

Abstract
Human cytomegalovirus (HCMV) encodes at least 25 membrane glycoproteins that are found in the viral envelope1. While gB represents the fusion protein, two glycoprotein complexes control the tropism of the virus: the gHgLgO trimer is involved in the infection of fibroblasts, while the gHgLpUL128L pentamer is required for infection of endothelial, epithelial and myeloid cells2–5. Two reports suggested that gB binds to ErbB1 and PDGFRα6,7, however these results do not explain the tropism of the virus and were recently challenged8,9. Here we provide a 19Å reconstruction for the gHgLgO trimer and show that it binds with high affinity through the gO subunit to PDGFRα, which is expressed on fibroblasts but not on epithelial cells. We also provide evidences that the trimer is essential for viral entry in all cell types. Furthermore, we identified the pentamer as a trigger for the ErbB pathway, which is essential for infection of epithelial cells. These findings help explain the broad tropism of HCMV and indicate that PDGFRα and the viral gO subunit could be targeted by novel anti-viral therapies.
Human cytomegalovirus (HCMV) is a widespread pathogen that establishes lifelong latency in healthy individuals10 and can cause severe disease in immunodeficient individuals and in the fetus, being the leading viral cause of congenital birth defects11. VR1814 is a clinical HCMV isolate that can infect both fibroblasts and epithelial cells and expresses both the gHgLgO trimer and the gHgLpUL128L pentamer2–5. In contrast, AD169 is a laboratory adapted virus that infects fibroblasts, but not epithelial cells3,12,13 and expresses the trimer, but not the pentamer due to a point mutation in UL131 ORF2,4. Antibodies that bind to the shared gH glycoprotein neutralize infection of both fibroblasts and epithelial cells14, while antibodies to the pUL128 gene products are extremely potent in neutralizing infection of epithelial, endothelial and myeloid cells, but fail to neutralize infection of fibroblast14,15. While it is well established that trimer and pentamer determine the tropism of the virus16, the cellular receptors involved remain a matter of debate1.
To identify the mechanisms that underline the cellular tropism of different HCMV strains, we investigated the activation of receptor tyrosine kinases (RTK) upon exposure of epithelial cells (ARPE-19) or fibroblasts (MRC-9) to VR1814 or AD169 viruses (Fig. 1a and Supplementary Fig. 1). Consistent with a previous report7, the pentamer-deficient AD169 virus led to the phosphorylation of PDGFRα and PDGFRβ in fibroblasts, while it failed to activate RTK in epithelial cells (Fig. 1a), a finding that is consistent with the inability of AD169 to infect this cell type4,12. Interestingly, the clinical isolate VR1814 led to the phosphorylation of PDGFRα, PDGFRβ and ErbB1 in fibroblasts, while in epithelial cells induced phosphorylation of ErbB1, ErbB2 and, to a lower extent, of PDGFRβ (Fig. 1a). These findings are consistent with the expression of ErbB1 on both cell types and PDGFRα on fibroblasts only (Supplementary Fig. 2a,b and Supplementary Fig. 3). Thus, the PDGFRα pathway is triggered only in fibroblasts by both pentamer-deficient and pentamer-sufficient viruses, while the ErbB pathway is triggered only by the pentamer-sufficient VR1814 virus in both fibroblasts and epithelial cells. These data are inconsistent with a previous report that suggested that gB, which is expressed by both viruses, is a ligand for ErbB6, but rather suggest that the pentamer might be the trigger of the ErbB pathway.
Figure 1. Evidence that trimer-PDGFR and pentamer-ErbB interactions determine cellular tropism for fibroblasts or epithelial cells.
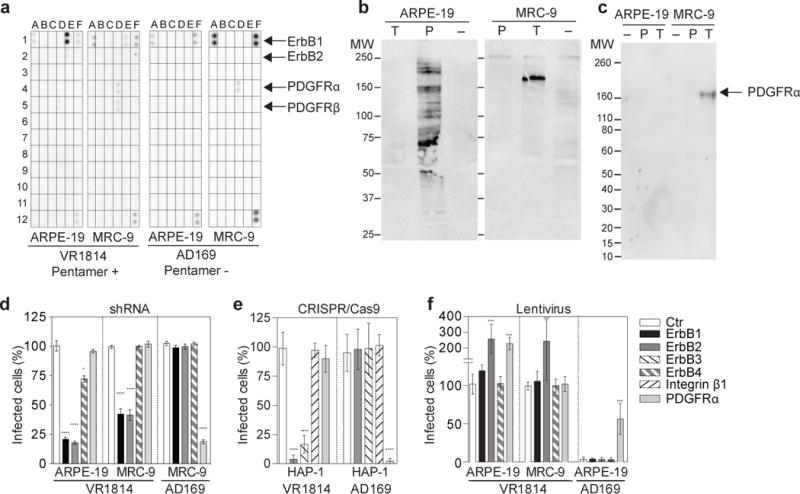
(a) Human epithelial cells (ARPE-19) or fibroblasts (MRC-9) were exposed to pentamer-sufficient virus (VR1814) or pentamer-deficient virus (AD169). After 10 minutes, the cells were lysed and the lysate was captured by antibodies specific for tyrosine kinase receptors and developed using an anti-phospho-tyrosine antibody. Coordinates of the Phospho-Receptor Tyrosine Kinase array are reported in Supplementary Fig. 1. (b) Soluble trimer and pentamer were used for immunoprecipitation of cell surface biotinylated proteins from MRC-9 or ARPE-19 cells followed by immunoblotting with streptavidin-HRP conjugate. (c) Pull down from ARPE-19 or MRC-9 cells with soluble trimer or soluble pentamer immobilized on streptactin beads. Precipitated material was submitted to western blot analysis with an anti-PDGFRα polyclonal antibody. (d) The indicated receptors were downregulated on ARPE-19 and MRC-9 by shRNA. Cells were then infected with VR1814 or AD169 viruses for 48 hours. Shown is the percentage of infected cells. (e) The indicated receptors were knocked out on HAP-1 cells by CRISPR/Cas9. Cells were infected with VR1814 or AD169 viruses and analyzed as above. (f) The indicated receptors were overexpressed in ARPE-19 or MRC-9 cells using lentiviral vectors. Cells were infected and analyzed as above. Data shown are mean ± s.d. of three independent experiments. Statistical significance was evaluated by Mann-Whitney U test *P < 0.05; ****P < 0.0001.
