

Abstract
Many Gram-negative pathogens such as Shigella and Salmonella assemble the type III secretion system (T3SS) to inject virulence proteins directly into eukaryotic cells to initiate infectious diseases. The needle apparatus of the T3SS consists of a base, an extracellular needle, a tip protein complex, and a translocon. The atomic structure of the assembled tip complex and the translocon is unknown. Here, we show by NMR paramagnetic relaxation enhancement (PRE) that the mixed α–β domain at the distal region of the Shigella and Salmonella tip proteins interact with the N-terminal ectodomain of their major translocon proteins. Our results reveal the binding surfaces involved in the tip-translocon protein-protein interaction and provide insights about the assembly of the needle apparatus of the T3SS.
Keywords: type III secretion system, protein-protein interactions, tip protein, translocon protein, NMR spectroscopy, paramagnetic relaxation enhancement
Graphical Abstract
Introduction
The T3SS plays a critical role in the pathogenesis of many Gram-negative bacteria [1-2] such as Shigella and Salmonella, the causative agents of bacillary dysentery and typhoid. These pathogens assemble the T3SS to inject virulence proteins directly into their target host cells to initiate infectious diseases.[3-4] The needle apparatus of the T3SS [5-6] consists of a base that spans the bacterial membranes, an external needle,[7-8] and at the needle tip, a complex of an estimated 4-5 copies of the tip protein.[9-10] Upon host cell contact, the tip complex serves as a platform for the assembly of the translocon, which forms a pore in the host cell membrane that allows the passage of effector proteins directly into the host cell cytoplasm.[10-11] The atomic structure of the assembled tip complex and the translocon is currently unknown and how the tip protein interacts with the translocon proteins is poorly understood.
IpaD and SipD are the tip proteins of the Shigella and Salmonella T3SS, respectively. Although the sequence identity between these proteins is less than 40% (Figure S1A), they share structural similarity. The crystal structures of IpaD[12-13] and SipD[14-15] show an overall oblong shape with a long central coiled-coil juxtaposed to a N-terminal α-helical hairpin and a distal domain consisting of mixed α–helices and β–sheets (mixed α–β domain). The translocon is assembled from the major and minor translocon proteins (the qualifiers refer to their relative sizes; both are essential in type III secretion). IpaB and IpaC are the major and minor translocon proteins of Shigella, respectively; whereas SipB and SipC are the major and minor translocon proteins of Salmonella, respectively. The major translocon proteins IpaB and SipB share similar structural features such as an N-terminal cytosolic ectodomain, a central hydrophobic region with two predicted transmembrane helices, and a C-terminal amphipathic region.[16-17] The atomic structure for any full length major translocon protein is currently unknown, however, crystal structures of the N-terminal ectodomains of IpaB and SipB show similar α–helical antiparallel coiled-coil motifs despite low sequence identity of ~ 25% (Figure 1 and S1B).[18]
Figure 1. Crystal structure of IpaB74-224 and SipB82-226.

(A) Crystal structure of IpaB74-224 (PDB ID 3U0C) and (B) SipB82-226 (PDB ID 3TUL) with residues 227-312 modeled based on the predicted secondary structure. The positions of the spin labels are shown (black spheres). Loop regions of SipB82-226 (residues 123-126 and 174-182) lacking electron density are shown with dotted lines.
Dickenson et al.[19] showed by FRET and fluorescence polarization the interaction between IpaD and the N-terminal ectodomain of IpaB (residues 1-226). Here, we used NMR paramagnetic relaxation enhancement (PRE) method to identify the surfaces involved in the IpaD-IpaB and SipD-SipB protein-protein interaction. PRE results indicate that the surface around the mixed α–β domain of the tip protein is involved in the interaction with the N-terminal domain of the major translocon protein, and that the tiptranslocon protein-protein interaction surfaces are conserved between Shigella and Salmonella.
Results
Expression and purification of protein constructs for PRE studies
IpaD (residues 38-332 with a C322S point mutation) and SipD (residues 39-343, C244S) were expressed and purified under native conditions as described before.[15, 20] Both IpaD and SipD were well-behaved in solution and have been previously characterized by NMR.[20-21] The IpaB and SipB constructs were designed based on the proteolysis data,[22] crystal structures,[18] and the known chaperone binding sites.[23-25] Four constructs of the IpaB and SipB ectodomains (IpaB76-308, IpaB74-224, SipB82-312, and SipB82-226) were subcloned, expressed and purified under native conditions. Purified IpaB76-308 showed limited stability and solubility, whereas the construct IpaB74-224 showed better behavior in solution and was more amenable for NMR studies and thus was used here for PRE studies. SipB82-312 showed higher protein expression level and stability than SipB82-226 and thus, SipB82-312 was used for the PRE studies. The crystal structures of IpaB74-224 and SipB82-226 have been reported as antiparallel α-helical coiled-coil motifs (Figure 1).[18] Our SipB PRE construct is 87 residues longer than the reported crystal structure SipB82-226. Secondary structure predictions of the N-terminal domain of SipB suggested residues 82-312 to be primarily α-helical with coils between residues 171-181 and 230-250 (Figure S1B). In agreement with the predictions, Circular Dichroism (CD) spectrum of SipB82-312 showed well-defined minima at 208 nm and 222 nm, characteristic of predominantly α-helical secondary structure (Figure S2A). Thermal denaturation curves displayed SipB82-312 having a Tm value of 54 °C compared to 50.2 °C for the shorter SipB82-226 construct (Figure S2B). For the purposes of representation of SipB82-312, we have modeled residues 227 to 312 as a α-helix (helix α4) with a flexible loop region around residues 227-270 (Figure 1B) based on the secondary structure prediction and results of CD spectroscopy.
