

Abstract
RNAi is a powerful tool to achieve suppression of a specific gene expression and therefore it has tremendous potential for gene therapy applications. A number of vector systems have been developed to express short-hairpin RNAs (shRNAs) to produce siRNAs within mammalian T-cells, primary hematopoietic stem/progenitor cells (HSPC), human peripheral blood mononuclear cells, and in animal model systems. Among these, vectors based on lentivirus backbones have significantly transformed our ability to transfer shRNAs into nondividing cells, such as HSPC, resulting in high transduction efficiencies. However, delivery and long-term expression of shRNAs should be carefully optimized for efficient knock down of target gene without causing cytotoxicity in mammalian cells. Here, we describe our protocols for the development of shRNA against a major HIV co-receptor/chemokine receptor CCR5 and the use of lentiviral vectors for stable shRNA delivery and expression in primary human PBMC and HSPC.
Keywords: shRNA, Lentiviral vectors, Transfection, Transduction, 293-T cells, PBMC, CD34+ cells, CCR5
1 Introduction
Lentivirus vectors are powerful tool to efficiently transduce and express shRNAs in mammalian cells and to knock down specific target gene expression through RNA interference. Initially, shRNA was expressed from a transcriptionally strong U6 pol III promoter as an internal RNA polymerase promoter from a lentiviral vector. The U6 promoter was frequently used in order to effectively knock down a target mRNA. However, we and others recognized that continuous high level of shRNA over expression from the U6 promoter could cause cytotoxicity in vector transduced cells [1]. High level of shRNA expression might interfere the function of endogenous microRNA since maturation of shRNA to siRNA utilizes the endogenous microRNA processing pathway and miRNAs plays a vital role in normal cellular functions. We learned from our experience of knocking down CCR5 expression in human peripheral blood mononuclear cells that a potent shRNA must be identified and expressed at lower level using transcriptionally weaker H1 RNA polymerase III promoter to stably knockdown CCR5 gene expression and to avoid cytotoxic effects [2]. Here, we describe our detailed protocol of a lentiviral vector construction for stable shRNA expression in human PBMC and CD34+ HSPC in vitro.
2 Materials
2.1 Construction of shRNA-Expressing Lentivirus Vector
pBS-hH1-3 plasmid DNA: The pBluescript II SK (−) plasmid DNA that contains a human H1 RNA polymerase III promoter, a RNA polymerase III promoter termination signal (5Ts), and restriction enzyme sites for a shRNA cloning (Fig. 1a, see Note 1). The plasmid DNA sequence data are available upon request.
FG12 HIV-1-based lentiviral vector plasmid DNA that contains restriction enzyme sites for an H1 promoter shRNA unit cloning. The plasmid DNA sequence data are available upon request (Fig. 2, see Note 2).
Restriction enzyme s: Afe I enzyme, BamH I enzyme, Xba I enzyme, Xho I enzyme.
Enzyme buffer 4.
TE buffer: 10 mM Tris-Cl, pH 7.5, 1 mM EDTA.
T4 DNA Ligase.
10× T4 DNA ligase buffer.
DNA gel extraction kit.
DNA mini prep kit.
Electroporation cuvettes (2 mm gap).
XL-1 blue competent cells.
2× YT medium.
2× YT medium with 50 μg/mL ampicillin.
LB agar plate containing 50 μg/mL ampicillin.
Sterilized toothpicks (see Note 3).
Sequencing primers: M13 rev/M13 fwd.
Fig. 1.