To study the molecular interactions that lead to the triggering of the PDGFR and ErbB pathways, we produced recombinant trimer and pentamer15 and used them in immunoprecipitation experiments. The trimer immunoprecipitation performed on cell surface biotinylated fibroblasts showed a clear band above 150 KDa (Fig. 1b), which was identified as PDGFRα by mass spectrometry analysis (Supplementary Table 1). In contrast, no specific band was obtained using pentamer on cell surface biotinylated fibroblasts, while multiple bands were obtained using pentamer on epithelial cells (Fig. 1b and Supplementary Table 2). Recombinant trimer and pentamer were also used for pull-down experiments followed by western blot analysis. Pull-downs confirmed a specific interaction between trimer and PDGFRα from fibroblasts, but not from epithelial cells (Fig. 1c). These findings indicate that the HCMV trimer can bind to PDGFRα, which is selectively expressed on fibroblasts but not on epithelial cells, thus explaining the limited tropism of the pentamer-deficient AD169 virus17. In contrast, immunoprecipitation experiments failed to demonstrate a direct high affinity interaction between the pentamer and ErbB receptors, leaving the possibility that this interaction may have low affinity or requires additional molecules.
To further investigate the role of the ErbB and PDGFR pathways in the infection process, we used a genetic approach based on down-regulation and knockout of specific receptors (Supplementary Fig. 4a,b). Down-regulation of ErbB1 and ErbB2 by shRNA reduced infection of epithelial cells and fibroblasts by the pentamer-sufficient VR1814 virus, while it did not affect infection of fibroblasts by the pentamer-deficient AD169 virus (Fig. 1d). In contrast, downregulation of PDGFRα inhibited by 80% the infection of fibroblasts by AD169, but did not affect their infection by VR1814. To further validate these data we used a gene knockout approach in the haploid cell line HAP-1, which expresses PDGFRα and ErbB2, as well as low levels of ErbB3 (Supplementary Fig. 2c). CRISPR-Cas9-mediated knockout of ErbB2 and ErbB3, but not PDGFRα, strongly reduced infection of HAP-1 cells by VR1814 virus (Fig. 1e). Reciprocally, knockout of PDGFRα, but not ErbB2 or ErbB3, completely inhibited infection by AD169. Of note, knockout of integrin β1, which was shown to play a role as co-receptor in HCMV entry18,19, did not affect infection by AD169 or VR1814 viruses (Fig. 1e), a finding which suggests that multiple integrins may play a role as co-receptors in HCMV entry18,19.
Consistent results were obtained using a lentiviral-mediated over-expression approach (Fig. 1f and Supplementary Fig. 4c,d). ErbB2 over-expression in both epithelial cells and fibroblasts led to a 2–3 fold higher infection by VR1814, while over-expression of PDGFRα in epithelial cells rendered the cells permissive to infection by the pentamer-deficient AD169 virus and increased infection by VR1814 virus by 2 fold (Fig. 1f). Collectively, the above results indicate that pentamer-sufficient VR1814 virus, can trigger PDGFR and ErbB receptor tyrosine kinases pathways onto fibroblasts, while the pentamer-deficient AD169 virus relies only on PDGFRα
To address viral entry mechanisms into fibroblasts, we measured the capacity of antibodies and soluble ligands to inhibit infection. Fibroblast infection by the AD169 virus was inhibited by all ligands that interfere with the trimer-PDGFRα interaction, namely soluble PDGFRα, soluble gHgLgO trimer, anti-PDGFRα, anti-gH and anti-gO monoclonal antibodies (Fig. 2a and Supplementary Fig. 5a–d). Infection of fibroblasts by the VR1814 virus was inhibited by an anti-gH antibody that targets both trimer and pentamer14,20 and a monoclonal anti-gO antibody (Fig. 2b and Supplementary Fig. 5a–d). Furthermore, while infection of fibroblasts by VR1814 was not affected by soluble trimer or pentamer when used alone, the combination of soluble trimer and pentamer was strongly inhibitory (Fig. 2c). Similarly, fibroblasts infection by VR1814 was potently inhibited by a combination of anti-pUL128 antibody and either soluble PDGFRα or anti-gO antibody (Fig. 2d and Supplementary Fig. 5a–c).
Figure 2. HCMV trimer is required for both fibroblasts and epithelial cells infection, while HCMV pentamer is required for epithelial cells only.

(a–d) MRC-9 cells were exposed to VR1814 or AD169 viruses that had been pre-incubated with different concentrations of antibodies or soluble ligands, alone or in different combinations: neutralizing antibodies specific for gH (13H11) or UL128/130/131 (7I13) or gO (CVB234); blocking antibody to PDGFRα (αR1), soluble trimer, soluble pentamer or soluble PDGFRα. (e) ARPE-19 cells were exposed to VR1814 virus that had been pre-incubated with different concentrations of antibodies to pUL128, gH or gO, soluble trimer or soluble pentamer. (f) ARPE-19 cells were pre-incubated with ErbB ligands (EGF, HB-EGF or Nrg-1) and subsequently infected with VR1814. Shown is the percentage of infected cells relative to untreated controls. Data shown are mean ± s.d. of four independent experiments.
Similar experiments using antibodies and soluble ligands were performed to address the mechanisms of viral entry into epithelial cells. Infection by VR1814 was strongly inhibited by anti-pUL128 antibody and to a lower extent, by anti-gH antibody or soluble pentamer, as previously reported14,15,21 (Fig. 2e). Remarkably, a monoclonal anti-gO was also able to reduce ARPE-19 infection (Fig. 2e). This surprising result suggests that the trimer might have a dual function of viral ligand and activator of gB fusogenic activity, as recently proposed22,23. Since we did not find evidence for binding of pentamer to ErbB receptors, we tested the inhibitory capacity of ErbB ligands, which down-regulate the cognate receptors (Supplementary Fig. 6). Interestingly, infection of epithelial cells was inhibited by pre-incubation of the cells with HB-EGF (which binds to ErbB1 and ErbB4), EGF (which binds to ErbB1) or Nrg-1 (which binds to ErbB4 on ARPE-19 cells) (Fig. 2f). These results support the notion that entry into epithelial cells relies on ErbB receptors.
To investigate the molecular interactions involved in the HCMV target cell tropism, we probed trimer and pentamer with PDGFRα and ErbB1 using surface plasmon resonance (SPR) and biolayer interferometry (BLI). A high affinity interaction between trimer and PDGFRα was detected using both SPR and BLI, with a KD value of 2 × 10−9 M (Fig. 3a,c). The trimer/PDGFRα complex was still accessible to two neutralizing antibodies specific for gH (Fig. 3d). Interestingly, soluble gHgL dimer did not bind to PDGFRα (Fig. 3d), indicating that the gO component of the trimer is essential for binding. Similar SPR experiments performed with soluble pentamer and ErbB1-4 receptors or PDGFRα did not reveal any interactions (Supplementary Fig. 7).