NMR spectroscopy on the major translocon proteins IpaB and SipB
NMR spectroscopy was used to gain further insights into the solution behavior of IpaB and SipB constructs. The 2D 1H-15N TROSY spectrum of IpaB74-224 at 37 °C displayed overall good dispersion, but peaks were broad and poorly resolved (Figure 2A). Increasing the acquisition temperature to 47 °C significantly improved the NMR spectrum of IpaB74-224 (Figure 2B). The peaks were distinct, sharp, and well resolved, however, only ~65-70% of the residues from the protein were represented in 2D 1H-15N TROSY spectrum of IpaB74-224, thus backbone assignment of IpaB74-224 by 3D NMR would have been challenging and were not carried out.
Figure 2. NMR spectroscopy on the major translocon proteins IpaB and SipB.

(A-D) 2D 1H-15N TROSY spectra of IpaB74-224 at 37 °C, IpaB74-224 at 47 °C, SipB82-312 at 30 °C, and SipB82-226 at 30 °C, respectively.
The 2D 1H-15N TROSY spectrum of SipB82-312 at 30 °C showed poor dispersion and peak broadening (Figure 2C). We reasoned that the poor NMR data quality of SipB82-312 could be due to the predicted coil regions between residues 230-250. Removal of these flexible loops from SipB82-312 construct improved the overall quality of the 2D 1H-15N TROSY spectrum of SipB82-226 (Figure 2D), however, the presence of overlapping peaks and poor resolution in the middle of the spectrum rendered the SipB82-226 construct unsuitable for further NMR characterization. Consistent with the reported high conformational flexibility of SipB N-terminal polypeptides,[18] varying buffer conditions and acquisition temperatures did not improve the NMR data, suggesting the poor solution behavior of SipB constructs is due to its intrinsic flexibility.
Because the backbone resonances of the tip proteins IpaD[20] and SipD[21] have already been assigned, and the translocon protein constructs IpaB and SipB gave less than ideal NMR spectra as shown above (Figure 2), we used 15N-labeled IpaD/SipD with spin-labeled IpaB/SipB to characterize the tip-translocon protein-protein interaction by PRE.
Generation of IpaB74-224 and SipB82-312 cysteine mutants for spin labeling
PRE measurements require attachment of paramagnetic spin labels at various positions. The site-directed spin labeling was achieved by engineering cysteine residues in specific locations on IpaB74-224 and SipB82-312. A total of eight cysteine point mutations in IpaB74-224 and six in SipB82-312 were created for the attachment of the MTSL spin label (Figure 1, black spheres). CD spectroscopy was used to assess the folding of the cysteine mutants and to ensure that the MTSL conjugation did not alter the overall secondary structure of the proteins. All the cysteine mutants before and after the attachment of MTSL spin label displayed CD spectra similar to the wild type construct (Figure S3). The extent of the spin labeling was confirmed by ElectroSpray Ionization Mass Spectrometry (ESI-MS) to be 100% with the expected mass increase of 185 Da upon MTSL conjugation (Figure S4).
