

Abstract
Tumor necrosis factor receptor‐associated factors (TRAFs) constitute a family of adapter proteins that act in numerous signaling pathways important in human biology and disease. The MATH domain of TRAF proteins binds peptides found in the cytoplasmic domains of signaling receptors, thereby connecting extracellular signals to downstream effectors. Beyond several very general motifs, the peptide binding preferences of TRAFs have not been extensively characterized, and differences between the binding preferences of TRAF paralogs are poorly understood. Here we report a screening system that we established to explore TRAF peptide‐binding specificity using deep mutational scanning of TRAF‐peptide ligands. We displayed single‐ and double‐mutant peptide libraries based on the TRAF‐binding sites of CD40 or TANK on the surface of Escherichia coli and screened them for binding to TRAF2, TRAF3, and TRAF5. Enrichment analysis of the library sequencing results showed differences in the permitted substitution patterns in the TANK versus CD40 backgrounds. The three TRAF proteins also demonstrated different preferences for binding to members of the CD40 library, and three peptides from that library that were analyzed individually showed striking differences in affinity for the three TRAFs. These results illustrate a previously unappreciated level of binding specificity between these close paralogs and demonstrate that established motifs are overly simplistic. The results from this work begin to outline differences between TRAF family members, and the experimental approach established herein will enable future efforts to investigate and redesign TRAF peptide‐binding specificity.
Keywords: protein–protein interactions, deep mutational scanning, bacterial surface display, interaction specificity
Introduction
Tumor necrosis factor receptor‐associated factors (TRAFs) were originally identified in the mid‐1990s as proteins that interacted with the cytoplasmic tails of TNFR superfamily members in yeast two‐hybrid assays.1, 2, 3, 4 The TRAF family in humans has since been extended to include seven members, TRAFs 1–7.5 TRAFs mediate interactions downstream of a diverse array of signaling receptors including TNFR super family members, Toll‐like receptors, the T cell receptor, interleukin receptors, NOD‐like receptors, RIG‐I‐like receptors, IFN receptors, and TGFβ receptors.6 These pathways control inflammation, adaptive and innate immunity, and apoptosis, and they are important in many human diseases. Deletion studies in mice have revealed effects varying from embryonic lethality to impaired immune function for the different TRAFs.1, 3, 5, 7, 8, 9, 10, 11, 12
The multiple domains present in TRAFs allow them to connect and regulate components of signaling pathways. TRAFs 2–6 contain an N‐terminal RING domain, followed by 5–7 zinc‐finger domains, a coiled‐coil domain, and a MATH (meprin and TRAF homology) domain. Together, the coiled‐coil and MATH domains make up the TRAF domain, which mediates the homo‐ and hetero‐trimerization of TRAFs.13 TRAF1 does not have a RING domain and only contains one zinc finger, whereas TRAF7 has both RING and zinc‐finger domains, but contains WD40 repeats in place of the TRAF domain. The MATH domain (also known as the TRAF‐C domain), which is the subject of this study, binds short linear motifs in the cytoplasmic tails of receptors or their downstream adapter proteins. These interactions are weak (dissociation constants of tens‐to‐hundreds of micromolar) for monomeric MATH‐peptide interactions and generally require the avidity conferred by receptor and TRAF oligomerization. Dependence on oligomerization allows control of pathway activation by extracellular ligands, which oligomerize receptors upon binding to them. The zinc‐finger and RING domains of TRAF proteins mediate interactions with downstream effectors and are important for activating kinase cascades. The RING finger domains possess E3 ligase activities, and TRAFs are known to mediate K63‐linked poly‐ubiquitination of themselves and other proteins to recruit and activate effectors.12 Degradative ubiquitination is also important in TRAF function and regulation, as TRAFs 1 and 2 recruit the E3 ubiquitin ligases cIAP1 and 2, which mediate K48‐linked ubiquitination of TRAFs themselves and other associated proteins.12 The cIAP proteins interact with the coiled‐coil domains of TRAFs 1 and 2.14 Thus, TRAFs posses multiple protein‐protein interaction interfaces with which to scaffold signaling complexes, and they utilize their E3 ubiquitin ligase activities to further modulate signaling.
TRAFs can have overlapping or distinct functions related to their differential expression and binding preferences and a significant component of TRAF functional specificity arises from the interaction specificity of their MATH domains. An example is the best‐studied pathway of TRAF function, that downstream of the TNFR superfamily member CD40. CD40 contains two TRAF interaction motifs (TIMs) in its cytoplasmic tail, one that can be bound by TRAFs 1, 2, 3, and 5, and another that can bind TRAF6.13 TRAF3, by competing for binding to the same site on CD40, can block activation mediated by TRAFs 2 and 5, but not by TRAF6.15
Evolutionary analysis of TRAFs suggests that TRAFs 4 and 6 are the more ancient homologs, with examples of TRAF6 present in insects and TRAF4 in cnidaria and early chordates.16 In contrast, TRAFs 1, 2, 3, and 5 are largely found in vertebrates.16 TRAFs 1 and 2 and TRAFs 3 and 5 share the highest sequence similarity to one another and may have evolved through gene duplication. In support of this, these pairs are also known to hetero‐oligomerize.13 Different peptide binding motifs have been defined for TRAFs 1, 2, 3, and 5 versus TRAF6. TRAFs 1, 2, 3, and 5 share many of the same binding partners, recognizing a major ((P/S/A/T)x(Q/E)E) and minor (PxQxxD) motif.17 A number of TRAF 1, 2, 3, and 5 binding partners also fit the motif PxQxT, suggesting that these TRAFs can accommodate a variety of sequences C‐terminal to the critical central Q/E position (we refer to this position as position 0 below).18 See Figure 1(A) for a structure of TRAF3 bound to a peptide from TANK, with the PIQCT core motif of TANK highlighted in Figure 1(B). TRAF6 recognizes a different motif (PxExx(Aromatic/Acidic)). Binding partners of TRAF4 have not been directly characterized, although TRAF4 may bind some of the same sites as TRAF6.20, 21, 22 However, at least one example of a TIM exists that binds both TRAFs 1/2/3/5 and TRAF6. TRAF6 association with the Epstein‐Barr virus protein LMP1 was shown to occur at the same PQQATD site used by TRAFs 1/2/3/5.23 This site fits the minor TIM motif of PxQxxD identified for TRAF2, and is close to the TRAF6 motif PxExx(Ar/Ac), suggesting that peptides that fit the more general motif Px(Q/E)xx(Ac) might be accessible to both groups of TRAFs. These results suggest that there is considerable plasticity in the established motifs, and a more detailed examination of binding capabilities is needed to create more accurate binding models.
Figure 1.
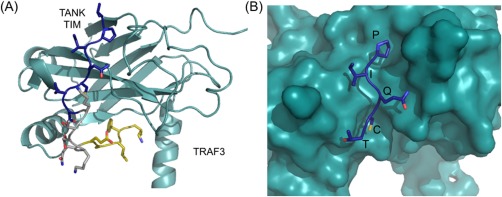
Structure of a TRAF‐peptide complex (PDB ID 1L0A). (A) Structure of TRAF3 (teal) bound to the TRAF interaction motif (TIM) from TANK (shown in sticks) with the peptide core site and exosite‐binding region used in the SiteMAP analysis highlighted in blue and gold, respectively.19 (B) The five residues corresponding to the core motif (PIQCT) of TANK are shown in stick representation (blue) in the TRAF3 binding groove (teal).
The classification of TRAF binding specificity has focused on the core motifs described above, but the literature contains numerous hints that peptide regions outside of the core motif can influence affinity and specificity. A number of these examples support the idea that TRAF3, especially, makes interactions outside of the peptide core. An early report by Devergne et al. showed that mutation of either the core motif proline or glutamine to alanine in LMP1 abrogated TRAF1 and TRAF2 binding, but both mutations were necessary to lose TRAF3 binding.18 This suggested that TRAF3 might make use of additional interactions to bind LMP1. Indeed, this was shown to be the case for CD40, the human receptor that LMP1 mimics. Substitution SPOT arrays (arrays of peptide mutants synthesized on membranes) that were used to test binding of short (core binding motif) and C‐terminally extended CD40 peptides showed more relaxed binding preferences in the core for TRAF3 binding when the C‐terminal extension was present, relative to the shorter peptides.24 Structures of TRAF3 bound to long peptides from TANK and BAFF‐R also show extended interactions C‐terminal to the peptide core, with the peptide wrapping around the MATH domain [Fig. 1(A)].19, 25 However, the relative contributions of core versus extended peptide regions to TRAF3 binding affinity are unknown. Little is known about the importance of peptide sequence N‐terminal to the core motif, but one recent study found that a histidine to tyrosine mutation three residues N‐terminal to the core proline in BAFF‐R increases binding affinity to TRAFs 2, 3, and 6.26 Signaling through BAFF‐R prevents apoptosis, and this mutation was found in a subset of patients with non‐Hodgkin lymphoma. As more interactions are characterized, further examples of interactions mediated by peptide regions outside the core are likely to be discovered. Interaction preferences outside the core could vary widely due to lower sequence identity between the TRAFs outside of the core‐binding groove. Therefore, these interactions may be an important source of specificity.
