

Abstract
Propofol is one of the most widely used intravenous anesthetics. However, repeated exposure to propofol may cause neurodegeneration in the developing brain. Dexmedetomidine (Dex), an α2 adrenoceptor agonist, has been previously demonstrated to provide neuroprotection against neuroapoptosis and neurocognitive impairments induced by several anesthetics. Thus, the current study aimed to investigate the effect of Dex on neonatal propofol-induced neuroapoptosis and juvenile spatial learning/memory deficits. Propofol (30 mg/kg) was intraperiotoneally administered to 7-day-old Sprague Dawley rats (n=75) three times each day at 90 min intervals for seven consecutive days with or without Dex (75 µg/kg) treatment 20 min prior to propofol injection. Following repeated propofol exposure, reduced Akt and GSK-3β phosphorylation, increased cleaved caspase-3 expression levels, an increased Bax/Bcl-2 ratio, and increased terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)-positive cells in the CA1 hippocampal subregion were observed. Morris Water Maze testing at postnatal day 29 also demonstrated spatial learning and memory deficits following propofol treatment compared with the control group. Notably, these changes were significantly attenuated by Dex pretreatment. The results of the current study demonstrated that Dex ameliorates the neurocognitive impairment induced by repeated neonatal propofol challenge in rats, partially via its anti-apoptotic action and normalization of the disruption to the PI3K/Akt/GSK-3β signaling pathway. The present study provides preliminary evidence demonstrating the safety of propofol on the neonatal brain and the potential use of dexmedetomidine pretreatment in pediatric patients.
Keywords: dexmedetomidine, propofol, neuronal toxicity, neonate, GSK-3β, Akt
Introduction
It is estimated that more than 6 million children receive anesthesia every year (1). In modern anesthesiology, general anesthesia is the most common practice for surgery or relief from procedural pain, particularly in children (2). A previous study suggested that anesthetic exposure can lead to neurotoxicity in the developing brain (3). Similarly, children exposed to anesthetics during early life have been reported to exhibit a higher incidence of learning deficits at adolescence (4). These results prompt concerns regarding the possible detrimental effects of commonly used anesthetics in the pediatric population.
An alkyl phenol derivative, 2,6-di-isopropylphenol (propofol), was introduced as a sedative and anesthetic agent in 1977 due to its rapid onset and recovery (5). It demonstrated neuroprotective effects in the adult brain in vivo (6) and in vitro (7). However, in the developing brain, propofol is able to cause neurotoxicity by inducing apoptosis (8), disturbing neuronal proliferation (9) and disrupting synapse formation (10). Additionally, long-term behavioral deficits in pups exposed to propofol have been previously observed (8). The cellular mechanisms and signaling pathways that underlie propofol-induced neurotoxicity are poorly understood. The phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/v-akt murine thymoma viral oncogene homolog 1 (Akt) signaling pathway is pivotal for cell growth, proliferation and survival (11). Activation of PI3K/Akt signaling is widely recognized to provide protection against cerebral ischemia/reperfusion injury (12). Phosphorylation of the downstream protein, glycogen synthase kinase-3β (GSK-3β), also improves long-term memory in hippocampal-associated tasks (13) and inhibits propofol-induced lysosome/mitochondrial apoptosis in macrophages (14). Whether this signaling pathway is associated with propofol-induced neurotoxicity remains to be determined.
Several drugs have been demonstrated to exhibit neuroprotective effects in different cerebral injury models. Among them, dexmedetomidine (Dex), a potent and highly selective α2-adrenoceptor agonist and a widely used auxiliary anesthetic, has previously been reported to reduce isoflurane- and ketamine-induced neuroapoptosis in vivo and in vitro (15,16). The neuroprotective effect of Dex may be associated with its anti-oxidant (17) and anti-apoptotic (15) activity, its positive impact on astrocyte brain-derived neurotrophic factor expression (18), or inhibition of the PI3K/Akt/GSK-3β pathway (19). However, the effect of Dex on propofol-induced neurotoxicity in the developing brain has not been determined. Thus, the current study aimed to examine whether Dex has an effect on apoptosis and neurobehavioral changes elicited by repeated propofol exposure in neonatal rats, and whether the PI3K/Akt/GSK-3β pathway is involved.
Materials and methods
Animals and experimental groups
All experiments were conducted in accordance with protocols approved by the Peking University Biomedical Ethics Committee Experimental Animal Ethics Branch (Beijing, China) and the National Institutes of Health guide for the Care and Use of Laboratory Animals. In total, 75 male postnatal day 7 (P7) Sprague Dawley rats (obtained from the Department of Laboratory Animal Science, Peking University Health Science Centre, Beijing, China) weighing 12–16 g were used in the current study. Pups were bred and maintained under standard housing conditions (24±2°C; 12 h:12 h, light/dark), and had access to food and water ad libitum. Rats were randomly divided into three groups (25 animals per group) using the random table method. Groups were defined as control (CON), propofol administration (PRO), and Dex preconditioning prior to propofol administration (Dex + PRO).