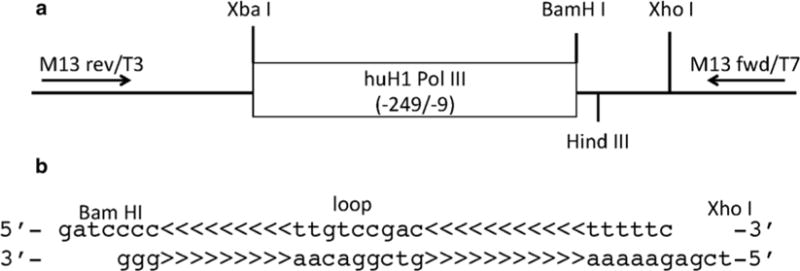
(a) The map of pBS-hH1-3 plasmid DNA. A DNA fragment of human H1-RNA polymerase III promoter (−241/−9, amplified from HEK293T genomic DNA by PCR) was cloned between Xba I and Hind III sites of pBS-SKII(−) (Stratagene). BamH I and Xho I are designed for cloning oligo DNAs for a shRNA. XbaI and XhoI sites are designed for subcloning the shRNA expression cassette into FG12 lentivirus vector. huH1 Pol III: human H1 RNA polymerase III promoter. M13 rev/T3 and M13 fwd/T7: sequence primer-binding sites. (b) The design of siRNA oligo DNAs. A sense and antisense DNA including BamH1 restriction enzyme site at the 5′ prime, a 19–21mer antisense strand of the shRNA targeting sequence <<<<<<<<<, a loop, a corresponding reverse complementary sense sequence <<<<<<<<<, a RNA polymerase III promoter termination signal (ttttt) and a XhoI restriction enzyme site are required in the siRNA oligo DNAs
Fig. 2.
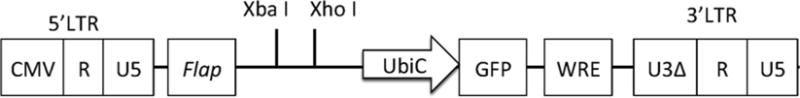
The map of FG12 lentivirus vector. FG12 lentivirus vector is derived from FGUW lentivirus vector with modified multiple restriction sites. A shRNA expression cassettes cloned into pBS-hH1-3 plasmid DNA can be subcloned at XbaI and XhoI sites in FG12 vector. LTR long terminal repeat. CMV CMV promoter. R repeat sequence in HIV-1 LTR: Flap: UbiC: Human Ubiquitin C promoter. GFP green fluorescent protein. WRE woodchuck hepatitis virus posttranscriptional regulatory element. U3Δ nucleotide deletions in the U3 region for self-inactivation
2.2 Transfection of 293-T Cells for Production of VSV-G Pseudotyped Lentiviral Vectors
293-T cell line.
Culture medium: Iscove’s modified Dulbecco’s medium (IMDM) with 8 % Bovine serum, 2 % fetal calf serum, and 1 % glutamine/penicillin/streptomycin.
10 mM Chloroquine.
2 M Calcium chloride solution.
2× HBS, pH 7.0: Dissolve 8 g NaCl, 0.2 g Na2HPO4·7H2O, 6.5 g HEPES in deionized water to a total volume of 500 ml.
phHCMV-G plasmid DNA for VSV-G expression.
pMDLg_pRRE plasmid DNA for HIV-1 gag and pol expression.
pRSV REV plasmid DNA for HIV-1 rev expression.
FG12 plasmid DNA for a HIV-1-based lentiviral vector.
2.3 Concentration of VSV-G Pseudotyped Lentiviral Vector, Resuspension, and Storage
Steriflip Filter Units; pore size 0.22 μm.
1× Hanks’ Balanced Salt Solution (HBSS).
50 % Sucrose buffer: 125 g Sucrose dissolved in 245 ml HBSS supplemented with 10 mM EDTA.
10 % Sucrose buffer: 50 % Sucrose buffer diluted five times with HBSS such that the final working concentration is 10 % sucrose, 2 mM EDTA.
Ultraclear centrifuge tubes.
SW32 Ti rotor with swinging bucket in Beckman Coulter Optima L-90k Ultracentrifuge.
2.4 Titration of shRNA Lentivirus Vector
293-T cell line.
2/8 IMDM: Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 8 % bovine serum (BS) and 2 % fetal calf serum (FCS) and 1 % glutamine/penicillin/streptomycin (GPS).
1 mg/ml Polybrene: Hexadimethrine bromide dissolved in double-distilled H2O.
FACS buffer: 2 % FCS in 1× Phosphate buffer saline (PBS).
10F RPMI: RPMI-1640 supplemented with 10 % FCS and 1 % GPS.
Trypsin-EDTA solution: 2.5 % Trypsin and 0.5 M ethylenediaminetetraacetic acid (EDTA).
