

Abstract
Von Hippel–Lindau disease is an autosomal dominant syndrome which occurs secondary to germline mutations in the VHL tumor suppressor gene, located on chromosome 3. Clinically von Hippel–Lindau disease is characterized by an increased risk of developing simple visceral cysts, most commonly in the pancreas and kidneys, in addition to an increased risk of developing neoplasms, often with clear cell features, in a multitude of organ systems. The most common neoplasms are cerebellar and retinal hemangioblastomas, adrenal pheochromocytomas, clear cell renal cell carcinomas, pancreatic neuroendocrine tumors, pancreatic serous cystadenomas, and endolymphatic sac tumors. These lesions most commonly present during adulthood; however, screening and surveillance for the development of these lesions should begin in the pediatric years for patients with von Hippel–Lindau disease. In this review article, the genetics and most common neoplasms of von Hippel–Lindau disease are reviewed, with an eye towards implications for the pediatric patient.
Keywords: von Hippel–Lindau, pathology, pediatric
Introduction
Von Hippel–Lindau (VHL) disease (MIM #193300) occurs as the result of germline mutations in the VHL tumor suppressor gene, located on chromosome 3p25–26. Patients with VHL disease are at risk of developing visceral cysts and tumors throughout the body. VHL disease is eponymously named after the German ophthalmologist Eugene von Hippel, who described retinal angiomatosis, and the Swedish pathologist Arvid Lindau, who studied cerebellar tumor.1 2 3 The prototypical lesions of VHL disease include simple cysts, hemangioblastomas (HBs) of the retina and central nervous system, clear cell renal cell carcinomas (RCCs), pheochromocytomas, pancreatic neuroendocrine tumors (PNETs), pancreatic serous cystadenomas, and endolymphatic sac tumors (ELSTs). Simple cysts are most commonly encountered in the pancreas, liver, and kidney, and cystadenomas have been described in the epididymis and broad ligament.4 While VHL disease typically presents in early adulthood, in patients with established VHL disease screening and surveillance for the development of neoplastic lesions is recommended to begin starting by the age of 2 years.5 This article serves as a review of VHL disease, including the genetic alterations underlying the syndrome, clinical features, and the pathology of the visceral cysts and tumors that are common to VHL disease.
Genetic Basis of VHL Disease
The VHL gene is a tumor suppressor gene which was mapped to chromosome 3 in 1988 and isolated in 1993.6 7 8 The VHL gene is composed of three exons coding for two isoforms of the protein pVHL.9 10 Since the initial discovery of the VHL gene, it has been elucidated that pVHL is part of a multiprotein complex including elongin B, elongin C, and Cullin 2 (CUL2).11 This multiprotein complex is normally responsible for ubiquitination and degradation of the α subunits of hypoxia-inducible factors (HIFs) 1 and 2.11 Inactivation of the VHL tumor suppressor gene and subsequent loss of function of the pVHL complex (VBC-CUL2) result in accumulation of HIFs with subsequent increase in the transcription and expression of tumorigenic factors including vascular endothelial growth factor and platelet derived growth factor. Increased expression of these tumorigenic factors are subsequently associated with the development of highly vascular tumors, as is seen in several VHL disease–related neoplasms.10
VHL disease–associated tumors occur by the classic Knudson two-hit model where both VHL alleles are inactivated due to mutation. Individuals with VHL disease typically inherit a VHL gene mutation from a parent and only cells that undergo a second-hit somatic mutation of the wild-type allele will develop tumors.6 Somatic mutations in VHL tumor suppressor gene have been shown to sporadically occur in clear cell RCCs and central nervous system HBs, suggesting the VHL tumor suppressor gene also plays a role in the development of these sporadic tumors.12 13 A clinical diagnosis of VHL disease can be made in a simplex case, or with no family history, in an individual with two or more VHL disease–related tumors, or by finding a single VHL-related tumor in an at-risk relative of an individual with known VHL disease.14 The detection of germline mutations by sequence analysis and deletion/duplication analysis in individuals with clinical VHL disease is 100%.15 The detection of a mutation in a proband with VHL allows for identification of at-risk asymptomatic family members through targeted mutation analysis.16
Missense, frameshift, nonsense, in-frame deletions/insertions, large/complete deletions, and splice mutations have been described as pathogenic in families with VHL.17 Genotype-phenotype correlations allow for targeted medical management particularly related to risk for pheochromocytoma;5 however VHL disease is highly variable and therefore current genotype-phenotype studies have limited clinical value.9 18 VHL disease has been separated into two clinical types based on the presence or absence of pheochromocytoma, with VHL type 1 associated with a lower risk of developing pheochromocytomas.19 20 21 VHL type 1 typically results from loss of function mutations or mutations that affect protein folding.22 In contrast, the majority of VHL type 2 results from missense mutations.17 VHL type 2 has been further subdivided into type 2A, associated with a lower risk for renal cell carcinoma, type 2B, associated with a high risk for renal cell carcinoma, and type 2C, associated with isolated pheochromocytoma.21 23 Specific missense mutations have been correlated with subtypes of VHL Type 2 including the C505T missense mutation correlating with low risk of renal cell carcinoma, missense mutations occurring at codon 167 correlated with a high risk of pheochromocytoma and the “Black Forest” founder mutation Y98H correlated with a VHL Type 2A phenotype.23 24 It has also been suggested that individuals with VHL deletions that include the C3orf10 gene in exon 1 are at a lower risk of renal cell carcinoma.25 26 Germline mutations in the VHL tumor suppressor gene have been identified in individuals with familial isolated pheochromocytoma. This finding has led some to suggest genetic testing for VHL disease in patients with isolated and apparently sporadic pheochromocytoma.27 28 Additionally, it is estimated that up to 30% of those with apparently isolated CNS hemangioblastomas have a germline mutation in VHL and therefore clinical screening or genetic testing for VHL is recommended.29 In contrast, epididymal cysts, although frequent in VHL, are not sufficient for clinical diagnosis of VHL and therefore it is not recommended to perform VHL gene testing when present without other indications.7