Dex preconditioning and propofol administration
Dex + PRO group rats were intraperiotoneally (i.p.) administered with Dex (Jiangsu Hengrui Medicine Co., Ltd., Jiangsu, China) at 75 µg/kg (1 µg/ml) in saline every day for seven consecutive days from P7 to P13. Following 20 min of Dex preconditioning, 30 mg/kg of propofol (Diprivan; Astra-Zeneca, London, UK) was administered (i.p.) to pups from PRO and Dex + PRO groups every 90 min up to a cumulative dose of 90 mg/kg each day (n=25). Pups in the CON group were injected with an equal volume of intralipid (Huarui Pharmaceutical Co., Wuxi, China) under the same conditions. For the anesthesia procedure, pups were placed in a temperature-controlled incubator (28°C) until they could successfully perform the righting reflex. To avoid the rebreathing of CO2, the inspired CO2 concentration was continuously monitored and maintained at <1% by adjusting the fresh gas flow. During anesthesia, respiratory frequency and skin color were observed to detect apnea and hypoxia. If apnea occurred, rats received a pain stimulus; if breathing did not restart or resuscitation efforts were necessary, rats were excluded from further processing and analysis. The animals were observed for a further 90 min until they were awake and were returned to their mothers following the last injection.
Blood gas analysis and body weight
To determine whether propofol anesthesia would cause physiological side effects, including hypoxia, hypercapnia, or hypoglycemia, five rats in each group were selected as cardiorespiratory control animals (n=5) following the last injection of propofol at P7, as described in a previous study (20). Cardiorespiratory control rats were not used for any other part of the study. Every pup from each group was weighed at P7, P10, P13, P17, P21, P24 and P27 to monitor body development.
Tissue preparation
For western blot studies, at P29, five rats per group were sacrificed by decapitation. Hippocampi were isolated immediately on ice and stored at −80°C until used. For terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assays, five rat pups per group were deeply anesthetized with sodium pentobarbital (45 mg/kg, i.p.; Sigma-Aldrich, St. Louis, MO, USA), perfused transcardially with 0.1 M phosphate-buffered saline (PBS; pH=7.4) and then perfused with 4% (w/v) paraformaldehyde in 0.1 M PBS. The brains were removed and post-fixed with the identical fixative overnight at 4°C. Formalin-fixed hippocampal tissue was dehydrated, embedded in paraffin, and sliced into 5 µm-thick sections.
TUNEL fluorescent assay
According to the protocol of our previous study (21), hippocampal apoptosis was detected by TUNEL using an in situ cell death detection kit (Roche Applied Science, Mannheim, Germany). Apoptosis was quantified by calculating the percentage of TUNEL-positive nuclei out of total nuclei in an average of 20 high-power fields for each section in a blinded manner.
Western blotting
Following experimental exposure, rat pups (n=5 per group) were sacrificed and western blot analysis was performed as previously described (20). In brief, hippocampus tissues were homogenized in cold radioimmunoprecipitation assay buffer (Applygen Technologies Inc., Beijing, China), and the quantity of protein in the supernatants was determined using a bicinchoninic acid protein assay kit (Applygen Technologies Inc.). Protein samples (60 mg protein/lane) were separated by 8 or 10% SDS-PAGE. Following transfer to nitrocellulose membranes, the blots were probed using the following primary antibodies: Rabbit anti-Akt (Ser473; cat. no. 9272), anti-phosphorylated (p)-Akt (Ser473; cat. no. 9271), anti-GSK-3β (Ser9; cat. no. 9315), and anti-p-GSK-3β (Ser9; cat. no. 9336) antibodies (1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA); and rabbit anti-B-cell CLL/lymphoma 2 (Bcl-2; cat. no. sc-783), anti-Bcl-2-associated X protein (Bax; cat. no. sc-526) and anti-caspase-3 (cat. no. 7148) antibodies (1:200; Santa Cruz Biotechnology, Inc., Dallas, TX, USA). Fluorescent secondary antibodies (1:10,000; cat. nos. 926–32211 and 926–32210; LI-COR, Inc., Lincoln, NE, USA) were used to detect the binding of primary antibodies. Proteins were visualized by scanning the membrane on an Odyssey Infrared Imaging System (version 3.0; LI-COR, Inc.).
Behavioral studies
As described in a previous study (20), the Morris water maze (MWM) test, including place and probe trials, was used to evaluate spatial learning and memory of rats (P29–P33) in the three groups (n=10) by investigators blinded to the group conditions. At P28, a day before the formal MWM test, rats were checked for their ability to swim to a visible platform to determine whether non-cognitive impairment had occurred (e.g. visual or swimming deficits). In the MWM test, rats received four training trials daily for four consecutive days.
Statistical analysis
Data are expressed as the mean ± standard error and were analyzed using SPSS software version 20 for Windows (IBM SPSS, Armonk, NY, USA). Body weight, average swimming speed and escape latency of pups were analyzed by two-way repeated-measures analysis of variance (ANOVA), with Bonferroni post-hoc analysis. The remaining data were analyzed with one-way ANOVA, followed by Bonferroni post-hoc analysis. P<0.05 was considered to indicate a statistically significant difference.
Results
Physiological parameters following propofol exposure
There were no significant differences in arterial blood gas values, blood glucose concentrations (Table I) or body weights (Fig. 1) among the three treatment groups. These results reduce the possibility that the anesthesia protocol of the current study caused hypoxia and hypercapnia, which may result in neuronal apoptosis, hypoglycemia and delayed puberty.