Fix Buffer: 1 % Formaldehyde in 1× PBS.
Concentrated VSV-G pseudotyped lentiviral vector stocks.
2.5 Transduction of T-Cell Line with Lentiviral Vector Carrying shRNA
CCR5 expressing Molt-4/CCR5 T-cell line.
1 mg/ml Polybrene (hexadimethrine bromide dissolved in double-distilled H2O).
10F RPMI: RPMI-1640 supplemented with 10 % FCS and 1 % glutamine/penicillin/streptomycin (GPS).
Titrated VSV-G pseudotyped lentiviral vector stocks.
1× PBS.
FACS buffer: 2 % FCS in 1× PBS.
Fix buffer: 1 % Formaldehyde in 1× PBS.
2.6 Transduction of PBMC with Lentiviral Vector Carrying shRNA
Peripheral blood mononuclear cells (PBMC) can be isolated from whole blood from healthy donors. The PBMC can be isolated using Ficoll by centrifugation.
Anti-human CD8 mAb magnetic beads.
Magnetic box.
FACS buffer: 2 % FCS in 1× PBS.
1 mg/ml Polybrene (hexadimethrine bromide dissolved in double-distilled H2O).
5 μg/ml PHA.
200 units/ml Interleukin-2 human hIL-2 (IL-2).
10F RPMI: RPMI-1640 supplemented with 10 % FCS and 1 % glutamine/penicillin/streptomycin (GPS).
20F RPMI: RPMI-1640 supplemented with 20 % FCS and 1 % glutamine/penicillin/streptomycin (GPS).
Growth medium: 10 units/ml IL-2 in 20F RPMI.
Titrated lentiviral vector.
Fix buffer: 1 % Formaldehyde in 1× PBS.
2.7 Transduction of Human Fetal Liver-Derived CD34+ Cells with Lentiviral Vector Carrying shRNA
Human fetal liver for the source of CD34+ cells. The fetal liver can be provided from the UCLA CFAR Gene and Cellular Core laboratory or purchased through Advanced Bioscience Resources, Inc, Alameda, California.
CD34 microbead kit.
Cytokines: IL-6, IL-3, SCF.
1 mg/ml Recombinant Fibronectin (RetroNectin) stock solution.
FACS Buffer: 2 % FCS in 1× PBS.
10F RPMI: RPMI-1640 supplemented with 10 % FCS and 1 % glutamine/penicillin/streptomycin (GPS).
20F RPMI: RPMI-1640 supplemented with 20 % FCS and 1 % glutamine/penicillin/streptomycin (GPS).
Infection medium: Yssel’s Serum-Free T-cell Medium supplemented with 2 % bovine serum albumin (BSA), 2.5 μg/ml Fungizone and 0.45 mg/ml Piperacillin and Tazobactram (PT, also known as Zocyn).
Cytokine medium: 20F RPMI supplemented with 50 ng/ml IL-6, 50 ng/ml IL-3, 50 ng/ml SCF.
Titrated VSV-G pseudotyped lentiviral vector stocks.
Fix buffer: 1 % Formaldehyde in 1× PBS.
2.8 CCR5 Detection in Humanized Mouse Blood
10× Red blood cell lysis buffer: 82.9 g of Ammonium Chloride, 10.0 g of potassium bicarbonate, 0.37 g of ethylenediamine tetraacetic acid (EDTA) disodium salt in 1L water (see Note 4).
1× Red blood cell lysis buffer (see Note 5).
FACS buffer: 2 % FCS in 1× PBS.
Fix buffer: 1 % formaldehyde in 1× PBS.
Master Mix1: 0.5 μl of CD45-eFluore 450, 0.5 μl of CD3-APCH7, 0.5 μl of CD4-APC, 0.5 μl of CD8-PerCPCy5.5, 0.5 μl of CD19-V500, 0.5 μl of CCR5 -PC7 in 100 μl of FACS buffer (see Note 6).
3 Methods
3.1 Construction of shRNA Expressing Lentivirus Vector
Synthesize a forward and a reverse oligonucleotide DNAs for a shRNA based on the design described in Fig. 1b.