Figure 3. The gHgLgO trimer, but not the gHgL dimer, binds with high affinity to PDGFRα.
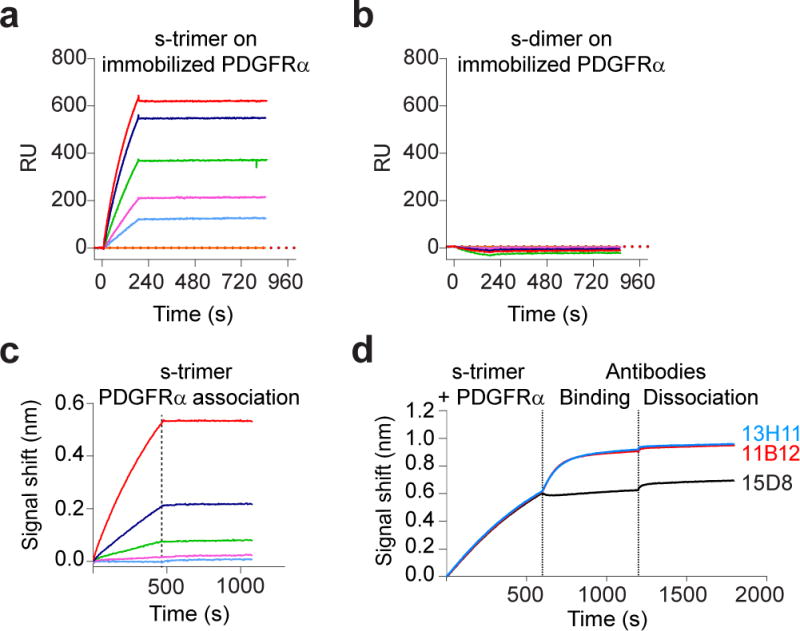
(a,b) Protein-protein interaction determined by Surface Plasmon Resonance (SPR) analysis. PDGFRα–Fc was immobilized to a GLM sensor chip and various concentrations (12.5 to 100 nM) of soluble trimer (a) or soluble dimer (b) were injected. Response units (RU) were recorded using ProteOn. (c,d) Protein-protein interactions were determined by Biolayer interferometry (BLI). PDGFRα-Fc was loaded on Protein G tip, dipped into various concentrations (ranging from 0.3 to 25 nM) of trimer and response units recorded. (c). The immobilized PDGFRα/Trimer complex was dipped into a solution of neutralizing anti-gH (13H11 or 11B12) or anti-pUL128 (15D8) antibodies and response units recorded (d). SPR and BLI sensorgrams are representative examples of experiments repeated three times.
To further investigate the interaction between the gHgLgO trimer and PDGFRα, we studied complex formation in solution using size-exclusion chromatography and visualized the complexes by electron microscopy. Addition of PDGFRα to soluble trimer shifted the chromatography peak to a lower retention time, indicating formation of a complex (Fig. 4a,b). Addition of a Fab fragment of the anti-gH antibody 13H11 to the purified complex further decrease the complex’s retention time (Fig. 4c). SDS-PAGE analysis confirmed that the purified trimer-PDGFRα and 13H11-trimer-PDGFRα complexes contained all indicated components (Fig. 4d). The images of the complexes formed were visualized by negative staining and transmission electron microscopy with 2D averaging (Fig. 4e–h and Supplementary Fig. 8). The gHgLgO trimer alone presented a boot shaped structure, wherein gHgL formed the elongated structure and gO the angled extension (Fig. 4e, h), similar to what was recently reported26. Interestingly, and in agreement with the BLI results, the image of gHgLgO trimer-PDGFRα complex revealed that only the gO subunit interacts with PDGFRα, it appeared that three Ig-like domains of the PDGFRα were visible in the 2D-averaged density, resembling the mode of interaction between platelet-derived growth factor and its receptor (Supplementary Fig. 9). However, not all domains of PDGFRα were well-defined due to its flexibility27(Fig. 4f). This result was further confirmed by the observation that 13H11 Fab bound to a site of gH away from the gO interacting face and, therefore, did not interfere with binding of gO to PDGFRα (Fig. 3c and Fig. 4g). Consequently, we conclude that the neutralizing activity of 13H11 is not due to the inhibition of trimer-PDGFRα interaction but may be dependent on its ability to interfere with the capacity of the complex to trigger gB and consequently viral fusion20.
Figure 4. Visualization of the gHgLgO-PDGFRα complex.
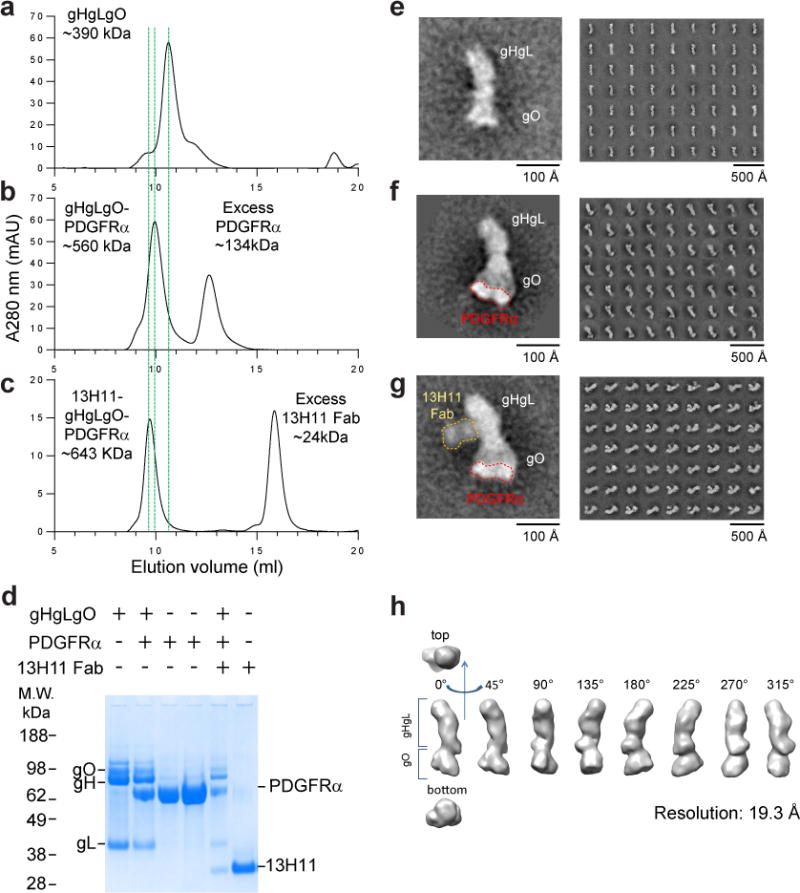
(a–c) Size exclusion chromatography profiles of gHgLgO alone (a) or in complex with soluble PDGFRα (b) or with PDGFRα and a Fab fragment of the anti-gH antibody 13H11 (c). The shifts of the elution time caused by formation of binary and ternary complexes were marked with green dashed lines. SDS-PAGE analysis under reducing conditions of individual protein and complexes containing gH/gL/gO, PDGFRα and 13H11 Fab in different combinations isolated by size-exclusion (d). (e–g) Negative-stain electron microscopy and reference free 2D class averages of gHgLgO and its complexes purified by size exclusion chromatography. One representative image from each purification step was zoomed in to show the shape of the molecules and addition of volume. Trimer alone (e) or with receptor PDGFRα bound to gO. Of note, for the gHgLgO-PDGFRα complex, PDGFRα is only well resolved in some averages (f) or with the 13H11 Fab bound to the twisted region of gH in the gHgLgO-PDGFRα complex (g). Three-dimensional map of gHgLgO at 19.3 Å of resolution.