Table I.
Dex preconditioning and PRO administration have no effect on arterial blood gas values of neonatal rats.
| Group | pH | PO2 (mmHg) | PCO2 (mmHg) | Lactate (mmol/l) | Glucose (mmol/l) |
|---|---|---|---|---|---|
| CON | 7.408±0.177 | 88.6±1.75 | 40.2±1.43 | 2.96±0.27 | 6.0±0.71 |
| PRO | 7.398±0.171 | 89.8±1.80 | 40.8±1.46 | 3.02±0.11 | 6.6±0.75 |
| Dex+PRO | 7.396±0.221 | 90.0±1.95 | 39.6±1.08 | 3.00±0.15 | 5.8±0.97 |
Results are presented as the mean ± standard error (n=5). CON, control; PRO, propofol; Dex, dexmedetomidine.
Figure 1.
Dex preconditioning and PRO administration exhibit no side effects on the body weight of neonatal rats. Results are presented as the mean ± standard error (n=5). CON, control; PRO, propofol; Dex, dexmedetomidine.
Dex preconditioning attenuates propofol-induced hippocampal apoptosis
The number of TUNEL-positive cells per mm2 in the hippocampal CA1 region was higher in the propofol-treated animals compared with control animals (P=0.0007; Fig. 2). Pretreatment with 75 µg/kg Dex 20 min prior to propofol exposure significantly reduced the number of TUNEL-positive cells in the CA1 region compared with the PRO group (P=0.0005; Fig. 2).
Figure 2.
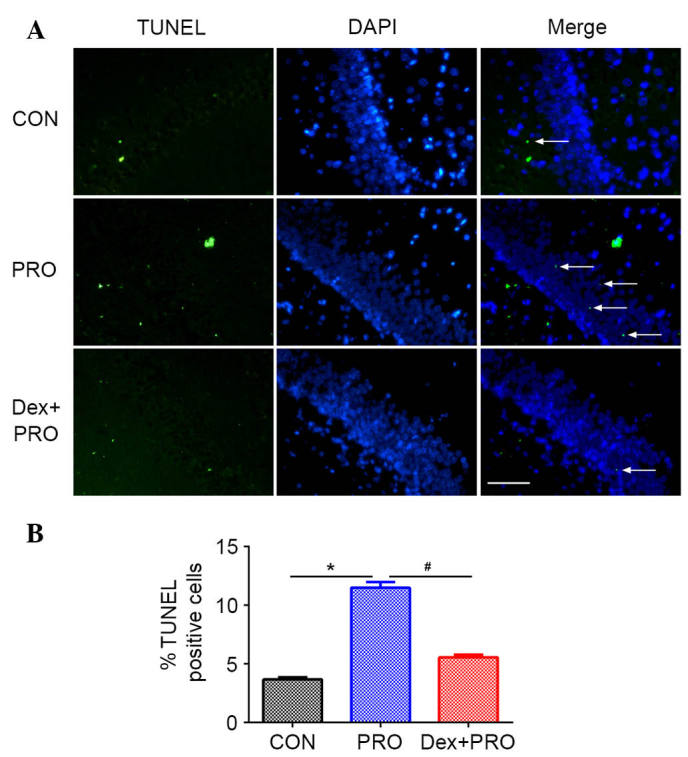
Dex preconditioning ameliorated PRO-induced hippocampal apoptosis. (A) Representative images (magnification, ×400) of hippocampus TUNEL assay from rats in each group. (B) Results are presented as the mean ± standard error (n=5). *P<0.05 vs. CON group; #P<0.05 vs. PRO group. CON, control; PRO, propofol; Dex, dexmedetomidine; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling; DAPI, 4′,6-diamidino-2-phenylindole.
Dex preconditioning regulates the ratio of Bax/Bcl-2 and caspase-3 activation
Western blot analysis (Fig. 3A) demonstrated that, compared with controls, rats subjected to repeated propofol exposure exhibited a significantly higher ratio of Bax/Bcl-2 (P=0.0006; Fig. 3B) and caspase-3 activation (P=0.0008; Fig. 3C). However, compared with the PRO group, Dex pretreatment alleviated the change in Bax/Bcl-2 ratio and caspase-3 activation induced by propofol (P=0.0009).
Figure 3.