A better understanding of TRAF binding preferences could be applied to interactome prediction and the design of specific peptide inhibitors. Given the diversity in TRAF binding preferences, it is not straightforward to identify the TIM in an interaction partner identified by pull‐down or other methods. Because TRAFs are often present in multi‐protein assemblies, and themselves have more than one interaction interface, it is not always clear whether interactions are with the MATH peptide binding groove. Better models of TRAF‐peptide binding preferences would allow identification of TIMs on known partners, as well as prediction of new partners. Knowledge of the interaction preferences of each TRAF protein would allow prediction of the relative affinity of TRAFs for a given TIM, providing hypotheses about signaling mechanisms. Specific inhibitors of TRAFs would provide the means to test such hypotheses. It is a common practice to swap cytoplasmic domains of TNFR super family members in order to put the downstream effects under control of a different extracellular domain/ligand pair.26, 27 It is possible that TRAF binding sites specific for binding one TRAF could be swapped into an interaction partner of interest to examine pathway requirements for individual TRAFs.
Because of the importance of TRAFs in many disease states, specific peptide inhibitors would be useful therapeutic leads. Several groups have demonstrated this idea for TRAF6. TRAF6 signaling downstream of RANK is responsible for osteoclast differentiation, which can lead to osteoporosis and cancer‐induced bone lesions when it occurs aberrantly.28 Ye et al. fused peptides corresponding to the TRAF6 binding sites on RANK to a cell penetrating peptide and showed that treating osteoclast precursor cells with these peptides reduced NF‐κB activation and associated osteoclast differentiation.20 Inhibiting TRAF1 is also of interest, as its overexpression is associated with several B‐cell malignancies. Its role in these leukemias and lymphomas may be to prevent apoptosis by recruiting anti‐apoptotic proteins like the cIAPs to activated TNFRs.29 The roles of TRAF3 and TRAF6 in autoimmunity and inflammation make them attractive targets for the treatment of related diseases.
This study presents an initial characterization of the peptide binding preferences of TRAFs 2, 3, and 5 using deep mutational scanning.30, 31, 32, 33 Because the TRAFs are close homologs, the possibility of differences in binding preferences was of interest, and to investigate this we compared the sequence identity and physicochemical characteristics of the different TRAF binding sites. Then, to generate sequences for many TRAF‐binding peptides, we developed a bacterial surface display protocol and used it to screen single and double mutant libraries of peptides based on TIM sequences from CD40 and TANK. This screen yielded interesting insights into differences between TRAFs 2, 3, and 5, as well as leads for the development of specific peptide inhibitors.
Results
Comparison of TRAFs using sequence identity of the MATH domain and peptide‐binding site
Sequence identity in the TRAF MATH domains and their peptide‐binding sites illustrates which TRAF family members are most similar to each other (Fig. 2). The peptide‐binding site considered includes the 18 TRAF residues within 7 Å of the 5‐mer peptide core (‘PVQET’) in a structure of CD40 bound to TRAF2. TRAF6 has the lowest sequence identity to the other TRAFs, with ∼30% identity over the full MATH domain and only ∼20% identity in the core‐ binding site. TRAF4 has ∼10% higher sequence identity to TRAFs 1, 2, 3, and 5 than to TRAF6 over both the MATH domain and the peptide‐binding site. TRAFs 1, 2, 3, and 5 share high identity (>52%) within the MATH domain, and very high identity in the core‐binding site (>78%). Therefore, the core TRAF 1/2/3/5‐binding motif in TRAF interaction partners contacts a nearly identical set of residues when binding these four homologs. The lower sequence identities for the full MATH domains suggest that differences between these four homologs outside the core‐binding site could influence interaction specificity.
Figure 2.
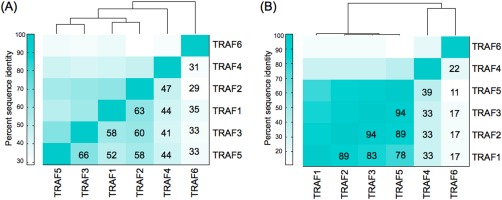
Sequence identities show similarities between TRAFs 1, 2, 3, and 5. (A) Sequence identities over the entire TRAF MATH domain. (B) Sequence identities in the core peptide binding site. Residues included are within 7 Å of the core ‘PVQET’ peptide sequence in a structure of CD40 bound to TRAF2 (PDB ID 1D00).17
Comparison of TRAFs based on physicochemical properties of the peptide‐binding sites
To compare peptide‐binding environments on the surfaces of the TRAFs, we looked at the physicochemical properties around the core‐binding site and around an exosite [illustrated in Fig. 1(A)]. Here we define an exosite as a binding site on the TRAF surface outside of the region bound by the core peptide motifs. We aligned all available TRAF structures to a structure of TRAF3 bound to a long peptide from TANK and used SiteMAP to characterize binding potential on the TRAF surface for hydrophobic, hydrogen bond donor, and hydrogen bond acceptor groups. SiteMAP evaluates the physicochemical characteristics of binding grooves and generates maps, constructed of points, where hydrophobic or hydrogen bonding groups are predicted to have a propensity to bind.34, 35 Using such maps, we computed an intersection score, which we defined in prior work to provide a metric of similarity.36 The intersection score for two binding grooves is the sum of points of a specific type (e.g., hydrophobic) that fall within overlapping regions of both proteins' maps, as further described in Materials and Methods. We used intersection scores to quantify the similarity of pairs of binding sites. The core‐binding site was defined as the region within 6 Å of the Cβ residues of the “VPIQCTD” sequence of TANK. Although TRAF6 binds peptides in a somewhat different orientation than TRAF3 does, the core‐binding motif residues fall within this same region. The core‐binding site comparison [Fig. 3(A)] reflects the sequence identity results in that TRAFs 2, 3, and 5 have highly similar binding sites, and TRAF4 and TRAF6 are each unique and not very similar to any other TRAFs. Two TRAF3 structures, 1FLL and 1RF3 [the bottom rows in Fig. 3(A)], have lower similarity, but these structures are of low quality and have long peptides bound in unique hairpin conformations that may be influenced by crystal contacts.37, 38
Figure 3.
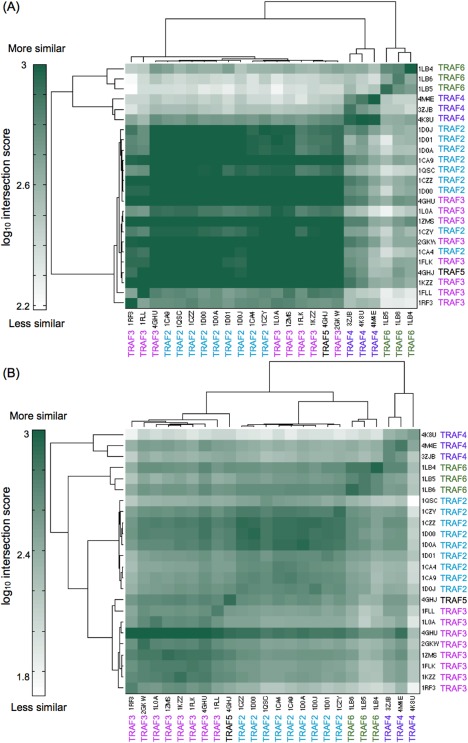
Comparison of the physicochemical characteristics of core and exosite regions of TRAF binding grooves by SiteMAP. The intersection score is a measure of the similarity of binding potential for hydrophobic, hydrogen bond acceptor and hydrogen bond donor groups between structures. (A) Comparison of the core peptide‐binding groove. (B) Comparison of an exosite that is bound by a C‐terminal extension of TANK in a structure of TRAF3 (PDB ID 1L0A).19
The exosite region we examined was the area surrounding the “EALF” residues of TANK in structure 1L0A, which are C‐terminal to the core motif in the peptide [gold in Fig. 1(A)]. This exosite is functionally significant for TRAF3 and TRAF2.19 SiteMAP comparisons of this exosite for the different TRAFs show that each TRAF has unique surface features in this region [Fig. 3(B)]. TRAF2, 3, and 5 structures segregate into their own clusters, contrasting with the groupings based on core‐binding site similarity, in which these three proteins are intermingled. This demonstrates that outside of the core binding site, TRAFs 2, 3, and 5 have differences that may influence peptide binding specificity.