This study demonstrates that HCMV infection of fibroblasts relies on the direct interaction of the gHgLgO trimer with PDGFRα and identifies gO as the essential component for this interaction. In contrast, we could not find evidence of a direct interaction between the gHgLpUL128L pentamer and the ErbB receptors. However, using wild-type pentamer-deficient virus and cellular knockdown, we reported that the ErbB pathway activation might be involved in pentamer-dependent tropism. It is therefore tempting to suggest that HCMV pentamer induces ErbB activation, which induces macropinocytosis, as this endocytic mechanism was already proposed to mediate HCMV entry in epithelial cell9,24.
The broad distribution of the PDGFRα and ErbB receptors is consistent with the ability of HCMV to infect different cell types, ranging from epithelial, endothelial, myeloid, hematopoietic precursors, neuronal, astrocytes and glial cells expressing either PDGFRα and/or the ErbB receptors25. The role of PDGFRα as a HCMV receptor was recently challenged by Vanarsdall and colleagues9 showing that PDGF-AA and anti-PDGFRα antibodies did not block infection of fibroblasts by a HCMV clinical isolate. These results are in agreement with our finding. Pentamer-sufficient virus might alter the nature of the entry pathway by inducing viral endocytosis.
Two previous studies, suggested that gB binds to ErbB1 or PDGFRα6,7. However, this model is inconsistent with the differential tropism of wild type and pentamer-deficient viruses, since both viruses express gB. In addition, the recent experiment showing that gB expressed on target cells can promote the fusion of gB- deficient trimer-expressing virus might suggest that gB lacks a cellular receptor26. It is therefore tempting to suggest that the interaction of gB with PDGFRα and ErbB1 might be mediated by pentamer and trimer, which might have the dual function of cellular receptor binding and of triggering the fusogenic activity of gB as proposed recently for the trimer22,23 and similarly to what described for gD in HSV-127.
Importantly, we also identified an anti-gO monoclonal antibody isolated from a HCMV seropositive donor. Surprisingly, this antibody neutralized HCMV VR1814 with the same potency on fibroblasts and epithelial cells, indicating that gHgLgO trimeric complex is necessary for viral entry on all cell types. This finding is consistent with recent studies reporting gHgLgO as the only promoter of gB fusion22,23. Nonetheless, gB-mediated fusion occurs at the plasma membrane in fibroblast28, while viral fusion happens in the endosome of epithelial, endothelial and myeloid cells9,29,30 that do not express PDGFRα. It will be interesting to determine the mechanism of trimer-induced endosomal fusion in absence of PDGFRα.
In conclusion, here we demonstrated that the HCMV trimer cellular receptor is PDGFRα. We also showed that HCMV can use PDGFR and ErbB receptor for entry into fibroblast involving pentamer and trimer (Supplementary Fig. 10). The identification of multiple pathways may explain the broad HCMV tropism and assists in the development of anti-viral agents and vaccines effectively controlling viral infection and dissemination.
Methods
Cells and viruses
ARPE-19 retinal pigmented epithelium cell line (ATCC CRL-2302) was cultured in D-MEM/F-12 (Gibco, Invitrogen) supplemented with 10% FBS and penicillin/streptomycin. MRC-9 fibroblast cell line (ATCC CCL-212) was cultured in Minimum Essential Medium Eagle (Sigma-Aldrich) supplemented with 10% FBS, glutamine, penicillin/streptomycin. HAP-1 cells, parental line and HAP-1 CRISPR knockouts, were obtained from Horizon Genomics GmbH and cultured in Iscove’s Modified Dulbecco’s Medium (IMDM) in the presence of 10% FBS and penicillin/streptomycin. All cell lines were confirmed to be free of Mycoplasma. HCMV clinical isolate VR1814 and laboratory-adapted strain AD169 were kindly provided by Virologia e Microbiologia, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy. HCMV VR1814 and AD169 were propagated on HUVEC cells and fibroblasts respectively. ARPE-19, MRC-9 and HAP-1 cells are not listed according to ICLAC 3/10/2014. ARPE-19, MRC-9 and HAP-1 cells31 were authenticated by analysis of Short tandem repeat (STR) loci at ATCC (ARPE-19, MRC-9) and Horizon Genomics GmbH (HAP-1).
Cell surface biotinylation, immunoprecipitation, pull down and western blotting
ARPE-19 and MRC-9 cells were grown to confluency in 10 cm culture dishes (Nunc, Thermo Scientific). Before biotinylation cells were washed twice with ice-cold PBS and incubated with 2 ml of 0.5 mg/ml sulfo-NHS-biotin in PBS per plate for 15 minutes (Thermo Scientific). Reactions were quenched by addition of 10% FBS for 10 minutes on ice. Cells were washed twice with PBS and lysed with 1 ml/per plate of ice-cold lysis buffer (1% n-Dodecyl-β-D-maltoside, 50 mM Tris pH 7.2, 150 mM NaCl, 1X Complete protease inhibitor from Roche). Centrifuged cell lysates were incubated with 30 μg of trimer or pentamer (pre-complexed with 30 μg of 13H11 antibody) for 3 hours with continuous shaking. Protein A beads were subsequently added to the cell lysates for 90 minutes. Beads were washed three times with lysis buffer and one time with PBS, then boiled in reducing SDS-PAGE loading buffer and subjected to SDS-PAGE and western blotting. Detection was performed with streptavidin-HRP conjugate (Upsala) in 1% BSA for 1 hour at RT. For pull down experiment MRC-9 or ARPE-19 cell lysates were incubated with 30 μg of trimer or pentamer for 3 hours with continuous shaking. StrepTactin sepharose beads (GE Healthcare) were subsequently added to the cell lysates for 90 minutes. Beads were washed three times with lysis buffer and one time with PBS, then boiled in reducing SDS-PAGE loading buffer and subjected to SDS-PAGE and western blotting. Detection was performed with rabbit polyclonal anti-PDGFR-α antibody (C-20, Santa Cruz Biotechnology, Inc) in 1% BSA for 2 hours at RT. Uncropped images of western blot are shown in Supplementary Fig. 11.