VHL disease is inherited in an autosomal dominant pattern.8 30 Individuals with VHL disease have a 50% chance of passing the mutation to their offspring. VHL disease is almost completely penetrant, with most individuals with a pathogenic mutation in the VHL tumor suppressor gene having VHL disease–related symptoms by the age of 65 years.30 Most individuals with VHL disease have a positive family history; however, some report no family history of VHL-related tumors due to either a de novo, or sporadic, mutation or a parent who is a mosaic for the VHL gene mutation.31 The estimated incidence of sporadic mutations in VHL disease is 1 in 36,000 live births with no known parental age affect.30 32 Mosaicism is described as more than one genetically or cytogenetically distinct cell lines within an organism.33 Individuals with clinical VHL disease and no identifiable VHL gene mutation in peripheral blood lymphocytes have been documented, suggesting they are mosaic for the VHL gene mutation and the VHL gene mutation is not present in the peripheral blood.31 Mosaicism should be considered in any individual with clinical manifestations of VHL disease and negative molecular genetic testing.31 Mosaicism can be either present in the somatic cells and gonadal cells or confined to the gonadal, or germline, cells. An individual who is mosaic for a mutation in the VHL tumor suppressor gene has up to a 50% chance of passing on the mutation to an offspring, depending on the gonadal generation in which the VHL gene mutation occurred.34 While individuals who are germline mosaics are unaffected, individuals with somatic mosaicism may be mildly affected or unaffected, depending on the distribution of the mutation within the tissue.35 Therefore, it is reasonable to test parents of a presumably simplex case, and if no mutation is identified in the peripheral blood lymphocytes, testing of siblings may be considered due to the chance of gonadal mosaicism.31 Presymptomatic testing for at-risk relatives is available with a known familial mutation and allows for presymptomatic screening for VHL-related tumors.27 36
Hemangioblastomas
HBs and VHL disease have a well-studied relationship. HBs are benign vascular neoplasms of the central nervous system (World Health Organization [WHO] grade I of IV).37 Patients with VHL disease are at increased risk of developing HBs, often in the cerebellum or the retina, with less common occurrence in the spinal cord or brainstem.13 38 In VHL patients, HBs often present in the third decade, with a mean age of diagnosis of 29 years, although they have been reported in the pediatric population as well.13 39 40 In addition, these tumors are most often multiple. Irrespective of location, HBs have a common histology, characterized by a vascular proliferation with thin-walled capillaries admixed with a variable number of larger stromal cells with clear to eosinophilic foamy cytoplasm (Fig. 1). The capillaries are typically closely packed together and are variably sized. The stromal cells have hyperchromatic nuclei and, on special staining, demonstrate PAS-positive, diastase-resistant cytoplasmic globules. There should be no necrosis and only rare mitotic figures identified. Immunohistochemical staining demonstrates that inhibin, S-100 protein, and neuron-specific enolase are variably expressed in the stromal cells, with VEGF always positively expressed in the stromal cells. The differential diagnosis includes metastatic RCC, however the combination of inhibin-positivity and CD10-negativity in HBs should result in the appropriate diagnosis.41
Fig. 1.
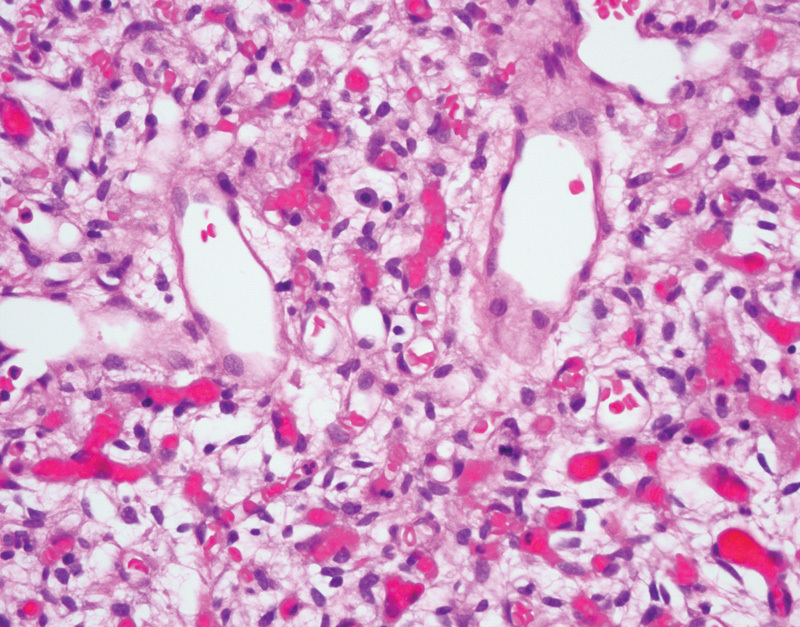
Hemangioblastoma of the cerebellum, demonstrating a rich proliferation of thin-walled capillaries and large stromal cells with foamy cytoplasm (hematoxylin and eosin, 400×).
Cerebellar HBs will initially present with headaches, nausea, and vomiting which, in the pediatric population, may be confused with the more common medulloblastomas.39 Cerebellar HBs can be subdivided into four categories based upon morphology.42 It has been suggested that the development of cystic features (types 1, 2, or 4) is associated with the development of symptoms which often necessitates intervention.42 The growth pattern of cerebellar HBs has been described as stuttering, and patients may be asymptomatic for long periods of time.43 In one study, the cumulative probability of VHL patients developing cerebellar HB was 0.84 by 60 years of age.44 Treatment is surgical resection and although curative, patients do not necessarily have an improvement in their long-term outcomes, due to the nature of VHL disease.45
The connection between cerebellar and retinal capillary hemangioblastomas (RCHBs) was described by Lindau nine decades ago in 1926.46 RCHBs are often the first manifestation of VHL disease.44 The mean age of presentation is during the early third decade (ages 22–26 years), although the age range of presentation varies widely, with reports of RCHBs in infants.44 46 47 Despite the genetic underpinnings and mechanism of VHL disease, recent evidence fails to demonstrate a significant association between increasing patient age and the number of RCHBs.48 Treatment of RCHBs is either laser photocoagulation, cryotherapy, or vitreoretinal surgery, depending on the severity of the lesion, although recurrence is not uncommon and vision loss is possible.46 49
Brainstem HBs are far less likely to occur than cerebellar HBs. However, in younger populations (<40 years old), a HB of the brainstem or spinal cord has a high likelihood of multiple occurrences.13 50 Similar to cerebellar HBs, surgical resection of brainstem HBs is the typical treatment once they are symptomatic, with microsurgery preferred over radiosurgery.51 52 For both spinal cord and brainstem HBs, the neurological status before surgery is the best predictor for subsequent neurological function.52 53 If the patient is asymptomatic, frequent neurological monitoring and imaging should be performed.52 53
Renal Lesions of VHL disease: Renal Cysts and Renal Cell Carcinoma
Renal lesions are a common manifestation of VHL disease, with approximately one-third of patients developing kidney lesions with an increased likelihood for the development of multiple bilateral tumors.1 9 44 54 The cumulative probability of a VHL patient developing RCC by the age of 60 years is 0.69.44 RCCs typically present in patients with VHL disease in the fourth decade, however VHL disease–associated RCC has been reported occurring as young as 16 years old.1 55 In a study of 328 renal lesions from 23 patients with VHL disease, 58% of the lesions were carcinomas, all with clear cell features.56 The remaining lesions were either simple cysts (31%) or atypical cysts (7%).56 Patients with RCC may develop hematuria, flank pain, or a palpable mass as the presenting sign or symptom.
