanistic dissection of reconstructed ancestral proteins and their trajectories can tell us how epistasis shaped the pathway that evolution did follow. By characterizing the sequence space around ancestral proteins, we can begin to address key questions about evolutionary processes: How did epistasis shape the sequence space of an evolving protein? How many pathways could have been followed to the same or similar outcomes? How many different outcomes could have been achieved under a given selection pressure, and how did epistasis influence their accessibility? To what extent were mutational trajectories transiently opened and closed by permissive and restrictive substitutions? What are the roles and particular mechanisms of specific and nonspecific epistasis that mediated these effects? To date, only one study has sought to characterize an ancestral sequence space—the study of specific epistasis in the steroid receptors and this study was very limited in the number of mutational combinations assessed.75 The extent to which the strong evolutionary contingency observed in that case pertains to the evolution of other proteins—and what biophysical and genetic factors contribute to the ensuing evolutionary dynamics—remains to be determined.
Second, higher‐order interactions are probably important in evolution, but they have just begun to be investigated. Most of the studies of epistasis in protein evolution discussed here focus on pairwise epistasis (the interaction between mutations at two sites). However, there is no reason why the impact of epistasis exposed for the interactions between two sites cannot extend to higher‐order combinatorial interactions (e.g., the joint effect of two mutations varies with the state at a third site,72 etc.). A detailed understanding of higher‐order epistasis requires very large numbers of variants to be characterized.88 However, the technological innovations underlying high‐throughput mutational scanning techniques,31, 89 coupled with quantitative formalisms for higher‐order epistasis88, 90 now make it possible to explore how the importance and mechanisms of epistasis inferred at the pairwise level extend to higher‐order interactions among mutations
Third, epistasis between molecules is a ripe matter for evolutionary biochemical analysis. Proteins interact with other proteins, nucleic acids, and small molecules. Epistasis between mutations in different molecules should also have important evolutionary ramifications, but it is unclear how the relative prevalence and evolutionary implications compare to those associated with intramolecular epistasis. Recent experimental dissections,72, 91 high‐throughput mutational scans,92 and systems‐level approaches93 have begun to address this question, revealing molecular mechanisms and evolutionary implications of epistasis that are unique to interactions between molecules. Further work in this area has the potential to broaden our understanding of the impact of epistasis on protein biochemistry and evolution.
Finally, we would like to highlight a conceptual issue that frames our understanding of epistasis and protein evolution. Epistasis is frequently discussed as a “constraint” in molecular evolution. This view may reflect the role of epistasis in constraining the outcomes of protein engineering efforts, in confounding genetic predictions from single‐site data, or in structuring sequence space to produce local optima. But epistasis is not only a brake on evolution: dissecting the dense network of genetic interactions in multidimensional sequence space has revealed how epistasis can also make possible the evolution of new genotypes, functions, and phenotypes. Permissive mutations can relieve constraints that would otherwise make potentially beneficial mutations inaccessible,34, 36 allowing proteins to evolve new functions in a very small number of mutational steps. Thus, more functional diversity may exist within the local sequence landscape of any given protein than is typically appreciated, and epistasis may allow proteins to travel along connected paths among these functionally distinct regions of sequence space.
Because of the size of sequence space and the ways that epistasis structures it, even very ancient proteins have not yet fully explored the boundaries of their networks of neutral divergence.45 As we develop a more complete picture of these sequence spaces, our understanding of present‐day proteins, their histories, and their possible futures should become deeper and more precise.
Acknowledgments
The authors thank members of the Thornton laboratory past and present for insightful comments. The authors declare no conflict of interest.