PRE of15 N-amino acid-specifically labeled IpaD and SipD with spin-labeled IpaB and SipB
IpaD (298 amino acids) and SipD (308 amino acids) are large proteins making PRE analysis of their ~300 amide peaks complicated. To reduce the complexity of the NMR datasets and enable the determination of subtle reductions in peak intensities upon protein-protein interaction, 15N-amino acid-specifically labeled IpaD and SipD were used in this study. IpaD and SipD were labeled with 15N-leucine. Another SipD sample used was 15N-labeled altogether at alanine, isoleucine, lysine, and methionine (AIKM) residues. The leucine residues of IpaD and SipD are distributed throughout the protein and provide coverage around all three domains: the N-terminal hairpin, central coiled-coils, and mixed α–β region. Corresponding to the 35 leucines present in the IpaD construct, selective 15N-leucine labeling resulted in an increase of mass by 35 Da (33151 + 35 = 33186 Da, Figure S4C). An optimal molar titration ratio of 1:1 for Shigella IpaD:IpaB and 1:0.5 for Salmonella SipD:SipB was chosen for the PRE experiments on the basis of the extent of peak broadening observed at higher titration ratios (data not shown). PRE measurements were carried out by single time-point method.[26] IpaD and SipD residues that lie close to the spin label will experience high PRE effect and show reduction in peak intensity in the paramagnetic datasets when compared to the control diamagnetic datasets (Ipara/Idia < 1). In contrast, IpaD and SipD residues residing away from the spin label will remain unaffected (Ipara/Idia ~1). Representative 2D NMR spectra used in the PRE determination for 15N-leucine IpaD titrated with IpaB74-224 and 15N-leucine SipD titrated with SipB82-312 are shown in Figure 3. In both cases, spin labels resulted in the reduction of the peak intensity for specific IpaD and SipD residues (Figure 3).
Figure 3. Representative 2D 1H-15N TROSY spectra of IpaD and SipD used in the PRE determination.

15N-Leu IpaD complexed with (A) diamagnetic and (B) paramagnetic IpaB74-224 N85C. 15N-Leu SipD complexed with (C) diamagnetic and (D) paramagnetic SipB82-312 D207C. Arrows point to residues that showed reduction in peak intensity in the presence of spin label. Peaks marked with an asterisk are visible at a lower contour level.
PRE of Shigella IpaD-IpaB74-224 interaction
Spin labels attached on IpaB74-224 at positions Asn-156, Asn-142, Gln-201, Ser-205, and Ser-77 essentially resulted in weak to moderate PRE effect on IpaD (Figure 4). With the exception of Leu-70 of IpaD, majority of the residues gave an Ipara/Idia ratio above 0.7 (Figure 4A). Of these spin label positions, position Asn-156 displayed weakest PRE with a large number of peaks showing an Ipara/Idia ratio of ~1. Spin labels positioned at Asn-85, Gln-109, and Lys-115 (clustered towards the top N-terminal region of IpaB in the crystal structure, Figure 4B) produced strongest PRE effect on specific IpaD residues. Leu-70, Leu-202, Leu-211, and Leu-260 of IpaD experienced strongest PRE with Ipara/Idia values between 0.3-0.55 and Leu-199, Leu-227, Leu-257, and Leu-311 experienced moderate PRE with Ipara/Idia of ~0.6 (Figure 4A). Spin labels close to the N-termini of IpaB74-224 produced highest PRE effect and as the labels were moved down towards the end of the coiled-coil, PRE effect was minimized (Figure 4). Irrespective of the spin label position, Leu-70 of IpaD gave highest PRE that could be due to its location on a highly flexible loop region. The affected residues mapped largely near the mixed α–β region of IpaD (Figure 4C). A detailed analysis of PRE effects with individual spin-label sites has been shown in Figure S5A. Our PRE results indicate that region around the mixed α–β domain of IpaD is the binding site for the N-terminal domain of IpaB.
Figure 4. PRE results of 15N-Leu IpaD and spin-labeled IpaB74-224.

(A) Each panel corresponds to one spin label position of IpaB74-224. Circles in each panel represent Ipara/Idia intensity ratio for leucine residues of IpaD. The gray line at Ipara/Idia intensity ratio of 1.0 is taken as having no PRE effect. L70, L202, L211, and L260 of IpaD (labeled in red) experienced the strongest PRE effect. (B) The position of the spin labels (spheres) in IpaB74-224 are colored based on the strength of the PRE as strong (red), moderate (yellow), and weak or none (gray). Spin label positions yielding strong PRE on IpaD (N85, Q109, K115) are clustered on the top N-terminal region of IpaB. (C) Results of PRE mapped onto the structure of IpaD (PDB ID 2J0O) with IpaD colored as follows: N-terminal region (gray), central coiled-coil (cyan), and mixed α–β region (green). IpaD leucine residues that experienced the strongest PRE effect (Ipara/Idia < 0.6) are colored red, residues showing moderate PRE (Ipara/Idia ~ 0.6-0.69) are orange, weak (Ipara/Idia ~ 0.7-0.79) are yellow, and almost no effect (Ipara/Idia > 0.8-1.0) are white. Unassigned leucines are colored light blue. The affected leucine residues clustered near the distal region of IpaD.