On gross examination, clear cell RCCs are typically well-circumscribed masses with pushing borders. These tumors are often orange-yellow with a variegated appearance due to interspersed red hemorrhage and necrosis. Tumors may be confined to the renal parenchyma, or may bulge the Gerota fascia, extend into the renal sinus, or involve the renal vein. Microscopically these tumors demonstrate neoplastic cells with clear cytoplasm and prominent vascular component interspersed between the clear cells (Fig. 2). The nuclear features of the neoplastic epithelial cells can range from small and round nuclei without nucleoli to large, pleomorphic, and irregular nuclei with prominent nucleoli, and it is these nuclear changes which are evaluated with the Fuhrman nuclear grading system.57 In cystic RCCs the tumors are composed of variably sized cysts which demonstrate variable amounts of neoplastic clear cells in the cyst walls. RCC is the most common cause of death in patients with VHL disease, accounting for 50% of deaths.44 Patients with RCC may develop metastatic disease, with a predilection for metastases to occur in the bone, lung, and liver.1 Surgical resection is the mainstay of treatment for RCC, however targeted surgical therapy such as cryoablation and drug therapy targeting VHL transcription products are being employed, especially in advanced disease.54 58
Fig. 2.
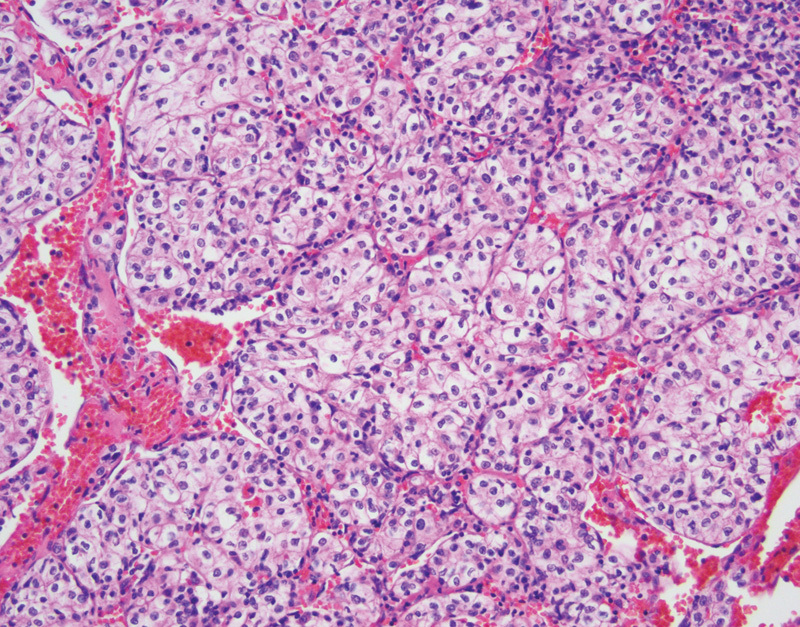
Clear cell renal cell carcinoma with cytoplasmically clear epithelial cells and anastomosing vascular network (hematoxylin and eosin, 200×).
Pheochromocytomas and Paragangliomas
Pheochromocytomas and paragangliomas are neoplasms of the autonomic nervous system which are uncommon tumors in the general population but develop in approximately one-fifth of patients with VHL disease.59 Pheochromocytomas arise from the chromaffin cells in the medulla of the adrenal gland. Paragangliomas are similar to pheochromocytomas, however they arise from extra-adrenal chromaffin cells. These extra-adrenal chromaffin cells are present in the sympathetic nervous system as well as the parasympathetic ganglia, leading to the potential to develop paragangliomas throughout the body, including in the thorax, abdomen, pelvis, or head and neck regions. Symptoms may be secondary to mass effect or catecholamine production, resulting in secondary hypertension or stroke, although paragangliomas arising in the parasympathetic nervous system are nonfunctional.60 61 In VHL disease the pheochromocytomas and paragangliomas are almost universally of the nonadrenergic phenotype.
As outlined earlier, the phenotypic presentation of VHL disease (i.e., type 1 vs. type 2) is based upon the presence or absence of pheochromocytomas. Pheochromocytomas tend to present at a younger age in VHL patients as compared with nonsyndromic patients, with a mean age of diagnosis of 28 years.62 63 Conservative surgical resection of adrenal lesions has been advocated, especially when lesions are identified at an early stage.59
The histologic features of pheochromocytomas and paragangliomas are similar, only differing in their location of origin. Histologically these tumors are classically characterized by polygonal neoplastic cells arranged in small nests or “zellballen” pattern (Fig. 3). The neoplastic cells have round to oval nuclei with a prominent nucleolus and finely granular amphophilic cytoplasm. Surrounding the nest of neoplastic cells are sustentacular cells which are highlighted on S-100 protein immunohistochemical staining. Mitotic figures and necrosis should not be prominent features.
Fig. 3.
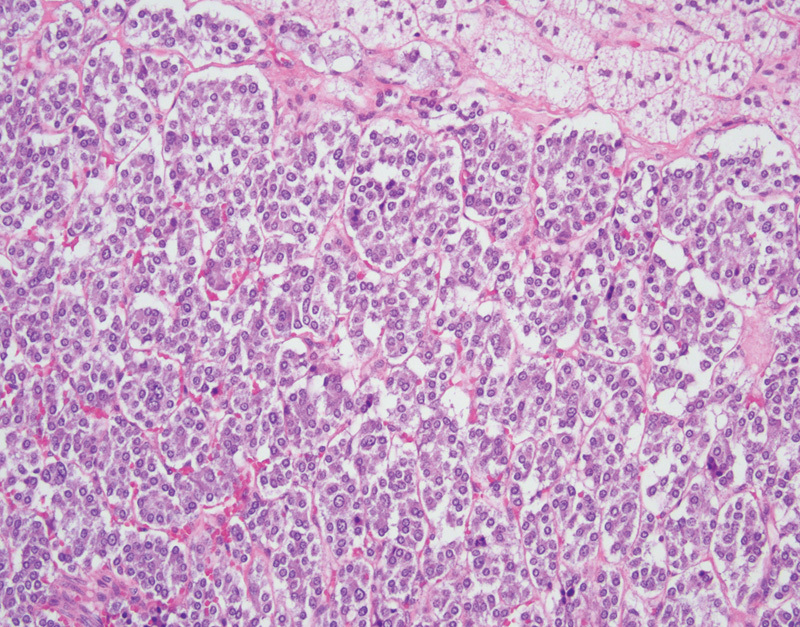
Pheochromocytoma with large polygonal epithelial cells adjacent to normal adrenal cortical tissue (hematoxylin and eosin, 200×).