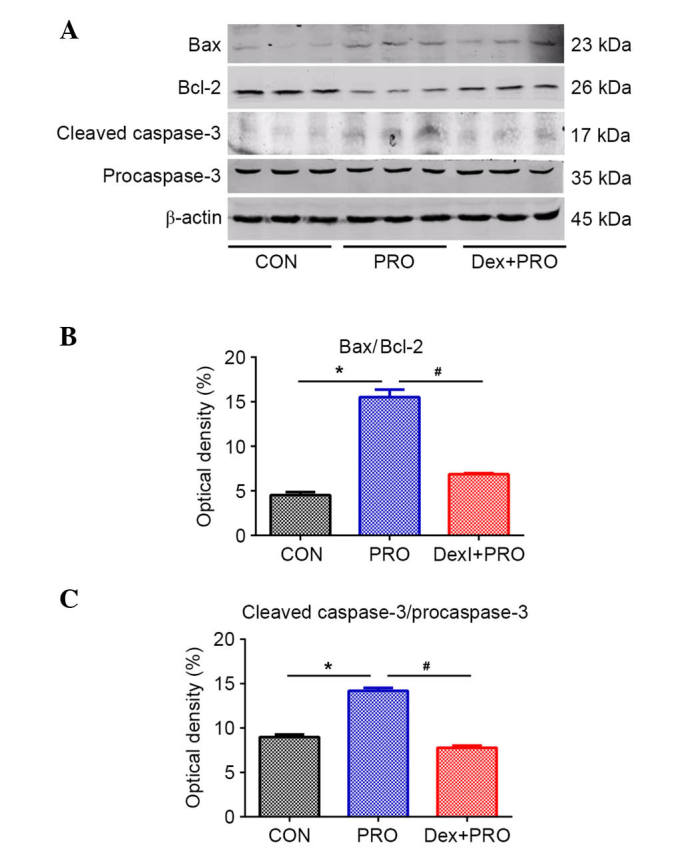
Dex preconditioning regulates the Bcl-2/Bax ratio and cleaved caspase-3 protein expression. (A) Representative gel images of western blot analysis of hippocampal Bcl-2, Bax and cleaved caspase-3 protein expression. Statistical analysis of (B) Bax/Bcl-2 and (C) cleaved-caspase-3/procaspase-3 from rats in each group. Results are presented as the mean ± standard error (n=5). *P<0.05 vs. CON group; #P<0.05 vs. PRO group. Bax, Bcl-2-associated X protein; Bcl-2, B-cell CLL/lymphoma 2; CON, control; PRO, propofol; Dex, dexmedetomidine.
Dex preconditioning activates the PI3K/Akt/GSK-3β signaling pathway
PRO group rats exhibited a significantly lower ratio of p-Akt/Akt (P= 0.03) and p-GSK-3β/GSK-3β (P=0.04) compared with controls. However, compared with the PRO group, the ratios were significantly reduced by Dex pretreatment (P=0.0008; Fig. 4).
Figure 4.
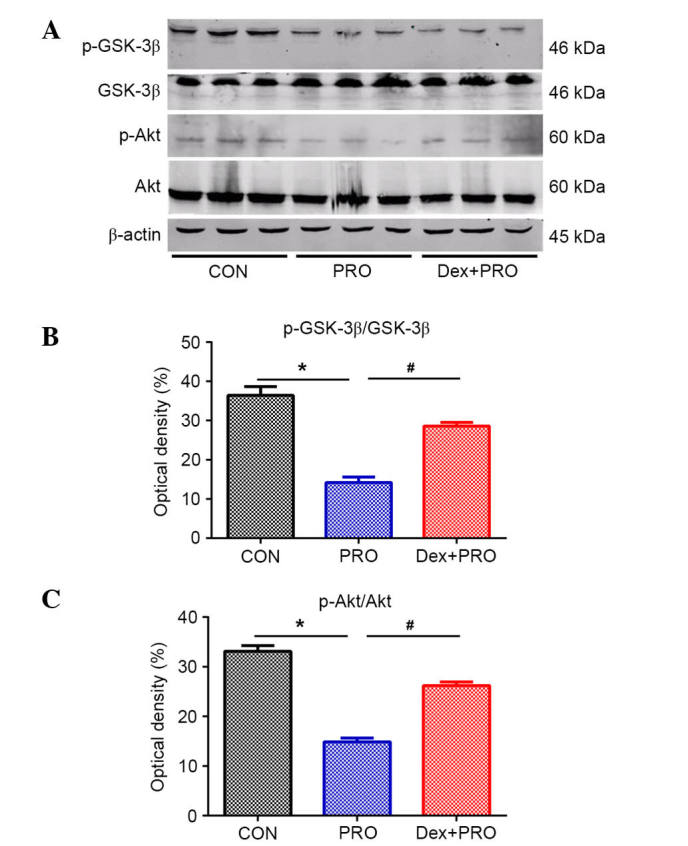
Dex preconditioning activates the phosphatidylinositol-4,5-bisphosphate 3-kinase/Akt/GSK-3β signaling pathway. (A) Representative gel images of western blot analysis of hippocampal Akt, GSK-3β, p-Akt and p-GSK. Statistical analysis of (B) p-Akt/Akt and (C) p-GSK-3β/GSK-3β from rats in each group. Results are presented as the mean ± standard error (n=5). *P<0.05 vs. CON group; #P<0.05 vs. PRO group. p, phosphorylated; GSK-3β, glycogen synthase kinase-3β; Akt, v-akt murine thymoma viral oncogene homolog 1; CON, control; PRO, propofol; Dex, dexmedetomidine.
Dex preconditioning attenuates propofol-induced cognitive deficits
During the MWM test, the escape latency to locate the submerged platform was significantly higher in the PRO group rats compared with controls (P=0.0005) and the Dex + PRO group (P=0.0007) at P33 (Fig. 5A), and there were no significant differences in swimming speeds among the three groups (Fig. 5B). In the probe test, compared with controls, the time spent on the platform area (P=0.0009; Fig. 5C) and the number of platform crossings (P=0.0006; Fig. 5D) were significantly reduced in the PRO group rats. Furthermore, Dex significantly increased the time spent on the platform area and platform crossing compared with the PRO group rats (P=0.02).