ELISA
We performed an ELISA to ensure that the particles used for the experiment were similar in term of gH derived glycoprotein content (trimer and pentamer). AD169 and VR1814 particles were coated on 96-well MaxiSorp plates (Nunc), blocked with 1% bovine serum albumin (BSA) in PBS and incubated with titrated antibodies (13H11), followed by AP-conjugated goat anti-human IgG antibody (Southern Biotech, catalogue no. 2040-04). Plates were then washed, substrate (p-NPP, Sigma) was added and plates were read at 405 nm.
Flow cytometry staining
For cell surface staining, adherent cells (ARPE-19, MRC-9 and HAP1) were gently detached with a cell scraper and washed twice with PBS + 2% FBS, incubated with PDGFRα-specific mouse monoclonal antibody (BD Biosciences, clone αR1 and Sino biological, clone 5A10H9), mouse monoclonal anti-ErbB1 (Sigma, clone 225), mouse monoclonal anti-ErbB2, anti-ErbB3, anti-ErbB4 (R&D systems, Clone 191924, Clone 66223, Clone 182818) or mouse IgG1 anti-CMV pp72 (clone 6E1; Santa Cruz Biotechnology), mouse IgG2a H1P73, mouse IgG2b (MA5-14447; Invitrogen) at 2 μg/ml for 30 minutes on ice for isotype control. After two washes, cells were incubated with the following secondary antibodies: goat anti-Mouse IgG (H+L) AlexaFluor 594 conjugated (Invitrogen) at 2 μg/ml for 30 minutes on ice, washed twice and acquired with a FACSFortessa (BD Biosciences) flow cytometer. For intracellular staining, 293Expi cells were fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences), according to the manufacturer’s instructions, and then stained with the anti-gO antibodies or 13H11 mab (as isotype control) at 2 μg/ml for 30 minutes on ice, washed and incubated with goat anti-Human IgG (H+L) AlexaFluor 594 conjugated (Invitrogen) at 2 μg/ml for 30 minutes on ice, washed twice and acquired with a FACSFortessa flow cytometer. Analysis was performed with FlowJo software.
Gene over expression and down regulation
Human ErbB1, ErbB2, ErbB4 and PDGFRα were ordered from GenEZ™ ORF database (Genscript). Lentiviral vectors were generated by cloning ErbB1, ErbB2, ErbB4 and PDGFRα in pCDH-EF1-MCS (System Biosciences). Lentiviral particles were generated by PEI transfection of HEK293FT (Invitrogen) with pCDH, pMD2.G (addgene), psPAX (addgene) and purified on a sucrose cushion. ARPE-19 and MRC-9 were transduced at 60% confluency and expression was accessed after two days. Down regulations of ErbB1, ErbB2 and PDGFRα were done by lentiviral transduction of cells using 4 unique 29mer shRNA constructs in pGFP-C-shLenti and 29-mer scrambled shRNA cassette in pGFP-C-shLenti Vector (Origene) using manufacturer’s instructions. Best constructs were selected by puromycin selection and effective down regulation of the target gene without decrease in cell viability. Sequence of the shRNAs used are listed thereafter.
ErbB1: 5′_ATGGAGGAAGACGGCGTCCGCAAGTGTAA_3′,
ErbB2: 5′_TGTTGGATGATTGACTCTGAATGTCGGCC_3′
ErbB4: 5′_TGTAAGGCAATGCTGCCTACTATCAAACT_3′
PDGFRα: 5′–AGTTCCACCTTCATCAAGAGAGAGGACGA_3′
Scrambled: 5′_GCACTACCAGAGCTAACTCAGATAGTACT_3′
HAP-1 CRISPR/Cas9 KO
HAP1-PDGFRα KO was obtained by causing a frameshift mutation (insertion of 176bp in exon 2) in PDGFRα (NM-006206). As a result the defective transcript has the following amino acid sequence: MGTSHPARSHVISVCSAT*. The guide RNA has the following sequence: 5′-CAGCCTAAGACCAGGAACGC-3′. Clone was verified by PCR and Sanger sequencing and the following primers were used:
PCR_fwd: TGTAAAACGACGGCCAGTCAGAAGGTTTTGGCTTCAGG
PCR_bwd: AACTGCCACTGGAGAGCATT
Sequencing primer: TGTAAAACGACGGCCAGTCAGAAGGTTTTGGCTTCAGG
Sequencing result was as follow: 5′_ATGGGGACTTCCCATCCGGCGCGCAGTCACGTGATCAGTGTATGTTCAGCCACGTGATCAGTGCATGATAGCAAACTGATCAGTGCACATTAAGGAACGTGATTAGTGCATGATCAGCCACGTGATAAGTGTACAATGAGACACGTGATCAGTGCATGATCAGCCACGTAATCAGTGCATGATAGCCACCTGATCAGT TCCTGGTCTTAGGCTGTC_3′.
HAP1-ErbB2 KO was obtained by causing a frameshift mutation (10bp deletion in exon 6) in ErbB2 (NM_001005862). As a result the defective transcript has the following amino acid sequence: MKLRLPASPETHLDMLRHLYQGCQVVGNLELTYLPTNASLSFLQDIQEVQGYVLIAHNQVRQVPLQRLRIVRGTQLFEDNYALAVLDNGDPLNNTTPVTGASPGGLRFEASQRS*.
The guide RNA has the following sequence: 5′_TTCGAAGCTGCAGCTCCCGC_3′. Clone was verified by PCR and Sanger sequencing and the following primers were used:
PCR_fwd: CTCATCGCTCACAACCAAGTGA
PCR_bwd: GAATTATATGTGCTGACGCAAGCTA
Sequencing primer: CTCATCGCTCACAACCAAGTGA
Sequencing result was as follow: 302: 5′_GGCCTCCCCAGGAGGCCTGC----------GCTTCGAAGCCTCACAG_3′ HAP1-ErbB3 KO was obtained by causing a frameshift mutation (4bp deletion in exon 3) in ErbB3 (NM_001982).