PRE of Salmonella SipD-SipB82-312 interaction
Spin labels on SipB82-312 at positions Ala-109, Lys-127, and Glu-303 produced weak to moderate PRE effect on SipD with Ipara/Idia ratio above 0.7 for most of the residues (Figure 5). Out of these, spin label at Lys-127 showed almost no PRE with an Ipara/Idia of ~1. On the other hand, spin labels positioned at Asp-207, Lys-211, and Asn-283 induced strong PRE effect on certain SipD residues. In the crystal structure of SipB82-226, Asp-207 and Lys-211 lie close to each other on helix α3, and Asn-283 can be expected to lie on helix α4 as shown (Figure 5C). Ala-115, Leu-116, Ala-123, Leu-214, Ala-217, and Leu-280 of SipD experienced the strongest PRE effect (Figure 5A, 5B) with Ipara/Idia ratio between 0.2-0.5. Most of the affected residues cluster close to the mixed α–β domain of SipD (Figure 5D). A detailed analysis of PRE effects with individual spin-labeling sites has been shown in Figure S5B. Our PRE results indicate that the mixed α–β domain of SipD interacts with a surface along helix α3/α4 of the N-terminal domain of SipB.
Figure 5. PRE results of 15N-AIKM and 15N-Leu SipD in complex with spin-labeled SipB82-312.

Results of PRE using (A) 15N-AIKM SipD and (B) 15N-Leu SipD. (A,B) Each panel shows Ipara/Idia intensity ratio as a function of SipD residue number for one spin label position in SipB. The gray line at Ipara/Idia ratio of 1.0 indicated no PRE effect. A115, L116, A123, L214, A217, and L280 of SipD (red) experienced strongest PRE effect. (C) SipB spin labels (spheres) are color-coded based on their strength of the PRE effect on SipD as strong (red), moderate (yellow), and weak/none (gray). Positions of spin label that induced strong PRE on SipD (at residues 207, 211, 283) lie along helix α3 and α4 of SipB. (D) Results of PRE mapped onto the structure of SipD (PDB ID 3NZZ) with SipD colored as follows: N-terminal region (gray), central coiled-coil (cyan), and mixed α–β region (green). A 22-residue loop absent in the crystal structure is shown as dotted line. Spheres on SipD are colored according to the strength of PRE effect as follows: strong (Ipara/Idia < 0.6, red); moderate (Ipara/Idia ~ 0.6-0.69, orange); weak (Ipara/Idia ~ 0.7-0.79, yellow), and unaffected (Ipara/Idia > 0.8-1.0, white). SipD residues A115, L116, and A123 displaying strong PRE belong to the missing loop region (labeled in red). The stronger affected residues cluster at the distal region of SipD.
Discussion
An important step in type III secretion is the assembly of the translocon on the tip complex. The protein-protein interactions between the tip and the translocon proteins are crucial for the attachment of the bacterium to the host cell membrane and the delivery of effector proteins through the translocon pore.[10,27-28] However, the atomic detail of the protein-protein interactions governing the assembly of the tiptranslocon structure remains poorly understood. The current hypothesis in the literature is that IpaD and IpaB form a heteropentameric complex consisting of 4 copies of IpaD and 1 copy of IpaB at the needle tip[29-31], and this IpaD:IpaB interaction is essential for host cell sensing, assembly of the translocon pore, and regulation of effector secretion into the host cell cytoplasm.[13, 27, 32]
There are conflicting results regarding the role of the bile salt deoxycholate in triggering the presentation of IpaB at the tip complex. While several studies indicate exposure of IpaD to the bile salt deoxycholate is needed for the recruitment of IpaB at the tip,[19, 32-33] others have shown the association of IpaD and IpaB at the needle tip without the need for deoxycholate.[13, 29, 31, 34] Dickenson et al.[19] used fluorescence polarization and FRET assays to show that IpaD interacts with the N-terminal fragment of IpaB, and that the IpaD-IpaB interaction was detected only in the presence of high concentrations of deoxycholate. Our PRE results presented here (Figure 3-6) indicate that IpaB and SipB ectodomains interact with their cognate tip protein IpaD and SipD, respectively, in the absence of deoxycholate.
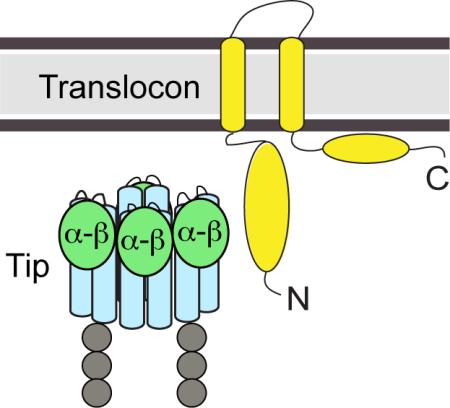
Figure 6. Model of tip-translocon interaction.

(A) Predicted membrane topology for the major translocon protein IpaB and SipB (yellow). (B) Proposed model for the interaction between the distal region of IpaD (or SipD) with the N-terminal ectodomains of IpaB (or SipB) in the assembly of T3SS needle apparatus. The tip complex is represented based on the EM model of Shigella tip with the central coiled-coil domain shown in light blue and the mixed α–β region in light green.