References
- 1. Phillips PC (2008) Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet 9:855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lunzer M, Golding GB, Dean AM (2010) Pervasive cryptic epistasis in molecular evolution. PLOS Genet 6:e1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Breen MS, Kemena C, Vlasov PK, Notredame C, Kondrashov FA (2012) Epistasis as the primary factor in molecular evolution. Nature 490:535–538. [DOI] [PubMed] [Google Scholar]
- 4. Doud MB, Ashenberg O, Bloom JD (2015) Site‐specific amino acid preferences are mostly conserved in two closely related protein homologs. Mol Biol Evol 11:2944–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pollock DD, Thiltgen G, Goldstein RA (2012) Amino acid coevolution induces an evolutionary Stokes shift. Proc Natl Acad Sci U S A 109:E1352–E1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shah P, McCandlish DM, Plotkin JB (2015) Contingency and entrenchment in protein evolution under purifying selection. Proc Natl Acad Sci U S A 112:E3226–E3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gong LI, Suchard MA, Bloom JD (2013) Stability‐mediated epistasis constrains the evolution of an influenza protein. eLife 2:e00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ashenberg O, Gong LI, Bloom JD (2013) Mutational effects on stability are largely conserved during protein evolution. Proc Natl Acad Sci U S A 110:21071–21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Risso VA, Manssour‐Triedo F, Delgado‐Delgado A, Arco R, Barroso‐delJesus A, Ingles‐Prieto A, Godoy‐Ruiz R, Gavira JA, Gaucher EA, Ibarra‐Molero B, Sanchez‐Ruiz JM (2015) Mutational studies on resurrected ancestral proteins reveal conservation of site‐specific amino acid preferences throughout evolutionary history. Mol Biol Evol 32:440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW (2007) Crystal structure of an ancient protein: evolution by conformational epistasis. Science 317:1544–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lynch VJ, May G, Wagner GP (2011) Regulatory evolution through divergence of a phosphoswitch in the transcription factor CEBPB. Nature 480:383–386. [DOI] [PubMed] [Google Scholar]
- 12. Natarajan C, Inoguchi N, Weber RE, Fago A, Moriyama H, Storz JF (2013) Epistasis among adaptive mutations in deer mouse hemoglobin. Science 340:1324–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maynard Smith J (1970) Natural selection and the concept of a protein space. Nature 225:563–564. [DOI] [PubMed] [Google Scholar]
- 14. Kondrashov DA, Kondrashov FA (2015) Topological features of rugged fitness landscapes in sequence space. Trends Genet 31:24–33. [DOI] [PubMed] [Google Scholar]
- 15. Peisajovich SG, Tawfik DS (2007) Protein engineers turned evolutionists. Nat Methods 4:991–994. [DOI] [PubMed] [Google Scholar]
- 16. Wagner A (2008) Neutralism and selectionism: a network‐based reconciliation. Nat Rev Genet 9:965–974. [DOI] [PubMed] [Google Scholar]
- 17. Romero PA, Arnold FH (2009) Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol 10:866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gumulya Y, Reetz MT (2011) Enhancing the thermal robustness of an enzyme by directed evolution: least favorable starting points and inferior mutants can map superior evolutionary pathways. Chembiochem 12:2502–2510. [DOI] [PubMed] [Google Scholar]
- 19. Salverda MLM, Dellus E, Gorter FA, Debets AJM, van der Oost J, Hoekstra RF, Tawfik DS, de Visser JAGM (2011) Initial mutations direct alternative pathways of protein evolution. PLOS Genet 7:e1001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dickinson BC, Leconte AM, Allen B, Esvelt KM, Liu DR (2013) Experimental interrogation of the path dependence and stochasticity of protein evolution using phage‐assisted continuous evolution. Proc Natl Acad Sci U S A 110:9007–9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaltenbach M, Jackson CJ, Campbell EC, Hollfelder F, Tokuriki N (2015) Reverse evolution leads to genotypic incompatibility despite functional and active site convergence. eLife 4:e06492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bridgham JT, Carroll SM, Thornton JW (2006) Evolution of hormone‐receptor complexity by molecular exploitation. Science 312:97–101. [DOI] [PubMed] [Google Scholar]
- 23. Bloom JD, Romero PA, Lu Z, Arnold FH (2007) Neutral genetic drift can alter promiscuous protein functions, potentially aiding functional evolution. Biol Direct 2:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fasan R, Meharenna YT, Snow CD, Poulos TL, Arnold FH (2008) Evolutionary history of a specialized P450 propane monooxygenase. J Mol Biol 383:1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bershtein S, Goldin K, Tawfik DS (2008) Intense neutral drifts yield robust and evolvable consensus proteins. J Mol Biol 379:1029–1044. [DOI] [PubMed] [Google Scholar]