As a result the defective transcript has the following amino acid sequence: MRANDALQVLGLLFSLARGSEVGNSQAVCPGTLNGLSVTGDAENQYQTLYKLYERCEVVMGNLEIVLTGHNADLSFLQWIREVTGYVLVAMNEFSTLPLPNLRCEGPRSTMGSLPSSSC*
The guide RNA has the following sequence: 5′_ACCATTGCCCAACCTCCGCG_3′. Clone was verified by PCR and Sanger sequencing and the following primers were used:
PCR_fwd: TGTAAAACGACGGCCAGTGGCAACAAGAGCGTAACTCC
PCR_bwd: TTGCTTAGGGAGCAATGGACCT
Sequencing primer: TGTAAAACGACGGCCAGTGGCAACAAGAGCGTAACTCC
Sequencing result was as follow: 287: 5′_TCTACCATTGCCCAACCTCC----GGTGCGAGGGACCCAGGTCT_3′
HAP1- Integrinβ1 KO was obtained by causing a frameshift mutation (10bp deletion in exon 4) in ITGBI (NM_002211).
As a result the defective transcript has the following amino acid sequence: MNLQPIFWIGLISSVCCVFAQTDENRCLKANAKSCGECIQAGPNCGWCTNSTFLQEGMPTSARCDDLEALKKKGCPPDDIENPRGSKDIKKNKNVAKEQQRSSSQRILLRSNHSSWFCD*
The guide RNA has the following sequence: 5′-TGCTGTTCCTTTGCTACGGT-3′. Clone was verified by PCR and Sanger sequencing and the following primers were used:
PCR_fwd: TGTAAAAACGAGCCTTCAACAGAAA
PCR_bwd: ACTTCTGCACGATGTGATGATTTAG
Sequencing primer: TGTAAAAACGAGCCTTCAACAGAAA
Sequencing result was as follow: 517:_5′CTGTGGTTGGATCTGAGTAATATCCTCTGGCTTGAGCTTCTCTGCTGTTCCTTTGCTACATTTTTATTTTTCTTTATATCTTTGGAGCCTCTGGGATTTTCTATGTCATCTGGA_3′
Human phospho-RTK array
Human phospho-RTK array was performed with a kit (R&D systems, catalogue number ARY-001B). ARPE-19 and MRC-9 were grown in T75 plate (4 million cells total) and infected with the different virus strains for 10 minutes with a MOI of 1. Both viruses were purified by ultracentrifugation on a sucrose gradient and dialyzed against PBS with a membrane cut off 100 kDa to remove proteins and small molecules that could co-sedimented with the viruses. Both viruses were considered similar for the overall content of gH derived glycoprotein and gO content. Glycoprotein O content for AD169 and VR1814 viruses was analyzed by western blot (Supplementary Fig. 5c). Glycoprotein H quantity was analyzed by both mass spectrometry (Supplementary Table 3, PRIDE repository dataset identifier PXD003810) and ELISA (Supplementary Fig. 5d).
Monoclonal antibody isolation from memory B cells
Memory B cells were isolated from cryopreserved PBMCs using CD22 microbeads or anti-FITC (fluorescein isothiocyanate) microbeads (Miltenyi Biotec) after staining of PBMCs with CD22-FITC, and were immortalized with Epstein–Barr virus (EBV) and CpG in multiple wells as described previously32. Supernatants of immortalized memory B cells collected after 2 weeks were screened for their ability to bind gO or neutralize HCMV virus (AD169) infections in high-throughput FACS and micro-neutralization assays, respectively. Positive EBV-B cell cultures were expanded in complete RPMI medium. The antibodies were affinity purified by protein A chromatography (GE Healthcare).
Negative-stain electron microscopy and single particle analysis
gHgLgO, gHgLgO-PDGFR and 13H11-gHgLgO-PDGFR complexes were diluted to about 0.01 mg/ml, adsorbed to freshly glow-discharged carbon-film grids for 15 s, washed with a buffer containing 20 mM HEPES, pH 7.5, and 150 mM NaCl, and stained with 0.7% uranyl formate, pH 5. Images were collected semi-automatically with SerialEM33 on an FEI Tecnai T20 operating at 200 kV and equipped with a 2,000 × 2,000 Eagle CCD camera at a pixel size of 0.22 nm/px. Particles were picked automatically and manually using e2boxer from the EMAN2 software package34. Initial reference-free 2D class averages were obtained in EMAN2. Further 2D classification was performed using reference-free alignment and correspondence analysis in SPIDER35. Numbers of particles used for 2D was analysis: 10,356 (gHgLgO); 43,000 (gHgLgO-PDGFR) and 12,947 (13H11-gHgLgO-PDGFR).
To obtain the 3D map of gHgLgO, the dataset containing 10,356 particles was subjected to iterative stable alignment and clustering (ISAC)36 in SPARX37. The ISAC classes were then used to generate an initial 3D map in EMAN2, which was used as the initial model for three-dimensional reconstruction and refinement using reference projections in SPIDER38. The resolution of the final map was 19.3 Å when a Fourier shell correlation (FSC) threshold of 0.5 was used. The map was visualized in UCSF Chimera39. The reconstruction data have been deposited in the Electron Microscopy Data Bank (Accession Code: EMD-3391).
PDGFRα, Pentamer, Trimer and Dimer expression and purification
Extracellular domain of PDGFRα with C-terminal encoding eight Histidine and a twin strep tags was produced in HEK 293 GnTI−/− cells via transient transfection. Cell supernatant was batch-incubated with 5 ml complete His-Tag Purification Resin (Roche) at room temperature for 30 minutes with continuous inversion mixing. 6 mM imidazole was added to reduce non-specific binding. The resin was packed into a column and sequentially washed with 3 column volume of buffers containing 6 mM and 15 mM imidazole in 400 mM NaCl, 20 mM Tris/Cl− pH7.5. The protein was eluted with the same buffer containing 300 mM imidazole. Expression and purification of the soluble pentamer was previously described by our group15. Soluble trimer complex was generated using a single polycistronic vector encoding three genes (gH, gL, and gO). The gH subunit lacking the transmembrane domain and modified at the C-terminus by addition of a TEV cleavage site, six histidine and a twin strep tags was separated from gL by a self-cleaving 2A peptide. The gHgL and gO subunits were cloned, respectively, in pEE6.4 and pEE12.4 vectors (Lonza Biologics). Stable cell line secreting the HCMV trimer, was generated by nucleofection of CHO-K1 SV line (GS-System, Lonza). For protein expression, CHO cells were seeded at 1×106/ml in ProCho5 (Lonza); after 10 days, supernatants were harvested and loaded on IMAC column eluted with imidazole and further purified by affinity column StrepTactin II (Qiagen). Histidine and twin strep tags were removed by treatment with the TEV protease. Protease and uncleaved proteins were removed by negative chromatography. Finally, proteins were further purified by size-exclusion chromatography (Superdex 200 from GE Healthcare). Soluble dimer was generated by transient transfection of gH2AgL in pEE6.4 vectors (Lonza Biologics). Dimer protein purification was performed as described above.