Pancreatic Lesions of VHL Disease
Patients with VHL disease are at risk for developing multiple different pancreatic lesions, including simple cysts within the pancreatic parenchyma, serous cystadenomas, and PNETs.64 Approximately 75% of patients with VHL disease will develop simple pancreatic cysts.4 These simple cysts are benign and typically asymptomatic, unless they cause compressive or obstructive symptoms.65 Histologically these cysts are composed of uniform cuboidal epithelial cells virtually identical to that of the serous cystadenoma discussed below. In comparison to the serous cystadenoma, however, the cysts are distributed irregularly throughout the pancreatic parenchyma.66 Surgical treatment is typically only indicated when patients are symptomatic.
Approximately one-fifth of patients with VHL disease will develop a PNET, with approximately half of these tumors occurring in the head of the pancreas.67 68 Patients with PNETs are often asymptomatic, as PNETs in VHL disease are nonfunctioning.69 PNETs often present in VHL disease in the fourth decade, as compared with two decades later in nonsyndromic patients.70 Radiographically PNETs are typically well demarcated when they are small, and as they increase in size the borders become less well defined.71 These tumors are highly vascular and will show enhancement on contrast enhanced imaging. Grossly PNETs are typically small (less than 3 cm), tan to pink, well-circumscribed lesion, although infiltrative edges may be identified grossly. Histologically these tumors are composed of neuroendocrine cells, characterized by moderately sized uniform cells with pale to eosinophilic cytoplasm and round to oval nuclei with the characteristic neuroendocrine “salt-and-pepper” granular chromatin pattern (Fig. 4). In patients with VHL disease, PNETs may also demonstrate clear cell features, with cleared-out cytoplasm secondary to the presence of glycogen or lipid in the cytoplasm.71 The neuroendocrine cells are often arranged in trabecular, solid, or glandular architectural configurations.72 The histologic grading, as defined by the WHO, involves assessment of the mitotic rate and Ki-67 proliferation index.73 Given the potential for metastatic disease regardless of the histologic grade, surgical resection is typically recommended for all PNETs greater than 2 cm, however, in VHL disease this must be weighed against the likelihood of the development of multiple or recurrent PNETs.74 Nearly half of VHL patients with PNETs develop metastatic disease, with patients more likely to develop metastases if the primary tumor was greater than 3 cm in size or harbors an exon 3 mutation.67
Fig. 4.
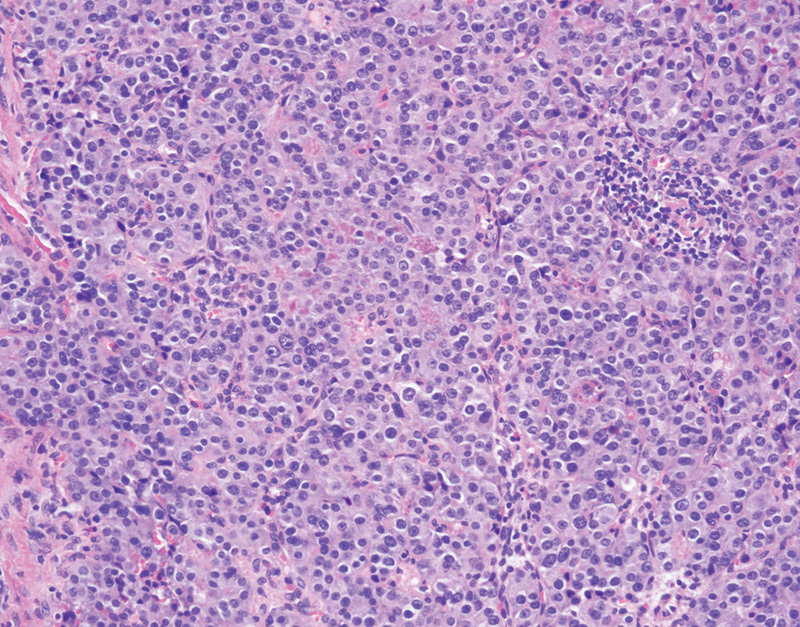
Pancreatic neuroendocrine tumor with nest of neoplastic neuroendocrine cells (hematoxylin and eosin, 200×).
In addition to the development of simple cysts and PNETs, patients with VHL disease are at risk of developing pancreatic serous cystadenomas. The serous cystadenoma is typically a benign neoplasm which is most commonly located in the body or tail of the pancreas, however malignant transformation is possible.66 Abdominal pain is the most common associated symptom.75 Grossly the serous cystadenoma is composed of numerous small cysts with a stellate scar. Histologically these cysts are lined by relatively uniform cuboidal epithelial cells with a round nucleus (Fig. 5). The cytoplasm of the epithelial cells is rich in glycogen, thus imparting a clear appearance on standard hematoxylin-and-eosin–stained slides and positivity on PAS staining. On cytology these cysts contain serous fluid. Observation is considered appropriate for smaller asymptomatic lesions, while it has been recommended that resection is undertaken for larger serous cystadenomas irrespective of symptomatic status.75
Fig. 5.
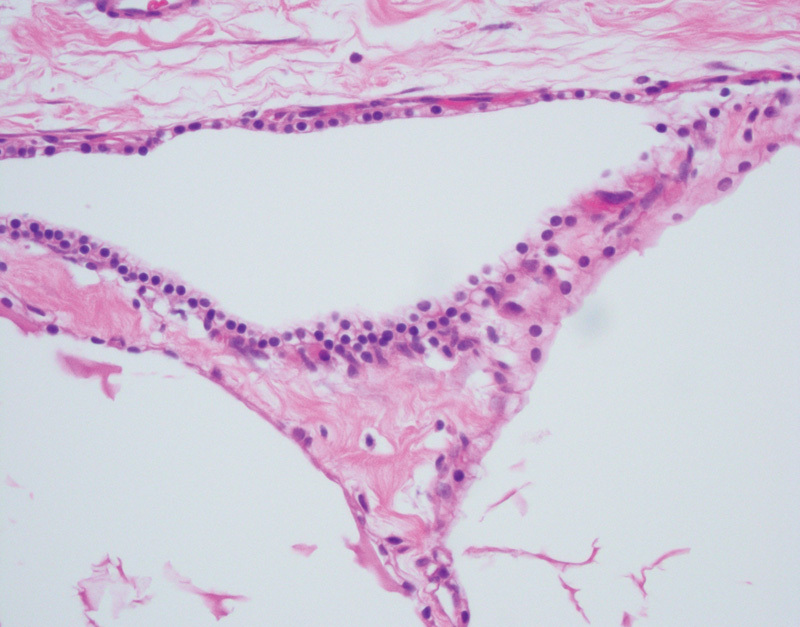
Pancreatic microcystic serous cystadenoma with variably sized cystic spaces lined by uniform, cytoplasmically clear, cuboidal epithelial cells (hematoxylin and eosin, 400×).