Screening of single and double mutant libraries of CD40 and TANK
To explore the binding preferences of TRAFs 2, 3, and 5, we created libraries of single and double point mutants of CD40 and TANK. Peptide libraries were displayed on the C‐terminus of eCPX, on the surface of E. coli, and were screened for binding to the MATH domain of each TRAF using fluorescence‐activated cell sorting (FACS) [Fig. 4(A)]. A native cysteine was mutated to serine in both TANK and CD40 to reduce background binding signal due to disulfide formation between the TRAFs and the peptides. See Supporting Information for a detailed description of the optimization of the TRAF and bacterial display constructs. The libraries included all single mutants in the 21‐mer (CD40) or 18‐mer (TANK) colored regions in Figure 4(B). Figure 4(B) also defines our peptide position numbering convention. All double mutants in three 7‐mer segments of different colors (or underlined for the middle TANK section) were also included. The libraries were sorted in three rounds, or cycles of growth and sorting, for binding to 10 μM TRAF2, TRAF3 or TRAF5. Sorting gates were set to include ∼1% of a negative threonine‐to‐alanine control for each TRAF (see Methods). See Figure 4(C) for representative FACS distributions for 10 μM TRAF2 binding to a CD40_C6S positive control, a TANK_C‐4S_T2A negative control, and the naïve CD40 library. The final pools (from round 3) were analyzed for binding to 10 μM of each TRAF. The FACS distributions of events according to binding signal are shown in Figure 5. The enriched TANK library pools showed no binding differences between TRAFs 2, 3, and 5; all three TRAFs bound equally to pools enriched for binding the other TRAFs. In contrast, the enriched CD40 library pools showed varying degrees of specificity. The CD40 pool enriched for TRAF3 binding (blue traces) showed greatly reduced binding to TRAF2 and TRAF5. The CD40 pools enriched for binding to TRAFs 2 and 5 (red and green traces) showed modestly reduced binding to TRAF3. However, there was no specificity apparent between TRAFs 2 and 5, as both bound equally to CD40 pools enriched for binding either TRAF.
Figure 4.
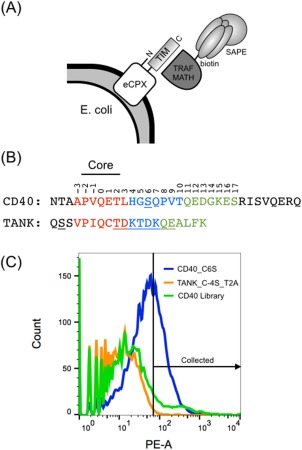
Cell surface display system, peptide constructs and example binding data. (A) Peptides encoding TRAF interaction motifs (TIMs) were displayed on the C‐terminus of eCPX. Binding of biotinylated TRAF MATH domains was detected with streptavidin‐phycoerythrin (SAPE). (B) TIMs from CD40 and TANK were displayed and mutated in the libraries. The core site is labeled. The three 7‐mer segments that are colored (and underlined for the middle TANK segment) were mutated to generate all single and double point mutants. The underlined serines were mutated from the wild‐type cysteine. Peptide positions are numbered in reference to the core glutamine as position zero. (C) 10 μM TRAF2 binding to a positive control (CD40_C6S, blue), a negative control (TANK_C‐4S_T2A, orange), and the naïve CD40 library (green). An example sorting gate is shown, which was set to include ∼1% of the negative control.
Figure 5.
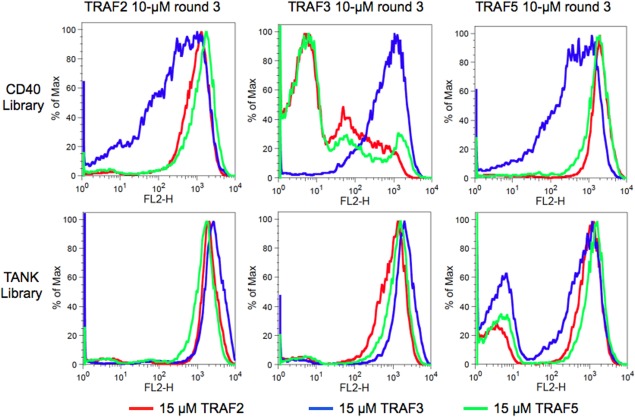
TRAFs 2, 3, and 5 binding to the final CD40 and TANK library pools. Columns are labeled with the sorting pool analyzed (round 3, done at 10 μM for each TRAF), with CD40 library pools on top and TANK library pools on bottom. The conditions used for analysis are colored: binding to 15 μM TRAF2 (red), TRAF3 (blue), or TRAF5 (green). The bi‐modal population in the TANK library sorted for binding to TRAF5 is due to incomplete enrichment for binders. The x axis is the fluorescence due to TRAFs bound to the cell surface, and the y axis is proportional to the number of events. Data are representative of three experiments performed on separate days.
Enrichment analysis of CD40 and TANK library sequences
To analyze the enrichment of variants in each sort, we Illumina sequenced each library pool and calculated a functional score for each peptide variant similarly to Starita et al.32 See Materials and Methods for details. Briefly, an enrichment ratio was calculated from the frequency of each variant in each round relative to the naïve library. A line was fit to the enrichment ratios across rounds, and the slope of this line was converted to a functional score. The functional scores were normalized by the wild‐type sequence functional score, such that variants enriched relative to wild type had functional scores >1, and variants reduced relative to wild type had scores <1. Note that under the fairly stringent sorting conditions we used, the wild‐type peptides typically decreased in frequency over three rounds of sorting.
We analyzed the relationship between functional score, counts, and the R 2 of the line fit to the enrichment ratios across rounds for each variant. In Supporting Information Figure 3 (CD40 library) and 4 (TANK library), each variant is plotted with counts shown for the input round, average in the selected rounds, or the last round. These plots, which, due to their appearance, we refer to as butterfly plots, show a distinctive central band of variants with poor R 2 values (0–0.3). The binding signal distributions of these variants likely lie with their centers near the cutoff used in sorting, such that in sequential rounds these variants were randomly enriched or de‐enriched due to inherent noise. The functional scores of variants with poor R 2 values varied for different TRAFs. This is expected, because the proportion of the wild‐type controls that fell into the collection gate differed for different TRAFs, due to their different affinities (the sorting gate was defined using a negative control, as described above). The majority of variants showed functional scores below 1, indicating that most mutations decreased binding. For variants with high functional scores, counts generally increased from the input library to the selected rounds.
To identify clones that were consistently enriched or depleted, we used the R 2 value as a quality metric. Because each round of sorting was performed under equal stringency, we expect the enrichment in each round to be roughly equivalent. Following others who have similarly observed this behavior, we used the R 2 value for a line fit to the data to identify strong deviations from the expected enrichment pattern.33 We examined several peptide variants with known binding behavior to define an R 2 value cutoff by which to filter the data. The wild‐type peptide, four single mutants with stop codons in the first four peptide positions, the threonine‐to‐alanine mutation at position 2 (T2A) and, in CD40, Q0W, E1Y, and D13T_G14N, are highlighted with different colored edges on the butterfly plots (Fig. 6). The wild‐type peptides consistently had high counts and also high R 2 values. The T2A and stop codon mutations, expected to weaken or abrogate binding, had functional scores clustered around 1. Our ability to resolve non‐binders and binders with affinity below that of the wild‐type peptides is poor in this dataset, as the sorting conditions were sufficiently stringent that even the wild‐type clones typically decreased in frequency. The CD40 mutants Q0W and E1Y were demonstrated to increase binding to TRAF3 and decrease binding to TRAF2 and TRAF5 (see below). As seen in Figure 6, these mutations had high counts in the last round of the CD40 library sorted for binding to TRAF3, and lower counts for TRAFs 2 and 5. To include such variants with demonstrable binding effects, we set a relatively generous cutoff of a R 2 value of 0.4, because CD40_Q0W had a R 2 value of 0.42 in the TRAF3‐sorted library. Single mutants passing this cutoff, with their functional scores, counts and R 2 values, are provided in the Supporting Information.
Figure 6.
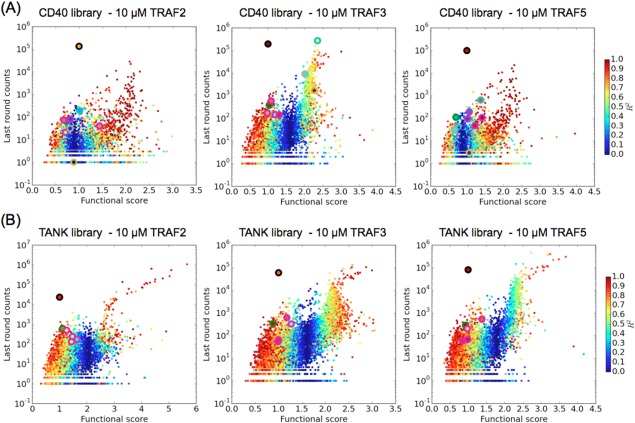
Library variants plotted according to last‐round sequencing counts, functional score, and R 2 value. (A) CD40 library variants sorted for binding to TRAF2, 3, or 5. (B) TANK library variants sorted for binding to TRAF2, 3, or 5. The following variants are highlighted with a colored outline: wild‐type (black), T2A (green), the four single mutants with stop codons in peptide positions −3 to 0 (magenta), and for CD40: Q0W (gray), E1Y (cyan), and D13T_G14N (yellow).