Results of fluorescence spectroscopy show weak binding affinity at μM range between IpaD and IpaB.[19] Such weak interactions probably provide these proteins sufficient conformational flexibility to perform their multi-functional roles in host environment sensing, translocon assembly, or for switching the system to an ‘actively secreting’ state. To gain further insights into this weak interaction, we used a sensitive NMR method based on PRE, which requires paramagnetic spin labels that are conjugated to the protein through cysteine residues. Nuclear dipoles within ~15-20 Å of the spin label undergo increased relaxation, resulting in the reduction of the peak intensities of the residues lying close to the spin label. We engineered eight IpaB74-224 and six SipB82-312 cysteine mutants to attach the spin label and confirmed by CD spectroscopy that the cysteine mutation and the MTSL spin label did not alter the global fold of the proteins (Figure S3). Our PRE results showed that the distal region of IpaD is the primary binding site for N-terminal ectodomain of IpaB74-224. Likewise, for the Salmonella SipD-SipB82-312 interaction, SipD residues experiencing the highest PRE effect upon interaction with SipB82-312 were also concentrated around the distal region of SipD.
Our results on the tip-translocon interaction are consistent with the previous results of Johnson et al.[13] showing that deletion of the mixed α–β domain of IpaD (residues 192 to 267) abolishes the localization of IpaB on the needle tip. The recent EM model of the Shigella tip-translocon complex, albeit at low resolution, shows that the mixed α–β domain of IpaD faces outside and lie close to the observed density for IpaB on the assembled tip.[31] Additionally, the recent crystal structure of AopB (the homolog of IpaB/SipB in Aeromonas) indicates that the N- and C-terminal domains of AopB remain on the extracellular portion upon membrane insertion, making these domains potential candidates for protein-protein interaction with the tip protein.[35] Incorporating these with our previous results that the needle protein binds at the proximal end (or lower portion of the coiled-coil domain as depicted in Figure 4 and Figure 5) of the tip protein,[36-37] we propose a model where the N-terminal ectodomain of IpaB (or SipB) acts as an anchor to attach to the distal end of the tip protein IpaD (or SipD) at the mixed α–β domain, while the proximal end of the tip protein interacts with the needle (Figure 6).
In summary, our PRE results confirm a direct protein-protein interaction between the major translocon protein of Shigella and Salmonella and their cognate tip proteins in the absence of deoxycholate. Further, we have localized the interaction interface to the N-terminal cytosolic domain of the major translocon protein and the distal end of the tip protein. Our findings provide new insights into the interactions involved in the assembly of the T3SS needle apparatus.
Experimental Section
Protein expression and purification
The plasmid for protein expression of SipD residues 39-343 C244S from Salmonella typhimurium strain SL1344 in pET-21a has been described before.[20] We previously reported that the point mutation C244S did not alter the crystal structure of SipD and Salmonella was fully functional in assembling the T3SS and invading eukaryotic cells.[15] Analogous to SipD, IpaD residues 38-332 C322S from Shigella flexneri was subcloned in the NdeI/SalI sites of pET-21a vector. The SipD and IpaD constructs contained an N-terminal His6-tag followed by a tobacco etch virus (TEV) protease cleavage site. DNA corresponding to SipB82-312, SipB82-226, IpaB76-308, and IpaB74-224 were PCR amplified and subcloned into the NdeI/XhoI sites of pET-22b, which introduced a C-terminal His6-tag for protein purification. Plasmids were freshly transformed in E. coli BL21 (DE3) and cells were grown in 1 L culture media containing 100 μg/ml carbenicillin. Unlabeled proteins were expressed in LB broth and 15N-labeled proteins were obtained by cell growth in M9 minimal media supplemented with 1 g of 15N-ammonium chloride. Bacteria were grown at 37 °C, induced with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at OD600 ~ 0.7-0.8, and cell growth was continued overnight at 15 °C. Cells were harvested by centrifugation and cell pellets were resuspended in 30 ml binding buffer (500 mM NaCl, 20 mM Tris-HCl pH 8.0, 5 mM imidazole). Cells were lysed by sonication in the presence of 0.1 mM phenylmethanesulfonylfluoride (PMSF). Cellular debris was removed by centrifugation at 13,000 rpm for 10 min, and to the supernatant was added 600 μl of 5% (v/v) polyethyleneimine to precipitate the nucleic acids. Following centrifugation (13,000 rpm, 10 min), the supernatant was loaded on a ~5 mL Ni2+-affinity chromatography resin (Gold Biotechnology) and the Ni2+ column was washed with 100 ml binding buffer, followed by elution with 50 ml elution buffer (500 mM NaCl, 20 mM Tris-HCl pH 8.0, 250 mM imidazole). Fractions containing IpaD and SipD, were incubated overnight at room temperature with 250 μl of 0.04 mM recombinant TEV protease[38] in buffer (20 mM NaCl, 20 mM Tris-HCl pH 8.0, 0.5 mM EDTA, and 1 mM DTT) and the digest was passed through a Ni2+ column to separate the protein from the His6-tag. IpaD and SipD retained a 3-residue ‘GHM’ cloning artifact whereas the IpaB and SipB constructs a C-terminal LEH6 affinity tag. Purified proteins were dialyzed in NMR buffer (100 mM NaCl, 20 mM sodium phosphate pH 7.0) and concentrated using Amicon Ultra 10K centrifugal filter (Millipore) and protein concentrations were estimated by absorbance at A280.