Endolymphatic Sac Tumor
ELSTs are rare tumors that are most commonly associated with VHL disease, with 10 to 15% of patients with VHL disease developing ELSTs.76 ELSTs arise in the endolymphatic sac, a portion of the membranous labyrinth of the inner ear that resides within the petrous temporal bone. While VHL disease–associated ELSTs had been sporadically reported since 1921, Heffner's report of a series of 20 cases of low-grade adenocarcinoma of probably endolymphatic sac origin in 1989 solidified this as a distinct entity.76 77 78 79 80 It has been demonstrated that VHL deficiency induces cellular proliferations along the epithelium of the endolymphatic sac and duct, with preferential involvement of the intraosseous portion of this epithelium.81 These typically slow-growing tumors tend to destroy a large portion of the posterior temporal bone and extend into the posterior cranial fossa.80 ELSTs may be present bilaterally and a majority of patients report subjective hearing loss, tinnitus, and vertigo.82 When performed early, surgical resection of ELSTs can preserve hearing and reduce audiovestibular dysfunction.83
ELSTs demonstrate either papillary, cystic, or epithelioid clear cell patterns. The majority of tumors demonstrate highly vascular papillary structures lined by a single row of cuboidal epithelial cells (Fig. 6). These tumors have low-grade morphology, with limited pleomorphism and rare mitotic figures. Approximately half of ELSTs have cystic features containing proteinaceous material and a single layer of epithelial cells lining the cyst wall.81 Additional histologic features include the presence of hemosiderin and degenerative features, including fibrosis, inflammation, and cholesterol clefting.81 In keeping with their underlying pathogenesis, these tumors are highly vascular, with strong immunohistochemical expression of HIF-1 and HIF-2.81
Fig. 6.
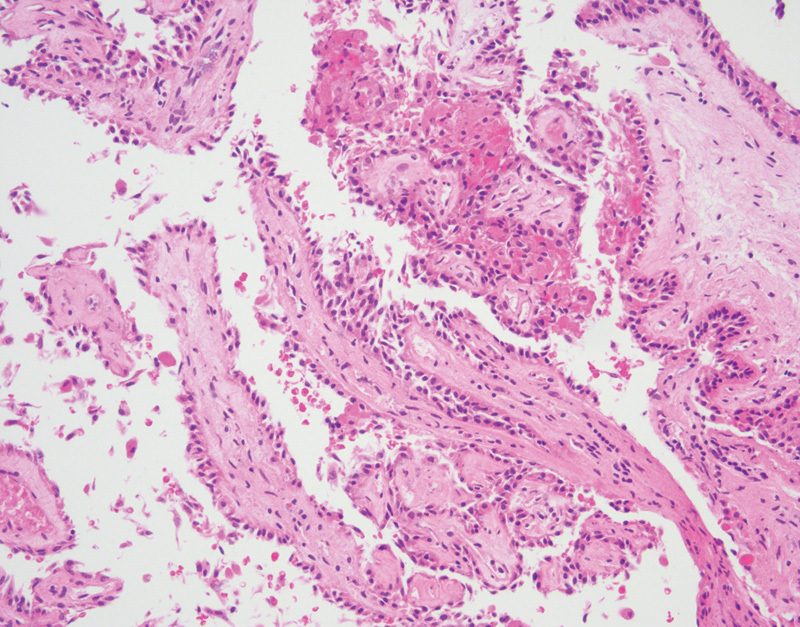
Endolymphatic sac tumor with papillary architecture and uniform cuboidal epithelial cells (hematoxylin and eosin, 200×).
Surveillance
Surveillance guidelines have been developed by the VHL Family Alliance and are recommended for individuals with known VHL disease, individuals who have known VHL disease by presymptomatic testing, and at-risk family members of individuals with VHL disease who have not undergone molecular genetic testing.84 VHL Family Alliance surveillance recommendations for the evaluation of newborns with VHL, or at risk for VHL, to include screening for signs of neurological symptoms, nystagmus, strabismus, and white pupil, in addition to routine newborn hearing evaluation. Starting at one year of age, annual ophthalmology evaluation with indirect ophthalmoscope and physical examination including signs of neurological symptoms, nystagmus, strabismus, and white pupil are recommended in addition to annual blood pressure monitoring and examination for vision and hearing. Starting at five years, testing for fractioned metanephrines through blood or 24-hour urine test is recommended to screen for pheochromocytoma. An audiology examination is recommended starting at five years of age and occurring every two to three years, or annually in those with hearing loss, tinnitus, or vertigo. Screening for visceral lesions using abdominal ultrasound is recommended at age eight years or earlier if indicated, and abdominal MRI only if biochemical abnormalities are found. Starting at sixteen years, abdominal ultrasound, and at least every other year abdominal MRI with and without contrast to assess kidneys, pancreas, and adrenals and MRI of the brain and total spine every two years to screen for CNS lesions is recommended.84
Conclusion
VHL disease is an autosomal dominant disease characterized by visceral cysts and neoplasms, including clear cell RCC, HBs of the central nervous system and retina, pheochromocytoma, PNETs, pancreatic serous cystadenomas, and ELSTs. Although a majority of VHL disease–associated tumors develop during adulthood, it has been recommended that vigilant screening of VHL patients should start during the pediatric years to identify, monitor, and control neoplasms before they become symptomatic.5
References
- 1.Neumann H PH, Zbar B. Renal cysts, renal cancer and von Hippel-Lindau disease. Kidney Int. 1997;51(1):16–26. doi: 10.1038/ki.1997.3. [DOI] [PubMed] [Google Scholar]
- 2.von Hippel E. Über eine sehr seltene Erkrankung der Netzhaut. Graefe Arch Ophthalmol. 1904;59:83–106. [Google Scholar]
- 3.Lindau A. Zur Frage der Angiomatosis Retinae und Ihrer Hirncomplikation. Acta Ophthalmol. 1927;4:193–226. [Google Scholar]
- 4.Maher E R, Neumann H P, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet. 2011;19(6):617–623. doi: 10.1038/ejhg.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedrich C A. Genotype-phenotype correlation in von Hippel-Lindau syndrome. Hum Mol Genet. 2001;10(7):763–767. doi: 10.1093/hmg/10.7.763. [DOI] [PubMed] [Google Scholar]
- 6.Knudson A G. Cancer genetics. Am J Med Genet. 2002;111(1):96–102. doi: 10.1002/ajmg.10320. [DOI] [PubMed] [Google Scholar]
- 7.Seizinger B R, Rouleau G A, Ozelius L J. et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature. 1988;332(6161):268–269. doi: 10.1038/332268a0. [DOI] [PubMed] [Google Scholar]
- 8.Latif F, Tory K, Gnarra J. et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260(5112):1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 9.Linehan W M, Lerman M I, Zbar B. Identification of the von Hippel-Lindau (VHL) gene. Its role in renal cancer. JAMA. 1995;273(7):564–570. [PubMed] [Google Scholar]