Dilute the forward and reverse oligonucleotides in TE at a concentration of 10 pmol/μl each. Mix each 10 μl of forward and reverse oligo DNA into 0.5 mL tube and incubate at 95 °C for 10 min and slowly cool down to room temperature.
Digest 1 μg of pBS-hH1.3 with 1 U of BamH I and Xho I at 37 °C for 1 h in NEBuffer4. Run the product on 1 % agarose gel, cut the 3130 bp DNA fragment and purify using the gel extraction kit.
Incubate purified vector and 7 μl annealed oligos with T4 DNA ligase at 16 °C for 1 h in 10 μl of 1× T4 DNA polymerase buffer (see Note 7).
Transform competent cells with the product by electroporation. Thaw competent cells on ice. Chill an electroporation cuvette and ligation sample on ice. Add 1 μl of ligation sample into 55 μl of competent cells and transfer into electroporation cuvette gently, and apply one pulse.
Immediately add 250 μl of 2× YT medium (see Note 8).
Plate all of them into LB agar plate containing 50 μg/mL ampicillin using a spreader.
Incubate the plate at 37 °C overnight.
Pick 5–10 single colonies with toothpick and culture in 2 mL of 2× YT containing 50 μg/mL ampicillin medium overnight with constant shaking at 200 rpm (see Note 9).
Carry out Mini prep following the manufacturer’s instructions.
Confirm sequence using M13 fwd/M13 rev primers.
Insert: Digest 1 μg of pBS-hH1-3 plasmid DNA with 1 U of Xho I and Xba I at 37 °C for 1 h in NEBuffer4 then, run the product on 1 % agarose gel, cut the 294 bp band and purify using gel extraction kit.
Vector: Digest 1 μg of FG12 vector with 1 U of Xho I and Xba I at 37 °C for 1 h in NEBuffer4. Run the product on 1 % agarose gel, isolate the 9865 bp band and purify using gel extraction kit.
Incubate purified vector (at least 50 ng) and insert with 1 μl of T4 DNA Ligase at 16 °C for 1 h in 10 μl of 1× T4 DNA polymerase buffer.
Transform competent cells with the product by electroporation. Thaw competent cells on ice. Chill an electroporation cuvette and ligation sample on ice. Add 1 μl of ligation sample into 55 μl of competent cells and transfer into electroporation cuvette gently, and apply one pulse.
Immediately add 250 μl of 2× YT medium into the cells.
Plate all of them into LB agar plate containing 50 μg/mL ampicillin using a spreader.
Incubate the plate at 37 °C overnight.
Pick 5–10 single colonies with toothpick and culture in 2 mL of 2× YT medium containing 50 μg/mL ampicillin overnight with constant shaking at 200 rpm.
Carry out Mini prep following the manufacturer’s instructions.
Digest 5 μl of miniprep sample with 1 U of Afe I and Xba I enzyme in NEBuffer4 and confirm ~380 bp insert (see Note 10).
3.2 Transfection of 293-T Cells for Production of VSV-G Pseudotyped Lentiviral Vectors
Culture 15 × 106 293-T cells into T-175 flasks in 25 ml culture medium 1 day before.
On the day of transfection, change the medium into chloroquine medium (40 μl of 10 mM chloroquine + 10 ml culture medium).
Make a master mix of the viral DNA in a screw cap tube (viral DNA + 2 M calcium chloride + ddH2O such that the total volume is 1100 μl.
Add 10 μg of vector DNA to the master mix. This is solution A.
Incubate solution A on ice for 5 min.
For solution B, add 1110 μl 2× HBS into a 50 ml conical tube.
Adding solution A to solution B: Using a p200 micropipette, Add solution A dropwise by the sides of the tube containing solution B. Shake tube to mix drops. The mixture will collect on the bottom of the tube. This is solution C.
Incubate solution C on ice for 20 min.
After 20 min, add solution C dropwise to the T-175 flask containing cells. Solution C should be added to the culture media in the flask and should not touch the cells.
Slowly swirl the media in the flask to mix the solution C.
Slowly lay the flask flat so that the media touched the cells.
Incubate for 8 h at 37 °C, 5 % CO2.