Recombinant proteins
Recombinant EGF, HB-EGF, Nrg-1, extracellular domain of PDGFRα-Fc conjugated, extracellular domain of ErbB1-Fc conjugated, extracellular domain of ErbB2-Fc conjugated, extracellular domain of ErbB3-Fc conjugated, extracellular domain of ErbB4-Fc conjugated were obtained from R&D systems.
Reverse Transcription and PCR
For RT-PCR, 100 cells per reaction were used. Reverse transcription was performed with the SuperScript III RT (Invitrogen) and random hexamer. The following, PCR was performed with specific primer listed below using the Q5 polymerase (NEB).
ErbB1:
Fwd: 5′_CGGGCTCTGGAGGAAAAGA_3′
Rev: 5′_CTGACTATGTCCCGCCACT_3′
ErbB2:
Fwd: 5′_AACCTGGAACTCACCTACCT_3′
Rev: 5′_ATCCTCAGAACTCTCTCCCC_3′
ErbB3:
Fwd: 5′_CTGAATGGCCTGAGTGTGAC_3′
Rev: 5′_CCCCTTGCAAACCTCATGAC_3′
ErbB4:
Fwd: 5′_AGGACTTTGGGTCTGGGTG_3′
Rev: 5′_AAGTTGGAAGGCCATGGGT_3′
GAPDH:
Fwd: 5′_GGAAGGTGAAGGTCGGAGT_3′
Rev: 5′_GTGATGGCATGGACTGTGG_3′
PDGFRα:
Fwd:5′_TCCTGGTCTTAGGCTGTCTT_3′
Rev: 5′_GGTCCCATTAAAGCCCTGTC_3′
PDGFRβ:
Fwd:5′_CTGCTGTTGCTGTCTCTCCT_3′
Rev: 5′_TGATCATAGGGGACAGGCAG_3′
Biolayer interferometry experiment
FortéBio Octet Red384 instrument was used to measure binding of gHgLgO trimer to immobilized PDGFRα-Fc receptor. PDGFRα-Fc receptor at 10 nM was loaded during 8 minutes on Protein G tips and Biosensor tips were then equilibrated for 2 minutes prior to measuring association for 8 minutes and dissociation for 10 minutes of the gHgLgO trimer. Protein G sensors loaded with PDGFRα-Fc complexes were blocked and saturated for 20 minutes in a solution containing an unspecific human IgG1 at 60 nM prior to be dipped in a new well containing the gHgLgO trimer at 10 nM to form the complex. In a following step, the sensors were dipped into anti-gH neutralizing antibodies (13H11 and 11B12 at 14 nM) or anti-pUL128 antibody (15D8 at 14 nM) as control. Assays were performed with agitation set to 1,000 rpm in phosphate-buffered saline (PBS) supplemented with 0.01% bovine serum albumin (BSA) and 0.002% Tween-20. Data analysis and curve fitting were carried out using Octet software, version 8.0.
Surface plasmon resonance (SPR)
The experiments were carried out at 25°C on a ProteON XPR-36 instrument (Bio-Rad Laboratories) in a PBS buffer (Gibco, Invitrogen), 0.05% Tween-20. The indicated proteins were immobilized on a GLM sensor chip surface through amine coupling at 1000 response units (RU) and a blank surface with no protein was created under identical coupling conditions for use as a reference. Analyte proteins (gHgL dimer, gHgLgO trimer and gHgLpUL128L pentamer) were injected at a flow rate of 100 μl/min, at concentrations of 100, 75, 50, 25 and 12.5 nM in different sensor channels. The data were processed using Proteon software and double referenced by subtraction of the blank surface and buffer only injection before local fitting of the data. Curve fitting were locally fitted, to perform KD calculation.
Virus neutralization assay and microscopy analysis
Confluent layers of ARPE-19, MRC-9 or HAP1 cells in 96-well flat-bottom plates were infected with a fixed amount of virus. MOI of infection (0.5) was chosen to give 500 to 1000 infected cells per microscopic field. 48 hours post-infection the cells were fixed with 5% acetic acid in methanol for 7 minutes on ice, washed four times with PBS and then stained with 0.5 μg/ml of mouse anti-pp72 antibody for 2 hours on ice. After four PBS washes, cells were incubated with 1 μg/ml of a goat anti-Mouse IgG (H+L) AlexaFluor 594 conjugated (Invitrogen). After four PBS washes, cells were counterstained with DAPI, images were acquired on BD Pathway bioimaging system and percentage of the infected cells was automatically calculated by BD AttoVision software. The number of infected cells was plotted as histograms and dose response curves were generated by plotting the relative infected cells against the ligand or antibodies dilutions.
Statistical analysis
No statistical methods were used to predetermine sample size. Data were analyzed with Prism 6 (GraphPad Software) using the two-tailed non-parametric Mann-Whitney U test for two groups’ comparison, or Kruskall-Wallis test (and Dunn’s posttest) when three or more groups were compared.
Data and materials availability
The reagents described in the manuscript can be obtained through a MTA.
Supplementary Material
Acknowledgments
The authors thank A. von Heyl for graphical representation, the Virologia e Microbiologia, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy, for the HCMV clinical isolate and deficient virus, Horizon Genomics GmbH for the HAP-1 parental line and CRISPR/Cas9 KO cells and Oxford University, Central Proteomics Facility, Oxford, UK, for mass spectrometry analysis. The Institute for Research in Biomedicine is supported by the Helmut Horten Foundation. This project has been funded in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract no. HHSN261200800001E, and by the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases, National Institutes of Health. Leidos Biomedical Research provided support in the form of a salary for author Y.T.
Footnotes
Author contributions
A.K., J.M., S.B.,D.L. and R.G. performed and analysed the experiments. C.S.-F. and B.M.F.-R. generated new reagents. B.M.F.-R., M.F. and M.P. performed and provided analysis on the gene expression of ARPE-19 and MRC-9 cells. T.Z., U.B., Y.T., A.D., B.Z. and P.D.K. designed, performed and analysed electron microscopy data and expressed and purified the PDGFRα-trimer complex. F.S. provided intellectual input and wrote the manuscript. D.C. designed experiments, analysed the data and wrote the manuscript. A.L. designed experiments, analysed the data, wrote the manuscript and provided supervision. L.P. designed experiments, analysed the data, provided supervision and wrote the manuscript
Competing interests.