The functional scores for single mutants are shown as heat maps in Figure 7, with variants with R 2 values below 0.4 shaded gray. In the CD40 libraries [Fig. 7(A)], the TRAF2 and TRAF5 preferences are similar, and different from the TRAF3 preferences. This agrees with the FACS analysis performed on the final pool of these libraries, which showed that TRAF2 and TRAF5 bound to peptides in each other's pools equally, but bound weakly to peptides in the TRAF3 pool; the opposite was true for the pool selected for binding to TRAF3 (Fig. 5). For TANK library peptide enrichment [Fig. 7(B)], the overall patterns were similar for all three TRAFs, in agreement with all three TRAFs binding each other's pools equally in Figure 5.
Figure 7.
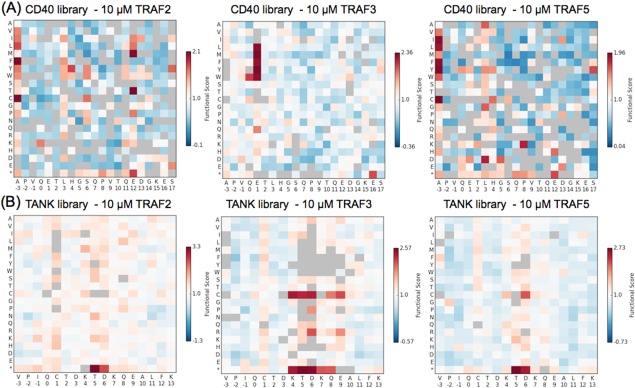
Functional scores of single mutants. (A) CD40 library single mutants. (B) TANK library single mutants. Scores are normalized to the wild‐type peptide (score of 1), with scores greater than 1 indicating enrichment over the wild type. Variants with gray squares had a R 2 value of <0.4 for the fit of a line to the enrichment ratios across rounds. Asterisks stand for stop codons.
In addition to our sorting experiment using 10 μM of each TRAF protein, the TANK library was sorted for three consecutive rounds using 3 μM TRAF2, TRAF3, or TRAF5. The single mutant functional scores for the 3 μM sorts are shown in Supporting Information Figure 5. Comparison to the 10 μM sorts in Figure 7(B) shows very similar trends for all three TRAFs. The Pearson correlation coefficient between the 3 μM and the 10 μM single mutant functional scores, for clones that passed the R 2 value cutoff of 0.4, was 0.39 for TRAF2, 0.85 for TRAF3, and 0.70 for TRAF5. The correlation roughly corresponds to the observed affinity of each TRAF for the wild‐type TANK peptide (TRAF2<TRAF5<TRAF3), suggesting that the lower correlation for the TRAF2 results may arise in part from a greater difference in the degree of binding observed between 3 and 10 μM.
Analysis of strongly enriched peptide mutants
For the TANK library, all three TRAFs demonstrated relaxed preferences at peptide position 1, and in a section of peptide spanning positions 4–8, with greatest permissiveness at positions 5 and 6. This agrees well with the structure of a long TANK peptide bound to TRAF3 (PDB ID 1L0A) in which this middle section of the peptide does not make close contacts with the TRAF surface and has higher B‐factors than the rest of the peptide, indicating flexibility [gray section of peptide in Fig. 1(A)].19 The most enriched variants in this middle region included cysteines and stop codons. Although experiments were done in the presence of DTT, it seems likely that the preference for cysteines resulted from disulfide‐formation between the peptide cysteine and the cysteines present in the TRAF binding groove. Redox‐dependent interaction signals were observed for the wild‐type peptides, prior to introducing a cysteine‐to‐serine mutation into the CD40 and TANK backgrounds to reduce background (Supporting Information Fig. 1).
To assess the effect of the stop codons, we titrated TRAF3 against cells displaying TANK or TANK with stop codons at positions 5, 6, 7, or 8 (Supporting Information Fig. 6). The binding signal saturated at different levels for the different‐length TANK variants, indicating that truncating peptides can have significant effects on expression. TANK with stop codons at positions 5 and 6 had the highest binding signal saturation level and also had the highest functional scores. When fit to the Hill equation, TANK‐5stop and 6stop had dissociation constants of 3.6 and 3.8 μM for TRAF3 binding, respectively (Supporting Information Table I). The dissociation constant for TRAF3 binding TANK was 9.6 μM, and TANK‐7stop and 8stop had intermediate K D values of 7.3 and 7.8 μM, respectively. These values agree well with the trends seen in the functional scores [Fig. 7(B)]. Thus, in addition to expression differences, the higher functional scores for truncated TANK variants also reflect tighter binding. This could be due to an interaction between a negatively charged C‐terminus and a conserved arginine in the TRAF binding groove, which is best positioned to interact with negative charges near positions 4–6 in the structure of TANK bound to TRAF3 (see arginine 393 in TRAF3 in structure 1L0A).19 Notably, the Hill equation was fit with a Hill coefficient of ∼3 for all peptides, indicating that the TRAF3 MATH monomers form trimers on the E. coli surface, even though our constructs did not include the coiled‐coil region that contributes to trimerization of full‐length TRAFs (Supporting Information Table I).
The CD40 library functional score trends show similarities to previously published SPOT arrays for TRAF2 and TRAF3 binding to CD40 single mutants.24 Figure 8 shows a representation of the SPOT array data in a qualitative gray‐scale, which can be compared with the TRAF2 and TRAF3 enrichment data for the CD40 library in Figure 7(A). TRAF2 binding demonstrates very little tolerance of mutation in the core “PVQET” sequence in either the SPOT arrays or the library results. In contrast, TRAF3 shows moderate tolerance at “PVQ,” and greater tolerance at position 1 in the SPOT arrays. The most strongly enriched single mutants are also those that vary at position 1 in the TRAF3 library results. Outside of the core positions (−2 to 2), it is difficult to interpret the smaller variations in binding signal seen on the SPOT arrays, which could result from differences in peptide synthesis or accessibility. The library results indicated that the TRAFs have sequence preferences in the C‐terminal peptide region, with TRAF2 showing strong enrichment of non‐wild‐type residues at several positions.
Figure 8.
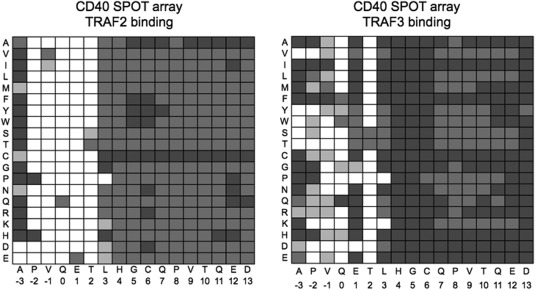
Representation of CD40 SPOT arrays binding to TRAF2 and TRAF3 from Pullen et al.26 Boxes are shaded proportional to binding, with darker gray indicating more binding and white indicating no observed binding.
CD40 mutants demonstrate binding specificity between TRAF paralogs
To further investigate differences in paralog binding preferences observed in the CD40 library background, we tested two single mutants, E1Y and Q0W, and one double mutant, D13T_G14N, that were greatly enriched in TRAF3 screening but not in screening for binding to TRAF2 or TRAF5. The E1Y and Q0W mutations are part of the core binding motif, whereas positions 13 and 14 are located near the C‐terminus of the library variable region. Binding was tested to peptides displayed on the surface of E. coli at two TRAF concentrations (Fig. 9 and Supporting Information Fig. 7). Although differences in expression levels may contribute to differences in binding signals, we can compare binding of the same peptide across the three TRAF proteins.
Figure 9.
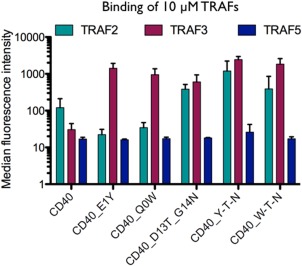
TRAF binding to CD40 mutants illustrates specificity between paralogs. Interaction data are for 10 μM TRAF binding to peptides displayed on the surface of E. coli. The median binding fluorescence signal is shown with error bars indicating the standard deviation of experiments performed on three separate days.