Figure 5.

Dex preconditioning attenuates PRO-induced cognitive deficits. (A) The latency of rats to reach the platform in the three groups. (B) Mean swimming speed during 5 days of training in three groups. (C) Time spent in the target quadrant and (D) number of platform crossings in the probe trial. (E) Representative searching swimming paths of three juvenile rats in the probe tests. Results are presented as the mean ± standard error (n=10). *P<0.05 vs. CON group; #P<0.05 vs. PRO group. CON, control; PRO, propofol; Dex, dexmedetomidine.
Discussion
The present study demonstrated that repeated propofol-induced juvenile spatial learning/memory impairments in neonatal rats were ameliorated by pretreatment with 75 µg/kg Dex 20 min prior to challenge. Dex pretreatment alleviated hippocampal apoptosis, regulated the imbalance of Bax/Bcl-2 expression and caspase-3 activation, and normalized the phosphorylation of Akt and GSK-3β. These results strongly suggested that Dex preconditioning protects the developing brain against propofol-induced neurotoxicity and hippocampal apoptosis, a process that may be mediated by the PI3K/Akt/GSK-3β signaling pathway.
Almost all general anesthetics have dual effects of neuroprotection and neurotoxicity, depending on the concentration, duration and the age of exposure (22,23). During early postnatal life, which is termed the brain growth-spurt period, neuronal proliferation, migration, differentiation and synapse formation occur. Increasing experimental data in pre-clinical settings suggest that, during this period, brains are more vulnerable to anesthetics (6,8). The current study used P7 rats in the experimental model, as during this time neurogenesis is still occurring, thus neurons have a high degree of plasticity and are vulnerable to internal and external environment.
As one of the most widely used general anesthetics, propofol has previously been demonstrated to induce neuronal apoptosis, which may contribute to further memory impairment, in several studies (8). However, there is some controversy regarding the appropriate dose of propofol to use in P7 rats. Gao et al (24) demonstrated that 75 mg/kg propofol i.p. once a day at regular 24-h intervals for 7 days in rats from P7 to P13 impairs learning and memory of pups without visible signs of cyanosis. However, Pesić et al (25) observed that 50 mg/kg propofol immediately induces loss of the righting reflex and promotes cyanotic changes in skin color. In a preliminary experiment, it was observed in our laboratory that 75 mg/kg propofol could lead to respiratory depression and cyanosis over a long time (data not shown). Thus, the anesthesia scheme was adjusted to the protocol used by Bercker et al (26) (30 mg/kg propofol every 90 min up to a cumulative dose of 90 mg/kg every day for seven consecutive days), and no clearly discernible anoxic events occurred.
Several protein families contribute to apoptotic cell death regulation. Among these proteins, Bcl-2 is anti-apoptotic and Bax is pro-apoptotic, thus, their relative expression determines cell viability. Activation of the downstream protein caspase-3 was previously demonstrated to be induced by propofol in postnatal brains during maturation (25). In the present study, cognitive impairments associated with propofol exposure were accompanied by significant increases in the Bax/Bcl-2 ratio, caspase-3 expression and the number of TUNEL-positive cells. These results further suggested that apoptosis in the hippocampus is important for propofol neurotoxicity.
Dex, a potent and highly selective α2-adrenoceptor agonist, is commonly administered by the continuous infusion method in pediatric patients for anesthesia and sedation, and occasionally it is also received as a bolus. In addition to its hypnotic and analgesic effect in several brain injury models, it has previously been demonstrated to exhibit anti-apoptotic properties in both pre-clinical and clinical studies (15,18). A previous report demonstrating that the expression of Bcl-2 and Mdm-2 proto-oncogene was greater following Dex treatment compared with controls suggests that the neuroprotective properties of Dex involve ultra-early modulation of the balance between pro- and anti-apoptotic proteins (27). In the current study, 75 µg/kg Dex was chosen as the experimental dose based on a previous study, which demonstrated that pretreatment with 75 µg/kg prior to anesthesia exposure significantly reduced isoflurane-induced apoptosis, whereas Dex alone did not induce a change (19). The addition of Dex potentially increases the depth of anesthesia, however, the gas blood results measured in the present study demonstrated no evident physiological parameter changes. The results of the current study are similar to the report by Sanders et al (16), which demonstrated that, following Dex preconditioning, the number of TUNEL-positive cells were reduced, the expression of apoptotic markers (Bax/Bcl-2, cleaved caspase-3/caspase-3) was reduced and spatial learning/memory was improved. These results demonstrate that Dex exerts an important anti-apoptotic effect against propofol-induced injury.
However, the underlying mechanism of the effects of Dex in the central nervous system remains to be clearly delineated. It is suggested that Dex provides its neuroprotective effect via the α2-adrenergic receptor subtype (28). However, as the α2-adrenoceptor antagonist, atipamezole, only partially reverses the neuroprotective effects of Dex following isoflurane-induced neurotoxicity in rats (16), other mechanisms may be involved.