The authors declare no financial or commercial conflict of interests.
References
- 1.Vanarsdall AL, Johnson DC. Human cytomegalovirus entry into cells. Current opinion in virology. 2012;2:37–42. doi: 10.1016/j.coviro.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hahn G, et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol. 2004;78:10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang D, Shenk T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci U S A. 2005;102:18153–18158. doi: 10.1073/pnas.0509201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang D, Shenk T. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol. 2005;79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryckman BJ, et al. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J Virol. 2008;82:60–70. doi: 10.1128/JVI.01910-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Huong SM, Chiu ML, Raab-Traub N, Huang ES. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature. 2003;424:456–461. doi: 10.1038/nature01818. [DOI] [PubMed] [Google Scholar]
- 7.Soroceanu L, Akhavan A, Cobbs CS. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature. 2008;455:391–395. doi: 10.1038/nature07209. [DOI] [PubMed] [Google Scholar]
- 8.Isaacson MK, Feire AL, Compton T. Epidermal growth factor receptor is not required for human cytomegalovirus entry or signaling. J Virol. 2007;81:6241–6247. doi: 10.1128/JVI.00169-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanarsdall AL, Wisner TW, Lei H, Kazlauskas A, Johnson DC. PDGF receptor-alpha does not promote HCMV entry into epithelial and endothelial cells but increased quantities stimulate entry by an abnormal pathway. PLoS Pathog. 2012;8:e1002905. doi: 10.1371/journal.ppat.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mocarski ES., Jr Immune escape and exploitation strategies of cytomegaloviruses: impact on and imitation of the major histocompatibility system. Cellular microbiology. 2004;6:707–717. doi: 10.1111/j.1462-5822.2004.00425.x. [DOI] [PubMed] [Google Scholar]
- 11.Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol. 2007;17:253–276. doi: 10.1002/rmv.535. [DOI] [PubMed] [Google Scholar]
- 12.Gerna G, et al. Rescue of human cytomegalovirus strain AD169 tropism for both leukocytes and human endothelial cells. The Journal of general virology. 2003;84:1431–1436. doi: 10.1099/vir.0.19055-0. [DOI] [PubMed] [Google Scholar]
- 13.Murphy E, et al. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci U S A. 2003;100:14976–14981. doi: 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macagno A, et al. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J Virol. 2010;84:1005–1013. doi: 10.1128/JVI.01809-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kabanova A, et al. Antibody-driven design of a human cytomegalovirus gHgLpUL128L subunit vaccine that selectively elicits potent neutralizing antibodies. Proc Natl Acad Sci U S A. 2014;111:17965–17970. doi: 10.1073/pnas.1415310111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li G, Nguyen CC, Ryckman BJ, Britt WJ, Kamil JP. A viral regulator of glycoprotein complexes contributes to human cytomegalovirus cell tropism. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1419875112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cha TA, et al. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol. 1996;70:78–83. doi: 10.1128/jvi.70.1.78-83.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feire AL, Koss H, Compton T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc Natl Acad Sci U S A. 2004;101:15470–15475. doi: 10.1073/pnas.0406821101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feire AL, Roy RM, Manley K, Compton T. The glycoprotein B disintegrin-like domain binds beta 1 integrin to mediate cytomegalovirus entry. J Virol. 2010;84:10026–10037. doi: 10.1128/JVI.00710-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciferri C, et al. Antigenic Characterization of the HCMV gH/gL/gO and Pentamer Cell Entry Complexes Reveals Binding Sites for Potently Neutralizing Human Antibodies. PLoS Pathog. 2015;11:e1005230. doi: 10.1371/journal.ppat.1005230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loughney JW, et al. Soluble Human Cytomegalovirus gH/gL/pUL128-131 Pentameric Complex, but Not gH/gL, Inhibits Viral Entry to Epithelial Cells and Presents Dominant Native Neutralizing Epitopes. The Journal of biological chemistry. 2015;290:15985–15995. doi: 10.1074/jbc.M115.652230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou M, Lanchy JM, Ryckman BJ. Human Cytomegalovirus gH/gL/gO Promotes the Fusion Step of Entry into All Cell Types, whereas gH/gL/UL128-131 Broadens Virus Tropism through a Distinct Mechanism. J Virol. 2015;89:8999–9009. doi: 10.1128/JVI.01325-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schultz EP, Lanchy JM, Ellerbeck EE, Ryckman BJ. Scanning mutagenesis of human cytomegalovirus gH/gL. J Virol. 2015 doi: 10.1128/JVI.01875-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haspot F, et al. Human cytomegalovirus entry into dendritic cells occurs via a macropinocytosis-like pathway in a pH-independent and cholesterol-dependent manner. PLoS One. 2012;7:e34795. doi: 10.1371/journal.pone.0034795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sinzger C, Digel M, Jahn G. Cytomegalovirus cell tropism. Current topics in microbiology and immunology. 2008;325:63–83. doi: 10.1007/978-3-540-77349-8_4. [DOI] [PubMed] [Google Scholar]
- 26.Wille PT, Wisner TW, Ryckman B, Johnson DC. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. MBio. 2013;4:e00332–00313. doi: 10.1128/mBio.00332-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campadelli-Fiume G, Menotti L, Avitabile E, Gianni T. Viral and cellular contributions to herpes simplex virus entry into the cell. Current opinion in virology. 2012;2:28–36. doi: 10.1016/j.coviro.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Compton T, Nepomuceno RR, Nowlin DM. Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology. 1992;191:387–395. doi: 10.1016/0042-6822(92)90200-9. [DOI] [PubMed] [Google Scholar]
- 29.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol. 2006;80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang D, Yu QC, Schroer J, Murphy E, Shenk T. Human cytomegalovirus uses two distinct pathways to enter retinal pigmented epithelial cells. Proceedings of the National Academy of Sciences. 2007;104:20037–20042. doi: 10.1073/pnas.0709704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Essletzbichler P, et al. Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome research. 2014;24:2059–2065. doi: 10.1101/gr.177220.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Traggiai E, et al. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nat Med. 2004;10:871–875. doi: 10.1038/nm1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. Journal of structural biology. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 34.Tang G, et al. EMAN2: an extensible image processing suite for electron microscopy. Journal of structural biology. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 35.Shaikh TR, et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nature protocols. 2008;3:1941–1974. doi: 10.1038/nprot.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure. 2012;20:237–247. doi: 10.1016/j.str.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hohn M, et al. SPARX, a new environment for Cryo-EM image processing. Journal of structural biology. 2007;157:47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 38.Frank J, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. Journal of structural biology. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 39.Pettersen EF, et al. UCSF Chimera–a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.