In these experiments using single clones, TRAF3 bound better than TRAF2 or TRAF5 to both CD40_E1Y and CD40_Q0W, consistent with the library enrichment results (Fig. 9 and Supporting Information Fig. 7). Double mutant CD40_D13T_G13N bound to TRAF3 more than to TRAF2, although this preference was quite modest and to TRAF2 and TRAF3 more than to TRAF5, which showed low binding signal similar to wild type. Interestingly, mutation E1Y was well tolerated in the context of the two C‐terminal mutations D13T and G14N for binding to TRAF2 (Fig. 9, CD40_Y‐T‐N). This mutation did not increase the difference in TRAF2 vs. TRAF3 binding compared to what was observed for CD40_D13T_G13N, which contrasts with the behavior observed when E1Y was made as a single mutation in CD40. In a departure from this pattern, mutation Q0W favored TRAF3 binding relative to TRAF2 binding in both the wild‐type CD40 and CD40_D13T_G14N contexts. These results clearly show that mutations in the core binding site (E1Y and Q0W), and well outside of the core (D13T_G14N) can have varying effects on binding to these three closely related TRAF paralogs.
Comparison of double and single mutant enrichment profiles
Library sorting also allowed us to evaluate double mutant preferences for all possible mutation pairs in three 7‐mer segments of each peptide, as shown in Figure 4(B). To measure the change in enrichment for each substitution in a double‐mutant versus wild‐type background, we subtracted the single‐mutant functional score from the double‐mutant functional score. The resulting difference heat maps are shown in Supporting Information Figure 8. There was little change observed between double‐ and single‐mutant functional scores for all three TANK library pools [Supporting Information Fig. 8(B)]. In contrast, many substitutions were significantly more enriched as double mutants in the CD40 libraries (red squares). Many of these substitutions occurred in the middle section of CD40, around positions 5–7.
Two single mutants of CD40 discussed above that were highly enriched by TRAF3, Q0W, and E1Y, are motif‐breaking mutations, in that they change two of the highly conserved positions in the TRAF1/2/3/5‐binding motif, (P/S/A/T)x(Q/E)E. As such, it is possible that they could lead to a change in binding conformation and thereby change preferences at other positions. To examine this possibility, we made functional score difference heat maps for the double mutants that occurred with E1Y or Q0W (Fig. 10). Because the E1Y and Q0W mutations were unfavorable for TRAF2 and TRAF5 binding, fewer double mutants made it through the library screen for these TRAFs. For TRAF3, many positions became more permissive of mutation in the presence of E1Y. In the presence of Q0W, an increase in functional score was observed for basic residues at positions −3, −2, and 1 in the TRAF3‐enriched pool. The TRAF2 and TRAF5 results for Q0W hint that they may also share this preference for basic residues in conjunction with Q0W, as the few enriched residues for these TRAFs include basic residues at positions −2 or 1. The significant changes in double mutant versus single mutant preferences in the CD40 library pools suggest that these mutant peptides can bind with alternate interaction modes. Future work will explore these differences and their structural basis.
Figure 10.
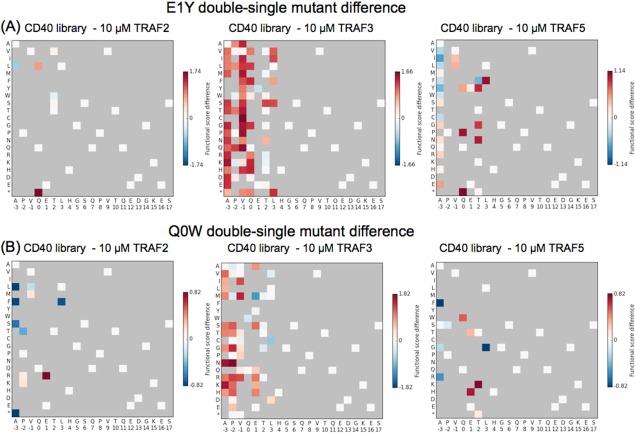
Changes in functional scores for single mutants in the wild‐type background vs. in the presence of E1Y or Q0W. For substitutions that appeared in the double mutant pools with (A) E1Y or (B) Q0W, a change in functional score was calculated as the double‐mutant functional score minus the single‐mutant functional score. Red indicates residues that were more enriched in the presence of E1Y or Q0W than in the wild‐type background.
Discussion
We have established a system to explore the peptide binding preferences of TRAF paralogs using bacterial surface display and deep mutational scanning.30 Our results agree well with the limited published data on this system. The CD40 single‐mutant library results showed similar trends as CD40 SPOT arrays published for TRAF2 and TRAF3, and we have expanded this mutational analysis to TRAF5.24 The TANK library results showed trends of permissivity consistent with solved structures: residues that can be substituted without disrupting binding lie in the middle section of the TANK TIM that does not contact the surface of TRAF3.19 Thus validated, this experimental system demonstrates the potential to rapidly increase our knowledge of TRAF‐peptide interaction preferences.
Though this simple approach demonstrated good agreement with expected binding results, we also identified areas in which the method could be improved. An analysis of stop codons near the binding site, which were frequently enriched, showed that peptide length influenced expression levels on the bacterial surface as well as binding. Optimizing a construct that contains an epitope tag for use as an expression control may make it possible to accurately determine relative binding affinities in high throughput. Additionally, the butterfly plot analysis of the relationship between counts, functional scores and R 2 values indicated sets of variants for which enrichment was not linear. We believe that these variants may have had distributions that centered on the gate cutoff used during FACS and were thus subject to random inclusion/exclusion from round to round, leading to noise in the functional scores. One way to address this problem may be to sort at more than one concentration of TRAF, to allow resolution of a greater proportion of the affinity range. Another strategy, given an expression system that includes an expression control, is to use multi‐gate sorting to resolve peptide binding affinity directly, as done by Reich et al.39
Several new insights into the peptide binding preferences of TRAFs 2, 3, and 5 were gained from this work. First, a comparison of the TANK and CD40 library results showed that binding preferences for each TRAF varied significantly depending on the peptide background. For example, TRAF2 showed broad permissivity at position 1 in TANK, but not in CD40 (Fig. 7). The stretch of permissive residues from positions 4–8 in the TANK library was not mirrored in the CD40 single mutant results. These results suggest different binding conformations for TANK and CD40. Variations among binding modes for different peptides are supported by solved structures, although the quality of many reported structures is poor, and it is also difficult to rule out the possibility that crystal packing influences peptide conformation in some complexes.19, 25, 37, 40 Second, we found that TRAFs 2, 3, and 5 have binding preferences outside of the core motif region. In particular, we observed sequence preferences in a peptide extension C‐terminal to the core motif. Finally, we found significant differences between the binding preferences of TRAFs 2, 3, and 5, which are very close homologs. Despite the high sequence identity between the TRAFs in the core‐binding region, mutations such as Q0W and E1Y in CD40 had different effects on binding to the three TRAFs. Results for the E1Y_D13T_G14N triple mutant for TRAF2 binding, compared to the single E1Y point mutant, provide evidence for interdependencies between core and C‐terminal regions of the peptide. Explanations of differences between different peptide backgrounds, the binding preferences of the TRAF paralogs, and communication between peptide regions will all benefit from further targeted structural exploration of this system.
This work establishes deep mutational scanning with bacterial display of peptide libraries as an effective means to investigate TRAF binding preferences. In particular, we highlight the conditional nature and variation of binding preferences of three closely related TRAFs. This approach and the current dataset will be valuable for the future design of specific peptide inhibitors of individual TRAF paralogs and for developing binding models to investigate TRAF binding sites in the proteome. The integral role of TRAFs in numerous signaling pathways necessitates a better understanding of their peptide interaction preferences.
Materials and Methods
SiteMAP and sequence identity analysis
SiteMAP analysis of the TRAF MATH domains and comparison of the TIM binding grooves was performed similarly to a previous analysis done on Bcl‐2 homologs.36 Given the relatively high sequence identity and similar structures of TRAF MATH domains, alignment was successfully performed with the “align” command in Pymol. All structures were aligned to the structure of TRAF3 bound to a long peptide from TANK (PDB ID 1L0A).19 For structures that included multiple MATH domain copies, one monomer with a peptide bound was chosen. All existing structures of TRAF MATH domains were analyzed: TRAF3:TANK (1L0A),19 TRAF3 apo (1FLK),37 TRAF3:CD40 (1FLL),37 TRAF3:TANK (1KZZ),19 TRAF3:LTβR (1RF3),38 TRAF3:LMP1 (1ZMS),41 TRAF3:BAFFR (2GKW),25 TRAF3:CARDIF (4GHU),40 TRAF2 apo (1CA4),42 TRAF2:TNFR2 (1CA9),42 TRAF2: LMP1 (1CZY),17 TRAF2:CD40 (1CZZ),17 TRAF2:CD40 (1D00),17 TRAF2:OX40 (1D0A),17 TRAF2:m4‐1BB (1D0J),17 TRAF2:CD30 (1D01),17 TRAF2:CD40 mutant (1QSC),43 TRAF6 apo (1LB4),20 TRAF6:RANK (1LB5),20 TRAF6:CD40 (1LB6),20 TRAF4 apo (3ZJB, 4K8U, 4M4E),44, 45, 46 TRAF5 apo (4GHJ).40
Briefly, aligned complex structures were prepared for SiteMAP analysis with Maestro (version 9.7) Prepwizard. SiteMAP (version 3.0, Schrödinger, LLC) was run on the prepared structures with the peptides removed using a 3 Å sitebox around the 1L0A TANK peptide to search for sites. Coordinates and potentials of sitemaps for individual sites on the surface of each TRAF were concatenated to create one inclusive sitemap for each structure. The binding‐site environments were compared by computing an intersection score between sitemaps for different structures as previously described.36 Briefly, the sitemap points of a particular type (hydrophobic, hydrogen bond acceptor or donor) that fell within 1 Å of a point in another receptor's sitemap were summed across all three map types to get the intersection score. The Cβ coordinates used as binding site locators were from TANK in 1L0A: VPIQCTD for the core binding site, and EALF for the TRAF3 exosite.