It is recognized that the PI3K/Akt/GSK-3β signaling pathway also participates in the protective effect of Dex against ischemia-reperfusion injury in the brain (29). Akt is a serine/threonine kinase and is activated by phosphorylation under normal physiological conditions. Activated Akt (p-Akt, Ser473) then phosphorylates GSK-3β at Ser9 to decrease the activity of GSK-3β. Activation of Akt is involved in anti-apoptotic signaling via phosphorylation of Bcl-2 associated agonist of cell death, an important member of the Bcl-2 family that can inhibit the inactivation of Bcl-2 (30). GSK-3β, a homologous mammalian isoform of GSK-3, is highly expressed in the brain, particularly in the hippocampus, striatum and thalamus (31). It phosphorylates a wide variety of cellular substrates and thereby regulates various cellular processes, ranging from glycogen metabolism to cell survival and neuronal polarity (32). Phosphorylation of GSK-3β at Ser9 negatively regulates pro-apoptotic activity (14), and has been previously demonstrated to improve long-term memory in hippocampal-associated tasks (13). In the current study, the expression of p-GSK-3β at Ser9 and p-Akt at Ser473 were downregulated by propofol, which was accompanied by an increase in apoptosis markers (Bax) and spatial deficits, and these changes were ameliorated by Dex, which is similar to the observations of Linseman et al (33). These results suggest that Dex can ameliorate propofol-induced disturbances to the PI3K/Akt/GSK-3β signaling pathway in the developing brain, alleviate hippocampal apoptosis and inhibit learning/memory deficits. However, the exact effect of Dex on the PI3K/Akt/GSK-3β signaling pathway requires further investigation.
Disruption of intracellular calcium homeostasis, particularly excessive calcium release from intracellular stores, mediates general anesthetic-induced neuronal apoptosis (34,35). Furthermore, calcium is also a regulator of PI3K/Akt/GSK-3β signaling (36), and Dex has been previously demonstrated to affect calcium release from intracellular stores in astrocytes (37). Thus, calcium release may act as an inhibitor of the PI3K/Akt/GSK-3β pathway and an inducer of apoptosis, which may be associated with the neuroprotective effect of Dex on propofol-induced neurotoxicity. However, this issue requires further investigation.
There are several limitations of the current study. Notably, a ʻDex aloneʼ group was not included in the study. Although a previous study has already shown that one dose of 75 µg/kg Dex alone does not increase neuronal apoptosis compared with controls (38), it would be beneficial to include a Dex alone group in future studies to further confirm these findings. Additionally, the effect of Dex on long-term hippocampal-dependent memory was not determined. Furthermore, although there are reports of propofol infusions being used for five days or more in pediatric intensive care units (ICUs), repeated administration of propofol for seven consecutive days in the present study is highly unlikely to occur in pediatric anesthesia. This dosing protocol was designed to simulate repeated propofol infusion in a pediatric ICU, and whether this dose of propofol (30 mg/kg, i.p.) in rodents is relevant to clinical practice is difficult to predict. However, the results of the current study provide a novel basis for exploring the activity of Dex in neurogenesis following propofol anesthesia in neonate brains. Further studies investigating this issue are required.
In conclusion, the in vivo data of the present study suggests that repeated exposure to propofol anesthesia impairs juvenile spatial learning and memory in neonatal rats, and this impairment may be associated with hippocampal apoptosis. Dex administration prior to propofol anesthesia improves cognitive dysfunction, partially by attenuating hippocampal apoptosis and regulating the PI3K/Akt/GSK-3β signaling pathway.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant no. 28342016).
References
- 1.Sun LS, Li G, DiMaggio CJ, Byrne MW, Ing C, Miller TL, Bellinger DC, Han S, McGowan FX. Feasibility and pilot study of the Pediatric Anesthesia NeuroDevelopment Assessment (PANDA) project. J Neurosurg Anesthesiol. 2012;24:382–388. doi: 10.1097/ANA.0b013e31826a0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu C, Gao J, Karlsson N, Li Q, Zhang Y, Huang Z, Li H, Kuhn HG, Blomgren K. Isoflurane anesthesia induced persistent, progressive memory impairment, caused a loss of neural stem cells, and reduced neurogenesis in young, but not adult, rodents. J Cereb Blood Flow Metab. 2010;30:1017–1030. doi: 10.1038/jcbfm.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun L. Early childhood general anaesthesia exposure and neurocognitive development. Br J Anaesth. 2010;105(Suppl 1):i61–i68. doi: 10.1093/bja/aeq302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilder RT, Flick RP, Sprung J, Katusic SK, Barbaresi WJ, Mickelson C, Gleich SJ, Schroeder DR, Weaver AL, Warner DO. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110:796–804. doi: 10.1097/01.anes.0000344728.34332.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kay B, Rolly G. I.C.I. 35868, a new intravenous induction agent. Acta Anaesthesiol Belg. 1977;28:303–316. [PubMed] [Google Scholar]