- 10.Kaelin W G Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2(9):673–682. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 11.Shuin T, Yamasaki I, Tamura K, Okuda H, Furihata M, Ashida S. Von Hippel-Lindau disease: molecular pathological basis, clinical criteria, genetic testing, clinical features of tumors and treatment. Jpn J Clin Oncol. 2006;36(6):337–343. doi: 10.1093/jjco/hyl052. [DOI] [PubMed] [Google Scholar]
- 12.Gnarra J R, Tory K, Weng Y. et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet. 1994;7(1):85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- 13.Kanno H, Kuratsu J, Nishikawa R. et al. Clinical features of patients bearing central nervous system hemangioblastoma in von Hippel-Lindau disease. Acta Neurochir (Wien) 2013;155(1):1–7. doi: 10.1007/s00701-012-1514-y. [DOI] [PubMed] [Google Scholar]
- 14.Melmon K L, Rosen S W. Lindau's disease. Review of the literature and study of a large kindred. Am J Med. 1964;36:595–617. doi: 10.1016/0002-9343(64)90107-x. [DOI] [PubMed] [Google Scholar]
- 15.Stolle C, Glenn G, Zbar B. et al. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 1998;12(6):417–423. doi: 10.1002/(SICI)1098-1004(1998)12:6<417::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 16.Glenn G M, Linehan W M, Hosoe S. et al. Screening for von Hippel-Lindau disease by DNA-polymorphism analysis. J Am Med Assoc. 1992;267:1226–1231. [PubMed] [Google Scholar]
- 17.Nordstrom-O'Brien M, van der Luijt R B, van Rooijen E. et al. Genetic analysis of von Hippel-Lindau disease. Hum Mutat. 2010;31:521–537. doi: 10.1002/humu.21219. [DOI] [PubMed] [Google Scholar]
- 18.Gossage L, Eisen T, Maher E R. VHL, the story of a tumour suppressor gene. Nat Rev Cancer. 2015;15(1):55–64. doi: 10.1038/nrc3844. [DOI] [PubMed] [Google Scholar]
- 19.Chen F, Kishida T, Yao M. et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat. 1995;5(1):66–75. doi: 10.1002/humu.1380050109. [DOI] [PubMed] [Google Scholar]
- 20.Maher E R, Webster A R, Richards F M. et al. Phenotypic expression in von Hippel- Lindau disease: Correlations with germline VHL gene mutations. J Med Genet. 1996;33:328–332. doi: 10.1136/jmg.33.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crossey P A Richards F M Foster K et al. Identification of intragenic mutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype Hum Mol Genet 1994. b:31303–1308. [DOI] [PubMed] [Google Scholar]
- 22.Stebbins C E, Kaelin W G Jr, Pavletich N P. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- 23.Brauch H, Kishida T, Glavac D. et al. Von Hippel-Lindau (VHL) disease with pheochromocytoma in the Black Forest region of GermanyL Evidence for a founder effect. Hum Genet. 1996;95:551–556. doi: 10.1007/BF00223868. [DOI] [PubMed] [Google Scholar]
- 24.Ritter M M, Frilling A, Crossey P A. et al. Isolated familial pheochromocytoma as a variant of von Hippel-Lindau disease. J Clin Endocrinol Metab. 1996;81:1035–1037. doi: 10.1210/jcem.81.3.8772572. [DOI] [PubMed] [Google Scholar]
- 25.McNeill A, Rattenberry E, Barber R, Killick P, MacDonald F, Maher E R. Genotype-phenotype correlations in VHL exon deletions. Am J Med Genet A. 2009;149A:2147–2151. doi: 10.1002/ajmg.a.33023. [DOI] [PubMed] [Google Scholar]
- 26.Maranchie J K, Afonso A, Albert P S. et al. Solid renal tumor severity in von Hippel Lindau disease is related to germline deletion length and location. Hum Mutat. 2004;23:40–46. doi: 10.1002/humu.10302. [DOI] [PubMed] [Google Scholar]
- 27.Woodward E R, Eng C, McMahon R. et al. Genetic predisposition to phaeochromocytoma: analysis of candidate genes GDNF, RET and VHL. Hum Mol Genet. 1997;6(7):1051–1056. doi: 10.1093/hmg/6.7.1051. [DOI] [PubMed] [Google Scholar]
- 28.Neumann H P, Bausch B, McWhinney S R. et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346(19):1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 29.Hes F J McKee S Taphoorn M J et al. Cryptic von Hippel-Lindau disease: germline mutations in patients with haemangioblastoma only J Med Genet 2000. b:37939–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maher E R, Iselius L, Yates J R. et al. Von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28(7):443–447. doi: 10.1136/jmg.28.7.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sgambati M T, Stolle C, Choyke P L. et al. Mosaicism in von Hippel-Lindau disease: lessons from kindreds with germline mutations identified in offspring with mosaic parents. Am J Hum Genet. 2000;66(1):84–91. doi: 10.1086/302726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richards F M, Payne S J, Zbar B, Affara N A, Ferguson-Smith M A, Maher E R. Molecular analysis of de novo germline mutations in the von Hippel-Lindau disease gene. Hum Mol Genet. 1995;4(11):2139–2143. doi: 10.1093/hmg/4.11.2139. [DOI] [PubMed] [Google Scholar]
- 33.Youssoufian H, Pyeritz R E. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. 2002;3(10):748–758. doi: 10.1038/nrg906. [DOI] [PubMed] [Google Scholar]
- 34.van der Meulen M A, van der Meulen M J, te Meerman G J. Recurrence risk for germinal mosaics revisited. J Med Genet. 1995;32(2):102–104. doi: 10.1136/jmg.32.2.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hall J G. Review and hypotheses: somatic mosaicism: observations related to clinical genetics. Am J Hum Genet. 1988;43(4):355–363. [PMC free article] [PubMed] [Google Scholar]