15N-amino acid-specific labeling of IpaD and SipD
15N-amino acid-specifically labeled IpaD and SipD were used in this study. IpaD was labeled with 15N-leucine and SipD was 15N-amino acid-specifically labeled at alanine, isoleucine, lysine, and methionine (AIKM) residues. Another SipD sample used was only labeled with 15N-leucine. The 15N-amino acid-specifically labeled IpaD and SipD were prepared following published protocol with minor modification.[20] Briefly, cells expressing IpaD or SipD from overnight 1 L LB culture were harvested and resuspended in 2X 500 ml M9 minimal media supplemented with 20 amino acids. To obtain 15N-leucine labeled protein, the minimal media was supplemented with 125 mg of 15N-leucine and 300 mg of the remaining 19 unlabeled amino acids. To obtain 15N-AIKM SipD, the minimal media was supplemented with 15N-alanine, 15N-isoleucine, 15N-lysine, and 15N-methionine plus 16 of the remaining unlabeled amino acids. The starting OD600 was kept around 0.6-0.8 and cells were grown at 37 °C for 10 minutes followed by induction with 1 mM IPTG. Cell growth was continued for 4 hours at 37 °C or until OD600 was ~ 2.6 to 3.0. Recombinant proteins were purified as described above.
Engineering of cysteine mutations in IpaB74-224 and SipB82-312 for spin labeling
Several nonconserved polar residues that are expected to be surface exposed in IpaB74-224 and SipB82-312 were mutated into cysteine for the attachment of the spin label for PRE measurements. QuikChange site-directed mutagenesis was used to generate eight cysteine mutants in IpaB74-224 residues Ser-77, Asn-85, Gln-109, Lys-115, Asn-142, Asn-156, Gln-201, and Ser-205 and six in SipB82-312 residues Ala-109, Lys-127, Asp-207, Lys-211, Asn-283, and Glu-303. All mutations were confirmed by DNA sequencing. The cysteine mutant proteins were expressed and purified following the protocol described above. The purified cysteine-containing IpaB74-224 and SipB82-312 proteins were dialyzed in NMR buffer containing 5 mM DTT to prevent the formation of intermolecular disulfide bonds.
Spin labeling of IpaB74-224 and SipB82-312
A nitroxide spin label, MTSL (1-Oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl) methanethiosulfonate (Toronto Research Chemicals) was conjugated to the cysteine mutants of IpaB74-224 and SipB82-312 following our published protocol.[37] Briefly, 10 mg of MTSL was dissolved in 250 μl of acetone to make a 150 mM stock solution that was added at 7-fold molar excess to the protein for labeling. The extent of MTSL labeling was confirmed by ESI-MS.
Circular dichroism spectroscopy
CD spectra were acquired with a JASCO J-815 spectropolarimeter. Proteins were diluted to a final concentration of 1-2 μM in buffer (5 mM NaCl, 5 mM sodium phosphate pH 7.0). Spectra were acquired in triplicate at 20 °C with a scan rate of 50 nm/min. Thermal denaturation curves were acquired by monitoring the molar ellipticity at 222 nm over a temperature range of 20 °C to 80 °C with a temperature ramp rate of 2 °C/min.