- 26. Field SF, Matz MV (2010) Retracing evolution of red fluorescence in GFP‐like proteins from Faviina corals. Mol Biol Evol 27:225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bloom JD, Gong LI, Baltimore D (2010) Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 328:1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McKeown AN, Bridgham JT, Anderson DW, Murphy MN, Ortlund EA, Thornton JW (2014) Evolution of DNA specificity in a transcription factor family produced a new gene regulatory module. Cell 159:58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Melero C, Ollikainen N, Harwood I, Karpiak J, Kortemme T (2014) Quantification of the transferability of a designed protein specificity switch reveals extensive epistasis in molecular recognition. Proc Natl Acad Sci U S A 111:15426–15431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harms MJ, Thornton JW (2010) Analyzing protein structure and function using ancestral gene reconstruction. Curr Opin Struc Biol 20:360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, Fields S (2010) High‐resolution mapping of protein sequence‐function relationships. Nat Methods 7:741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Araya CL, Fowler DM, Chen W, Muniez I, Kelly JW, Fields S (2012) A fundamental protein property, thermodynamic stability, revealed solely from large‐scale measurements of protein function. Proc Natl Acad Sci U S A 109:16858–16863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Melamed D, Young DL, Gamble CE, Miller CR, Fields S (2013) Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)‐binding protein. RNA 19:1537–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Olson CA, Wu NC, Sun R (2014) A comprehensive biophysical description of pairwise epistasis throughout an entire protein domain. Curr Biol 24:2643–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bank C, Hietpas RT, Jensen JD, Bolon DNA (2015) A systematic survey of an intragenic epistatic landscape. Mol Biol Evol 32:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Podgornaia AI, Laub MT (2015) Pervasive degeneracy and epistasis in a protein‐protein interface. Science 347:673–677. [DOI] [PubMed] [Google Scholar]
- 37. O'Maille PE, Malone A, Dellas N, Andes Hess B, Smentek L, Sheehan I, Greenhagen BT, Chappell J, Manning G, Noel JP (2008) Quantitative exploration of the catalytic landscape separating divergent plant sesquiterpene synthases. Nat Chem Biol 4:617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kondrashov AS, Sunyaev S, Kondrashov FA (2002) Dobzhansky‐Muller incompatibilities in protein evolution. Proc Natl Acad Sci U S A 99:14878–14883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kulathinal RJ, Bettencourt BR, Hartl DL (2004) Compensated deleterious mutations in insect genomes. Science 306:1553–1554. [DOI] [PubMed] [Google Scholar]
- 40. Soylemez O, Kondrashov FA (2012) Estimating the rate of irreversibility in protein evolution. Genome Biol E 4:1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu J, Zhang J (2014) Why human disease‐associated residues appear as the wild‐type in other species: genome‐scale structural evidence for the compensation hypothesis. Mol Biol Evol 31:1787–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ferrer‐Costa C, Orozco M, Cruz XdL (2007) Characterization of compensated mutations in terms of structural and physico‐chemical properties. J Mol Biol 365:249–256. [DOI] [PubMed] [Google Scholar]
- 43. Barešić A, Hopcroft LEM, Rogers HH, Hurst JM, Martin ACR (2010) Compensated pathogenic deviations: analysis of structural effects. J Mol Biol 396:19–30. [DOI] [PubMed] [Google Scholar]
- 44. Jordan DM, Frangakis SG, Golzio C, Cassa CA, Kurtzberg J, Davis EE, Sunyaev SR, Katsanis N (2015) Identification of cis‐suppression of human disease mutations by comparative genomics. Nature 524:225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Povolotskaya IS, Kondrashov FA (2010) Sequence space and the ongoing expansion of the protein universe. Nature 465:922–926. [DOI] [PubMed] [Google Scholar]