After 8 h, slowly remove all the media in the flask and add 35 ml of culture media.
Incubate at 37 °C, 5 % CO2.
3.3 Concentration of Vector, Resuspension, and Storage
Observe infected 293-T cells under the microscope for infection by checking for GFP expression.
Harvest supernatant in a 50 ml tube and filter the supernatant using the Steriflip filter unit.
Concentrate VSV-G-psuedotyped virions from the supernatant by ultracentrifugation. Transfer the filtrate (containing the vector) into an ultracentrifuge tube (Ultraclear 38.5 ml Beckman Coulter).
Slowly add 5 ml of 10 % sucrose buffer to the tubes by touching the pipette all the way to the bottom of the ultracentrifuge tube and releasing the sucrose buffer slowly. This will create a sucrose cushion and will help avoid the coprecipitation of unwanted particles, resulting in vector of significantly higher purity.
Use serum-free medium to balance the tubes to within 30 mg, and then load in an SW32 rotor.
Centrifuge at 67214 × g at 4 °C for 90 min. The brake must be inactivated to prevent disturbing the viral pellet during deceleration.
Discard the supernatant by inversion and leave the ultracentrifuge tube inverted for 90 s on absorbent paper towel.
Resuspend the viral pellet in 100 μl of HBSS and seal the ultracentrifuge tubes with parafilm. Store the tubes overnight at 4 °C.
Following overnight storage at 4 °C, carefully mix the vector by vigorous pipetting, and then store at −80 °C in small aliquots.
3.4 Titration of Lentivirus Vector Stocks
Plate 293-T cells at 0.5 × 105 cells/500 μl in 24-well plate 1 day before the infection.
Prepare titration medium containing 2/8 IMDM + 8 μl/ml polybrene.
Thaw an aliquot of the vector on ice, and prepare a serial dilution of vector with the titration media (see Note 11).
Dilution 1:1/300 diluted vector:1 μl of concentrated vector + 300 μl of titration media.
Dilution 2:1/3000 diluted vector:30 μl of 1/300 diluted vector + 270 μl of titration media.
Dilution 3:1/30,000 diluted vector:30 μl of 1/3000 diluted vector + 270 μl of titration media.
Remove media from plated 293-T cells and add 250 μl of the serially diluted vector to corresponding wells (see Note 12).
Incubate the infected cells for 2 h at 37 °C, 5 % CO2, incubator.
After 2 h, remove the supernatant and add 1 ml of 2/8 IMDM to each well.
Three days post-infection, remove all the supernatant from infected 293-T cells and wash cells with 1× PBS.
Add 100 μl of trypsin-EDTA solution to each well and incubate for 2 min at 37 °C, 5 % CO2 incubator.
Observe under the microscope to see if all the cells have detached from the well surface. If not then keep for an additional 2 min in the incubator.
Add 1 ml of 2/8 IMDM to wells to block trypsin-EDTA action and flush cells with the help of micropipette.
Count the cells and take 0.1 × 106 cells in 1.5 ml screw-cap tubes.
Centrifuge at 3500 × g for 1 min and aspirate supernatant.
Add 500 μl of fix buffer to each tube and transfer to FACS tubes.
Acquire samples by fluorescence-activated cell sorting (FACS) in order to measure % EGFP-positive cells.
- Based on % EGFP-positive cells, calculate the titer of the vector according to the formula indicated below.
- Select the dilution which shows closest to tenfold increase in % EGFP-positive cells as compared to previous dilution. For example: %EGFP+ cells with 1/30,000 dilution = 1.02; %EGFP+ cells with 1/3000 dilution = 10.8; %EGFP+ cells with 1/300 dilution = 63.7. Therefore, we will select 1/3000 as the dilution to calculate titer.
- Formula for calculating titer units (TU/ml): [%EGFP+ cells/100] × [number of cells] × [dilution factor] × [1000/volume of vector (ml)], wherein the number of cells refers to the number of cells taken in screw caps tubes after harvesting the cells. For example: %EGFP+ cells = 10.8, number of cells = 0.1 × 106 cells, dilution factor = 3000, volume of vector = 250 μl. Therefore, [10.8/100] × [0.1 × 106] × [3000] × [1000/250] = 1.296 × 108 TU/ml.