NMR spectroscopy
NMR data were acquired on a Bruker Avance 800 MHz spectrometer equipped with a cryogenic triple-resonance probe, processed by NMRPipe,[39] and analyzed using NMRView.[40] Two-dimensional 1H-15N TROSY spectra were acquired using 0.2 mM 15N-labeled IpaB74-224, SipB82-312, and SipB82-226 in buffer (100 mM NaCl, 10 mM sodium phosphate pH 7.0, 10% D2O). PREs were measured by single time-point method.[26] For PRE data acquisition, two 2D 1H-15N HSQC spectra were acquired on two NMR samples with identical NMR acquisition parameters. The first 2D 1H-15N TROSY spectrum was acquired using an 15N-amino acid-specifically labeled IpaD complexed with paramagnetic (or MTSL-spin labeled) IpaB74-224. A second 2D 1H-15N TROSY spectrum was acquired using 15N-amino acid-specifically labeled IpaD complexed with diamagnetic IpaB74-224. A similar PRE protocol was followed to determine the PREs of 15N-amino acid-specifically labeled SipD complexed with paramagnetic or diamagnetic SipB82-312. The protein concentrations, buffer conditions, acquisition and processing parameters were kept identical for the paramagnetic and diamagnetic NMR samples to ensure that the observed intensity changes were only due to the effect of the spin label on the tip protein. Typical PRE samples contained 0.4 mM 15N-leucine IpaD mixed with 0.4 mM (paramagnetic or diamagnetic) IpaB74-224 or 0.8 mM 15N-amino acid-specifically labeled SipD (AIKM-SipD and Leu-SipD) with 0.4 mM (paramagnetic or diamagnetic) SipB82-312 in PRE buffer (100 mM NaCl, 20 mM sodium phosphate pH 7.0, 10% D2O). 2D 1H-15N TROSY experiments on Leu-IpaD with IpaB were collected at 32 °C and 2D 1H-15N TROSY or HSQC experiments on Leu-SipD or AIKM-SipD with SipB were acquired at 30 °C and 25 °C, respectively. Published amide backbone assignments of IpaD[21] and SipD[20] were used to calculate the peak intensity ratio, Ipara/Idia for each non-overlapping peak.
Supplementary Material
Acknowledgements
We are grateful to Dr. Dalian Zhong for IpaD expression plasmid and Dr. Asokan Anbanandam for assistance with NMR spectroscopy. This research was supported by the National Institutes of Health through grants AI074856 (R.N.D) and P30GM110761 (University of Kansas Biomolecular NMR Facility).
Abbreviations
- NMR
nuclear magnetic resonance
- MTSL
(1-Oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl) methanethiosulfonate
- PRE
paramagnetic relaxation enhancement
- T3SS
type III secretion system
References
- 1.Hueck CJ. Microbiol. Mol. Biol. Rev. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coburn B, Sekirov I, Finlay BB. Clin. Microbiol. Rev. 2007 doi: 10.1128/CMR.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carayol N, Tran Van Nhieu G. Cold Spring Harb Perspect Med. 3:2013. doi: 10.1101/cshperspect.a016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galan JE. Annu. Rev. Cell. Dev. Biol. 2001;17:53–86. doi: 10.1146/annurev.cellbio.17.1.53. [DOI] [PubMed] [Google Scholar]
- 5.Cornelis GR. Nat. Rev. Microbiol. 2006;4:811–825. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee S, Chaudhury S, McShan AC, Kaur K, De Guzman RN. Biochemistry. 2013;52:2508–2517. doi: 10.1021/bi400160a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demers JP, Habenstein B, Loquet A, Kumar Vasa S, Giller K, Becker S, Baker D, Lange A, Sgourakis NG. Nature communications. 2014;5:4976. doi: 10.1038/ncomms5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loquet A, Sgourakis NG, Gupta R, Giller K, Riedel D, Goosmann C, Griesinger C, Kolbe M, Baker D, Becker S, Lange A. Nature. 2012;486:276–279. doi: 10.1038/nature11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Espina M, Olive AJ, Kenjale R, Moore DS, Ausar SF, Kaminski RW, Oaks EV, Middaugh CR, Picking WD, Picking WL. Infect. Immun. 2006;74:4391–4400. doi: 10.1128/IAI.00440-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lara-Tejero M, Galan JE. Infect. Immun. 2009;77:2635–2642. doi: 10.1128/IAI.00077-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mueller CA, Broz P, Cornelis GR. Mol. Microbiol. 2008;68:1085–1095. doi: 10.1111/j.1365-2958.2008.06237.x. [DOI] [PubMed] [Google Scholar]