- 46. Naumenko SA, Kondrashov AS, Bazykin GA (2012) Fitness conferred by replaced amino acids declines with time. Biol Lett 8:825–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zou Z, Zhang J (2015) Are convergent and parallel amino acid substitutions in protein evolution more prevalent than neutral expectations? Mol Biol Evol 32:2085–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goldstein RA, Pollard ST, Shah SD, Pollock DD (2015) Non‐adaptive amino acid convergence rates decrease over time. Mol Biol Evol 32:1375–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Usmanova DR, Ferretti L, Povolotskaya IS, Vlasov PK, Kondrashov FA (2015) A model of substitution trajectories in sequence space and long‐term protein evolution. Mol Biol E 32:542–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McCandlish DM, Rajon E, Shah P, Ding Y, Plotkin JB (2013) The role of epistasis in protein evolution. Nature 497:E1–E2. [DOI] [PubMed] [Google Scholar]
- 51. Socolich M, Lockless SW, Russ WP, Lee H, Gardner KH, Ranganathan R (2005) Evolutionary information for specifying a protein fold. Nature 437:512–518. [DOI] [PubMed] [Google Scholar]
- 52. Russ WP, Lowery DM, Mishra P, Yaffe MB, Ranganathan R (2005) Natural‐like function in artificial WW domains. Nature 437:579–583. [DOI] [PubMed] [Google Scholar]
- 53. Marks DS, Colwell LJ, Sheridan R, Hopf TA, Pagnani A, Zecchina R, Sander C (2011) Protein 3D structure computed from evolutionary sequence variation Sali A, editor. Plos One 6:e28766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, Zecchina R, Onuchic JN, Hwa T, Weigt M (2011) Direct‐coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci U S A 108:E1293–E1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hopf TA, Colwell LJ, Sheridan R, Rost B, Sander C, Marks DS (2012) Three‐dimensional structures of membrane proteins from genomic sequencing. Cell 149:1607–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ovchinnikov S, Kinch L, Park H, Liao Y, Pei J, Kim DE, Kamisetty H, Grishin NV, Baker D (2015) Large scale determination of previously unsolved protein structures using evolutionary information. eLife 4:e09248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Skerker JM, Perchuk BS, Siryaporn A, Lubin EA, Ashenberg O, Goulian M, Laub MT (2008) Rewiring the specificity of two‐component signal transduction systems. Cell 133:1043–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ovchinnikov S, Kamisetty H, Baker D (2014) Robust and accurate prediction of residue‐residue interactions across protein interfaces using evolutionary information. eLife 3:e02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hopf TA, Schärfe CPI, Rodrigues JPGLM, Green AG, Kohlbacher O, Sander C, Bonvin AMJJ, Marks DS (2014) Sequence co‐evolution gives 3D contacts and structures of protein complexes. eLife 3:e03430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Aakre CD, Herrou J, Phung TN, Perchuk BS, Crosson S, Laub MT (2015) Evolving new protein‐protein interaction specificity through promiscuous intermediates. Cell 163:594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Figliuzzi M, Jacquier H, Schug A, Tenaillon O, Weigt M (2016) Coevolutionary landscape inference and the context‐dependence of mutations in beta‐lactamase TEM‐1. Mol Biol Evol 33:268–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hopf TA, Ingraham JB, Poelwijk FJ, Springer M, Sander C, Marks DS (2015) Quantification of the effect of mutations using a global probability model of natural sequence variation. arXiv 1510.04612.
- 63. Süel GM, Lockless SW, Wall MA, Ranganathan R (2002) Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol 10:59–69. [DOI] [PubMed] [Google Scholar]
- 64. Morcos F, Jana B, Hwa T, Onuchic JN (2013) Coevolutionary signals across protein lineages help capture multiple protein conformations. Proc Natl Acad Sci U S A 110:20533–20538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sutto L, Marsili S, Valencia A, Gervasio FL (2015) From residue coevolution to protein conformational ensembles and functional dynamics. Proc Natl Acad Sci U S A 112:13567–13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Draghi JA, Plotkin JB (2013) Selection biases the prevalence and type of epistasis along adaptive trajectories. Evolution 67:3120–3131. [DOI] [PubMed] [Google Scholar]
- 67. Potapov V, Cohen M, Schreiber G (2009) Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng Des Sel 22:553–560. [DOI] [PubMed] [Google Scholar]