- Calculation of amount of vector needed to reach a certain Multiplicity of Infection (MOI).
After titrating the vector, in order to perform immune cell transductions, it is important to calculate the amount of vector that will need to be added to the cell cultures to infect them at a certain MOI. The formula of calculation the amount of vector for a certain MOI is indicated below, wherein the total number of cells refers to the number of cells seeded in each well before infected them. Total plaque-forming units (PFU) = [Total number of cells] × [desired MOI], followed by volume of vector needed to reach desired MOI (μl) = [Total PFU] × [TU/ml].
3.5 Transduction of T-Cell Line with Lentiviral Vector Carrying shRNA
Aliquot 0.1 × 106 cells into sterile screw cap tubes.
Centrifuge the cells at 3,500 × g for 1 min and carefully remove the supernatant.
Prepare 250 μl of polybrene/vector solution (248 μl of 10F RPMI + 2 μl of polybrene + calculated amount of vector). The final concentration of polybrene should be 8 μg/ml (see Note 13).
Add 250 μl of the polybrene/vector solution to each tube.
Loosen the caps of the tubes and incubate for 2 h at 37 °C, 5 % CO2.
After 2 h, add 1 ml of 10F RPMI to the tubes.
Centrifuge the cells at 3,500 × g for 1 min and carefully remove the supernatant.
Resuspend the cells in 1 ml 10F RPMI and plate cells in a 12-well plate.
Incubate at 37 °C, 5 % CO2 for 3 days. Transgene expression can be assessed in 72 h.
After 72 h, collect and count the cells. Centrifuge the cells at 3,500 × g for 1 min at room temperature.
Resuspend cells in 10F RPMI at 0.1 × 106 cells/well.
Centrifuge cells at 3,500 × g for 1 min.
Add 300 μl Fix buffer and acquire by flow cytometer and check for % EGFP.
3.6 Transduction of PBMC with Lentiviral Vector Carrying shRNA
We deplete CD8+ cells from PBMC for investigation of lentiviral vector transduction and CCR5 knock down in CD4+ cells. For every 10 × 106 PBMC, add 70 μl of anti-human CD8 mAb magnetic beads into a 15 ml.
Add 7 ml of FACS buffer. Place the 15 ml tube in the magnetic box and wait for 3 min until the red magnet beads attach to the side of the tube.
Remove the supernatant by decanting. Repeat the washing (step 2).
Resuspend the beads in 400 μl cold FACS buffer for every 10 × 106 cells and keep on ice.
Aliquot 10 × 106 PBMC in a 15 ml tube and centrifuge at 453 × g for 5 min. Aspirate the supernatant.
Mix the PBMC and washed anti-human CD8 mAb magnetic beads.
Incubate on ice for a total of 30 min, mixing every 10 min.
Add 7 ml of cold FACS buffer.
Place the 15 ml tube containing the cells and the beads in the magnet box and wait for 3 min until the red magnet beads attach to the side of the tube.
Decant supernatant (containing the cells of interest) into a fresh 15 ml conical tube.
Check percentage and purity of CD8+ depleted cells by staining 0.1 × 106 cells with antibodies against CD4, CD8, CD27, and CD45RA.
Spin down the CD8+ depleted cells and resuspend the cells in 20F RPMI. Count the cells.
Seed 2 × 106 CD8+ depleted cells per ml and activate the cells with PHA (Final concentration should be 2.5 μg/ml) in 20F RPMI for 3 days.
Post-activation, wash the CD8+ depleted cells with warm 10F RPMI and resuspend the cells in growth medium in a 96-well plate at 0.4 × 106 cells/100 μl/well.
Incubate cells for 4 h at 37 °C, 5 % CO2.
Prepare 50 μl of polybrene/vector solution (48.8 μl of 10F RPMI + 1.2 μl of polybrene + calculated amount of vector). The final concentration of polybrene should be 8 μg/ml.
After 4 h, add the polybrene/vector solution to the cells and mix well by pipetting.
Incubate cells for 2 h at 37 °C, 5 % CO2 incubator.