- 12.Barta ML, Guragain M, Adam P, Dickenson NE, Patil M, Geisbrecht BV, Picking WL, Picking WD. Proteins. 2012;80:935–945. doi: 10.1002/prot.23251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson S, Roversi P, Espina M, Olive A, Deane JE, Birket S, Field T, Picking WD, Blocker AJ, Galyov EE, Picking WL, Lea SM. J. Biol. Chem. 2007;282:4035–4044. doi: 10.1074/jbc.M607945200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lunelli M, Hurwitz R, Lambers J, Kolbe M. PLoS Pathog. 2011;7:e1002163. doi: 10.1371/journal.ppat.1002163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatterjee S, Zhong D, Nordhues BA, Battaile KP, Lovell SW, De Guzman RN. Protein Sci. 2011;20:75–86. doi: 10.1002/pro.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hume PJ, McGhie EJ, Hayward RD, Koronakis V. Mol. Microbiol. 2003;49:425–439. doi: 10.1046/j.1365-2958.2003.03559.x. [DOI] [PubMed] [Google Scholar]
- 17.McGhie EJ, Hume PJ, Hayward RD, Torres J, Koronakis V. Mol. Microbiol. 2002;44:1309–1321. doi: 10.1046/j.1365-2958.2002.02958.x. [DOI] [PubMed] [Google Scholar]
- 18.Barta ML, Dickenson NE, Patil M, Keightley A, Wyckoff GJ, Picking WD, Picking WL, Geisbrecht BV. J. Mol. Biol. 2012;417:395–405. doi: 10.1016/j.jmb.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickenson NE, Arizmendi O, Patil MK, Toth R. T. t., Middaugh CR, Picking WD, Picking WL. Biochemistry. 2013;52:8790–8799. doi: 10.1021/bi400755f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Nordhues BA, Zhong D, De Guzman RN. Biochemistry. 2010;49:4220–4226. doi: 10.1021/bi100335u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dickenson NE, Zhang L, Epler CR, Adam PR, Picking WL, Picking WD. Biochemistry. 2011;50:172–180. doi: 10.1021/bi101365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayward RD, McGhie EJ, Koronakis V. Mol. Microbiol. 2000;37:727–739. doi: 10.1046/j.1365-2958.2000.02027.x. [DOI] [PubMed] [Google Scholar]
- 23.Kim BH, Kim HG, Kim JS, Jang JI, Park YK. Microbiology. 2007;153:2998–3008. doi: 10.1099/mic.0.2007/007872-0. [DOI] [PubMed] [Google Scholar]
- 24.Lokareddy RK, Lunelli M, Eilers B, Wolter V, Kolbe M. J. Biol. Chem. 2010;285:39965–39975. doi: 10.1074/jbc.M110.135616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adam PR, Patil MK, Dickenson NE, Choudhari S, Barta M, Geisbrecht BV, Picking WL, Picking WD. Biochemistry. 2012;51:4062–4071. doi: 10.1021/bi300243z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gillespie JR, Shortle D. J. Mol. Biol. 1997;268:158–169. doi: 10.1006/jmbi.1997.0954. [DOI] [PubMed] [Google Scholar]
- 27.Menard R, Sansonetti PJ, Parsot C, Bacteriol J. 1993;175:5899–5906. doi: 10.1128/jb.175.18.5899-5906.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collazo CM, Galan JE. Mol. Microbiol. 1997;24:747–756. doi: 10.1046/j.1365-2958.1997.3781740.x. [DOI] [PubMed] [Google Scholar]
- 29.Veenendaal AK, Hodgkinson JL, Schwarzer L, Stabat D, Zenk SF, Blocker AJ. Mol. Microbiol. 2007;63:1719–1730. doi: 10.1111/j.1365-2958.2007.05620.x. [DOI] [PubMed] [Google Scholar]
- 30.Blocker AJ, Deane JE, Veenendaal AK, Roversi P, Hodgkinson JL, Johnson S, Lea SM. Proc. Natl. Acad. Sci. U.S.A. 2008;105:6507–6513. doi: 10.1073/pnas.0708344105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheung M, Shen DK, Makino F, Kato T, Roehrich AD, Martinez-Argudo I, Walker ML, Murillo I, Liu X, Pain M, Brown J, Frazer G, Mantell J, Mina P, Todd T, Sessions RB, Namba K, Blocker AJ. Mol Microbiol. 2015;95:31–50. doi: 10.1111/mmi.12843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olive AJ, Kenjale R, Espina M, Moore DS, Picking WL, Picking WD. Infect. Immun. 2007;75:2626–2629. doi: 10.1128/IAI.01599-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stensrud KF, Adam PR, La Mar CD, Olive AJ, Lushington GH, Sudharsan R, Shelton NL, Givens RS, Picking WL, Picking WD. J. Biol. Chem. 2008;283:18646–18654. doi: 10.1074/jbc.M802799200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen DK, Saurya S, Wagner C, Nishioka H, Blocker AJ. Infect. Immun. 2010;78:4999–5010. doi: 10.1128/IAI.00470-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen VS, Jobichen C, Tan KW, Tan YW, Chan SL, Ramesh K, Yuan Y, Hong Y, Seetharaman J, Leung KY, Sivaraman J, Mok YK. Structure. 2015 doi: 10.1016/j.str.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 36.Rathinavelan T, Lara-Tejero M, Lefebre M, Chatterjee S, McShan AC, Guo DC, Tang C, Galan JE, De Guzman RN. J. Mol. Biol. 2014;426:2958–2969. doi: 10.1016/j.jmb.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rathinavelan T, Tang C, De Guzman RN. J. Biol. Chem. 2011;286:4922–4930. doi: 10.1074/jbc.M110.159434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geisbrecht BV, Bouyain S, Pop M. Protein Expr. Purif. 2006;46:23–32. doi: 10.1016/j.pep.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 39.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, Biomol J. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 40.Johnson BA. Methods Mol. Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.