Sequence identity was calculated from alignments built using Clustal Omega.47 Hierarchical clustering of TRAFs by sequence identity or SiteMAP similarity score was performed with the clustergram function in Matlab (version R2012b), based on correlation.
Protein constructs and purification
The TRAF constructs used contained only the MATH domain, which was inserted into the pDW363 biotinylation vector after the biotin acceptor peptide‐tag, a His6‐tag and a linker (N‐terminal tag sequence: MAGGLNDIFEAQKIEWHE DTGGSSHHHHHHGSGSGS). The TRAF sequences are shown in Supporting Information Table II. TRAF proteins were expressed in BL21 (DE3) pLysS Rosetta cells. 10 mL overnights were started from a fresh colony and grown at 37 °C with rotation, and 5 mL of the overnight cultures were used to inoculate 1 L of LB including 100 μg mL−1 ampicillin, 25 μg mL−1 chloramphenicol, and 12–15 mg of D‐(+)‐biotin. Cells were grown at 37 °C with shaking to an optical density at 600 nm (O.D. 600) of 0.5–0.6 and then moved to 18 °C and induced with 0.5 mM IPTG overnight before harvesting.
Purification of TRAFs was performed as follows. 1‐L cell pellets were resuspended in 25 mL of 5 mM imidazole, 500 mM NaCl, 1 mM DTT, 20 mM Tris pH 8.0, 0.1% v/v Tween‐20, and 0.2 mM phenylmethylsulfonyl fluoride (PMSF) protease inhibitor. Cells were sonicated ten times for 30 s followed by 30 s of rest. The supernatant from the centrifuged lysis product was filtered through 0.2 μm filters before application to 3 mL of Ni‐nitrilotriacetic acid agarose resin equilibrated in 20 mM Tris pH 8.0, 500 mM NaCl. After the supernatant was applied, the resin was washed three times with 8 mL of 20 mM imidazole, 500 mM NaCl, 20 mM Tris pH 8.0. The protein was eluted with 8 mL 20 mM Tris pH 8.0, 500 mM NaCl, 300 mM imidazole. The protein was applied to a S75 26/60 size exclusion column equilibrated in 20 mM Tris pH 8.0, 150 mM NaCl, 5% glycerol, 1 mM DTT. Purity was verified by SDS‐PAGE, and proteins were frozen at −80 °C in the final buffer at a concentration of 100–700 μM.
Bacterial surface display constructs and library assembly
CD40 and TANK peptides were expressed on the C‐terminus of eCPX. The constructs are shown in Supporting Information Table II. A cysteine in each peptide was mutated to serine to reduce background binding of TRAFs. A threonine to alanine mutation (PVQEA or PIQCA) was used as a negative control in library sorting experiments, as this mutation is reported to reduce TRAF binding.24 The single and double point mutant libraries were originally constructed in an eCPX construct that contained a FLAG‐tag C‐terminal to the peptide. Sorting these libraries led to stop codons after the core binding sequence, which removed the FLAG‐tag. Therefore, the FLAG‐tag was removed by PCR on the assembled library plasmid DNA, leaving CD40 at the C‐terminus and a ‘GGSGGS’ cloning artifact C‐terminal to TANK.
The libraries were assembled in three pieces, with each 7‐mer mutated segment covered by a different primer design. All single and double point mutants in 3, 7‐mers were encoded by NNK (N=A/C/G/T, K=G/T) in oligonucleotides made by machine mixing of nucleotides (Integrated DNA Technologies, Coralville, IA). The sections mutated in the CD40 library were: APVQETL, HGSQPVT, and QEDGKES. The sections mutated in the TANK library were: VPIQCTD, TDKTDKQ, KQEALFK. Underlined residues in the middle TANK section overlap with the first and third section; the TANK library contained all double mutants within each of these 7‐mers, but single mutants were not duplicated for the underlined residues. The libraries were assembled analogously to those in Foight & Keating, with the exception that the piece of insert DNA included the peptide and ∼150 bp of vector backbone 3′ to the peptide.41 The 3′ SfiI site was introduced further into the vector to allow for ∼200 bp between the SfiI sites, which SfiI needs for efficient cleavage (according to New England Biolabs). The SfiI site located between the signal peptide and the N‐terminus of eCPX in the original eCPX construct was removed. An empty vector (no peptide) was digested and treated with alkaline phosphatase to serve as the vector for library insertion. Inserts were assembled by overlap PCR with the library primer on a 3′ segment corresponding to the DNA from immediately 3′ of the last codon in each 7‐mer segment through the 3′ SfiI site. Each single and double mutant insert PCR was done individually, and PCR products were pooled at equimolar ratios before insert digestion with SfiI. For the second and third 7‐mer segment of each library, an additional primer was used to extend the insert DNA to the 5′ SfiI site. All PCRs were performed with Phusion high‐fidelity polymerase (New England Biolabs). Each 7‐mer library was electroporated into MC1061 E. coli individually and grown and harvested for glycerol stocks as in Foight and Keating.36
To create the libraries without a FLAG tag, plasmid DNA from the original libraries (assembled as described above) was miniprepped from overnight cultures started from glycerol stocks. The FLAG tag was removed from the CD40 libraries (parts 1, 2, and 3) by amplification of the plasmid with primers that excluded the FLAG‐tag and linker after the CD40 peptide, but had a 40 bp overlap covering the 3′ end of the peptide sequence and the vector after the stop codon (primers CL_GA_fwd and CL_GA_rev in Supporting Information Table II). The PCR was performed with Phusion HiFi polymerase (New England Biolabs) according to standard procedures, with an annealing temperature of 64 °C and 30 cycles. Three reactions were performed per library part, with 50 ng of original library template used in each reaction. The PCR products were digested with DpnI at 37°C for 1 h and then run on a 0.65% agarose gel with GelGreen dye (Biotium, Hayward, CA). Vector‐sized bands were extracted with Zymo Gel Extraction Kit (Zymo Research, Irvine, CA) and each library part was purified over two columns and eluted with 25 μL water. Gibson Assembly was performed to connect the overlap and ligate the plasmid. Three 20 μL Gibson Assembly reactions were performed per library part, with ∼150 ng library DNA in each reaction.
The FLAG‐tag was removed from the TANK library by PCR amplification of the vector with primers that excluded the FLAG‐tag and blunt end ligation. Part of the linker was retained for PCR due to the fact that the mutated region of the TANK peptide went to the end of the peptide. FLAG_cut_fwd and TL_rth_rev primers (Supporting Information Table II) were phosphorylated with PNK (New England Biolabs) using T4 ligase buffer. Phosphorylated primers were used for PCR with Phusion HiFi polymerase, with an annealing temperature of 69 °C and 30 cycles. Nearly 50 ng of TANK library template was used in each reaction, and two reactions were performed per library part. PCR products were digested and purified as for the CD40 library. Ligations were performed overnight at 4 °C with T4 DNA ligase. Two, 20 μL reactions were performed for each library part, with ∼350 ng DNA/reaction.
DNA for the libraries without the FLAG‐tag (from Gibson Assebmly reactions or ligation reactions) was desalted on sterile MilliQ water on a 0.025 μm Millipore filter for 20 min. The DNA for each library part was split equally between two, 250 μL MC1061 E. coli competent cell stocks. Each cell stock plus DNA was electroporated in 2‐mm cuvettes at 2.5 kV, 100 Ω, 50 μF and recovered in 10 mL warm SOC for an hour at 37 °C with rotation. Each library part was then added to 150 mL LB with 0.2% w/v glucose and 25 μg mL−1 chloramphenicol and grown at 37 °C for 8–20 h, to an O.D.600 of 1.0–1.8. Cells were then pelleted and resuspended in 20% glycerol and frozen at −80 °C.