- 68. Harms MJ, Thornton JW (2013) Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat Rev Genet 14:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Alexander PA, He Y, Chen Y, Orban J, Bryan PN (2009) A minimal sequence code for switching protein structure and function. Proc Natl Acad Sci U S A 106:21149–21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Halabi N, Rivoire O, Leibler S, Ranganathan R (2009) Protein sectors: evolutionary units of three‐dimensional structure. Cell 138:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yokoyama S, Xing J, Liu Y, Faggionato D, Altun A, Starmer WT (2014) Epistatic adaptive evolution of human color vision. PLOS Genet 10:e1004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Anderson DW, McKeown AN, Thornton JW (2015) Intermolecular epistasis shaped the function and evolution of an ancient transcription factor and its DNA binding sites. eLife 4:e07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bridgham JT, Ortlund EA, Thornton JW (2009) An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature 461:515–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dellus‐Gur E, Elias M, Caselli E, Prati F, Salverda MLM, de Visser JAGM, Fraser JS, Tawfik DS (2015) Negative epistasis and evolvability in TEM‐1 β‐lactamase—the thin line between an enzyme's conformational freedom and disorder. J Mol Biol 427:2396–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Harms MJ, Thornton JW (2014) Historical contingency and its biophysical basis in glucocorticoid receptor evolution. Nature 512:203–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Huang W, Palzkill T (1997) A natural polymorphism in beta‐lactamase is a global suppressor. Proc Natl Acad Sci U S A 94:8801–8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sideraki V, Huang W, Palzkill T, Gilbert HF (2001) A secondary drug resistance mutation of TEM‐1 beta‐lactamase that suppresses misfolding and aggregation. Proc Natl Acad Sci U S A 98:283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bloom JD, Silberg JJ, Wilke CO, Drummond DA, Adami C, Arnold FH (2005) Thermodynamic prediction of protein neutrality. Proc Natl Acad Sci U S A 102:606–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bloom JD, Labthavikul ST, Otey CR, Arnold FH (2006) Protein stability promotes evolvability. Proc Natl Acad Sci U S A 103:5869–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS (2006) Robustness–epistasis link shapes the fitness landscape of a randomly drifting protein. Nature 444:929–932. [DOI] [PubMed] [Google Scholar]
- 81. Tokuriki N, Tawfik DS (2009) Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature 459:668–673. [DOI] [PubMed] [Google Scholar]
- 82. Jacquier H, Birgy A, Le Nagard H, Mechulam Y, Schmitt E, Glodt J, Bercot B, Petit E, Poulain J, Barnaud G, Gros PA, Tenaillon O (2013) Capturing the mutational landscape of the beta‐lactamase TEM‐1. Proc Natl Acad Sci U S A 110:13067–13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lunzer M, Miller SP, Felsheim R, Dean AM (2005) The biochemical architecture of an ancient adaptive landscape. Science 310:499–501. [DOI] [PubMed] [Google Scholar]
- 84. Tokuriki N, Tawfik DS (2009) Stability effects of mutations and protein evolvability. Curr Opin Struc Biol 19:596–604. [DOI] [PubMed] [Google Scholar]
- 85. Brown NG, Pennington JM, Huang W, Ayvaz T, Palzkill T (2010) Multiple global suppressors of protein stability defects facilitate the evolution of extended‐spectrum TEM β‐lactamases. J Mol Biol 404:832–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bloom JD, Nayak JS, Baltimore D (2011) A computational‐experimental approach identifies mutations that enhance surface expression of an oseltamivir‐resistant influenza neuraminidase. PLOS One 6:e22201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bridgham JT, Keay J, Ortlund EA, Thornton JW (2014) Vestigialization of an allosteric switch: genetic and structural mechanisms for the evolution of constitutive activity in a steroid hormone receptor Zhang J, editor. PLOS Genet 10:e1004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Weinreich DM, Lan Y, Wylie CS, Heckendorn RB (2013) Should evolutionary geneticists worry about higher‐order epistasis? Curr Opin Genet Dev 23:700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hietpas RT, Jensen JD, Bolon DNA (2011) Experimental illumination of a fitness landscape. Proc Natl Acad Sci U S A 108:7896–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Poelwijk FJ, Krishna V, Ranganathan R (2015) The context‐dependence of mutations: a linkage of formalisms. arXiv 1502.00726. [DOI] [PMC free article] [PubMed]
- 91. Sorrells TR, Booth LN, Tuch BB, Johnson AD (2015) Intersecting transcription networks constrain gene regulatory evolution. Nature 523:361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. McLaughlin RN, Poelwijk FJ, Raman A, Gosal WS, Ranganathan R (2012) The spatial architecture of protein function and adaptation. Nature 491:138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kachroo AH, Laurent JM, Yellman CM, Meyer AG, Wilke CO, Marcotte EM (2015) Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 348:921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]