After 2 h, remove 140 μl of the culture medium carefully. Try not to disturb the cells.
Add 250 μl of growth medium and Incubate cells overnight at 37 °C, 5 % CO2.
Incubate cells overnight at 37 °C, 5 % CO2.
Collect all cells per well. Make sure that there are no cells left in the well by observing the wells under the microscope.
Wash the cells with 1 ml of pre-warmed 10F RPMI (optional).
Transfer cells to a 24-well plate and add 1 ml of growth medium into each well.
Incubate at 37 °C, 5 % CO2. Transgene expression can be assessed within 48–72 h.
After 72 h, collect and count the cells. Centrifuge the cells at 3,500 × g for 1 min at room temperature.
Resuspend cells in growth media at 1 × 106 cells/ml.
Stain remaining cell from each sample with 50 μl of FACS buffer + 1 ml of antibody against CD4, CD8, CD27 and CD45RA for 20 min at room temperature in the dark.
Add 1 ml 1× PBS and centrifuge at 3,500 × g for 1 min.
Add 300 μl fix buffer and acquire cell events by flow cytometer and assess EGFP expression.
3.7 Transduction of CD34+ Cells with Lentiviral Vector Carrying shRNA
Prepare 20 μg/ml RetroNectin-PBS solution (working solution) for coating the 24-well plate.
Dispense 500 μl of RetroNectin-PBS solution into each well. Incubate for 2 h at room temperature in the dark.
Remove the RetroNectin-PBS solution. This can be reused if stored at 4 °C.
Dispense 500 μl of FACS buffer into each well for blocking. Incubate for 30 min at room temperature in the dark.
Aspirate FACS buffer and wash wells once with 500 μl 1× PBS. Do not remove PBS from wells.
Quick thaw frozen CD34+ cells into 5 ml pre-warmed infection medium.
Centrifuge the CD34+ cells at 314 × g for 5 min.
Resuspend cell pellet in 1 ml infection medium.
Count the cells. Bring concentration of cells to 1 × 106 cells/ml.
Remove PBS from the wells. CD34+ cells should be cultured at 0.25 × 106 cells per well; therefore add 250 μl of cells to each well.
Incubate cells for 1 h at 37 °C, 5 % CO2, incubator.
Add 250 μl infection medium to each well such that there is 500 μl medium in each well.
A MOI of 0.3 is generally used to obtain transduction efficiencies of >50 %. CD34+ cells does not require the addition of polybrene and is not augmented by the presence of RetroNectin. Add calculated amount of vector to the designated well.
Incubate cells overnight at 37 °C, 5 % CO2.
The next day, add 800–1000 μl of 10F RPMI to each well.
Collect all cells per well. Make sure that there are no cells left in the well by observing the well under the microscope.
Centrifuge tubes at 453 × g for 5 min at room temperature.
Resuspend cell pellet again in 800–1000 μL OF 10F RPMI. Repeat step 17.
Resuspend the cell pellet in 1 ml of cytokine medium. Transfer cells into a 24 well plate.
Incubate at 37 °C, 5 % CO2. Transgene expression can be assessed within 48 h (see Note 14).
Collect and count the cells. Centrifuge the cells at 453 × g for 5 min at room temperature.
Resuspend cells in cytokine media at 0.1 × 106 cells/well.
Centrifuge remaining cell from each sample at 3,500 × g for 1 min.
Add 300 μl fix buffer and acquire cell events by flow cytometer and assess EGFP expression.
3.8 Detection of shRNA Expression by shRNA-Specific Real-Time RT-PCR
Real-time stem-loop RT-PCR method can be used to quantify the levels of the antisense strand of siRNA. For example, we successfully quantified antisense strand of siRNA directed to rhesus CCR5 [3].
Isolate 500 ng of total RNA from rhesus PBLs using TRIzol reagent (Invitrogen).
Subject the RNA to reverse transcription (RT) reaction (16 °C, 30 min; 42 °C, 30 min; 85 °C, 5 min).