Library sorting and FACS analysis
The general protocol for FACS sample preparation was as follows: Enough glycerol stock to oversample the library by at least 10‐fold was used to inoculate 5 mL LB plus 0.2% glucose and 25 μg mL−1 chloramphenicol and grown overnight at 37 °C on a rotator wheel. Fresh 5 mL LB plus 25 μg mL−1 chloramphenicol cultures were started from a volume equivalent to 100 μL of an O.D.600 6.0 overnight culture. If the overnight culture was dilute such that a large inoculum volume was needed, the inoculum was first pelleted at 3,000 relative centrifugal force (rcf) for 5 min and the pellet was resuspended in 100 μL LB to serve as the inoculum. Cultures were grown at 37 °C to an O.D.600 of 0.5–0.6 (∼2 h) and then induced to 0.04% w/v arabinose for 1.5 h. 7 × 107 cells were pelleted at 3,000 rcf for 5 min for library sorting samples (using the rule that a culture at an O.D.600 of 1.0 contains a concentration of E. coli of ∼5 × 108 cells mL−1) and resuspended in 100 μL PBS plus 0.5% w/v bovine serum albumin (BSA). A 100 μL 2X TRAF protein in PBS + 4 mM DTT was added to the cells and incubated at ∼23 °C for 1 h. Samples were pelleted, washed with 200 μL PBS plus 0.1% w/v BSA (PBSA), pelleted again, and then 200 μL streptavidin‐PE (Molecular Probes) at a 1:100 dilution in PBSA was added. Samples were incubated with the labeling reagent for 15 min on ice, and the wash step was repeated. Final samples for sorting were resuspended in 1.3 mL PBSA. FACS sorting was performed on a BD FACSAria, and analysis was performed on a BD FACSCalibur. Cells were collected in SOC and put on ice until all sorting was finished. Samples were then recovered in 15‐20 mL 37 °C SOC and incubated at 37 °C for ∼30 min before 25 μg mL−1 chloramphenicol was added and then grown over night in 125 mL flasks with 250 rpm shaking. When possible, sorts were done on consecutive days, with the previous day's recovery culture serving as the inoculum for the next day's sort. This served to minimize extra growth steps and introduction of growth biases. The day after sorting, adequate volume of the recovery culture to oversample the library was pelleted for glycerol stocks and for Illumina sample prep.
Sorting was performed with relaxed gates to allow enrichment of moderate binders. The gates were set such that 0.5–1.0% of the negative control population fell in the gate. For TRAF2, TANK_T2A mutation was used as the negative, and CD40_T2A was used for TRAF3 and TRAF5. Three sequential rounds of sorting on each library were performed at 3 μM (TANK library only) or 10 μM TRAF2, 3, and 5 (TANK and CD40 library). The same naïve TANK library was used for both the 3 and 10 μM TRAF sorts, and the same naïve TANK library sequences were used for input sequence frequencies for the calculation of functional scores for both concentrations.
Illumina sample preparation
Library pools were prepared for Illumina sampling as in Foight and Keating.36 Briefly, cell pellets from recovery cultures were mini prepped, and 50–100 ng of DNA was used for the first PCR, which added on the Illumina 3′ adapter region and the 5′ MmeI site. The primers used for Illumina sample preparation and sequencing are shown in Supporting Information Table II. Primers for the first PCR were MmeI_fwd_TRAF and 3prime_rev_TANKLib or 3prime_rev_CD40Lib. PCR products were digested with MmeI and then double stranded adapters with five base barcodes were ligated on as described. Twenty‐four barcodes were used for multiplexing each library with the naïve TANK library pool receiving three barcodes, the naïve CD40 library pool two barcodes, and each sorted pool one barcode. Barcoded products were amplified in a final round of PCR using primers FinalPCR_fwd and FinalPCR_rev_TANKLib or FinalPCR_rev_CD40Lib. Illumina sequencing was performed in the MIT Biomicro Center on an Illumina HiSeq2000 with each library in one lane. Paired‐end reads of 85 bases were produced with the universal Illumina forward read primer and the reverse primers rev_seq_CD40Lib or rev_seq_TANKLib. A PhiX lane was run as a control for accurate base calling.
Sequencing data processing
Illumina sequencing yielded 2.48 × 108 reads for the TANK library and 1.80 × 108 reads for the CD40 library. Reads were split into their sorting pool by using an in‐house Python script written by Vincent Xue, which required an exact match to the barcode and an average Phred score of 20 for the barcode. Paired‐end reads were fused using Enrich 2.0.31 For the TANK library, all peptide positions were covered by both the forward and reverse reads. For the CD40 library, the last base of the last variable peptide position was only included in the reverse read. Enrich required an average Phred score of 20, no “N” bases, and assigned the higher scoring base where the forward and reverse reads disagreed.
We calculated functional scores similarly to Starita et al.32 First, we filtered the sequence files to consider only those DNA sequences coding for protein sequences included in the intended library design (which would include non‐theoretical DNA sequences, i.e., synonymous mutations not included in the NNK codon). We then calculated the frequencies of each sequence in these pools. Enrich was used to calculate enrichment ratios (log2 (F selected/F input)) for each protein and DNA sequence. All enrichment ratios used the frequency of the sequence in the naïve library as F input, and F selected was the frequency of the sequence in the selected pool. We extracted enrichment ratios for each sequence from sequential rounds (e.g., naïve library, CD40 Library TRAF2 round 1, CD40 Library TRAF2 round 2, CD40 Library TRAF2 round 3). We then used the Scipy linear regression method to fit lines to the enrichment ratios across rounds, calculating the slope (S var) and R 2 value for each line. Finally, a functional score was calculated by taking the inverse log of the slope of each variant divided by the slope of the wild‐type sequence (S wt):
Variants enriched over wild type are assigned a functional score >1, and variants de‐enriched get a functional score <1. All scripts were written in Python, and plots were made with Matplotlib.
Supporting information
Supporting Information
Acknowledgment
The authors thank the MIT Biomicro Center for assistance with Illumina sequencing and V. Xue for Illumina demultiplexing code.
References
- 1. Rothe M, Wong SC, Henzel WJ, Goeddel DV (1994) A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell 78:681–692. [DOI] [PubMed] [Google Scholar]
- 2. Cheng G, Cleary AM, Ye ZS, Hong DI, Lederman S, Baltimore D (1995) Involvement of CRAF1, a relative of TRAF, in CD40 signaling. Science 267:1494–1498. [DOI] [PubMed] [Google Scholar]
- 3. Ishida TK, Tojo T, Aoki T, Kobayashi N, Ohishi T, Watanabe T, Yamamoto T, Inoue J (1996) TRAF5, a novel tumor necrosis factor receptor‐associated factor family protein, mediates CD40 signaling. Proc Natl Acad Sci USA 93:9437–9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ishida T, Mizushima SI, Azuma S, Kobayashi N, Tojo T, Suzuki K, Aizawa S, Watanabe T, Mosialos G, Kieff E, Yamamoto T, Inoue J (1996) Identification of TRAF6, a novel tumor necrosis factor receptor‐associated factor protein that mediates signaling from an amino‐terminal domain of the CD40 cytoplasmic region. J Biol Chem 271:28745–28748. [DOI] [PubMed] [Google Scholar]
- 5. Ha H, Han D, Choi Y (2009) TRAF‐mediated TNFR‐family signaling. Curr Protoc Immunol. 11.9D.1–11.9D.19. [DOI] [PubMed] [Google Scholar]
- 6. Xie P (2013) TRAF molecules in cell signaling and in human diseases. J Mol Signal 8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsitsikov EN, Laouini D, Dunn IF, Sannikova TY, Davidson L, Alt FW, Geha RS (2001) TRAF1 is a negative regulator of TNF signaling enhanced TNF signaling in TRAF1‐deficient mice. Immunity 15:647–657. [DOI] [PubMed] [Google Scholar]
- 8. Nakano H, Sakon S, Koseki H, Takemori T, Tada K, Matsumoto M, Munechika E, Sakai T, Shirasawa T, Akiba H, Kobata T, Santee SM, Ware CF, Rennert PD, Taniguchi M, Yagita H, Okumura K (1999) Targeted disruption of Traf5 gene causes defects in CD40‐ and CD27‐mediated lymphocyte activation. Proc Natl Acad Sci USA 96:9803–9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakano H, Oshima H, Chung W, Williams‐Abbott L, Ware CF, Yagita H, Okumura K (1996) TRAF5, an activator of NF‐kappaB and putative signal transducer for the lymphotoxin‐beta receptor. J Biol Chem 27:14661–14664. [DOI] [PubMed] [Google Scholar]