- 6.Harman F, Hasturk AE, Yaman M, Arca T, Kilinc K, Sargon MF, Kaptanoglu E. Neuroprotective effects of propofol, thiopental, etomidate, and midazolam in fetal rat brain in ischemia-reperfusion model. Childs Nerv Syst. 2012;28:1055–1062. doi: 10.1007/s00381-012-1782-0. [DOI] [PubMed] [Google Scholar]
- 7.Rossaint J, Rossaint R, Weis J, Fries M, Rex S, Coburn M. Propofol: Neuroprotection in an in vitro model of traumatic brain injury. Crit Care. 2009;13:R61. doi: 10.1186/cc7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Creeley C, Dikranian K, Dissen G, Martin L, Olney J, Brambrink A. Propofol-induced apoptosis of neurones and oligodendrocytes in fetal and neonatal rhesus macaque brain. Br J Anaesth. 2013;110(Suppl 1):i29–i38. doi: 10.1093/bja/aet173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krzisch M, Sultan S, Sandell J, Demeter K, Vutskits L, Toni N. Propofol anesthesia impairs the maturation and survival of adult-born hippocampal neurons. Anesthesiology. 2013;118:602–610. doi: 10.1097/ALN.0b013e3182815948. [DOI] [PubMed] [Google Scholar]
- 10.Sanchez V, Feinstein SD, Lunardi N, Joksovic PM, Boscolo A, Todorovic SM, Jevtovic-Todorovic V. General anesthesia causes long-term impairment of mitochondrial morphogenesis and synaptic transmission in developing rat brain. Anesthesiology. 2011;115:992–1002. doi: 10.1097/ALN.0b013e3182303a63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 12.Gray JJ, Bickler PE, Fahlman CS, Zhan X, Schuyler JA. Isoflurane neuroprotection in hypoxic hippocampal slice cultures involves increases in intracellular Ca2+ and mitogen-activated protein kinases. Anesthesiology. 2005;102:606–615. doi: 10.1097/00000542-200503000-00020. [DOI] [PubMed] [Google Scholar]
- 13.Dewachter I, Ris L, Jaworski T, Seymour CM, Kremer A, Borghgraef P, De Vijver H, Godaux E, Van Leuven F. GSK3beta, a centre-staged kinase in neuropsychiatric disorders, modulates long term memory by inhibitory phosphorylation at serine-9. Neurobiol Dis. 2009;35:193–200. doi: 10.1016/j.nbd.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 14.Hsing CH, Chen YH, Chen CL, Huang WC, Lin MC, Tseng PC, Wang CY, Tsai CC, Choi PC, Lin CF. Anesthetic propofol causes glycogen synthase kinase-3β-regulated lysosomal/mitochondrial apoptosis in macrophages. Anesthesiology. 2012;116:868–881. doi: 10.1097/ALN.0b013e31824af68a. [DOI] [PubMed] [Google Scholar]
- 15.Duan X, Li Y, Zhou C, Huang L, Dong Z. Dexmedetomidine provides neuroprotection: Impact on ketamine-induced neuroapoptosis in the developing rat brain. Acta Anaesthesiol Scand. 2014;58:1121–1126. doi: 10.1111/aas.12356. [DOI] [PubMed] [Google Scholar]
- 16.Sanders RD, Xu J, Shu Y, Januszewski A, Halder S, Fidalgo A, Sun P, Hossain M, Ma D, Maze M. Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology. 2009;110:1077–1085. doi: 10.1097/ALN.0b013e31819daedd. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, Kou D, Li Z, He Y, Yu W, Du H. Effects of propofol-dexmedetomidine combination on ischemia reperfusion-induced cerebral injury. Neuro Rehabilitation. 2014;35:825–834. doi: 10.3233/NRE-141177. [DOI] [PubMed] [Google Scholar]
- 18.Degos V, Charpentier TL, Chhor V, Brissaud O, Lebon S, Schwendimann L, Bednareck N, Passemard S, Mantz J, Gressens P. Neuroprotective effects of dexmedetomidine against glutamate agonist-induced neuronal cell death are related to increased astrocyte brain-derived neurotrophic factor expression. Anesthesiology. 2013;118:1123–1132. doi: 10.1097/ALN.0b013e318286cf36. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Zeng M, Chen W, Liu C, Wang F, Han X, Zuo Z, Peng S. Dexmedetomidine reduces isoflurane-induced neuroapoptosis partly by preserving PI3K/Akt pathway in the hippocampus of neonatal rats. PLoS One. 2014;9:e93639. doi: 10.1371/journal.pone.0093639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li ZQ, Rong XY, Liu YJ, Ni C, Tian XS, Mo N, Chui DH, Guo XY. Activation of the canonical nuclear factor-κB pathway is involved in isoflurane-induced hippocampal interleukin-1β elevation and the resultant cognitive deficits in aged rats. Biochem Biophys Res Commun. 2013;438:628–634. doi: 10.1016/j.bbrc.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 21.Liang Y, Li Z, Mo N, Li M, Zhuang Z, Wang J, Wang Y, Guo X. Isoflurane preconditioning ameliorates renal ischemia-reperfusion injury through antiinflammatory and anti-apoptotic actions in rats. Biol Pharm Bull. 2014;37:1599–1605. doi: 10.1248/bpb.b14-00211. [DOI] [PubMed] [Google Scholar]