- 36.Lamiell J M, Salazar F G, Hsia Y E. von Hippel-Lindau disease affecting 43 members of a single kindred. Medicine (Baltimore) 1989;68(1):1–29. doi: 10.1097/00005792-198901000-00001. [DOI] [PubMed] [Google Scholar]
- 37.Louis D N Ohgaki H Wiestler O D Cavenee W K, eds. WHO Classification of Tumours of the Central Nervous System Lyon, France: IARC Press; 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niemelä M, Lemeta S, Sainio M. et al. Hemangioblastomas of the retina: impact of von Hippel-Lindau disease. Invest Ophthalmol Vis Sci. 2000;41(7):1909–1915. [PubMed] [Google Scholar]
- 39.Neumann H PH, Eggert H R, Weigel K, Friedburg H, Wiestler O D, Schollmeyer P. Hemangioblastomas of the central nervous system. A 10-year study with special reference to von Hippel-Lindau syndrome. J Neurosurg. 1989;70(1):24–30. doi: 10.3171/jns.1989.70.1.0024. [DOI] [PubMed] [Google Scholar]
- 40.Vougioukas V I, Gläsker S, Hubbe U. et al. Surgical treatment of hemangioblastomas of the central nervous system in pediatric patients. Childs Nerv Syst. 2006;22(9):1149–1153. doi: 10.1007/s00381-005-0018-y. [DOI] [PubMed] [Google Scholar]
- 41.Jung S M, Kuo T T. Immunoreactivity of CD10 and inhibin alpha in differentiating hemangioblastoma of central nervous system from metastatic clear cell renal cell carcinoma. Mod Pathol. 2005;18(6):788–794. doi: 10.1038/modpathol.3800351. [DOI] [PubMed] [Google Scholar]
- 42.Slater A, Moore N R, Huson S M. The natural history of cerebellar hemangioblastomas in von Hippel-Lindau disease. AJNR Am J Neuroradiol. 2003;24(8):1570–1574. [PMC free article] [PubMed] [Google Scholar]
- 43.Ammerman J M, Lonser R R, Dambrosia J, Butman J A, Oldfield E H. Long-term natural history of hemangioblastomas in patients with von Hippel-Lindau disease: implications for treatment. J Neurosurg. 2006;105(2):248–255. doi: 10.3171/jns.2006.105.2.248. [DOI] [PubMed] [Google Scholar]
- 44.Maher E R, Yates J RW, Harries R. et al. Clinical features and natural history of von Hippel-Lindau disease. Q J Med. 1990;77(283):1151–1163. doi: 10.1093/qjmed/77.2.1151. [DOI] [PubMed] [Google Scholar]
- 45.Jagannathan J, Lonser R R, Smith R, DeVroom H L, Oldfield E H. Surgical management of cerebellar hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2008;108(2):210–222. doi: 10.3171/JNS/2008/108/2/0210. [DOI] [PubMed] [Google Scholar]
- 46.Singh A D, Shields C L, Shields J A. von Hippel-Lindau disease. Surv Ophthalmol. 2001;46(2):117–142. doi: 10.1016/s0039-6257(01)00245-4. [DOI] [PubMed] [Google Scholar]
- 47.Kuo M-T, Kou H-K, Kao M-L, Tsai M-H, Chen Y-J, Lin S-A. Retinal capillary hemangiomas: clinical manifestations and visual prognosis. Chang Gung Med J. 2002;25(10):672–682. [PubMed] [Google Scholar]
- 48.Wong W T, Agrón E, Coleman H R. et al. Clinical characterization of retinal capillary hemangioblastomas in a large population of patients with von Hippel-Lindau disease. Ophthalmology. 2008;115(1):181–188. doi: 10.1016/j.ophtha.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaudric A, Krivosic V, Duguid G, Massin P, Giraud S, Richard S. Vitreoretinal surgery for severe retinal capillary hemangiomas in von hippel-lindau disease. Ophthalmology. 2011;118(1):142–149. doi: 10.1016/j.ophtha.2010.04.031. [DOI] [PubMed] [Google Scholar]
- 50.Browne T R, Adams R D, Roberson G H. Hemangioblastoma of the spinal cord. Review and report of five cases. Arch Neurol. 1976;33(6):435–441. doi: 10.1001/archneur.1976.00500060041009. [DOI] [PubMed] [Google Scholar]
- 51.Na J H, Kim H S, Eoh W, Kim J H, Kim J S, Kim E-S. Spinal cord hemangioblastoma : diagnosis and clinical outcome after surgical treatment. J Korean Neurosurg Soc. 2007;42(6):436–440. doi: 10.3340/jkns.2007.42.6.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weil R J, Lonser R R, DeVroom H L, Wanebo J E, Oldfield E H. Surgical management of brainstem hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98(1):95–105. doi: 10.3171/jns.2003.98.1.0095. [DOI] [PubMed] [Google Scholar]
- 53.Lonser R R, Weil R J, Wanebo J E, DeVroom H L, Oldfield E H. Surgical management of spinal cord hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98(1):106–116. doi: 10.3171/jns.2003.98.1.0106. [DOI] [PubMed] [Google Scholar]
- 54.Barrisford G W Singer E A Rosner I L Linehan W M Bratslavsky G Familial renal cancer: molecular genetics and surgical management Int J Surg Oncol 2011;2011, Article ID 658767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keeler L L III, Klauber G T. Von Hippel-Lindau disease and renal cell carcinoma in a 16-year-old boy. J Urol. 1992;147(6):1588–1591. doi: 10.1016/s0022-5347(17)37636-x. [DOI] [PubMed] [Google Scholar]
- 56.Paraf F, Chauveau D, Chrétien Y, Richard S, Grünfeld J P, Droz D. Renal lesions in von Hippel-Lindau disease: immunohistochemical expression of nephron differentiation molecules, adhesion molecules and apoptosis proteins. Histopathology. 2000;36(5):457–465. doi: 10.1046/j.1365-2559.2000.00857.x. [DOI] [PubMed] [Google Scholar]
- 57.Fuhrman S A, Lasky L C, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol. 1982;6(7):655–663. doi: 10.1097/00000478-198210000-00007. [DOI] [PubMed] [Google Scholar]
- 58.Yang B, Autorino R, Remer E M. et al. Probe ablation as salvage therapy for renal tumors in von Hippel-Lindau patients: The Cleveland Clinic experience with 3 years follow-up. Urol Oncol. 2013;31:686–692. doi: 10.1016/j.urolonc.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 59.Baghai M Thompson G B Young W F Jr Grant C S Michels V V van Heerden J A Pheochromocytomas and paragangliomas in von Hippel-Lindau disease: a role for laparoscopic and cortical-sparing surgery Arch Surg 20021376682–688., discussion 688–689 [DOI] [PubMed] [Google Scholar]