- 10. Shiels H, Li X, Schumacker PT, Maltepe E, Padrid PA, Sperling A, Thompson CB, Lindsten T (2000) TRAF4 deficiency leads to tracheal malformation with resulting alterations in air flow to the lungs. Am J Pathol 157:679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Régnier CH, Tomasetto C, Moog‐Lutz C, Chenard MP, Wendling C, Basset P, Rio MC (1995) Presence of a new conserved domain in CART1, a novel member of the tumor necrosis factor receptor‐associated protein family, which is expressed in breast carcinoma. J Biol Chem 270:25715–25721. [DOI] [PubMed] [Google Scholar]
- 12. Häcker H, Tseng P‐H, Karin M (2011) Expanding TRAF function: TRAF3 as a tri‐faced immune regulator. Nat Rev Immunol 11:457–468. [DOI] [PubMed] [Google Scholar]
- 13. Pullen SS, Miller HG, Everdeen DS, Dang TT, Crute JJ, Kehry MR (1998) CD40‐tumor necrosis factor receptor‐associated factor (TRAF) interactions: regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero‐oligomerization. Biochemistry 37:11836–11845. [DOI] [PubMed] [Google Scholar]
- 14. Zheng C, Kabaleeswaran V, Wang Y, Cheng G, Wu H (2010) Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: affinity, specificity, and regulation. Mol Cell 38:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hauer J, Püschner S, Ramakrishnan P, Simon U, Bongers M, Federle C, Engelmann H (2005) TNF receptor (TNFR)‐associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5‐mediated activation of the noncanonical NF‐kappaB pathway by TRAF‐binding TNFRs. Proc Natl Acad Sci USA 102:2874–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zapata JM, Martínez‐García V, Lefebvre S (2007) Phylogeny of the TRAF/MATH domain. Adv Exp Med Biol 597:1–24. [DOI] [PubMed] [Google Scholar]
- 17. Ye H, Park YC, Kreishman M, Kieff E, Wu H (1999) The structural basis for the recognition of diverse receptor sequences by TRAF2. Mol Cell 4:321–330. [DOI] [PubMed] [Google Scholar]
- 18. Devergne O, Hatzivassiliou E, Izumi KM, Kaye KM, Kleijnen MF, Kieff E, Mosialos G (1996) Association of TRAF1, TRAF2, and TRAF3 with an Epstein‐Barr virus LMP1 domain important for B‐lymphocyte transformation: role in NF‐kappaB activation. Mol Cell Biol 16:7098–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li C, Ni C‐Z, Havert ML, Cabezas E, He J, Kaiser D, Reed JC, Satterthwait AC, Cheng G, Ely KR (2002) Downstream regulator TANK binds to the CD40 recognition site on TRAF3. Structure 10:403–411. [DOI] [PubMed] [Google Scholar]
- 20. Ye H, Arron JR, Lamothe B, Cirilli M, Kobayashi T, Shevde NK, Segal D, Dzivenu OK, Vologodskaia M, Yim M, Du K, Singh S, Pike JW, Darnay BG, Choi Y, Wu H (2002) Distinct molecular mechanism for initiating TRAF6 signaling. Nature 418:443–447. [DOI] [PubMed] [Google Scholar]
- 21. Marinis JM, Homer CR, McDonald C, Abbott DW (2011) A novel motif in the Crohn's disease susceptibility protein, NOD2, allows TRAF4 to down‐regulate innate immune responses. J Biol Chem 286:1938–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zepp JA, Liu C, Qian W, Wu L, Gulen MF, Kang Z, Li X (2012) Cutting edge: TNF receptor‐associated factor 4 restricts IL‐17‐mediated pathology and signaling processes. J Immunol 189:33–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arcipowski KM, Stunz LL, Graham JP, Kraus ZJ, Vanden Bush TJ, Bishop GA (2011) Molecular mechanisms of TNFR‐associated factor 6 (TRAF6) utilization by the oncogenic viral mimic of CD40, latent membrane protein 1 (LMP1). J Biol Chem 286:9948–9955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pullen SS, Dang TT, Crute JJ, Kehry MR (1999) CD40 signaling through tumor necrosis factor receptor‐associated factors (TRAFs): binding site specificity and activation of downstream pathways by distinct TRAFs. J Biol Chem 274:14246–14254. [DOI] [PubMed] [Google Scholar]
- 25. Ni C‐Z, Oganesyan G, Welsh K, Zhu X, Reed JC, Satterthwait AC, Cheng G, Ely KR (2004) Key molecular contacts promote recognition of the BAFF receptor by TNF receptor‐associated factor 3: implications for intracellular signaling regulation. J Immunol 173:7394–7400. [DOI] [PubMed] [Google Scholar]
- 26. Hildebrand JM, Luo Z, Manske MK, Price‐Troska T, Ziesmer SC, Lin W, Hostager BS, Slager SL, Witzig TE, Ansell SM, Cerhan JR, Bishop GA, Novak AJ (2010) A BAFF‐R mutation associated with non‐Hodgkin lymphoma alters TRAF recruitment and reveals new insights into BAFF‐R signaling. J Exp Med 207:2569–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arch RH, Thompson CB (1998) 4‐1BB and Ox40 are members of a tumor necrosis factor (TNF)‐nerve growth factor receptor subfamily that bind TNF receptor‐associated factors and activate nuclear factor kappaB. Mol Cell Biol 18:558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arron JR, Choi Y (2000) Bone versus immune system. Nature 408:535–536. [DOI] [PubMed] [Google Scholar]
- 29. Zapata JM, Lefebvre S, Reed JC (2007) Targeting TRAfs for therapeutic intervention. Adv Exp Med Biol 597:188–201. [DOI] [PubMed] [Google Scholar]
- 30. Araya CL, Fowler DM (2011) Deep mutational scanning: assessing protein function on a massive scale. Trends Biotechnol 29:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fowler DM, Araya CL, Gerard W, Fields S (2011) Enrich: software for analysis of protein function by enrichment and depletion of variants. Bioinformatics 27:3430–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Starita LM, Young DL, Islam M, Kitzman JO, Gullingsrud J, Hause RJ, Fowler DM, Parvin JD, Shendure J, Fields S (2015) Massively parallel functional analysis of BRCA1 RING domain variants. Genetics 200:413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Araya CL, Fowler DM, Chen W, Muniez I, Kelly JW, Fields S (2012) A fundamental protein property, thermodynamic stability, revealed solely from large‐scale measurements of protein function. Proc Natl Acad Sci USA 109:16858–16863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Halgren TA (2009) Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model 49:377–389. [DOI] [PubMed] [Google Scholar]
- 35. Halgren T (2007) New method for fast and accurate binding‐site identification and analysis. Chem Biol Drug Des 69:146–148. [DOI] [PubMed] [Google Scholar]
- 36. Foight GW, Keating AE (2015) Locating Herpes virus Bcl‐2 homologs in the specificity landscape of anti‐apoptotic Bcl‐2 proteins. J Mol Biol 427:2468–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ni CZ, Welsh K, Leo E, Chiou CK, Wu H, Reed JC, Ely KR (2000) Molecular basis for CD40 signaling mediated by TRAF3. Proc Natl Acad Sci USA 97:10395–10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li C, Norris PS, Ni C‐Z, Havert ML, Chiong EM, Tran BR, Cabezas E, Reed JC, Satterthwait AC, Ware CF, Ely KR (2003) Structurally distinct recognition motifs in lymphotoxin‐beta receptor and CD40 for tumor necrosis factor receptor‐associated factor (TRAF)‐mediated signaling. J Biol Chem 278:50523–50529. [DOI] [PubMed] [Google Scholar]
- 39. Reich L, Dutta S, Keating AE (2015) Sortcery‐A high‐throughput method to affinity rank peptide ligands. J Mol Biol 427:2135–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang P, Reichardt A, Liang H, Aliyari R, Cheng D, Wang Y, Xu F, Cheng G, Liu Y (2012) Single amino acid substitutions confer the antiviral activity of the TRAF3 adaptor protein onto TRAF5. Sci Signal 5:ra81–ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu S, Xie P, Welsh K, Li C, Ni C‐Z, Zhu X, Reed JC, Satterthwait AC, Bishop GA, Ely KR (2005) LMP1 protein from the Epstein‐Barr virus is a structural CD40 decoy in B lymphocytes for binding to TRAF3. J Biol Chem 280:33620–33626. [DOI] [PubMed] [Google Scholar]
- 42. Park YC, Burkitt V, Villa AR, Tong L, Wu H (1999) Structural basis for self‐association and receptor recognition of human TRAF2. Nature 398:533–538. [DOI] [PubMed] [Google Scholar]
- 43. McWhirter SM, Pullen SS, Holton JM, Crute JJ, Kehry MR, Alber T (1999) Crystallographic analysis of CD40 recognition and signaling by human TRAF2. Proc Natl Acad Sci USA 96:8408–8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rousseau A, McEwen AG, Poussin‐Courmontagne P, Rognan D, Nominé Y, Rio M‐C, Tomasetto C, Alpy F (2013) TRAF4 is a novel phosphoinositide‐binding protein modulating tight junctions and favoring cell migration. PLoS Biol 11:e1001726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yoon JH, Cho Y‐J, Park HH (2014) Structure of the TRAF4 TRAF domain with a coiled‐coil domain and its implications for the TRAF4 signalling pathway. Acta Cryst D70 2–10. [DOI] [PubMed] [Google Scholar]
- 46. Niu F, Ru H, Ding W, Ouyang S, Liu Z‐J (2013) Structural biology study of human TNF receptor associated factor 4 TRAF domain. Protein Cell 4:687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information