Subject RT product to PCR (95 °C, 10 min, 1 cycle; 95 °C, 15 s; 58 °C, 1 min, 50 cycles). Primer and probe sequences used for rhesus CCR5siRNA were as follows: reverse transcription stem-loop primer, 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAGAGCAA-3′; forward primer, 5′-GCGCGGTGTAAACTGAAC-3′; reverse primer, 5′-GTGCAGGGTCCGAGGT-3′; probe, 6-FAM-TGGATACGACAAGAGCAA-MGB. A set of serially diluted synthetic 22-nt antisense strand of siRNA against rhCCR5 (GGU GUA AAC UGA ACU UGC UC; Sigma-Proligo) was used as standard for quantification [3].
Footnotes
Human H1-RNA polymerase III promoter DNA (−241/−9) is amplified from HEK293T genomic DNA and cloned between Xba I and Hind III site of pBS-SKII(−) (Fig. 1a).
FG12 lentiviral vector is constructed based on FUGW lentiviral vector [4, 5] with useful restriction enzyme sites (XbaI and XhoI) for shRNA cloning.
Sterilized pipette tips can be used instead of toothpick.
10× Red lysis buffer is filtered with 45 μm filter and stored at 4 °C.
1× red blood cell lysis buffer should be used at room temperature. Dilute 10× RBC buffer with ddH2O and filter. Store at 4 °C.
We use clone 2D7 for anti-human CCR5 antibody for CCR5 cell surface detection by flow cytometry.
Make small aliquots (~10 μl) for T4 DNA ligase buffer to avoid repeated freeze and thaw cycles to preserve ATP.
S.O.C. can be used as a substitute to 2× YT medium.
Sometime lentiviral vector shows recombination during bacteria amplification; therefore bacteria should be cultured low speed (~100 rpm) and constant shaking.
For vector production, Midi prep sample is better than mini prep sample as it gives a high titer vector.
Though titers can be assessed using various techniques, titers based on transgene expression that can be monitored by flow cytometry are the simplest. It is best to evaluate titers at low dilutions or where the percentage of transduced cells is <30 %. This will help avoid issues of multiple integrations as at lower dilutions, the number of integrated copies is generally one.
293-T cells can easily detach from the well surface. Media must always be carefully removed and cells should never be allowed to dry up.
One should avoid picking up very small volumes with a micro-pipette as there are high chances of pipetting errors. If the volume of the vector solution is too small, dilute the vector 1:10 in 1× PBS so that a sufficient amount can be picked up by micropipettes. For the diluted vector, add ten times the calculated volume of the vector to the cells.
Transduction efficiencies can vary significantly with cell types. Molt4CCR5 T cell line show 30–40 % GFP+ at 3 days after transduction at M.O.I. = 0.1. However, at the same M.O.I in primary cells (PBMC or FL derived CD34+ cells), transduction efficiency becomes almost half. It is hard to predict transduction efficiency in primary cells which have more variation to susceptibility in transduction. You should also consider donor to donor variation in primary cells lines. The timing at which transgene expression and transduction efficiency is critical to access vector transduction efficiency. Transgene expression from lentiviral vector requires more than 48 h after transduction.
References
- 1.An DS, Qin FX, Auyeung VC, Mao SH, Kung SK, Baltimore D, Chen IS. Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol Ther. 2006;14:494–504. doi: 10.1016/j.ymthe.2006.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shimizu S, Hong P, Arumugam B, Pokomo L, Boyer J, Koizumi N, Kittipongdaja P, Chen A, Bristol G, Galic Z, Zack JA, Yang O, Chen IS, Lee B, An DS. A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood. 2010;115:1534–1544. doi: 10.1182/blood-2009-04-215855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.An DS, Donahue RE, Kamata M, Poon B, Metzger M, Mao SH, Bonifacino A, Krouse AE, Darlix JL, Baltimore D, Qin FX, Chen IS. Stable reduction of CCR5 by RNAi through hematopoietic stem cell transplant in non-human primates. Proc Natl Acad Sci U S A. 2007;104:13110–13115. doi: 10.1073/pnas.0705474104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 5.Qin XF, An DS, Chen IS, Baltimore D. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci U S A. 2003;100:183–188. doi: 10.1073/pnas.232688199. [DOI] [PMC free article] [PubMed] [Google Scholar]