- 60.Fishbein L, Merrill S, Fraker D L, Cohen D L, Nathanson K L. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol. 2013;20(5):1444–1450. doi: 10.1245/s10434-013-2942-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McNicol A M. Update on tumours of the adrenal cortex, phaeochromocytoma and extra-adrenal paraganglioma. Histopathology. 2011;58(2):155–168. doi: 10.1111/j.1365-2559.2010.03613.x. [DOI] [PubMed] [Google Scholar]
- 62.Opocher G, Conton P, Schiavi F, Macino B, Mantero F. Pheochromocytoma in von Hippel-Lindau disease and neurofibromatosis type 1. Fam Cancer. 2005;4(1):13–16. doi: 10.1007/s10689-004-6128-y. [DOI] [PubMed] [Google Scholar]
- 63.Havekes B, Romijn J A, Eisenhofer G, Adams K, Pacak K. Update on pediatric pheochromocytoma. Pediatr Nephrol. 2009;24(5):943–950. doi: 10.1007/s00467-008-0888-9. [DOI] [PubMed] [Google Scholar]
- 64.Neumann H P, Dinkel E, Brambs H. et al. Pancreatic lesions in the von Hippel-Lindau syndrome. Gastroenterology. 1991;101(2):465–471. doi: 10.1016/0016-5085(91)90026-h. [DOI] [PubMed] [Google Scholar]
- 65.van Asselt S J, de Vries E GE, van Dullemen H M. et al. Pancreatic cyst development: insights from von Hippel-Lindau disease. Cilia. 2013;2(1):3. doi: 10.1186/2046-2530-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Volkan Adsay N. Cystic lesions of the pancreas. Mod Pathol. 2007;20 01:S71–S93. doi: 10.1038/modpathol.3800706. [DOI] [PubMed] [Google Scholar]
- 67.Blansfield J A Choyke L Morita S Y et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs) Surgery 20071426814–818., discussion 818.e1–818.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marcos H B, Libutti S K, Alexander H R. et al. Neuroendocrine tumors of the pancreas in von Hippel-Lindau disease: spectrum of appearances at CT and MR imaging with histopathologic comparison. Radiology. 2002;225(3):751–758. doi: 10.1148/radiol.2253011297. [DOI] [PubMed] [Google Scholar]
- 69.Chen M, Van Ness M, Guo Y, Gregg J. Molecular pathology of pancreatic neuroendocrine tumors. J Gastrointest Oncol. 2012;3(3):182–188. doi: 10.3978/j.issn.2078-6891.2012.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lubensky I A, Pack S, Ault D. et al. Multiple neuroendocrine tumors of the pancreas in von Hippel-Lindau disease patients: histopathological and molecular genetic analysis. Am J Pathol. 1998;153(1):223–231. doi: 10.1016/S0002-9440(10)65563-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frankel W L. Update on pancreatic endocrine tumors. Arch Pathol Lab Med. 2006;130(7):963–966. doi: 10.5858/2006-130-963-UOPET. [DOI] [PubMed] [Google Scholar]
- 72.Asa S L. Pancreatic endocrine tumors. Mod Pathol. 2011;24 02:S66–S77. doi: 10.1038/modpathol.2010.127. [DOI] [PubMed] [Google Scholar]
- 73.Bosman F T Carneiro F Hruban R H Theise N D, eds. WHO Classification of Tumours of the Digestive System Lyon, France: IARC Press; 2010 [Google Scholar]
- 74.Tamura K, Nishimori I, Ito T, Yamasaki I, Igarashi H, Shuin T. Diagnosis and management of pancreatic neuroendocrine tumor in von Hippel-Lindau disease. World J Gastroenterol. 2010;16(36):4515–4518. doi: 10.3748/wjg.v16.i36.4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tseng J F Warshaw A L Sahani D V Lauwers G Y Rattner D W Fernandez-del Castillo C Serous cystadenoma of the pancreas: tumor growth rates and recommendations for treatment Ann Surg 20052423413–419., discussion 419–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wind J J, Lonser R R. Management of von Hippel-Lindau disease-associated CNS lesions. Expert Rev Neurother. 2011;11(10):1433–1441. doi: 10.1586/ern.11.124. [DOI] [PubMed] [Google Scholar]
- 77.Brandt R. Aur erage der angiomatosis retinae. Von Graefes Arch Ophthalmol. 1921;106:127–136. [Google Scholar]
- 78.Lindau A. Studies on cerebellar cysts: development, pathogenesis, and relationship with retinal angiomatosis. Acta Pathol Microbiol Scand Suppl. 1926;1:1–128. [Google Scholar]
- 79.Hassard A D, Boudreau S F, Cron C C. Adenoma of the endolymphatic sac. J Otolaryngol. 1984;13(4):213–216. [PubMed] [Google Scholar]
- 80.Heffner D K. Low-grade adenocarcinoma of probable endolymphatic sac origin A clinicopathologic study of 20 cases. Cancer. 1989;64(11):2292–2302. doi: 10.1002/1097-0142(19891201)64:11<2292::aid-cncr2820641119>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 81.Gläsker S, Lonser R R, Tran M G. et al. Effects of VHL deficiency on endolymphatic duct and sac. Cancer Res. 2005;65(23):10847–10853. doi: 10.1158/0008-5472.CAN-05-1104. [DOI] [PubMed] [Google Scholar]
- 82.Choo D, Shotland L, Mastroianni M. et al. Endolymphatic sac tumors in von Hippel-Lindau disease. J Neurosurg. 2004;100(3):480–487. doi: 10.3171/jns.2004.100.3.0480. [DOI] [PubMed] [Google Scholar]
- 83.Kim H J, Hagan M, Butman J A. et al. Surgical resection of endolymphatic sac tumors in von Hippel-Lindau disease: findings, results, and indications. Laryngoscope. 2013;123(2):477–483. doi: 10.1002/lary.23646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Family Alliance V HL The VHL Handbook: What You Need to Know about VHL. 4th ed. 2012 Available at: http://vhl.org/wordpress/library/Handbook/handbook40.pdf. Accessed July 11, 2015