- 22.Kawaguchi M, Furuya H, Patel PM. Neuroprotective effects of anesthetic agents. J Anesth. 2005;19:150–156. doi: 10.1007/s00540-005-0305-5. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Z, Ma D. Anaesthetics-induced neurotoxicity in developing brain: An update on preclinical evidence. Brain Sci. 2014;4:136–149. doi: 10.3390/brainsci4010136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao J, Peng S, Xiang S, Huang J, Chen P. Repeated exposure to propofol impairs spatial learning, inhibits LTP and reduces CaMKIIα in young rats. Neurosci Lett. 2014;560:62–66. doi: 10.1016/j.neulet.2013.11.061. [DOI] [PubMed] [Google Scholar]
- 25.Pesić V, Milanović D, Tanić N, Popić J, Kanazir S, Jevtović-Todorović V, Ruzdijić S. Potential mechanism of cell death in the developing rat brain induced by propofol anesthesia. Int J Dev Neurosci. 2009;27:279–287. doi: 10.1016/j.ijdevneu.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bercker S, Bert B, Bittigau P, Felderhoff-Müser U, Bührer C, Ikonomidou C, Weise M, Kaisers UX, Kerner T. Neurode-generation in newborn rats following propofol and sevoflurane anesthesia. Neurotox Res. 2009;16:140–147. doi: 10.1007/s12640-009-9063-8. [DOI] [PubMed] [Google Scholar]
- 27.Engelhard K, Werner C, Eberspächer E, Bachl M, Blobner M, Hildt E, Hutzler P, Kochs E. The effect of the alpha 2-agonist dexmedetomidine and the N-methyl-D-aspartate antagonist S (+)-ketamine on the expression of apoptosis-regulating proteins after incomplete cerebral ischemia and reperfusion in rats. Anesth Analg. 2003;96:524–531. doi: 10.1097/00000539-200302000-00041. [DOI] [PubMed] [Google Scholar]
- 28.Ma D, Hossain M, Rajakumaraswamy N, Arshad M, Sanders RD, Franks NP, Maze M. Dexmedetomidine produces its neuroprotective effect via the alpha 2A-adrenoceptor subtype. Eur J Pharmacol. 2004;502:87–97. doi: 10.1016/j.ejphar.2004.08.044. [DOI] [PubMed] [Google Scholar]
- 29.Zhu YM, Wang CC, Chen L, Qian LB, Ma LL, Yu J, Zhu MH, Wen CY, Yu LN, Yan M. Both PI3K/Akt and ERK1/2 pathways participate in the protection by dexmedetomidine against transient focal cerebral ischemia/reperfusion injury in rats. Brain Res. 2013;1494:1–8. doi: 10.1016/j.brainres.2012.11.047. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Luo QF, Peng AF, Long XH, Wang TF, Liu ZL, Zhang GM, Zhou RP, Gao S, Zhou Y, Chen WZ. Positive feedback regulation between Akt phosphorylation and fatty acid synthase expression in osteosarcoma. Int J Mol Med. 2014;33:633–639. doi: 10.3892/ijmm.2013.1602. [DOI] [PubMed] [Google Scholar]
- 31.Leroy K, Brion JP. Developmental expression and localization of glycogen synthase kinase-3beta in rat brain. J Chem Neuroanat. 1999;16:279–293. doi: 10.1016/S0891-0618(99)00012-5. [DOI] [PubMed] [Google Scholar]
- 32.Yuskaitis CJ, Jope RS. Glycogen synthase kinase-3 regulates microglial migration, inflammation and inflammation-induced neurotoxicity. Cell Signal. 2009;21:264–273. doi: 10.1016/j.cellsig.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA. Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci. 2004;24:9993–10002. doi: 10.1523/JNEUROSCI.2057-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao YL, Xiang Q, Shi QY, Li SY, Tan L, Wang JT, Jin XG, Luo AL. GABAergic excitotoxicity injury of the immature hippocampal pyramidal neurons' exposure to isoflurane. Anesth Analg. 2011;113:1152–1160. doi: 10.1213/ANE.0b013e318230b3fd. [DOI] [PubMed] [Google Scholar]
- 35.Sinner B, Friedrich O, Zink W, Zausig Y, Graf BM. The toxic effects of s (+)-ketamine on differentiating neurons in vitro as a consequence of suppressed neuronal Ca2+ oscillations. Anesth Analg. 2011;113:1161–1169. doi: 10.1213/ANE.0b013e31822747df. [DOI] [PubMed] [Google Scholar]
- 36.Wei H, Inan S. Dual effects of neuroprotection and neurotoxicity by general anesthetics: Role of intracellular calcium homeostasis. Prog Neuropsychopharmacol Biol Psychiatry. 2013;47:156–161. doi: 10.1016/j.pnpbp.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y, Zhao Z, Code WE, Hertz L. A correlation between dexmedetomidine-induced biphasic increases in free cytosolic calcium concentration and energy metabolism in astrocytes. Anesth Analg. 2000;91:353–357. doi: 10.1097/00000539-200008000-00022. [DOI] [PubMed] [Google Scholar]
- 38.Liao Z, Cao D, Han X, Liu C, Peng J, Zuo Z, Wang F, Li Y. Both JNK and P38 MAPK pathways participate in the protection by dexmedetomidine against isoflurane-induced neuroapoptosis in the hippocampus of neonatal rats. Brain Res Bull. 2014;107:69–78. doi: 10.1016/j.brainresbull.2014.07.001. [DOI] [PubMed] [Google Scholar]